

Abstract

We have used a subcellular spatial razor approach based on LC–MS/MS-based proteomics with SILAC isotope labeling to determine changes in protein abundances in the nuclear and cytoplasmic compartments of human IMR90 fibroblasts subjected to mild oxidative stress. We show that response to mild tert-butyl hydrogen peroxide treatment includes redistribution between the nucleus and cytoplasm of numerous proteins not previously associated with oxidative stress. The 121 proteins with the most significant changes encompass proteins with known functions in a wide variety of subcellular locations and of cellular functional processes (transcription, signal transduction, autophagy, iron metabolism, TCA cycle, ATP synthesis) and are consistent with functional networks that are spatially dispersed across the cell. Both nuclear respiratory factor 2 and the proline regulatory axis appear to contribute to the cellular metabolic response. Proteins involved in iron metabolism or with iron/heme as a cofactor as well as mitochondrial proteins are prominent in the response. Evidence suggesting that nuclear import/export and vesicle-mediated protein transport contribute to the cellular response was obtained. We suggest that measurements of global changes in total cellular protein abundances need to be complemented with measurements of the dynamic subcellular spatial redistribution of proteins to obtain comprehensive pictures of cellular function.
Keywords: quantitative proteomics, mass spectrometry, SILAC, oxidative stress, peroxide, DNA replication
1. INTRODUCTION
It has long been known that the oxidative state of cells contributes to various disease processes. There has been intense interest in recent years in the role of mitochondria and reactive oxygen species (ROS) in diverse diseases including diabetes, inflammatory diseases, neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease as well as cancer.1-5 Early efforts concentrated on protein damage caused by ROS and one branch of very active research has evolved toward protein damage processes and biological quality control mechanisms such as autophagy and mitophagy.6,7 In parallel it has become apparent that ROS are potent signaling systems under many physiological conditions.8,9 Much recent interest has centered on the intimate involvement of ROS in the special oxidative environment of mitochondria, where much of cellular ROS is generated10 and where mitochondrial proteins may show major dependence on ROS-related signaling, especially for cysteine-related systems.11 However, ROS are also generated at other locations, for example, by the NOX family of proteins,12-16 and some aspects of ROS signaling may be locally spatially restricted. Spatial localization is also apparent in redox compartmentalization in eukaryotic cells, where different subcellular compartments are at different redox potential and different sets of proteins may be involved in control of ROS-related signaling.17,18
At the same time there is increasing evidence that the subcellular distribution of many proteins and, consequently the proteome of any given subcellular organelle, are both highly dynamic.19-23 Response to oxidative stress and ROS signaling is no exception. For example, NRF2 is dissociated from its cytoplasmic inhibitor KEAP1 and translocated to the nucleus in response to ROS-related signaling involving lipid peroxidation products24-27 as well as phosphorylation signals.26,28 NRF2 controls expression of a wide variety of proteins related to oxidative stress29-31 and its activity as a transcription factor is dependent on the oxidation state of Cys residues.30,32,33
We have recently developed high-throughput proteomics subcellular spatial razor methods suitable for investigating dynamic aspects of protein subcellular distribution in the response of cells to stimulations.21,22 This approach, which combines global quantitative proteomics with the analysis of fractions enriched in target subcellular locations, has allowed measurement of the changes in total abundance and in the compartmental abundance/distribution between the nucleus and cytoplasm for several thousand proteins differentially expressed in IMR90 fibroblasts in response to mild tert-butyl peroxide (TBP) treatment. A highly selective subset of proteins shows appreciable response to TBP. We define a set of 121 proteins showing the most significant changes in total cellular abundance or in nucleocytoplasmic distribution and describe how these two mechanisms cooperate in achieving the changes in abundance in the nuclear and cytoplasmic compartments that are the basis of cellular response. Some of these proteins are involved in known ways with oxidative stress processes, but many are newly identified. Many proteins involved in iron homeostasis or with iron/heme as cofactors as well as mitochondrial proteins and proteins involved in subcellular protein trafficking are prominent in the nuclear response to TBP. The large variety of functional processes in very diverse subcellular locations in which these proteins are implicated emphasizes the need for global, spatially resolved monitoring of coordinated cellular response to stress stimulations like exposure to TBP.
2. MATERIAL AND METHODS
2.1. Cell Culture and SILAC Labeling
IMR-90 (ATCC CCL-186), a human diploid fibroblast adherent cell line, was obtained from LGC Standers (Middlesex, U.K.) at population doubling (PD) of 12. All experiments with IMR90 cells were done with a PD of less than 22 and tested for senescence using a readymade kit (KAA002) from Millipore Corporation (Billerica, MA). Cells were cultured under normal growth conditions at 37 °C and 5% CO2 in SILAC-DMEM media supplemented with 10% of SILAC dialyzed fetal bovine serum FBS. The SILAC kit (#89983) was used by following the manufacturer’s instructions (Thermo Scientific, U.K.).21,34,35
For SILAC quantitative analysis, cells were divided into two populations and cultured in either light media containing amino acids 12C6, 14N4-arginine, and 12C6-lysine (Arg0, Lys0) or heavy media containing amino acids 13C6, 15N4-arginine (Arg10), and 13C6-lysine (Lys6) by at least seven passages in parallel to achieve a full incorporation of SILAC amino acids. The full incorporation of heavy isotope was checked by mass spectrometry of samples of lysed cells.
2.2. Oxidative Stress
Mild oxidative stress was exerted upon IMR90 cells according to a published protocol36,37 with minor modifications. Heavy cells were treated with 50 μM tert-butyl hydrogen peroxide (TBP, Sigma, Poole, U.K.) in cell culture media for 2 h, while the light cells treated with 50 μM PBS were used as control. The treatment was stopped by replacing the cell culture media with fresh media without peroxide. On the next day, the heavy cells were again treated with TBP for 1 h. Four hours after the removal of media containing peroxide, heavy or light cells were detached from the surface of tissue culture flasks by use of Trypsin-EDTA (Sigma, Gillingham, U.K.), washed three times with PBS pH 7.4, and recovered by centrifugation at 60g. We collected and combined a total of four flasks each of heavy and light cells for sample preparation.
2.3. Sample Preparation
Protein concentration was measured by using a Bio-Rad protein assay kit (Bio-Rad, Hemel Hempstead, U.K.). Heavy and light cells were mixed in a ratio of 1:1 based on protein concentrations and the amount of cell pellet.35,38 Cell lysis and subcellular fractionation were performed to obtain cytoplasmic (C, nuclear-depleted) and nuclear fractions (N). Cells were lysed as described in Mulvey et al.34 in hypotonic buffer A containing 10 mM HEPES pH 7.9, 10 mM KCl, 0.1% Triton X-100, 2 mM EDTA, 2 mM DTT, 10% glycerol, 0.34 M sucrose, and protease inhibitors (Roche Diagnostics, Burgess Hill, U.K.). After 10 min of lysis, the salt concentration and viscosity of the buffer were adjusted by the addition of 100 mM NaCl and 200 mM sucrose (isotonic sucrose buffer B; IS buffer). Following a 5 min centrifugation at 2000g, the supernatant was collected as the crude cytoplasmic fraction. The nuclear fraction in the pellet was washed twice in IS buffer by an additional two centrifugation steps for 5 min at 2000g, with the washings restored to the crude cytoplasmic fraction. The crude cytoplasmic (nucleus-depleted) fraction was cleared by centrifugation at 13 000g for 15 min and the supernatant was used as the cytoplasmic fraction.
The cytoplasmic fraction was concentrated using the Aston precipitation method by adding 4:1 volumes of chilled acetone to the sample, which was kept overnight at −20 °C. On the next day, the sample was centrifuged and washed three times with chilled acetone at 13 000g for 10 min at 4 °C. The pellet was air-dried for 5 min at room temperature to eliminate any acetone residue and then dissolved in modified RIPA buffer (50 mM Tris-HCl pH 7.4, 300 mM NaCl, 1% sodium deoxycholate, 1% NP-40, 1 mM EDTA, 0.1% SDS, and protease inhibitors).
2.4. Protein Separation and In-Gel Digestion
For each of the nuclear (N) and cytoplasmic (C) samples, 60 μg of total protein was separately resolved by 10% SDS-PAGE under reducing conditions. Proteins were visualized by silver staining with a ProteoSilver Plus kit (Sigma-Aldrich, Poole, U.K.).39 At least 30 horizontal bands were excised from each gel lane and processed on a 96-well Progest plate (Digilab, Huntingdon, U.K.). Gel bands were processed with the Progest Investigator (Digilab) using established protocols for reduction and alkylation.40 Finally, gel plugs were rehydrated in 20 μg/mL sequencing-grade modified-trypsin (trypsin-gold) (Promega, Southampton, U.K.) that was prepared by adding (1:50 v/v) trypsin/25 mM ammonium bicarbonate and incubated overnight at 37 °C. Fifty microliters of 0.1% formic acid was added to stop the tryptic digestion, the extracted tryptic peptides were collected in siliconized Eppendorf tubes (Sigma-Aldrich, Poole, U.K.) and vacuum-dried to ~20 μL, the volume was adjusted to ~30 μL with 0.1% formic acid, and the sample was analyzed by Orbitrap LC–MS/MS.
2.5. Immunoblotting
Treated heavy (T) or untreated light cells (UT) were lysed in SDS-sample buffer for 3 min at 90 °C and 60 μg protein/lane resolved by 10–12% SDS-PAGE under reducing conditions. Proteins were transferred from gels onto Nitrocellulose membranes (Whatman, GE healthcare, Buckinghamshire, U.K.) by semidry electroblotting (15 V for 1 h). For oxidative stress markers, the membrane was blocked with 5% semi-fat milk powder TBS-Tween20 for 1 h, followed by three times washing with TBST; the antibodies were blocked in 5% semifat milk powder TBS-Tween20 overnight at 4 °C. The antibody concentrations were: 1:10 000 of superoxide dismutase 1 SOD1 (ab79390), 1:1000 of glutathione peroxidase 1 GPX1 (ab108429), and 1:500 of catalase (ab76110), all from Abcam, Cambridge, U.K. For the proteins Nrf2 and Keap, the membrane was blocked with 10% semi-fat milk powder PBS for 1 h or overnight at 4 °C, followed by 3× washing with PBS prior to the use of the antibodies. The antibody concentrations were: 1:300 Nrf2 (ab89443) and 1:500 Keap (ab66620) from Abcam.
The next day the membranes were washed three to five times with either TBS-Tween20 or PBS for 10 min each and incubated for 1.5 h at RT with the appropriate secondary antibodies dissolved in 5% semi-fat milk powder TBS-Tween20 or 10% semi-fat milk powder PBS. Secondary antibodies were 1:2000 antirabbit IgG HRP-linked antibody (7074S) from Cell Signaling, Hitchin, Hertfordshire, U.K. and 1:2500 goat for antimouse IgG2a HRP-linked antibody (ab 97245) from Abcam. Finally, the membranes were washed as previously described. However, the last washing step for TBS was without Tween20 to avoid interference with the ECL detection reagent. WB bands were visualized using ECL developing reagents (RPN2132) from GE Healthcare.
2.6. Flow Cytometry
Flow cytometry for cell cycle analysis followed the procedure described by Tudzarova et al., 2010.36 Heavy and light cells were collected and fixed overnight at −20 °C in 80% methanol in PBS. Cells were precipitated by centrifugation at 50g for 10 min, the supernatant was discarded, and the cells were suspended in a propidium iodide master mix in PBS containing 50 μg/mL RNase A, 50 μg/mL propidium iodide solution (Sigma-Aldrich, Poole, U.K.), and protease inhibitors (Roche Diagnostics) with a final cell density of 1 × 106 cells/mL. Cells were incubated at 37 °C for 30 min in darkness. Cells were sorted by use of a DAKO/Beckman Coulter MoFlo High speed sorter (Beckman Coulter, Orange County, CA). The forward scatter signal was used for detection of cells, and the propidium iodide signal fluorescence was linearly quantified to rationalize DNA content after excitation at 488 nm in the orange/red channel (613/20 nm bandpass filter).36
2.7. Immunofluorescence and Confocal Microscopy
To demonstrate that nuclei and mitochondria remained structurally intact before and after oxidative stress, we used immunofluorescence and confocal microscopy. The IMR90 cells were grown in glass-bottomed Petri dishes (12 mm, 1.5 thickness, GWSt-3512; WillCo Wells B.V., The Netherlands). Control and TBP-treated IMR90 cells were washed three times with PBS+ containing 1 mM CaCl2 and 0.5 mM MgCl2. Mitochondria were stained with 100 nM of MitoTracker green M-7514 or 200 nM MitoTracker deep-red FM (Life Technologies, Paisley, U.K.) in the cell culture growth medium for 30 min at 37 °C, followed by two washes for 5 min with medium under normal growth condition. Nuclear staining was performed by adding 1.5 drops of Hoechst 33342-Readymade R37605 (Life Technologies) to the medium for 15 min in the dark at room temperature.
Alternatively, prior to immunocytochemistry, cells were stained with 200 nM MitoTracker deep-red FM as before, directly fixed, permeabilized for 5 min at −20 °C with ice-cold 1:1 methanol/acetone, and washed three times with PBS for 2–5 min each. Primary antibodies for ALDH18A1 (#HPA008333) from Atlas Antibodies, Sigma, Gillingham, U.K. and ATP5A1 (#PA5-27504) from Thermo Fisher Scientific, Rockford, IL were respectively diluted 1:50 and 1:25 in 4% PBS/FBS and incubated with the fixed cells at 4 °C overnight. Cells were subsequently washed 4× for 10 min with PBS and incubated for 1.5 h in the dark with 1:1000 fluorescent secondary FITC antibody (#F0382) from Sigma, Gillingham, U.K., diluted in 4% FBS/PBS. Nuclei were stained, and cells were covered with VECTASHIELD mounting media with DAPI (#H-1200) from Vector Laboratories, Peterborough, U.K. Image acquisition was performed with a PerkinElmer spinning-disk confocal laser microscope (PerkinElmer, Cambridge, U.K.) and a 63×/1.4 numerical aperture oil immersion lens. The excitation maximum (nm) and emission maximum (nm) were the following: for DAPI, 358 and 461; for Hoechst 3342, 352 and 461; for Mitotracker Green FM, 490 and 516; for Mitotracker Deep red FM, 644 and 665; and for FITC, 490 and 525 respectively. 3D images were taken with technical assistance from Mr. Thomas Adejumo (Wolfson Institute for Biomedical Research and the UCL Cancer Institute, UCL, London, U.K.).
2.8. Mass Spectrometry
LC–MS/MS analysis was performed on a classic LTQ-Orbitrap (Thermo Fisher Scientific, U.K.) equipped with a SURVEYOR–MS pump and Thermo Micro AS-autosampler. Peptides were resolved or loaded using a fused silica capillary column (Nikkyo Technos, Japan), which is a part of the nanoelectrospray ion source with an initial desalting step using a MiChrom C18 Captrap for peptides. Liquid chromatography was carried out at ambient temperature at a flow rate of 500nL/min using a dual gradient of Buffer A: 0.1% formic acid and Buffer B: 100% acetonitrile (ACN) with 0.1% formic acid. Separation was achieved by a 5–23% buffer B gradient (95–77% Buffer A) for 65 min, followed by 23–40% Buffer B (77–60% Buffer A) gradient for 30 min and a step gradient to 60% Buffer B for 5 min. Full profile data were acquired on the LTQ-Orbitrap.
The measurements were done under positive ion mode. The tuning parameters were as follows: spray voltage 1.40 kV and a capillary temperature of 200 °C. A full scan was collected for eluted peptides in the range 450–1600 m/z with the Orbitrap portion of the instrument at a resolution of 60 000, followed by MS/MS using CID (collision-induced dissociation) with dynamic exclusion of 40s and a maximum number in the dynamic exclusion list of 500 in the LTQ portion of the instrument with a minimum count threshold of 500. An activation q value of 0.25 and activation time of 30 ms was applied for MS2 acquisitions. XCalibur software version 2.0.7 (Thermo Fisher Scientific, U.K.) was used for data acquisition.34 Homo sapiens species restriction with the number of protein entries searched: 218 357.
2.9. Data Analysis, Protein Identification, and Quantification
Raw MS files from all replicate SILAC experiments were uploaded into the MaxQuant software platform (version 1.3.0.3) for peak list generation, quantification of SILAC pairs, identification of individual peptides, protein identification, and assembly into protein groups. XCalibur raw files were processed and searched against a UniProt fasta (ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/proteomes/) human protein database (downloaded 5/10/2012). Selected MaxQuant analysis parameters included trypsin enzyme specificity, SILAC doublet measurements of Lys6 and Arg10, two missed cleavages, minimum peptide length of seven amino acids, minimum of two peptides (one of which is unique), top six MS/MS peaks per 100 Da, peptide mass tolerance of 10 ppm for precursor ions, and MS/MS tolerance of 0.5 Da. Oxidation of methionine and N-terminal protein acetylation were selected as variable modifications, and cysteine carbamidomethylation was selected as a fixed modification. All proteins were filtered according to a false discovery rate (FDR) of 1% applied at both peptide and protein levels. Proteins were automatically quantified by the MaxQuant software: A minimum of two peptide ratio counts from razor and unique peptides were necessary for protein quantification, and the “requantification” option was enabled. An experimental design template was used to specify individual experiments and reverse-labeling conditions within the analysis. The final protein groups and peptides text files were processed with Perseus (version 1.3.0.4). Peptides with a normalized Significance B score of p < 0.05 were included for downstream analysis.34,41,42
2.10. Correlation of Proteins across Different Samples
The MaxQuant software package41,42 was used to identify proteins for 12 data analysis sets: (a) each nucleus (N) sample replicate and the union of the three N samples, (b) each cytoplasm (C) sample replicate and the union of the three C samples, and (c) each C&N replicate and the union of the three C&N samples. C&N denotes that for each individual biological replicate, the MS data for the C and N samples were jointly processed with MaxQuant to estimate changes in total protein abundance. For these data, a correction for enrichment of nuclear proteins in the MS data analyses was applied during estimation of total protein abundance (see the Supplementary Text in the Supporting Information). Across these samples, a total of 4429 protein sequence groups were found. For the same underlying gene, slightly different protein sequence groups were sometimes observed, depending on the exact set of peptides detected in each sample. Using the principle that sequence groups in different samples that correspond to the same underlying protein(s) must have protein sequences in common, the union of the unique peptides for protein sequence groups with shared protein sequences was formed and used to requery the full UniProt human sequence data set to identify “consensus” sequence groups across the three sample types. This gave 3589 independent proteins (sequence groups), for which the MS data, including the individual replicates, are given in Supplementary Table S1 in the Supporting Information. All of the consensus sequence groups contained at least one consensus protein sequence that contained all unique peptides in the group, and all were independent; that is, there were no protein sequences shared between different consensus protein sequence groups.
2.11. Selection of Significantly Changed Proteins
To select a set of proteins showing the most significant changes in SILAC ratios for the oxidative stress experiments, we used the MaxQuant Significance B score41 (SigB hereafter) and procedures analogous to those applied previously for cell cycle arrest.21,34 A minimum of at least three ratio counts were required for inclusion of a SILAC ratio in the selection procedures. For SILAC ratios measuring changes in the nuclear (Sn), cytoplasmic (Sc), and total (St) abundances, we used the cutoff limits SigBunion < 0.002 and SigBreplicate < 0.006, where SigBunion refers to the union over the three replicates (e.g., the three nuclear replicates) and SigBreplicate refers to the individual replicates (e.g., each nuclear replicate). Proteins showing the most significant changes in Sn, Sc, and St were selected with the joint requirement SigBunion < 0.002 and SigBreplicate < 0.006 for at least two replicates. For the redistribution parameter Sn/Sc, which detects nucleus–cytoplasmic redistribution even in the absence of changes in total protein abundance21,22 (see the Supplementary Text in the Supporting Information), we used the joint limits |log2(Sn/Sc)|union > 0.9 (0.54 > Sn/Sc > 1.87) and |log2(Sn/Sc)|replicate > 0.8 for at least two replicates. This led to selection of 121 proteins (the 121-OxS set, see the text) for subsequent network analyses. A summary for these proteins and the selection procedures is given in Supplementary Table S2 in the Supporting Information. As previously described,21 the concomitant use of limits on the union and on individual replicates tended to select proteins with substantial numbers of ratio counts; that is, a minimum of 9 ratio counts (in a single sample) and a median of 120 ratio counts per protein were used in classifying the proteins in the 121-OxS set.
2.12. Bioinformatics Analysis of Functional Networks
For the identification of functional annotations, associations, interactions and networks within our data set, a combination of several data analysis tools was used. The consensus Gene Name identifiers for the 121-Oxs set of most significantly changed proteins were uploaded into STRING version 9.1 (Search Tool for the Retrieval of Interacting Genes/Proteins) to create a protein interaction network based on known and predicted protein–protein interactions.43,44 A threshold confidence score of 0.7 was used to ensure that only highly confident protein interactions were considered for inclusion in the network. Seven types of protein interaction information were used for network generation, including neighborhood, gene fusion, co-occurrence, coexpression, experimental, database knowledge, and text mining. Three networks from STRING were further analyzed: the 121-OxS set, a 240-OxS set containing an additional 119 “white nodes” densely connected to 121-OxS that were suggested by STRING and a 173-OxS set containing the 121-Oxs set and 52 of the 119 “white nodes” quantified in our experiments but not among the proteins showing the most significant changes. (See the text.) Densely connected clusters in these networks were identified with MCODE 1.32.45 BiNGO 2.4446 was used to obtain enriched GO BP (biological process) terms. DAVID47 was used to scan for other types of annotations. The combined STRING, BINGO, and DAVID results were used to select a set of 32 GO biological process terms to describe the cellular response. (See the text, Table 2, and Supplementary Table S3 in the Supporting Information.) These terms corresponded to groups of proteins with enriched GO BP terms, but we also included additional terns to cover other proteins selected as significant in the 121-OxS set (Supplementary Table S3 in the Supporting Information). The networks were imported into Cytoscape version 2.8.2 for further analysis and visualization.48,49 Other groups of proteins, for example, proteins annotated to glycolysis (see the text), were selected for comparison using the QuickGO50 facility at the European Bioinformatics Institute (http://www.ebi.ac.uk/QuickGO/) with appropriate sets of GO identifiers.
Table 2. Enrichment of GO Biological Process Terms.
| GO ID | GO Name | 240-OxSa | 173-OxSb | 121-OxSc | Quantifiedd | ||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| number genes | p-valuee | number genes | p-valuee | number genes | p-valuee | genes | number genes | ||
| GO:0042776 | mitochondrial ATP synthesis coupled proton transport | 11 | < 1.0E-17 | 10 | 1.20E-14 | 10 | < 1.0E-17 | ATP5A1, ATP5B, ATP5C1, ATP5D, ATP5F1, ATP5H, ATP5I, ATP5J, ATP5L, ATP5O | 11 |
| GO:0090382 | phagosome maturation | 11 | 3.05E-11 | 11 | 9.44E-13 | 11 | 7.11E-15 | ATP6V0A1, ATP6V0D1, ATP6V1A, ATP6V1B2, ATP6V1C1, ATP6V1D, ATP6V1E1, ATP6V1F, ATP6V1G1, ATP6V1H, TCIRG1 | 16 |
| GO:0033572 | transferrin transport | 11 | 2.71E-10 | 11 | 8.72E-12 | 11 | 6.83E-14 | ATP6V0A1, ATP6V0D1, ATPGV1A, ATP6V1B2, ATP6V1C1, ATP6V1D, ATP6V1E1, ATP6V1F, ATP6V1G1, ATP6V1H, TCIRG1 | 18 |
| GO:0008286 | insulin receptor signaling pathway | 13 | 1.53E-08 | 12 | 4.48E-09 | 11 | 5.32E-10 | ATP6V0A1, ATP6V0D1, ATP6V1A, ATP6V1B2, ATP6V1C1, ATP6V1D, ATP6V1E1, ATP6V1F, ATP6V1G1, ATP6V1H, TCIRG1 | 32 |
| GO:0055085 | transmembrane transport | 54 | 1.95E-10 | 45 | 1.56E-10 | 34 | 8.40E-10 | ABCC4, ATP5A1, ATP5B, ATP5C1, ATP5D, ATP5F1, ATP5H, ATP5I, ATP5J, ATP5L, ATP50, ATP6V0A1, ATP6V0D1, ATP6V1A, ATP6V1B2, ATP6V1C1, ATP6V1D, ATP6V1E1, ATP6V1F, ATP6V1G1, ATP6V1H, CCT8, FTH1, FTL, HK1, HMOX1, HSPA1A, HSPA4, L0NP1, PSMD6, RPS27A, SLC39A7, TCIRG1, THBS1 | 313 |
| GO:0051084 | ‘de novo’ posttranslational protein folding | 10 | 4.69E-06 | 10 | 2.58E-07 | 9 | 6.00E-08 | CCT2, CCT3, CCT4, CCT5, CCT6A, CCT7, CCT8, HSPH1, TCP1 | 30 |
| GO:0022904 | respiratory electron transport chain | 15 | 1.38E-04 | 12 | 2.93E-04 | 12 | 4.21E-06 | ATP5A1, ATP5B, ATP5C1, ATP5D, ATP5F1, ATP5H, ATP5I, ATP5J, ATP5L, ATP5O, SDHA, SDHB | 78 |
| GO:0043066 | negative regulation of apoptotic process | 41 | 1.65E-14 | 29 | 6.69E-10 | 15 | 1.77E-04 | AURKB, CEBPB, HMOX1, HSPA1A, HSPA9, POR, PSMD11, PSMD5, PSMD6, RPS27A, SERPINE1, SET, SQSTM1, THBS1, TIMP1 | 156 |
| GO:0007264 | small GTPase mediated signal transduction | 19 | 2.30E-04 | 15 | 5.10E-04 | 9 | 1.00E-02 | FARP1, HMOX1, IQGAP1, KIF14, RAB2A, RAB5A, RAB5B, RND3, SQSTM1 | 115 |
| GO:0055114 | oxidation-reduction process | 28 | 2.07E-01 | 22 | 1.64E-01 | 20 | 1.10E-02 | ABCC4, ALDH18A1, CYB5B, CYB5R3, ECH1, ETHE1, FTH1, GPD2, HADHA, HCCS, HK1, HMOX1, IQGAP1, POR, PTGS1, PYCR1, RPS27A, SDHA, SDHB, SQRDL | 334 |
| GO:0006936 | muscle contraction | 6 | 8.04E-02 | 5 | 6.18E-02 | 5 | 1.11E-02 | CALD1, CNN1, MYOF, TPM3, TPM4 | 51 |
| GO:0007155 | cell adhesion | 18 | 9.75E-03 | 13 | 2.92E-02 | 10 | 1.85E-02 | ATP5B, COL1A1, KIF14, LGALS1, MFGE8, RND3, SERPINE1, TGFBI, THBS1, TPM4 | 145 |
| GO:0006986 | response to unfolded protein | 8 | 3.75E-02 | 7 | 2.03E-02 | 5 | 2.71E-02 | ATP6V0D1, HSPA1A, HSPA4, HSPH1, THBS1 | 62 |
| GO:0000209 | protein polyubiquitination | 21 | 2.75E-10 | 17 | 4.42E-09 | 5 | 3.11E-02 | PSMD11, PSMD5, PSMD6, RPS27A, UBR5 | 64 |
Full set of 121-OxS nodes plus 119 suggested nodes from STRING.
Set of 121-OxS nodes plus 52 nodes suggested by STRING that were quantified.
Set of 121-OxS nodes.
Total number of quantified proteins annotated to the indicated process.
Hypergeometric p values calculated relative to the background of MS quantified proteins.
3. RESULTS
The proteomics subcellular spatial razor experiments described in the following are based on a model that envisages that in response to a cellular perturbation proteins may show changes both in total cellular abundance and in their subcellular spatial distribution between the nucleus and cytoplasm.21,22 To apply the model, we obtained experimental measurements of protein abundance ratios (SILAC ratios) between stimulated/unstimulated cells for three data sets: (1) a nucleus-enriched sample obtained by subcellular fractionation (N), (2) the corresponding nucleus-depleted sample (C), which we refer to as the “cytoplasm” in the following text, and (3) a total protein sample (T). The corresponding SILAC ratios provide measures for each protein of the overall change in total cellular abundance (St) or of the localized change in abundance in the nuclear (Sn) or cytoplasmic (Sc) subcellular compartments. A mathematical formulation of the model is given in the Supplementary Text in the Supporting Information. Stringent purification of organelles (e.g., nucleus) was not attempted in this study because we believe it is not feasible to purify to homogeneity organelles that are subject to dynamic changes in their protein content and that unnecessary protein loss is incurred during such organelle isolation methods. We preferred to use highly enriched fractions rather than highly purified organelles. This approach has previously been shown to successfully detect nucleocytoplasmic trafficking.21,22
We rigorously validated and checked the oxidative stress response for the IMR90 cells (a human diploid fibroblast adherent cell line) by the use of complementary cell biology and proteomics methods. Oxidative stress was generated by the use of tert-butyl hydrogen peroxide (TBP, see Materials and Methods). Cellular response to oxidative stress was monitored by flow cytometry to determine cell cycle profiles, inspection of cell viability by light microscopy, and Western blotting of oxidative stress markers. Optimal dosage with TBP for the IMR-90 cells was determined with these methods.
Response of the cells to oxidative stress was demonstrated by Western blotting. As expected,29 the oxidative stress-regulated protein NRF2 increased in abundance and accumulated in the nucleus, whereas its inhibitor KEAP1 was detected only in the cytoplasm and decreased in abundance (Figure 1A).
Figure 1.

Characterization of the cellular response to oxidative stress and the subcellular fractionation. (A) Western blotting of NRF2 and KEAP1 of the nuclear (N) and cytoplasmic (C) fractions for untreated (U) or THP-treated (Ox) cells showing cellular response to oxidative stress. (B) Flow cytometry determination of the cellular distribution over the cell cycle.
Flow cytometry analysis of DNA content (Figure 1B) shows that mild THP oxidative stress led to an accumulation of IMR90 cells at G2/M and in G1 with a concomitant decrease in the proportion of cells in S phase, indicative of checkpoint activation and cell cycle arrest. The lack of cell counts in the sub-G1 region together with a microscopically determined high degree of cell viability indicates that the IMR90 cells did not induce apoptosis in response to mild oxidative stress.
The subcellular fractionation protocols were optimized using the nuclear and cytosolic markers lamin and tubulin, respectively (data not shown). We adapted a published procedure using the nuclear protein Lamin A/C to test for nuclear leakage during fractionation. In the samples used for the quantitative analyses described below, the recorded cytoplasm/nucleus MS intensity ratio for Lamin A/C ranged over 0.2 to 0.8% for the three replicates, that is, for IMR90 cells with our protocols any nuclear leakage during fractionation was minimal and did not materially affect the measured SILAC ratios (1.185 ≤ Sn ≤ 1.197, 0.964 ≤ Sc ≤ 1.009 for Lamin A/C over the three replicates). The reproducibility of the preparations was tested by correlation of the SILAC ratios Sn, Sc and St between the three replicates for each sample type. High correlation (0.75 < R2 < 0.88) with most variance concentrated in a handful of readily recognizable outliers with few SILAC counts (Supplementary Figure S1 in the Supporting Information) provided confirmation of highly reproducible sample preparation. The purity of the nuclear and cytoplasmic fractions was evaluated from the MS data. (See section 3.2.)
3.1. Characterization of the MS data
The MaxQuant software package41,42 was used to identify proteins and determine SILAC ratios for 12 data sets: (1) each nucleus (N) sample replicate and the union of the three N samples, (2) each cytoplasm (C) sample replicate and the union of the three C samples, and (3) each C&N replicate and the union of the three C&N samples. C&N denotes that for each individual replicate the MS data for the C and N samples were jointly processed with MaxQuant to estimate changes in total protein abundance (T) (see Methods and the Supplementary Text in the Supporting Information). Correlation of these sequence groups across the samples (see Methods) gave the 3589 independent proteins (sequence groups) for which the full MS data, including the individual replicates, is given in Supplementary Table S1 in the Supporting Information. A total of 1929 C proteins and 2751 N proteins were detected. Their distribution over the nucleus and cytoplasm is shown in Figure 2: 783 proteins were detected only in the cytoplasm, 1605 proteins were detected only in the nucleus, and 1146 were detected in both locations. A further 35 proteins were detected only in the jointly processed C&N data set. Among the 783 C and 1605 N proteins, there were many with large numbers of sequenced peptides/ratio counts, which further confirmed that the subcellular fractionation was efficient and that there are many proteins characteristic of one or the other location. With the requirement of at least two peptides (1 unique) and three ratio counts in a single sample, SILAC ratios were obtained for 1623 cytoplasmic proteins (Sc), 2451 nuclear proteins (Sn), and 3124 total proteins (St). For 973 proteins, SILAC ratios (Sc, Sn, St) were obtained for all three data types.
Figure 2.
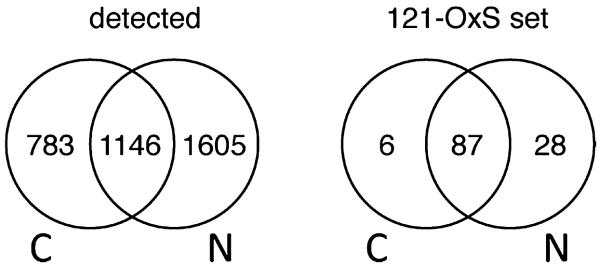
Distribution over the nucleus (N) and cytoplasm (C) for all identified proteins (detected) or for the set of proteins (121-OxS set) selected as showing the most significant changes in compartmentalized abundance (Sc, Sn), total abundance (St), or distribution between the nucleus and cytoplasm (Sn/Sc).
MaxQuant normalizes protein ratios for each data set and employs an algorithm within the Perseus program to calculate an outlier probability score (Significance B score, hereafter SigB) to find the most significantly altered proteins.41,42 SigB was calculated for the proteins in each of the 12 data sets using the MaxQuant quantitative analysis suite. These SigB values and the parameter log2(Sn/Sc), which detects redistribution between C and N even in the absence of total abundance changes21,22 (see the Supplementary Text in the Supporting Information), were used to select 121 proteins (3.4% of all identified proteins, referred to as the 121-OxS set hereafter) that showed the strongest, reproducible changes in compartmental abundance (Sn or Sc), in total abundance (St) or in redistribution between the cytoplasm and nucleus (Sn/Sc) in response to oxidative stress. The inclusion of reproducibility over the replicates in the selection criteria (see Material and Methods) meant that the selection of significant proteins was stringent and tended to reject proteins with few ratio counts: a minimum of 9 ratio counts (in a single sample type) and a median of 120 ratio counts per protein were used in classifying the proteins in the 121-OxS set. For the data sets in which they were quantified, Figure 2 shows the distribution of the proteins in the 121-OxS set over the cytoplasm and nucleus. Supplementary Table S2 in the Supporting Information contains a summary of the SILAC ratios Sc, Sn, St and Sn/Sc for these proteins. Overall, the number of significant changes for the 121-OxS set included 19 proteins (Sc), 63 proteins (Sn), 76 proteins (St) and 43 proteins (Sn/Sc), that is, some proteins satisfied more than one selection criterion.
3.2. Consistency of the Data with High Enrichment of the Nucleus Sample
There is increasing evidence that many proteins have multiple subcellular locations and may have different functions in different locations. (See the Discussion.) In keeping with this, many of the proteins that were quantified in the nucleus have GO CC (cellular component) annotations to other subcellular locations. For example, in the N sample we quantified 371 proteins with annotation to mitochondria, of which 147 were also annotated to the nucleus. There were also many proteins with annotations to other subcellular sites in the nuclear fraction (Figure 3, table inset). To further verify that the 121-OxS set of proteins was not distorted by cross-contamination of the nuclear fraction with other subcellular organelles, we looked at the distribution of the function Sn/St = fs/fu, where fs and fu are the fractions of a protein in the nucleus in stimulated or unstimulated cells respectively. (See the Supplementary Text in the Supporting Information.) The vast majority of the proteins with annotation to mitochondria had fs/fu ≈ 1, that is, there was no appreciable change in their nuclear fraction in response to oxidative stress (Figure 3A). A small number of proteins exhibited appreciable changes in fs/fu, including proteins such as HK151 and BAX52-54 which are known to show increases in nuclear abundance that are associated with important functional roles in response to oxidative stress. In agreement with previous studies, changes in total abundance for both of these proteins were small (St = 1.14/1.04 for HK1/BAX), that is, the dominant change was in subcellular location. Similar plots were obtained for proteins with GO annotations to plasma membrane, endoplasmic reticulum, Golgi apparatus, endosomes, lysosomes and peroxisomes (Supplementary Figure S2 in the Supporting Information). In all cases there were both increases and decreases in fs/fu involving a small percentage of the proteins (Figure 3, table). This pattern indicates that those proteins with appreciable changes in fs/fu are not a consequence of contamination of the nuclear fraction with other subcellular organelles. This conclusion is further stressed by looking at high abundance proteins that are present in multiple subcellular locations. There were many high abundance proteins annotated to mitochondria that showed no change in fs/fu, for example, PHB in Figure 3A. The same phenomenon was observed for numerous high abundance proteins annotated to other subcellular locations (Supplementary Figure S2 in the Supporting Information). For example, log2(fs/fu) = 0.009 for prohibitin (PHB) (Figure 3B), which is annotated to diverse functions including cell proliferation, differentiation and apoptosis.55-58 It has known functions in the nucleus,59 mitochondria60 and plasma membrane.61,62 Large numbers of high abundance proteins that show essentially no change in measured fs/fu, such as the examples in Figure 3B, are consistent both with high reproducibility of the subcellular fractionation and with very little cross-contamination of the nuclear fraction by other subcellular organelles, as has been observed for other cell types with other forms of analysis.23 Overall, these data patterns are very strong evidence that oxidative stress causes selective changes in nuclear abundance for a set of specific proteins. The data also suggest that current GO annotations underestimate the proportion of proteins that are shared between the nucleus and other subcellular locations such as mitochondria, a characteristic that we have also observed for other cell types.22,23,63
Figure 3.

Analysis of the enrichment/purity of the nuclear fraction. Table: The number of quantified proteins in the nuclear fraction that are annotated to other subcellular locations and the number showing appreciable change in their nuclear fraction (Sn/St = fs/fu). (A) Left: log2(fs/fu) as a function of the average number of ratio counts over the nucleus and total data sets for proteins with GO annotation to mitochondria and nucleus (red, 143 proteins) or to mitochondria but not nucleus (blue, 218 proteins). Right: number of proteins versus log2(fs/fu). (B) Log2(fs/fu) for selected high abundance proteins with at least 100 SILAC ratio counts, including at least 50 in the nucleus, that have GO annotation to multiple subcellular locations. The current GO cellular component annotations for these proteins are given in parentheses according to the letter code in the table.
3.3. Correlation of Changes in Abundance and Nucleus/Cytoplasmic Distribution
Because the abundance of a protein in the nucleus/cytoplasm can be altered by changes both in total cellular protein abundance and in distribution between the two compartments, the SILAC ratios Sn and Sc do not directly measure redistribution of a protein between the two compartments. A direct measure for redistribution to/from the nucleus that is independent of changes in total protein abundance is provided by Sn/Sc = fs(1 – fu)/fu(1 – fs). (See the Supplementary Text in the Supporting Information.) We used the 87 proteins with the most significant changes that were quantized in both the nucleus and cytoplasm to test for correlation between changes in total abundance (St) and changes in nucleus-cytoplasm distribution (Sn/Sc). There was very little correlation (Figure 4A), that is, the changes in abundance in the nuclear and/or cytoplasmic compartments do not simply mirror changes in total protein abundance. We further characterized the response to oxidative stress of these 87 proteins using the orthogonal 3D basis set {Sn/St, Sc/St, St} (Figure 4B,C). This “subcellular spatial razor” basis set expresses changes in the total abundance of proteins along a St axis and changes in their N/C distribution in a {Sn/St, Sc/St} distribution plane that is independent of changes in total abundance.21,22 Conservation of mass requires that in the distribution plane the data points appear in two allowed quadrants corresponding to N → C and C → N redistribution, respectively (Figure 4, see also the Supplementary Text in the Supporting Information).
Figure 4.

Subcellular characterization of the 87 proteins of the 121-OxS set for which Sn, Sc, and St were all measured. (A) Plot of changes in nucleocytoplasmic distribution (Sn/Sc) versus changes in total abundance (St). The red bounding box corresponds to 1.5-fold changes in total abundance and 0.57 > Sn/Sc > 1.74 (|log2(Sn/Sc)| > 0.9). A few proteins appear just inside the bounds because the combinations of basal distribution, changes in total abundance, and changes in distribution can lead to individual compartmental abundances (Sn or Sc) being selected as significant. (B) 3D spatial razor plot for the 87 proteins. The dotted green lines correspond to the bound |log2(Sn/Sc)| = 0.8. (C) Same as panel B for selected, labeled proteins or protein complexes. (See the text.) In panels B and C changes in total abundance St (perpendicular to the page) are color-coded according to the scale at the right.
The 3D subcellular spatial razor format is sensitive to changes in total protein abundance, to redistribution of proteins between the nucleus and cytoplasm, and also to the basal nuclear/cytoplasm distribution of proteins in unstimulated cells. For example, for the 121-OxS set of proteins classic oxidative stress response proteins such as the light and heavy subunits of ferritin (FTL, FTH1) showed substantial increases in total abundance but little or no C ↔ N redistribution (Figure 4C). In contrast, eight subunits of the CCT protein-folding complex showed little or no change in total abundance but substantial C → N redistribution, while proliferating cell nuclear antigen (PCNA) showed N → C redistribution with little change in total abundance. PCNA and the CCT proteins show log2(Sc/St) ≈ 0 that reflects basal abundance strongly skewed to the cytoplasm. The small proportion of the protein in the nucleus is increased (CCT) or expelled to the cytoplasm (PCNA) under oxidative stress. Such behavior might reflect “catalytic” transfer of information about oxidative stress between different subcellular locations. Proteins such as nucleoside diphosphate kinase A (NME1) have less strong basal skewing of abundance, show appreciable, coupled changes in abundance in both subcellular compartments in response to oxidative stress, and might represent more general “balancing” of function between different subcellular locations. Other protein groups appear to show some coupling between total abundance and subcellular distribution. For example, six subunits of ATP synthase (ATP5) show ca. two-fold decreases in total abundance accompanied by N → C redistribution, that is, reduced total abundance was accompanied by further depletion of these proteins in the nucleus. Conversely, eight subunits of V-type proton ATPase (ATP6) show about 1.8-fold increase in total abundance accompanied by a trend of C → N redistribution.
The data for Sn, Sc, St, and Sn/Sc for all proteins in the 121-OxS set are summarized in Supplementary Table S2 in the Supporting Information. Figure 5 shows a visual representation of the significant changes in Sc, Sn and Sn/Sc for the proteins in the 121-OxS set measured in both compartments. A general characteristic of many of the proteins is that they participate in many different functional processes as these are currently defined by GO ontology terms (see below). For example, proteins annotated to cell differentiation include proteins also annotated to ATP synthesis, lipid metabolism, heme/iron metabolism, cell adhesion, negative regulation of apoptosis and protein transport.
Figure 5.

Visual summary of the most significant changes in subcellular abundance (Sn or Sc) and in nucleocytoplasmic distribution (Sn/Sc) for the proteins of the 121-OxS set quantified in both the nuclear and cytoplasmic compartments. A selection of some of the GO biological process terms annotated to the proteins (see text) is indicated.
We used fluorescence imaging to verify several aspects of the MS analyses of the response to oxidative stress. First, imaging of mitochondria and the nucleus indicated structural integrity with no apparent nuclear breakage or major alteration of the mitochondria (Supplementary Figure S3 in the Supporting Information). This is in keeping with mild oxidative perturbation and no apparent increase in apoptotic cells detected by flow cytometry or light microscopy. Second, proteins such as the subunits of ATP synthase (ATP5) are well-known as part of the mitochondrial respiratory chain, but have not been regarded traditionally as nuclear proteins and we therefore investigated the presence of ATP5A1 in the nucleus of intact IMR90 cells. In both control cells and cells subjected to oxidative stress ATP5A1 was widely distributed over mitochondria, nuclei and generally over the cytoplasm (Supplementary Figure S3 in the Supporting Information). This is consistent with previous studies indicating that subunits of ATP synthase can be distributed to many subcellular locations and may have different functions at those locations.64-67 At present GO CC annotations for ATP5A1 include mitochondria, nucleus, plasma membrane and the extracellular region. Third, we also investigated the presence in the nucleus of Delta-1-pyrroline-5-carboxylate synthase (ALDH18A1), a constituent of the proline regulatory axis (see below), which showed >two-fold reduction in nuclear abundance for OxS (Supplementary Table S2 in the Supporting Information). Although this protein seems to preferentially redistribute from the cytoplasm to mitochondria under oxidative stress, there are focal points of the protein in the nucleus for both control and oxidatively stressed cells (Supplementary Figure S3 in the Supporting Information). It is currently annotated to the cytoplasm and mitochondria by GO.
Extensive dispersion of the proteins in the 121-OxS set over many subcellular locations is also evident in the current GO CC annotations for these proteins. Table 1 shows the number of annotations to four main top-level locations (nucleus, cytoplasm, plasma membrane, extracellular region) with a further breakdown of cytoplasmic locations. With the vocabulary shown in Table 1, 117 of the proteins had annotations. There was an average of 2.99 locations per protein and a maximum of eight locations per protein. The 45 proteins annotated to the nucleus included annotations to the cytoplasm (31 proteins), the extracellular region (18 proteins) and the plasma membrane (9 proteins). Similarly, among the 64 proteins annotated to the cytoplasm, 31, 20, and 37 were annotated to the nucleus, plasma membrane and extracellular region, respectively. The proteins are also annotated to a variety of different cytoplasmic locations. For example, proteins annotated to the nucleus include annotations to all of the cytoplasmic locations shown except the peroxisome. Conversely, we quantified 10 proteins in the nucleus and cytoplasm that were only annotated to nucleus and 33 proteins in the nucleus and cytoplasm that were only annotated to cytoplasm. This suggests that the current GO CC annotations underestimate the dispersion of proteins over multiple subcellular locations. This is a characteristic that we have also observed with other cell types.22,23,63
Table 1.
Distribution of GO CC Annotations for the 121-OxS Protein Seta
| subcellular location | nucleus | plasma membrane | extracellular region | cytoplasm | cytosol | cytoplasmic vesicle | mitochondrial | endoplasmic reticulum | Golgi apparatus | melanosome | peroxisome | endosome | lyssosome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| nucleus | 45 | 9 | 18 | 31 | 19 | 6 | 7 | 8 | 5 | 2 | 0 | 3 | 1 |
| plasma membrane | 32 | 24 | 20 | 14 | 11 | 8 | 3 | 1 | 5 | 0 | 7 | 8 | |
| extracellular region | 53 | 37 | 27 | 16 | 9 | 7 | 5 | 7 | 1 | 7 | 10 | ||
| cytoplasm | 64 | 34 | 12 | 13 | 11 | 7 | 7 | 0 | 7 | 10 | |||
|
|
|||||||||||||
| cytosol | 43 | 6 | 9 | 8 | 2 | 3 | 0 | 4 | 8 | ||||
| cytoplasmic vesicle | 18 | 1 | 3 | 4 | 3 | 0 | 7 | 4 | |||||
| mitochondrion | 41 | 4 | 0 | 0 | 1 | 2 | 3 | ||||||
| endoplasmic reticulum | 16 | 3 | 2 | 0 | 1 | 2 | |||||||
| Golgi appartus | 10 | 2 | 0 | 1 | 1 | ||||||||
| melanosome | 8 | 0 | 3 | 2 | |||||||||
| peroxisome | 1 | 0 | 0 | ||||||||||
| endossome | 8 | 4 | |||||||||||
| lysosome | 11 | ||||||||||||
|
| |||||||||||||
| GO CC ID | GO:0005634 | GO:0005886 | GO:0005576 | GO:0005737 | GO:0005829 | GO:0031410 | GO:0005739 | GO:0005783 | GO:0005794 | GO:0042470 | GO:0005777 | GO:0005768 | GO:0005764 |
Diagonal gives the number of proteins annotated to the indicated GO identifier. Off-diagonal terms are numbers of proteins jointly assigned to two different GO identifiers.
3.4. Functional Network Analyses
To further analyze the functional significance of the changes for proteins corresponding to the 121-OxS set, we used the STRING programs43 to look for functional networks, which were visualized with Cytoscape68 (see Materials and Methods). Interaction networks were obtained for three sets of proteins: (1) the 121-OxS set; (2) a 240-OxS set consisting of the 121-OxS set plus 119 “white nodes” suggested by STRING that correspond to other proteins that have the most dense interactions with the proteins in the 121-OxS set; and (3) a 173-OxS set consisting of the 121-OxS set plus the 52 additional “white node” proteins suggested by STRING and quantified in our experiments. Of the 119 white nodes suggested by STRING, 52 were in fact quantified in the MS data, but were not among the 121-OxS set of proteins selected as showing the most significant changes in response to oxidative stress. With these three data sets, the MCODE clustering algorithm was used to identify clusters of nodes with dense interactions. GO BP (biological process) enrichments calculated with BiNGO46 and other annotation identified with DAVID47 were used to identify functions associated with these clusters. The 240-OxS set defines the most likely main functional processes, the 173-OxS set provides a measure of how well these processes were monitored in our measurements, and the 121-OxS set provides a “pure” probability that may be small if only a few proteins in the network change.
Figure 6A provides a graphical overview of the STRING results and indicates that different behavior in response to oxidative stress is observed for different groups of proteins. For example, most of the subunits of ATP synthase (ATP5) were monitored (Table 2) and showed similar behavior (decrease in St with N → C redistribution). For V-type proton ATPase (ATP6), many subunits were monitored (Table 2) but showed inverse behavior (increase in St with C → N redistribution). In the full MS data set, 47 proteins that are included in proteasomal complexes were quantified. The 240-OxS network includes 16 proteasomal proteins, 14 of which were quantified, but only PSMD-5,6,11 (increased St and C → N redistribution) were in the 121-OxS set with the most significant changes. All three of these are annotated to GO:0071158, “positive regulation of cell cycle arrest”. From the nuclear COPS signalosome complex, nine proteins were quantified and are included in the 240-OxS network, but only COPS3, ATP5A1,69 and HSP1A1 were included among the proteins of the 121-OxS set with the most significant changes and COPS3/HSP1A1 showed different behavior than ATP5A1. Conversely, all eight proteins of the CCT complex involved in de novo post-translational protein folding (Table 2) were included in the 121-OxS set (St ≈ 1, C → N redistribution). There have long been indications of connections between oxidative status and the cell cycle and Figure 6A contains densely linked clusters for proteins (DNA polymerase delta subunits, replication factor C subunits) intimately linked to DNA replication (see the Discussion).
Figure 6.

Network analysis for the 240-OxS and 121-OxS data sets. (A) STRING interaction network for the 240-OxS set. Nodes corresponding to the 121-OxS set are colored in red, nodes suggested by STRING and quantified in the MS data but not included in the 121-OxS set are colored in yellow, and nodes suggested by STRING but not quantified in the MS data are colored in gray. Node sizes are mapped to the betweenness centrality values, while edge colors and sizes are mapped to edge betweenness values, as calculated by the Network Analysis plugin for the Cytoscape software. Betweenness centrality is a measure of a node’s centrality and importance in a network. Edge betweenness is a topological measure defined as a normalized number of shortest paths between two nodes. (B) Thumbnails of the interaction network: nodes colored in blue indicate significant changes in Sn, Sc, St, or Sn/Sc of proteins that are included in the 121-OxS set.
Despite these clear clusters of proteins associated with specific cellular functions, a striking feature of Figure 6A is that many classical oxidative stress proteins such as FTL, FTH1 and HMOX1 were only weakly connected by STRING and 35 proteins were unconnected. In part this seems to reflect incomplete inclusion of literature information in the STRING interaction database. For example, for POLDIP2 (unconnected) and PGRMC1 (unconnected) there is substantial literature about their functional roles (see the Discussion), but the interactions are not included in the STRING database. This is not a drawback of the STRING programs specifically. Similarly disconnected (in different ways) networks were also observed with preliminary GeneMania calculations and probably reflect the fact that interaction databases presently tend to be dominated by large-scale coexpression and physical interaction data sets.
There is another striking feature of the data that is summarized visually by Figure 6B. A protein may have the same or different functions in the nucleus or in the cytoplasm, but the contribution of the function(s) to cellular response should in both cases be dependent on its dynamic abundance in the subcellular location, as reflected in Sn and Sc. Changes in total abundance (St) and in nucleus/cytoplasm distribution (Sn/Sc) represent the mechanisms by which the changes in compartmental abundance are achieved. For the 121-OxS set, only 76 of the proteins were selected for inclusion on the basis of changes in total abundance and for 39 of these proteins SigB was smaller for changes in Sn or Sc than for St, that is, for these 39 proteins compartmental changes in abundance were more significant than changes in total abundance as a consequence of concomitant changes in total abundance and redistribution.
Conversely, for the 45 proteins for which St was not selected as being among the most significant features, 26 proteins had their most significant changes in the cytoplasmic (9 proteins) or in the nuclear (17 proteins) compartments as a consequence of concomitant changes in total abundance and redistribution. For 19 proteins, the redistribution parameter (Sn/Sc) was the most significant feature and most of these proteins had only very small changes in St. Polymerase delta-interacting protein 2 (POLDIP2), which is annotated to both the nucleus and mitochondria but for which GO presently has no biological process annotation, was of this type and seems to represent a modulator for the POLD-related cluster we detected (see the Discussion). Several RAB proteins (RAB2, RAB5A, RAB5B) that showed only very small changes in St were also included in this class. These might represent vesicle-mediated transport to/from the nucleus (see below). Figure 6B indicates that changes in both total protein abundance and in subcellular distribution are crucial in achieving the complex pattern of changes in compartmental abundances.
Finally, a third general characteristic apparent from the data is that the same protein may be involved in a variety of different functional processes that in many cases involve different subcellular locations. For example, plasminogen activator inhibitor 1 (SERPINE1) is annotated by GO to 10 biological processes ranging over extracellular matrix organization, cellular component movement, and DNA-templated transcription. Concomitantly the data suggest that any given biological process may be altered by changes in the compartmental abundance of only a limited number of proteins (Table 2). However, overall enrichment of current GO biological process terms was of moderate help in obtaining biologically meaningful interpretations of the data. Four proteins in the 121-OxS set lacked biological process annotations, much literature information seems not to have been incorporated in useful forms in the classifiers, and only 82 of the proteins in the 121-OxS set are covered by the enriched terms shown in Table 2. Furthermore, the enriched terms also seem to be conditioned by the contexts in which these proteins have previously been investigated. For example, IMR90 cells do not have phagosomes, and the processes “phagosome maturation” and “insulin receptor signalling pathway” almost certainly reflects lysosomal/endosomal processes (see the Discussion). There are strong indications for vesicle-mediated protein transport in the response to TBP treatment (see below), and the term “de novo post-translational protein folding” may be related to the role of the CCT complex in sperm/oocyte fusion, which involves membrane fusion of a specialized form of a lysosomal exocytotic vesicle (acrosome). Similarly “muscle contraction” may be an indication of relationships to endocytotic processes. Additional GO terms that cover other 121-OxS proteins and biological processes as well as the complete protein identities for the 240-OxS, 173-OxS, 121-OxS, and MS-quantified protein sets are given in Supplementary Table S3 in the Supporting Information.
3.5. Metabolic and Antioxidant Features Related to Nuclear Respiratory Factor 2 and the Proline Regulatory Axis
Nuclear factor-erythroid 2 p45-related factor 2 (NRF2, also called NFE2L2) is a crucial transcription factor for many aspects of intrinsic resistance and response to oxidative stress.31 There are substantial indications for involvement of NRF2 in intermediary metabolism including glycolysis, the TCA cycle, generation of NADPH, glutamine metabolism, and fatty acid oxidation.31 It translocates to the nucleus in our experiments (Figure 1), and we therefore examined the behavior in our data of proteins known to be regulated by NRF2. We used a recent compilation of genes reportedly subject to positive regulation by NRF2.31
NRF2 has been proposed to cause changes in the glycolysis and pentose phosphate pathways that generate NADPH used by antioxidant proteins. We monitored numerous proteins in this pathway (Figure 7A). There was little change in abundance for either of the isoforms of pyruvate kinase (St = 1.08, 0.99) or in the enzymes G6PD/PGLS/PGD (St = 1.12, 0.94, 1.04) in the oxidative arm of the pentose phosphate shunt. The TALDO/TKT arm of the pentose phosphate shunt showed modest increases in abundance (St = 1.08, 1.39). However, MTHDF2 showed an appreciable decrease (St = 0.58), suggesting that in our experiments either input to purine synthesis via the pentose phosphate shunt is not strongly modified or might augment modestly via MTHDF1/MTHDF1L1 (St = 0.96, 0.91). With the exception of the C → N translocation of HK1, none of the glycolytic proteins were included in the 121-OxS set of most significant changes. Although HK1 is totally disconnected in Figure 6A, subcellular translocation of HK1/HK2 is a known effect related to the metabolic fate of glucose and to glycolysis in cancer cells.51,70-72 We consider that the behavior observed for HK1 represents a prototype for a “selective messenger”, that is, a single protein displaced to a subcellular location distinct from a dense, spatially defined functional network. In this case, as a messenger from glycolysis to the nucleus, HK1 relies almost exclusively on redistribution of subcellular location (St = 1.14, Sn = 2.02, Sc = 1.10, log2(Sn/Sc) = 0.87).
Figure 7.

Three selected processes/networks. These dense networks are highly interrelated and were extensively quantified in the MS measurements (yellow nodes) but only a small subset of the proteins in each network (red nodes) was included in the most significant changes.
Similar results were obtained for the TCA cycle. For 12 enzymes in the core of this cycle (SUCLA2, SUCLG2, FH, MDH2, MDH1, IDH2, IDH3A, IDH3B, IDH3G, CS, ACO2, ACO1) there were only minor changes in total (0.83 < St < 1.11) or compartmental abundance (Figure 7B). Slightly more pronounced decrease in abundance was observed for the proteins with lipoamide E cofactor in the TCA cycle (DLD, DLST, OGDH; 0.70 < Sn, St < 0.76), with indication for moderate N → C redistribution (Supplementary Table S1 in the Supporting Information) and for SUCLG1 (St, Sc ≈ 0.75). This would be consistent with slightly reduced production of succinyl-CoA from 2-oxo-glutarate and its subsequent usage in the TCA cycle. The lipoamide E proteins of the pyruvate dehydrogenase complex (DLD, DLAT, PDHB) showed a similar moderate decrease in abundance (0.70 < St < 0.85). This would be consistent with reduced production of acetyl-CoA from pyruvate. There was a modest decrease in IDH2 (St, Sc ≈ 0.91) and a modest increase in cytoplasmic IDH1 (St, Sc ≈ 1.33). None of these proteins were included in the 121-OxS set of most significant changes (Figure 7B), where the outstanding change in the TCA cycle was decrease in SDHA and SDHB (Sn, St ≈ 0.37). SDHA and SDHB were connected by STRING to ATP synthase (Figure 6A), presumably as a consequence of well-studied mitochondrial interactions. However, there is very little information available on possible functional roles for SDHA, SDHB, and ATP5 subunits in the nucleus, even though SDHA and SDHB are known to be involved in various types of cancers and other diseases.73-77 SDHA and SDHB may be selective messengers to the nucleus about mitochondrial state.
NRF2 is thought to play an important role in NADPH homeostasis via regulation of the NADPH-generating enzymes G6PD, PGD, IDH1,2 and NADP-dependent malic enzyme (ME1), which provides an alternative source of pyruvate by decarboxylation of malate.31 We observed a modest increase in abundance for ME1 (St = 1.19). We also observed a modest change in mitochondrial NAD(P) transhydrogenase NNT (St = 0.71), which can couple production of NAPDH to the mitochondrial hydrogen ion gradient.78 Altogether, these results suggest that augmented production of NADPH via NRF2/glycolysis/TCA cycle is not a major response in our experiments. In keeping with this, we did not observe strong changes in the levels of proteins of glutathione- or thioredoxin-based antioxidant systems. Glutathione peroxidases GPX1 and GPX8 (St ≈ 0.7), glutaredoxin GLRX3 (St = 1.02), isoforms 1 and 3 of glutaminase GLS (St = 0.93, 0.83), and glutathione reductase GSR (St = 1.32) all showed only modest changes in abundance. Only small changes in abundance were also observed for thioredoxin TXN (St = 1.23), thioredoxin reductases TXNRD1 and TXNRD2 (St = 1.16, 0.93), and peroxiredoxins PRDX1,2,4,5,6 (0.93 < St < 1.15). Only PRDX3 showed slightly different behavior (St = 0.77, Sn = 0.63, Sc = 0.92). None of these antioxidant proteins were among the 121-OxS set.
We also monitored 24 proteins annotated to fatty acid oxidation (Supplementary Table S1 in the Supporting Information). For 21 of these proteins there was very little change in total (0.82 < St < 1.19) or compartmental abundance. Substantial changes in total and nuclear abundance were observed for subunits of the mitochondrial trifunctional enzyme HADHA/HADHB (St = 0.65/0.68; Sn = 0.61/0.60; Sc = 0.93/0.97). The electron-transfer flavoprotein–ubiquinone oxido-reductase ETFDH appeared to show similar behavior but with a small number of SILAC ratio counts (Supplementary Table S1 in the Supporting Information). We conclude that in our experiments any effects of NRF2 on fatty acid oxidation are limited to indirect effects associated with changes in the nuclear abundance of HADHA/HADHB and possibly ETFDH. Similarly, many proteins associated with detoxification of drugs whose abundance is controlled by NRF2,31 including oxidation of drugs (aldehyde dehydrogenases, aldo-keto reductases, carbonyl reductases CBR1 and CBR3, epoxide hydrolase EPHX1, prostaglandin reductase PTGR1), conjugation of drugs (glutathione S-transferases GSTM3, GSTP1, MGST1), and drug transport (ABCC1) showed very little change in abundance. From the many proteins associated with drug detoxification, there was one protein with modest changes, microsomal glutathione S-transferase MGST3 (St = 1.33), and one protein that was included in the 121-OxS set, multidrug resistance-associated protein 4, ABCC4 (St, Sn = 0.50).
Finally, it has been suggested that NRF2 may regulate glutamine metabolism via glutamine synthase (GLS).79 We monitored isoforms 1 and 3 of GLS, which showed only small changes in abundance (St = 0.93, 0.83). GLS is also part of a group of proteins termed the “proline regulatory axis” (PRA, Figure 7C) that are intimately connected to redox regulation, glutamine/proline metabolism, the urea cycle, the TCA cycle, and collagen and which have been proposed to be intimately involved in metabolic processes connected to cancer.80 The PRA represents another pathway in which levels of NADH/NADPH may determine response and subcellular distribution of proteins.80 We saw strong changes in abundance and subcellular distribution for pyrroline-5-carboxylate reductase (PYCR1), delta-1-pyrroline-5-carboxylate synthase (ALDH18A1), and collagen (COL1A1) that led to the inclusion of these proteins in the 121-OxS set (Figure 7C). GLS is also involved in the cell cycle81 and we have previously identified characteristic but different changes for proteins of the PRA associated with cell cycle arrest at the origin activation checkpoint for DNA replication.21 We therefore suggest that the apparent effects of NRF2 on glutamine metabolism may be indirect consequences of changes associated with the cell cycle or involvement of the proline regulatory axis, which has connections to oxidative stress, ROS signaling, p53 and, in the present context, antiproliferative effects.80
The previously described results would suggest that metabolic and antioxidant activities controlled by NRF2 are not a major contributor to the response of IMR90 cells that are preconditioned and then challenged with TBP. On the other hand, NRF2 seems to be active at antioxidant response elements (ARE) since among the transcription factors and accessory proteins controlled by NRF231 we detected a strong increase in the abundance of the CCAAT/enhancer-binding protein beta CEBPB (St, Sn = 2.23) and moderate increases for an accessory protein involved in NRF2 transcription MAFG (St = 1.48, but with only five SILAC ratio counts). This suggests that there may be modularity in the transcriptional activity of NRF2. This is also indicated by our results for proteins involved in heme and iron metabolism that are controlled by NRF2. Both subunits of ferritin FTL, FTH1 (St = 1.72, 1.84) and heme oxygenase HMOX1 (St = 7.11) showed strong increases in abundance, but biliverdin reductase (BLVRA) and flavin reductase NADPH (BLVRB) did not (St = 1.03, 1.00).
It may be that the features we observe are specific to TBP preconditioning and treatment and not necessarily identical to cellular response to other ROS since there is evidence for differences in cellular response to TBP and H2O282,83 and we also saw only moderate changes in catalase CAT (St = 0.89) and superoxide dismutases SOD1 and SOD2 (St = 1.34, 0.77). The present experiments suggest that NRF2 is intimately involved in the response to TBP, but via processes largely outside its “classic” metabolic, drug detoxification and antioxidant activities. Proteins containing heme/iron as cofactors are important in the response to TBP (see below), and it has recently been reported that NRF2 can act as a transcription factor for expression of hematopoietic proteins involved in heme metabolism and iron homeostasis (see the Discussion).84
3.6. Proteins Associated with Subcellular Localization and Transport
The 121-OxS set included a number of proteins associated with various aspects of subcellular protein localization and transport. We identified 448 proteins in the GO BP database with annotation to nuclear import/export/localization/maintenance or their regulation, of which 160 were quantified in the present study. There were 10 proteins with indications of possible changes in nuclear transport, of which five satisfied the more stringent conditions for inclusion in the 121-OxS set and were used in the network searches (Table 3). The GO terms suggest these proteins may be involved in different aspects of nucleocytoplasmic trafficking. For example, collagen COL1A1 is annotated to “protein localization to nucleus” (GO:0034504) and “regulation of canonical Wnt receptor signaling pathway” (GO:0060828), whereas karyopherin KPNA2 is annotated to “NLS-bearing substrate import into nucleus” (GO:0006607). Although the proteins in Table 3 are involved in many cellular processes (Supplementary Table S1 in the Supporting Information), only CDKN1A currently has annotation to processes related to oxidative stress (GO:2000377, “regulation of reactive oxygen species metabolic process”). Other proteins are best known for very different processes, for example, the involvement of HSPA9 in protein targeting to mitochondria.85-87
Table 3. Proteins Involved in Nuclear Import/Export/Localization/Maintenance or its Regulationa.
| Gene Name | Cytoplasm | Nucleus | Total | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Sc | SigB score | Ratio Count | Sn | SigB score | Ratio Count | St | SigB score | Ratio Count | |
| CDKN1A | 1.90 | 1.02E-03 | 3 | 2.07 | 1.40E-04 | 5 | |||
| COL1A1 | 0.51 | 9.26E-06 | 62 | 0.63 | 4.03E-02 | 50 | 0.57 | 1.68E-03 | 114 |
| G3BP2 | 0.86 | 4.97E-01 | 8 | 1.45 | 1.22E-02 | 48 | 1.44 | 3.06E-03 | 53 |
| HSPA9 | 1.01 | 7.31E-01 | 181 | 1.64 | 5.72E-05 | 476 | 1.58 | 2.27E-05 | 664 |
| JUP | 0.23 | 6.36E-08 | 5 | 0.25 | 3.64E-11 | 6 | |||
| KPNA2 | 0.53 | 1.18E-05 | 11 | 0.69 | 1.01E-01 | 62 | 0.67 | 2.90E-02 | 73 |
| RANBP17 | 0.18 | 7.10E-19 | 3 | ||||||
| SET | 1.65 | 6.31E-05 | 57 | 1.22 | 3.61E-01 | 11 | 1.55 | 4.03E-04 | 70 |
| TMEM173 | 0.54 | 2.78E-02 | 3 | 0.54 | 3.83E-03 | 3 | |||
| UBR5 | 1.78 | 1.36E-02 | 15 | 1.78 | 5.47E-04 | 17 | |||
Proteins with SigB < 0.005 and ≥3 ratio counts for at least one of Sn, Sc, or St. Bold gene names satisfied the more stringent conditions for selection of the 121-OxS set.
We identified 1175 proteins annotated to “vesicle-mediated transport” (GO:0016192) and 503 proteins annotated to “cytoplasmic vesicle” (GO:0031410) in the GO database. These included a significant number of common proteins. We therefore looked at the set of 1427 proteins that were annotated to one or both of these terms, of which 344 were quantified in the present experiments and 100 had annotations to the nucleus. We quantified 251 of these proteins in the nucleus and 70 were annotated to the nucleus. As with other subcellular organelles (Figure 3, Supplementary Figure S2 in the Supporting Information), a large majority of these proteins showed little or no change in their fraction in the nucleus and there was no apparent difference between proteins with/without annotation to the nucleus (Figure 8). That is, the data are consistent with trafficking of a specific subset of these proteins. Further evidence of selectivity in nucleocytoplasmic trafficking of these types of proteins is provided by the observation that 29 Ras-related RAB proteins were quantified in our experiments and 14 were annotated to vesicle mediated transport, but only 3 showed evidence of nucleocytoplasmic trafficking following TBP treatment. However, the 121-OxS set included 28 such proteins, of which 27 were quantified in the nucleus and 8 are presently annotated to the nucleus. Eleven of these proteins showed substantial changes in the fraction of the protein in the nucleus (|log2(Sn/St)| = |log2(fs/fu)| > 0.5, Figure 8), including RAB2A, RAB5A, and RAB5B. This suggests that vesicle trafficking involving the nucleus is an appreciable part of the response to oxidative stress, and it is striking that seven of these proteins are involved in iron metabolism (ATP6V0A1, ATP6V0D1, ATP6V1C1, ATP6V1H; FTL, FTH1, HMOX1). A further three proteins involved with iron/heme metabolism are currently annotated to the “vesicular fraction” (PGRMC1, POR, PTGS1, see below). It is also noteworthy that many of these 28 proteins were only modestly connected by STRING (Figure 6A), possibly because such subcellular trafficking is not well-represented in the STRING database (see the Discussion).
Figure 8.

Analysis of proteins annotated to vesicular trafficking. Left: log2(Sn/St) = log2(fs/fu) as a function of the average number of ratio counts over the nucleus and total data sets for proteins annotated to “vesicle-mediated transport” or to “cytoplasmic vesicle”. Proteins with (red, 70 proteins) or without (blue, 181 proteins) annotation to nucleus are indicated. Right: number of proteins versus log2(fs/fu). Table: Data for 28 proteins of the 121-OxS set.
3.7. Indications of Extensive Involvement of Iron/Heme Metabolism and Mitochondria
Proteins that involve iron homeostasis or heme/iron as a cofactor are substantially enriched in the 121-OxS set. In the full MS data set, 93 of the 3124 quantified proteins involve iron or heme in the GO biological process or GO molecular function terms and the 121-OxS set includes 20 of these proteins. Of these 20 proteins, 19 were quantified in the nucleus, 13 were among the most significant changes in Sn, and 6 were among the most significant changes for St or Sn/Sc. Only 3 of these 19 proteins are currently annotated to the nucleus (ATP6V0A1, HMOX1, PGRMC1) and although these three proteins participate in a multitude of biological processes (Supplementary Table S2 in the Supporting Information), their functions in the nucleus are poorly defined. The proteins not presently annotated to the nucleus include 10 subunits of V-type proton ATPase (ATP6) and both ferritin subunits (FTL, FTH1), which are known to play important roles in iron homeostasis88-90 but whose nuclear role(s) are ambiguous. Also included are three oxidoreductases (CYB5B, CYB5R3, POR) that are or involve heme proteins and the succinate dehydrogenase [ubiquinone] iron–sulfur subunit (SDHB) that relies on an iron–sulfur cluster for its activity. The heme protein prostaglandin G/H synthase 1 (PTGS1) was not quantified in the nucleus but showed significant decrease in abundance in the cytoplasmic compartment. Overall, the present data indicate that changes in both abundance and subcellular location of a specific set of proteins related to iron/heme metabolism are an important contributor to the cellular response to oxidative stress. Somewhat surprisingly, only 4 of the 20 proteins are currently annotated to stress responses, only 2 (HMOX1, PTSG1) are annotated to respond to oxidative stress, and very few of the 19 proteins we quantified in the nucleus seem to have annotated functional roles there (see the Discussion).
A similar situation appears to exist for mitochondrial proteins. The 121-OxS set includes 41 proteins annotated to mitochondria, of which 39 were quantified in the nucleus, 25 corresponded to most significant changes for Sn, 2 (CYB5B, ETHE1) corresponded to most significant change for Sc, and 12 corresponded to most significant changes for St and/or Sn/Sc. Only 7 of these proteins (ATP5A1, ETHE1, HSPA1A, HSPA9, NME1, POLDIP2, PTRF) are currently annotated to the nucleus. Polymerase delta-interacting protein 2 (POLDIP2, mitochondrial nucleoid91) and polymerase I and transcript release factor (PTRF, transcription from RNA polymerase I promoter92) potentially have similar roles in both locations and both are characterized by moderate increases in total abundance plus C → N redistribution. Of the other five proteins, ATP5A1, HSPA1A (colocalization with the COP9 signalosome69), NME1 (positive regulation of DNA binding93), and HSPA9 (poly(A) RNA binding;94,95 protein export from nucleus) presently have weak indications for possible nuclear functions in their GO biological process annotations. There seems to be little information on nuclear roles for the other 32 mitochondrial proteins detected in the nucleus, which include 27 proteins annotated to the mitochondrial matrix/inner membrane: 10 subunits of ATP synthase (ATP5), 2 proteins of the TCA cycle (SDHA, SDHB), 2 proteins of the proline regulatory axis (ALDH18A1, PYCR1), mitochondrial peptidases (AFG3L2, LONP1, PITRM1, PMPCB), other proteins involved in oxidation–reduction processes (CYB5B, CYB5R3, GPD2, HADHA, HCCS, POR, SQRDL), as well as LETM1 (a mitochondrial Ca2+ and/or K+ transporter96) and PTCD1. There were only a few proteins with clearly defined functions outside mitochondria (ATP6V1A, ATP6V1E1, CCT7, ECH1, HK1, HSPA4, POR, TCIRG1). Seven of the proteins annotated to mitochondria and detected in the nucleus were common to the set of iron/heme proteins previously described (ATP6V1A, ATP6V1E1, CYB5B, CYB5R3, POR, SDHB, TCIRG1). It is also striking that nine of the proteins in the 121-OxS set annotated to mitochondria (ATP5A1, ATP5B, ATP6V1A, ATP6V1E1, CCT7, ECH1, HSPA1A, HSPA4, HSPA9) are annotated to the extracellular vesicular exosome and one (TCIRG1) to the phagocytic vesicle membrane. Only four of these mitochondrial proteins are currently annotated to stress responses (HSPA1A, HSPA4, LONP1, PYCR1). Although further confirmatory experiments are clearly necessary, the observation that 39 mitochondrial proteins (of the 371 quantified in the nucleus) show such changes suggests that subcellular redistribution of specific mitochondrial proteins is also an important characteristic of response to oxidative stress.
4. DISCUSSION
The proteomics subcellular spatial razor offers a powerful framework to look at dynamic changes in protein distribution over defined locations. Our previous results on cell cycle arrest in IMR90 cells21,34 and on cellular response to estradiol in breast cancer cells22 as well as fluorescence-based results on genome-wide spatial response to hypoxia in yeast19 emphasize that the subcellular distribution of proteins is highly dynamic and context dependent. The present experiments confirm dynamic redistribution of numerous proteins in the context of oxidative stress and suggest that many aspects of cellular response to oxidative stress are still incompletely understood. Response to oxidative stress and ROS signaling is now known to have many ramifications in a wide variety of cellular processes and we have primarily viewed these processes from a nuclear-defined perspective. In this context, we discuss four topics in the following. (1) The general properties of the proteomics subcellular spatial razor, especially the selectivity of the compartmentalized changes in protein abundance. (2) An overview of proteins that are known to be involved processes such as cell cycle control, autophagy/mitophagy, and signaling systems. This overview is not and is not meant to be exhaustive. We anticipate that identifying those proteins that are most prominent in the nuclear response to TBP-induced oxidative stress (Table 2, Supplementary Tables S2 and S3 in the Supporting Information) will be useful to enthusiasts of these topics. (3) A more detailed analysis of two features that seem to be related and to be more novel in current understanding of response to oxidative stress: the involvement of proteins involved in heme/iron metabolism or with heme/iron as cofactors; and, links to nuclear hormone metabolism/receptors that are involved in cancer. (4) Finally, we briefly address the need to begin constructing models of cellular function that include functional systems spatially dispersed over different subcellular locations and dynamic changes in the subcellular spatial dispersion of proteins.
4.1. Properties of the Subcellular Spatial Razor
A striking general result of the present experiments is that both with and without TBP treatment large numbers of proteins are detected in the nucleus that are annotated by GO to other subcellular locations. However, in all of the subcellular locations tested (Figure 3, Supplementary Figure S2 in the Supporting Information) only a small subset of the proteins annotated to any given non-nuclear location respond to oxidative stress via changes in compartmental abundance. These changes in compartmental abundance are achieved by a combination of changes in total cellular abundance and in subcellular distribution, with redistribution at least equally important to changes in total abundance. The strong internal consistency of the results indicates they do not reflect artifacts of the nuclear enrichment and is consistent with results gradually being obtained and collated by large numbers of independent experiments by others, for example, that 36% of all human proteins in the GO database are annotated to multiple locations. A second line of evidence that indicates reliability is provided by our previous nuclear spatial razor experiments on IMR90 cells in the context of cell cycle arrest induced by depletion of the CDC7 kinase21 in which nuclear enrichment was achieved with the same protocol. Only 14 proteins are common to the 121-OxS set and the set of 124 most significant proteins detected following CDC7-depletion. That is, the changes in abundance/distribution detected with the spatial razor show high discrimination between different cellular stimulations. The TCA cycle, glycolysis, and the proline regulatory axis provide instructive examples. For glycolysis, the strong increase in HK1 abundance in the nucleus was observed only for oxidative stress but not for cell cycle arrest.21 For the proline regulatory axis (Figure 7C), PYCR1 and ALDH18A1 were in the 121-OxS set, but GLS, GLUD1, and OAT showed strong changes for CDC7-depletion-induced cell cycle arrest,21 and collagen (COL1A1) was substantially decreased/increased respectively. For the TCA cycle, with oxidative stress we detected changes in SDHA and SDHB, whereas for CDC7-depletion-induced cell cycle arrest, changes in ACO2, DLD, FH, IDH2, MDH2, and SUCLG1 were detected in the nucleus.21 Given that hypoxia also constitutes a form of oxidative stress, it is notable that a genome-wide scan of subcellular protein distribution in yeast cells in response to hypoxia19 also detected redistribution of SDHA. All three of these functional processes further corroborate that selective changes characteristic of the cellular stimulation are detected with the spatial razor. A third line of evidence for the selectivity/reliability of the subcellular spatial razor as applied to the nucleus is provided by direct comparison of the proteome of mitochondria and nuclei in MCF7 breast cancer cells, in which nuclei were enriched with similar protocols. We have previously shown that in MCF7 cells there are roughly 1000 proteins partitioned to both subcellular locations and their partitioning is highly reproducible.23 Stimulation with estrogen results in major nucleocytoplasmic redistribution of proteins in MCF7 cells, including over 100 proteins detected in mitochondria.22 The subcellular redistribution was in fact far more prominent than changes in total abundance, which led us to hypothesize that strong perturbation of subcellular spatial control may be a characteristic of cancer cells.22 We do not see such massive nucleocytoplasmic redistribution in IMR90 cells under oxidative stress or cell cycle arrest, but it is interesting that estrogen receptor alpha (ESR1) is prominent in the 240-OxS network (Figure 6A) and we did detect changes for proteins with direct connections to a progesterone receptor in the 121-OxS set (see below).
Overall, although there is probably still room for technical improvements in the proteomics experimental procedures22 (see also the Supplementary Text in the Supporting Information), there is now strong evidence that the nuclear spatial razor provides invaluable information on the role of nucleocytoplasmic subcellular redistribution in cellular function. An analogous approach should be applicable to other subcellular organelles such as mitochondria.
4.2. Overview of Cell Cycle Control, Autophagy/Mitophagy, Signaling Systems, and Mitochondrial Response
Cell Cycle Control
Given the substantial evidence for connections between oxidative status and the cell cycle,97,98 we were initially surprised to find only a few common proteins between our oxidative stress and cell cycle arrest sets of most strongly changed proteins in IMR90 cells. Deeper, joint analysis of the two data sets with inclusion of further proteins that show pronounced similarities or differences between the two responses indicates that there are a variety of nuclear processes, including DNA replication, that change in similar ways with both oxidative stress and cell cycle arrest. However, these changes are achieved by subtle variations in the identity, total abundance, and redistribution of multiple proteins involved in the process. This joint analysis will be published elsewhere. Here we give an example for a possibly critical protein, polymerase delta-interacting protein 2 (POLDIP2), whose homozygous deletion is mostly lethal in mice.99
POLDIP2 has well-studied functional roles involving binding to DNA polymerases and monoubiquitinated PCNA in the nucleus that seem to involve mechanisms for bypass of lesions such as 8-oxo-7,8-dihydroguanine during DNA replication.100-102 It has also been reported to be involved in mitotic spindles and chromatin segregation.103 Very recently it has been shown that POLDIP2 is involved in the spliceosome and is required for alternative splicing of MDM2 in response to UV irradiation, possibly as a consequence of ROS signaling.104 This could provide connections to p53,105 p53-induced transcription of HMOX1,106 and heme interactions with p53.107 Plasma membrane interactions with the homophilic cell–cell adhesion receptor CEACAM1 that result in nuclear import of POLDIP2 in coordination with quiescence/proliferation/cell cycle have also been studied.108 At the same time, POLDIP2 has other roles in interactions with the ROS-generating protein NADPH oxidase 4 (NOX4) in focal adhesions and stress fibers that are involved in ROS signaling, extracellular matrix composition, and vascular structure and function in mice, apparently by reductions in the generation of H2O2 and increased collagen secretion.99,109
We observed a substantial increase in nuclear abundance for POLDIP2 (Sn = 1.79) that is achieved by a combination of increased total abundance and C → N redistribution (St = 1.67, log2(Sn/Sc) = 1.18) and results in a moderate decrease in cytoplasmic abundance (Sc = 0.79). In our joint analysis with CDC7-depletion-induced cell cycle arrest, the nexus to DNA replication is POLD, PCNA, and the replication factor C complex, all of which are included in the 240-OxS set (Figure 6A, Supplementary Table S3 in the Supporting Information). Although there are ambiguities about the relative amounts of POLDIP2 in different subcellular locations,108 POLDIP2 has also been reported to have a mitochondrial targeting sequence, to be primarily in the mitochondria of some cell types,110,111 and to associate with TFAM and LONP1 in the mitochondrial nucleoid.110 Potentially POLDIP2 may be a link between DNA metabolism in the nucleus and mitochondria that is connected to ROS signaling systems. Interestingly, the POLDIP2 gene is part of a complex sense-antisense architecture that has been associated with ERBB2 and breast cancer.112
NRF2 and FOXO Signaling Systems
NRF2-related signaling is crucial to response to oxidative stress9,31,113 and is clearly engaged in our experiments (Figure 1). The influence of NRF2 on glycolysis, the TCA cycle, NADPH generation, and antioxidant systems seems not to be a major response in our experiments, but NRF2 is clearly active at antioxidant response elements (upregulation of CEBPB, MAGF) and heme/iron proteins under NRF2 transcriptional control (HMOX1, FTL, FTH) are among the strongest changes in total protein abundance. Connections from NRF2 signaling to p53 and cell cycle processes are thought to involve FOXO transcription factors.36,114,115 FOXO3 has recently been shown to be intimately involved in mitochondrial ROS signaling and apoptotic pathways.116 We quantified 296 proteins annotated to apoptosis and 12 were included in the 121-OxS set (Table 2). However, these proteins were roughly equally divided among antiapoptosis (SERPINE1, HSPA1A, HMOX1, HSPA9, POR), apoptosis (LGALS1, PSMD6, PSMD5, NME1, PSMD11), or both (RPS27A, SQSMT1). A similar mixed pattern between positive or negative regulation of apoptosis was found and there were no clear patterns of changes in total or compartmental abundances that differentiated between apoptosis/antiapoptosis. We also monitored 28 proteins annotated to caspase activity (GO:0004197) or regulation of cysteine-type endopeptidase activity involved in apoptotic process (GO:0043281), of which only thrombospondin-1 (THBS1) was included in the 121-OxS set. Overall, all of these proteins have numerous other activities (Supplementary Tables S1 and S3 in the Supporting Information) and the data are consistent with a modular response to TBP that does not include FOXO3-mediated connections to apoptosis. This is consistent with no perceptible increase in dead cells and only very small changes in Annexin A5 (St = 1.09). Our data suggested modularity in the NRF2-related response to TBP and other interactions involving NRF2, especially transcription of hematopoietic proteins and interaction with SQSMT1 (see below), may be crucial.
Autophagy and Sequestosome-1 (p62/SQSMT1)
Our experiments quantified 10 proteins that are currently annotated by GO to autophagy, including PARK7, FIS1, NPC1, and GABARAPL2 (Supplementary Table S1 in the Supporting Information). The only protein with a GO annotation to autophagy in the 121-OxS set is p62/SQSMT1, which has been reported to be important in the autophagy response to oxidative damage117 and in huntingtin-induced cell death.118 Other studies suggest that SQSMT1, together with Parkin, is involved in clustering of mitochondria in the perinuclear region119 but not in mitophagy.120 This is a protein for which apparently constitutive, rapid, nucleocytoplasmic trafficking via NLS import/export sequences under phosphorylation control has been established.121 A direct connection to oxidative stress is apparent in the binding of SQSMT1 to KEAP1 with concomitant release of NRF2 and creation of a feedback loop by transcription of SQSMT1.122,123 Under stress, SQSMT1 has been shown to participate in the formation of protein aggregates in the perinuclear region that are degraded by selective autophagy.124,125 Such protein aggregates do not seem adequate to explain the dynamic nucleocytoplasmic distributions that we observe. For example, we detected 371 mitochondrial proteins in the nucleus in unstimulated cells, but only 41 are included in the 121-OxS set. There are both increases and decreases in nuclear abundance for the 39 mitochondrial proteins in the 121-OxS set quantified in both cellular compartments and many of these proteins show redistribution between the nuclear and cytoplasmic compartments with little or no change in total abundance (Figure 3, Supplementary Table S2 in the Supporting Information). We suspect that in addition to its roles in aggregation of damaged proteins SQSMT1 might have other roles in nucleocytoplasmic trafficking of (ubiquitinated) proteins, for example, of PCNA, which binds both POLDIP2 and Flap endonuclease 1 (FEN1).126 Like many proteins associated with autophagy,127 SQSMT1 is involved in other processes (Table 2) and in our experiments it seems to be more directly associated with changes related to cancer and interactions with mTOR in lysosomes128 (see below). It may also be noted that clustering of mitochondria in the perinuclear region is not unique to autophagy since such clustering has been reported to produce an oxidant rich nuclear domain that is required for hypoxia-induced transcription.129 Furthermore, it has been reported that some conditions that favor mitophagy result in movement of mitochondria toward lysosomes.130
The 121-OxS set does include one further protein that has very recently been associated with autophagy (PGRMC1, see below), but in general our experiments seem to show limited direct connection to autophagy/mitophagy, with stronger indications for other processes (see below). As has been noted in the context of long-lived neuronal cells,131 the urgency attached to identifying disease-related processes may be emphasizing research into pathological extremes and obscuring the need to elucidate basic, underlying mechanisms of interorganelle trafficking and degradatory turnover of proteins in normal cellular function.
Mitochondria and the Retrograde Response
Because our experiments concentrate on the nucleus, they only indirectly bear on mitochondrial processes. However, the observation that of the 121 proteins showing the most significant changes, 39 proteins detected in the nucleus are annotated to mitochondria is consistent with extensive retrograde signaling from mitochondria to the nucleus in response to TBP-induced oxidative stress. Retrograde mitochondria to nucleus signaling is vital in many cellular processes and contexts.132
Detailed interpretation of our results is complicated by the fact that of these 39 mitochondrial proteins only a handful has known functions in the nucleus. For example, there has recently been considerable interest in KIAA1967 (also known as deleted-in-breast-cancer-1, DBC1) in the context of cancer mechanisms. While there is still discussion of whether KIAA1967 is really deleted/involved in cancer,133 clear evidence has been obtained for diverse functional roles in the nucleus, including as a regulator of histone deacetylase chromatin remodeling enzymes and transcription factors134 as well as in alternative mRNA splicing.135 These include direct interactions with several nuclear receptors including estrogen receptors α and β.136,137 Interestingly, KIAA1967 (DBC1) has been detected in mitochondria.138 We observed substantial changes for KIAA1967 (moderate change in Sn and St ≈ 1.25, Sc = 0.5, C → N redistribution with log2(Sn/Sc) = 1.3) in response to oxidative stress. It is tempting to speculate that KIAA1967 might also have mitochondrial functions related to oxidative stress, perhaps in connection with the metabolic changes associated with the presence of estrogen receptors and other nuclear hormone receptors in mitochondria.139
Conversely, we detected proteins in the nucleus that have defined function(s) in mitochondria but seem not to have established functions in the nucleus. For example, Lon protease (LONP1) is annotated to the mitochondrial nucleoid and has well-studied roles in proteolytic clearance of damaged proteins. Under acute stress within the mitochondrial matrix, Lon works as a protector protein in the degradation of oxidized proteins, while under chronic conditions the level of Lon declines and this function is lost.140 Lon can also act as a chaperone,141 attaches to promoter sequences in mDNA and to genomically important proteins such as mitochondrial transcription factors,142,143 and has been reported to bind to POLDIP2 in the mitochondrial nucleoid.110 We detected appreciable nuclear changes for LONP1 (Sc = 1.17, Sn = 0.55, St = 0.67, N → C redistribution with log2(Sn/Sc) = −1.08) that suggest that LONP1 might also have nuclear functions that are modulated by oxidative stress.
We detected many other mitochondrial proteins in the nucleus with tantalizing changes in response to oxidative stress. These are suggestive of extensive retrograde mitochondrial to nucleus signaling that might involve variable, dynamic partitioning of proteins between the nucleus and mitochondria. A prototype for this type of retrograde control is C. elegans ATFS-1 (activating transcription factor associated with stress-1), which senses mitochondrial stress and communicates with the nucleus during the mitochondrial unfolded protein response. Constitutively synthesized ATFS-1 is normally imported to mitochondria and degraded, but under stress changes in nuclear/mitochondrial distribution are caused by reduced mitochondrial import.144 In this context, we note that the 121-OxS set contains several proteins associated with mitochondrial import processes (HSPA9, PITRM1, PMPCB). NRF2 complexed with KEAP1 may be at least in part a similar retrograde system since the complex has been reported to tether to the mitochondrial outer membrane via interactions with PGAM5, placing it in proximity to sources of ROS from the respiratory chain.145
4.3. Heme/Iron Metabolism
The previous examples corroborate that we detect many proteins with established connections to oxidative stress, signaling systems and mitochondria. The information on dynamic spatial distribution is largely new and for many of these proteins clarification of possible nuclear functions is now desirable. In fact, our results suggest that many other subcellular locations besides mitochondria also have protein signaling to the nucleus under oxidative stress and may involve more novel systems. Iron metabolism is clearly associated with oxidative stress and has also been implicated in many diseases such as type-2 diabetes and obesity146 and Alzheimer’s and Parkinson’s diseases.147 It has long been known that iron metabolism is intimately connected to cancer,148-150 to the extent that iron chelators have been used in cancer treatment.151 Subcellular trafficking of proteins involved in iron homeostasis is clearly established for systems such as the transferrin cycle.152,153 In our experiments, 93/3569 proteins are currently annotated to heme/iron and 20 of these were included among the 121 proteins with the most significant changes. A striking feature is that while these proteins are annotated to many subcellular locations (10 to plasma membrane, 7 to mitochondria, 9 to lysosome, 5 to endoplasmic reticulum, 1 to Golgi apparatus, and 4 to endosome) only 4 are currently annotated to the nucleus. In response to TBP treatment cytochrome-b5 type B (CYB5B) and NADH-cytochrome-b5 reductase 3 (CYBR3), which are part of the mitochondrial outer membrane, both show moderate increases in abundance in the cytoplasmic compartment (Sc = 1.58/1.18, respectively) that is achieved by strong N → C redistribution that overweighs an appreciable decrease in total abundance (St ≈ 0.65); that is, both show substantially reduced abundance in the nucleus (Sn = 0.43/0.61 respectively). Ferritin heavy/light chains (FTL and FTH1) as well as hemoxygenase (HMOX1) show two- to seven-fold increases in total abundance that seem to be largely equilibrated over the two subcellular compartments within the 4 h following the TBP challenge. V-type ATP synthase, for which we observed increased abundance (St ≈ 1.8) and moderate C → N redistribution for many subunits (Figure 4), is involved in iron homeostasis by working to provide acidic conditions for iron metabolism within lysosomes and endosomes.154 PGRMC1 is a heme-containing protein with connections to both sterol receptors and autophagy (see below). The other outstanding feature of the group of iron/heme related proteins is that many are involved in vesicle-mediated subcellular trafficking or present in the extracellular vesicular exosome. In short, the present experiments provide strong evidence of an important role of trafficking of iron/heme proteins in the response to oxidative stress. More detailed interpretation of our results is complicated by the fact that many aspects of the subcellular trafficking of iron/heme and of proteins containing these cofactors remain ambiguous in both yeast155 and higher eukaryotes.156
At the same time evidence is accumulating that mitochondria are highly dynamic organelles that export proteins by vesicular trafficking to at least peroxisomes and lysosomes in what may be constitutive processes.157,158 Under acute oxidative stress mitochondria localize near the nucleus and fragment, leading to the damage-control process of mitoptosis in which many mitochondrial proteins appear in the vesicular exosome.159,160 These include a variety of proteins for which we detected changes in nuclear abundance. Recently evidence has been presented that in aging cells mitochondrial cristae vesicularize and mitochondria may bud vesicles containing ATP synthase.161 One of the mitochondrial proteins in the 121-OxS set (LETM1) is involved in the formation of mitochondrial cristae. Similarly, in mammalian cells it has recently been reported that reduced expression of subunits thought to be essential for oligomerization of ATP synthase dimers results in fission and altered organization of mitochondria.162 This is particularly intriguing because it has recently been proposed that dimers of ATP synthase distinct from those in cristae constitute the mitochondrial permeability transition pore,163,164 and TBP has been reported to lead to mitochondrial depolarization.165,166 This might provide mechanisms related to our observation of changes in nuclear abundance of many ATP synthase subunits. The occurrence of cyclic ROS bursts in mitochondria167-169 suggests that there may be mechanisms for such processes in normal physiology and not only under stress. Similarly, evidence has recently been obtained that under oxidative stress mitochondrial proteins can be transferred to lysosomes by vesicle-mediated transport that is independent of mitophagy.170
In short, on the basis of the present experiments and a body of suggestive literature evidence, the simplest interpretation of this part of our data would seem to be that vesicular trafficking occurs more widely in higher eukaryotic cells than is presently appreciated, which would be consistent with GO annotation of 1427 proteins to “vesicle-mediated transport” or to “cytoplasmic vesicle”. The present data suggest that this involves the nucleus and mitochondria in addition to other subcellular organelles and that heme/iron proteins are included in such vesicular trafficking. Whatever the mechanism(s) for trafficking of iron/heme proteins, on the basis of the present experiments we suggest three possibilities for further investigation of the possible role of proteins involved in iron/heme metabolism in subcellular trafficking in response to oxidative stress.
First, subcellular trafficking of the membrane-associated progesterone receptor (MAPR) family (PGRMC1, PGRMC2, neudesin, neuferricin) of cytochrome-b-related, heme-containing proteins171 could be investigated. The prototype of this family is PGRMC1. We observed strong N → C redistribution (log2(Sn/Sc) = −1.17) and no change in total abundance (St = 0.99) that results in strong decrease in nuclear abundance (Sn = 0.54) and moderate increase in cytoplasmic abundance (Sc = 1.22). PGRMC1 has been extensively studied as part of a plasma membrane receptor for progesterone that acts independently of the classical progesterone nuclear hormone receptor, including in neuronal cells.172,173 It has recently been proposed to be part of a protein complex containing the binding site for the somewhat enigmatic sigma-2-recptor that regulates lipid metabolism and hormone signaling and is a validated biomarker for tumor cell proliferation.174 There is substantial evidence of important roles of both the sigma-2-recptor and PGRMC1 in cancer.174 While much of the investigation of PGRMC1 has concentrated on its roles as a progesterone receptor in the plasma membrane, it has also been detected in the ER and mitochondria,174 the cellular vesicular fraction,175 in the perinuclear region and in the nucleus,176,177 where it has been reported to regulate genes associated with inhibition of apoptosis by progesterone.177,178 Very recently PGRMC1 was shown to promote autophagy179 and evidence has been presented that PGRMC1 may be an adaptor protein for membrane receptors beyond the progesterone receptor.180 There is some evidence that location of PGRMC1 to cytoplasmic vesicles is related to its carcinogenic activities.175,181-183 Finally, interaction of PGRMC1 with plasminogen activator inhibitor 1 RNA-binding protein (PAIRBP1) in nuclear transcriptional functions184 and colocalization with aurora kinase b (AURKB) at chromatin in oocyte maturation processes185 have been reported. Our 121-OxS set includes AURKB and plasminogen activator inhibitor 1 (SERPINE1). Overall, the present results suggest intimate connections between oxidative stress and diverse PGRMC1 functions, and PGRMC1 potentially has redox capacity that might be crucial to its multitude of functions. As a heme-containing protein with multiple known subcellular locations, interactions, and functions, PGRMC1 seems to be an excellent candidate for further elucidation of possible connections between heme protein trafficking and oxidative stress. We also detected that cytochrome b5 type B (CYB5B) and its partner NADH-cytochrome b5 reductase 3 (CYB5R3), which are mitochondrial outer membrane proteins, exhibited pronounced N → C redistribution, and these proteins might also be useful in such studies.
Second, subcellular trafficking of NADPH oxidase 4 (NOX4) might be studied. POLDIP2 and NOX4 can dynamically colocalize to the nucleus in a process that apparently involves H2O2 and produces collagen secretion.109 Although our experiments did not quantify NOX4, we did observe an increase in nuclear POLDIP2 and a reduction in cellular collagen (COL1A1) that may be mediated via the proline regulatory axis (Figure 7C). NOX4 has also been detected in the endoplasmic reticulum,186 specialized plasma membrane subcellular domains such as focal adhesions187 and invadopodia,188,189 as well as mitochondria.190-192 NOX4 generates ROS in diverse subcellular locations including the nucleus,193,194 endoplasmic reticulum195 and mitochondria.190,196 NOX4, and other NOX proteins, are associated with a wide diversity of cellular functions and diseases that include cancer, diabetic kidney disease, cardiovascular disease, Alzheimer’s disease, fibrosis, and atherosclerosis. (For reviews, see refs 15, 16, 197, and 198.) NOX4 is an integral membrane protein with six transmembrane helices containing two heme groups16 and its catalytic function is the transfer of electrons from NADPH across biological membranes to molecular oxygen. At present there seems to be limited information on how NOX4 partitions to such diverse subcellular locations. As an integral membrane protein it is unlikely to reach the nucleus by import/export involving the nuclear pore and it seems likely that vesicular trafficking of some kind is involved.
Third, further assessment of the possibility of vesicle trafficking in iron homeostasis might be provided by further studies of the subcellular trafficking of the V-type ATPases (ATP6). The best studied functional roles for V-type ATPases involve the regulation of the pH of the cytoplasm and acidic organelles,154,199 which protects the cell against defects such as iron misregulation and oxidative stress that appear if pH homeostasis is lost.200 V-type ATPases keep an acidic environment in endosomes and lysosomes that prevents cellular damage caused by interaction of iron with hydrogen peroxide.201 Recently, it has been reported that SQSMT1 interacts with V-type ATPase in lysosomes to activate the rapamycin mTOR pathway involved in cell growth and autophagy,202 senescence and amino acid metabolism.203 V-type ATPases also influence the insulin receptor signaling pathway: insulin is dissociated from its receptor by the acidic environment maintained in endosomes by V-type ATPase.204 We observed this pathway (Table 2). However, our experiments quantified 21 other proteins involved in the insulin receptor pathway that did not show significant changes in total or compartmental abundance, suggesting that we may have primarily detected consequences of changes in the acid–ification/depolarization of endosomes/lysosomes. In this context it is also interesting that endosomes and lysosomes show cyclic bursts of pH that depolarize these subcellular compartments154,201 and might provide normal physiological mechanisms for shedding of V-type ATPases. Various subunits of V-type ATPase are currently annotated by GO to mitochondria, the plasma membrane, endosomes, lysosomes, the Golgi apparatus, vacuoles, the nucleus, and cytoplasmic vesicles. V-type ATPases are known to be involved in vesicle trafficking199,205,206 and ATP6V0C has recently been reported to regulate the metabolism of autophagy proteins involved in age-related neurodegeneration.207 Interestingly, the 121-OxS set also included renin receptor (ATP6AP2, St = 1.59, Sn = 1.71), which is known to interact with V-type ATPases in Wnt/β-catenin signaling and also has (disputed) connections to the renin-angiotensin hormonal system and various other signaling systems.208 Overall, there seem to be numerous indications for involvement of subunits of V-type ATPases in a variety of processes/locations beyond those so far well studied: transfer to the nucleus under oxidative stress adds to the possibilities.
4.4. Conclusions
The present experiments contain extensive evidence that numerous proteins involved in cell cycle control, mitochondrial function, iron metabolism, signaling systems and other functional processes show substantial changes in nucleocytoplasmic compartmental abundance in response to oxidative stress. These changes involve many proteins that are also present in diverse non-nuclear subcellular locations. A protein can only exert a function in the locations where it exists and our results suggest that nucleocytoplasmic redistribution is at least equally important to changes in total protein abundance in achieving the appropriate changes in compartmental abundance. Changes in total protein abundance alone are inadequate to obtain a comprehensive picture of response to TBP. Similar characteristics have been observed for cell cycle arrest at the origin activation checkpoint for DNA replication21 and for response to hypoxia.19 Especially, pronounced nucleocytoplasmic trafficking of proteins has been observed in the response of breast cancer cells to estrogen simulation.22 There seems to be still much to be learned about the mechanisms that govern dynamic partitioning of proteins to different subcellular locations and many proteins for which their nuclear roles need to be investigated. A total of 115 proteins in the 121-OxS set were quantified in the nucleus, but only 45 of these proteins currently have GO annotations to the nucleus. That is, we detected 80 proteins whose possible nuclear functions are presently uncertain. Of the 63 proteins detected as having highly significant changes in Sn, there are 40 that are not currently annotated to the nucleus. These proteins are implicated in many functional processes (Table 2, Supplementary Table S2 in the Supporting Information) and the present results provide many interesting entry points in the context of oxidative stress. The development of heme was an ancient, crucial step in eukaryotic evolution209 and it should not be surprising to find iron/heme intimately connected to response to oxidative stress. Similarly, RAB proteins and kinesins are intimately involved in many kinds of vesicular trafficking210 and our observation of very selective changes for proteins of these types (RAB2A, RAB5A, RAB5B, KIF14, KIF20A) is a strong indication for a role of vesicular trafficking with the nucleus in response to TBP treatment.
More generally, it seems that correlated, dynamic, but very selective changes in the local abundance at diverse subcellular locations of substantial numbers of proteins is critical to cellular function, that we should begin to think of cellular function in terms of higher level processes spatially distributed over different subcellular locations and that we need to construct models of cellular function that explicitly take this into account. So far, apart from cancer cells, the proportion of total proteins that redistribute in response to a specific cellular perturbation seems to be small (2–5%) and the inclusion of spatial location as an explicit factor in models and databases should provide powerful ways to make sense of what may otherwise seem a bewildering array of interactions.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. S. Tudjarova, Dr. C. Mulvey, and Prof. L. R. Brown for valuable help and discussion. This study has been supported by the Wellcome Trust grant 081879/z/06/z (J.G.Z.), King Abdullah Al Saud Foreign Scholarship Program, and the Saudi Ministry of Higher Education (NB).
Footnotes
ASSOCIATED CONTENT
Supporting Information
Supplementary Text with the mathematical model for the subcellular spatial razor. Supplementary Table S1: Master table of MS data. Supplementary Table S2: Summary of SILAC ratios for the 121-OxS set of most significant proteins. Supplementary Table S3: Expanded version of Table 2 showing protein sets for each GO biological process. Supplementary Figure S1: Correlation of Sn, Sc, and St between the three replicates of each sample type. Supplementary Figure S2: Plots of the change in nuclear fraction for proteins with GO annotation to different subcellular organelles. Supplementary Figure S3: Immunofluorescence imaging results for ATP5A1 and ALDH18A1. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- (2).Finkel T, Serrano M, Blasco MA. The common biology of cancer and ageing. Nature. 2007;448:767–774. doi: 10.1038/nature05985. [DOI] [PubMed] [Google Scholar]
- (3).Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- (4).Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- (5).Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011;208:417–420. doi: 10.1084/jem.20110367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ryter SW, Choi AM. Autophagy: An Integral Component of the Mammalian Stress Response. J. Biochem. Pharmacol. Res. 2013;1:176–188. [PMC free article] [PubMed] [Google Scholar]
- (7).Lee J, Giordano S, Zhang JH. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem. J. 2012;441:523–540. doi: 10.1042/BJ20111451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Droge W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- (9).Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signalling. 2012;24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Park J, Lee J, Choi C. Mitochondrial network determines intracellular ROS dynamics and sensitivity to oxidative stress through switching inter-mitochondrial messengers. PLoS One. 2011;6:e23211. doi: 10.1371/journal.pone.0023211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Mailloux RJ, Jin X, Willmore WG. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2013;2:123–139. doi: 10.1016/j.redox.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gough DR, Cotter TG. Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell Death Dis. 2011;2:e213. doi: 10.1038/cddis.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Babior BM, Lambeth JD, Nauseef W. The neutrophil NADPH oxidase. Arch. Biochem. Biophys. 2002;397:342–344. doi: 10.1006/abbi.2001.2642. [DOI] [PubMed] [Google Scholar]
- (14).Chen K, Kirber MT, Xiao H, Yang Y, Keaney JF., Jr. Regulation of ROS signal transduction by NADPH oxidase 4 localization. J. Cell Biol. 2008;181:1129–1139. doi: 10.1083/jcb.200709049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Block K, Gorin Y. Aiding and abetting roles of NOX oxidases in cellular transformation. Nat. Rev. Cancer. 2012;12:627–637. doi: 10.1038/nrc3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- (17).Go YM, Jones DP. Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta. 2008;1780:1273–1290. doi: 10.1016/j.bbagen.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Hansen JM, Go YM, Jones DP. Nuclear and mitochondrial compartmentation of oxidative stress and redox signaling. Ann. Rev. Pharmacol. Toxicol. 2006;46:215–234. doi: 10.1146/annurev.pharmtox.46.120604.141122. [DOI] [PubMed] [Google Scholar]
- (19).Henke RM, Dastidar RG, Shah A, Cadinu D, Yao X, Hooda J, Zhang L. Hypoxia elicits broad and systematic changes in protein subcellular localization. Am. J. Physiol.: Cell Physiol. 2011;301:C913–C928. doi: 10.1152/ajpcell.00481.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Jung S, Smith JJ, von Haller PD, Dilworth DJ, Sitko KA, Miller LR, Saleem RA, Goodlett DR, Aitchison JD. Global analysis of condition-specific subcellular protein distribution and abundance. Mol. Cell. Proteomics. 2013;12:1421–1435. doi: 10.1074/mcp.O112.019166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Mulvey CM, Tudzarova S, Crawford M, Williams GH, Stoeber K, Godovac-Zimmermann J. Subcellular proteomics reveals a role for nucleo-cytoplasmic trafficking at the DNA replication origin activation checkpoint. J. Proteome Res. 2013;12:1436–1453. doi: 10.1021/pr3010919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Pinto G, Alhaiek AA, Amadi S, Qattan A, Crawford M, Radulovic M, Godovac-Zimmermann J. Systematic Nucleo-Cytoplasmic Trafficking of Proteins Following Exposure of MCF7 Breast Cancer Cells to Estradiol. J. Proteome Res. 2014;13(2):1112–1127. doi: 10.1021/pr4012359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Qattan AT, Radulovic M, Crawford M, Godovac-Zimmermann J. Spatial distribution of cellular function: the partitioning of proteins between mitochondria and the nucleus in MCF7 breast cancer cells. J. Proteome Res. 2012;11:6080–6101. doi: 10.1021/pr300736v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhu H, Jia Z, Zhang L, Yamamoto M, Misra HP, Trush MA, Li Y. Antioxidants and phase 2 enzymes in macrophages: regulation by Nrf2 signaling and protection against oxidative and electrophilic stress. Exp. Biol. Med. (Maywood, NJ, U. S.) 2008;233:463–474. doi: 10.3181/0711-RM-304. [DOI] [PubMed] [Google Scholar]
- (25).McMahon M, Itoh K, Yamamoto M, Hayes JD. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003;278:21592–21600. doi: 10.1074/jbc.M300931200. [DOI] [PubMed] [Google Scholar]
- (26).Numazawa S, Ishikawa M, Yoshida A, Tanaka S, Yoshida T. Atypical protein kinase C mediates activation of NF-E2-related factor 2 in response to oxidative stress. Am. J. Physiol.: Cell Physiol. 2003;285:C334–C342. doi: 10.1152/ajpcell.00043.2003. [DOI] [PubMed] [Google Scholar]
- (27).McMahon M, Lamont DJ, Beattie KA, Hayes JD. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc. Natl. Acad. Sci. U. S. A. 2010;107:18838–18843. doi: 10.1073/pnas.1007387107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lee SL, Son AR, Ahn J, Song JY. Niclosamide enhances ROS-mediated cell death through c-Jun activation. Biomed. Pharmacother. 2014;68(5):619–624. doi: 10.1016/j.biopha.2014.03.018. [DOI] [PubMed] [Google Scholar]
- (29).Baird L, Dinkova-Kostova AT. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011;85:241–272. doi: 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]
- (30).Kaspar JW, Niture SK, Jaiswal AK. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009;47:1304–1309. doi: 10.1016/j.freeradbiomed.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014;39:199–218. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- (32).Bloom D, Dhakshinamoorthy S, Jaiswal AK. Site-directed mutagenesis of cysteine to serine in the DNA binding region of Nrf2 decreases its capacity to upregulate antioxidant response element-mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase1 gene. Oncogene. 2002;21:2191–2200. doi: 10.1038/sj.onc.1205288. [DOI] [PubMed] [Google Scholar]
- (33).Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Mulvey C, Tudzarova S, Crawford M, Williams GH, Stoeber K, Godovac-Zimmermann J. Quantitative Proteomics Reveals a “Poised Quiescence” Cellular State after Triggering the DNA Replication Origin Activation Checkpoint. J. Proteome Res. 2010;9:5445–5460. doi: 10.1021/pr100678k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ong SE, Mann M. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC) Nat. Protoc. 2006;1:2650–2660. doi: 10.1038/nprot.2006.427. [DOI] [PubMed] [Google Scholar]
- (36).Tudzarova S, Trotter MWB, Wollenschlaeger A, Mulvey C, Godovac-Zimmermann J, Williams GH, Stoeber K. Molecular architecture of the DNA replication origin activation checkpoint. EMBO J. 2010;29:3381–3394. doi: 10.1038/emboj.2010.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Chen JH, Stoeber K, Kingsbury S, Ozanne SE, Williams GH, Hales CN. Loss of proliferative capacity and induction of senescence in oxidatively stressed human fibroblasts. J. Biol. Chem. 2004;279:49439–49446. doi: 10.1074/jbc.M409153200. [DOI] [PubMed] [Google Scholar]
- (38).Walther TC, Mann M. Mass spectrometry-based proteomics in cell biology. J. Cell Biol. 2010;190:491–500. doi: 10.1083/jcb.201004052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- (40).Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- (41).Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- (42).Cox J, Matic I, Hilger M, Nagaraj N, Selbach M, Olsen JV, Mann M. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 2009;4:698–705. doi: 10.1038/nprot.2009.36. [DOI] [PubMed] [Google Scholar]
- (43).Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, Jensen LJ, von Mering C. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinf. 2003;4:2. doi: 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- (47).Huang da W, Sherman BT, Stephens R, Baseler MW, Lane HC, Lempicki RA. DAVID gene ID conversion tool. Bioinformation. 2008;2:428–430. doi: 10.6026/97320630002428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Binns D, Dimmer E, Huntley R, Barrell D, O’Donovan C, Apweiler R. QuickGO: a web-based tool for Gene Ontology searching. Bioinformatics. 2009;25:3045–3046. doi: 10.1093/bioinformatics/btp536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).John S, Weiss JN, Ribalet B. Subcellular localization of hexokinases I and II directs the metabolic fate of glucose. PLoS One. 2011;6:e17674. doi: 10.1371/journal.pone.0017674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol. Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Jungas T, Motta I, Duffieux F, Fanen P, Stoven V, Ojcius DM. Glutathione levels and BAX activation during apoptosis due to oxidative stress in cells expressing wild-type and mutant cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 2002;277:27912–27918. doi: 10.1074/jbc.M110288200. [DOI] [PubMed] [Google Scholar]
- (54).Gill MB, Bockhorst K, Narayana P, Perez-Polo JR. Bax shuttling after neonatal hypoxia-ischemia: hyperoxia effects. J. Neurosci. Res. 2008;86:3584–3604. doi: 10.1002/jnr.21795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).McClung JK, Jupe ER, Liu XT, Dell’Orco RT. Prohibitin: potential role in senescence, development, and tumor suppression. Exp. Gerontol. 1995;30:99–124. doi: 10.1016/0531-5565(94)00069-7. [DOI] [PubMed] [Google Scholar]
- (56).Nuell MJ, Stewart DA, Walker L, Friedman V, Wood CM, Owens GA, Smith JR, Schneider EL, Dellorco R, Lumpkin CK, Danner DB, Mcclung JK. Prohibitin, an Evolutionarily Conserved Intracellular Protein That Blocks DNA-Synthesis in Normal Fibroblasts and Hela-Cells. Mol. Cell. Biol. 1991;11:1372–1381. doi: 10.1128/mcb.11.3.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Roskams AJ, Friedman V, Wood CM, Walker L, Owens GA, Stewart DA, Altus MS, Danner DB, Liu XT, McClung JK. Cell cycle activity and expression of prohibitin mRNA. J. Cell Physiol. 1993;157:289–295. doi: 10.1002/jcp.1041570211. [DOI] [PubMed] [Google Scholar]
- (58).Wang S, Nath N, Adlam M, Chellappan S. Prohibitin, a potential tumor suppressor, interacts with RB and regulates E2F function. Oncogene. 1999;18:3501–3510. doi: 10.1038/sj.onc.1202684. [DOI] [PubMed] [Google Scholar]
- (59).Fusaro G, Dasgupta P, Rastogi S, Joshi B, Chellappan S. Prohibitin induces the transcriptional activity of p53 and is exported from the nucleus upon apoptotic signaling. J. Biol. Chem. 2003;278:47853–47861. doi: 10.1074/jbc.M305171200. [DOI] [PubMed] [Google Scholar]
- (60).Nijtmans LGJ, de Jong L, Sanz MA, Coates PJ, Berden JA, Back JW, Muijsers AO, van der Spek H, Grivell LA. Prohibitins act as a membrane-bound chaperone for the stabilization of mitochondrial proteins. EMBO J. 2000;19:2444–2451. doi: 10.1093/emboj/19.11.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Sharma A, Qadri A. Vi polysaccharide of Salmonella typhi targets the prohibitin family of molecules in intestinal epithelial cells and suppresses early inflammatory responses. Proc. Natl. Acad. Sci. U. S. A. 2004;101:17492–17497. doi: 10.1073/pnas.0407536101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Mishra S, Murphy LC, Nyomba BL, Murphy LJ. Prohibitin: a potential target for new therapeutics. Trends Mol. Med. 2005;11:192–197. doi: 10.1016/j.molmed.2005.02.004. [DOI] [PubMed] [Google Scholar]
- (63).Qattan AT, Mulvey C, Crawford M, Natale DA, Godovac-Zimmermann J. Quantitative organelle proteomics of MCF-7 breast cancer cells reveals multiple subcellular locations for proteins in cellular functional processes. J. Proteome Res. 2010;9:495–508. doi: 10.1021/pr9008332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Moser TL, Kenan DJ, Ashley TA, Roy JA, Goodman MD, Misra UK, Cheek DJ, Pizzo SV. Endothelial cell surface F1-F0 ATP synthase is active in ATP synthesis and is inhibited by Angiostatin. Proc. Natl. Acad. Sci. U. S. A. 2001;98:6656–6661. doi: 10.1073/pnas.131067798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Panfoli I, Ravera S, Bruschi M, Candiano G, Morelli A. Proteomics unravels the exportability of mitochondrial respiratory chains. Expert Rev. Proteomics. 2011;8:231–239. doi: 10.1586/epr.11.1. [DOI] [PubMed] [Google Scholar]
- (66).Ravera S, Panfoli I, Calzia D, Aluigi MG, Bianchini P, Diaspro A, Mancardi G, Morelli A. Evidence for aerobic ATP synthesis in isolated myelin vesicles. Int. J. Biochem. Cell Biol. 2009;41:1581–1591. doi: 10.1016/j.biocel.2009.01.009. [DOI] [PubMed] [Google Scholar]
- (67).Yonally SK, Capaldi RA. The F(1)F(0) ATP synthase and mitochondrial respiratory chain complexes are present on the plasma membrane of an osteosarcoma cell line: An immunocytochemical study. Mitochondrion. 2006;6:305–314. doi: 10.1016/j.mito.2006.10.001. [DOI] [PubMed] [Google Scholar]
- (68).Kohl M, Wiese S, Warscheid B. Cytoscape: software for visualization and analysis of biological networks. Methods Mol. Biol. 2011;696:291–303. doi: 10.1007/978-1-60761-987-1_18. [DOI] [PubMed] [Google Scholar]
- (69).Fang L, Wang X, Yamoah K, Chen PL, Pan ZQ, Huang L. Characterization of the human COP9 signalosome complex using affinity purification and mass spectrometry. J. Proteome Res. 2008;7:4914–4925. doi: 10.1021/pr800574c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Mathupala SP, Rempel A, Pedersen PL. Aberrant glycolytic metabolism of cancer cells: a remarkable coordination of genetic, transcriptional, post-translational, and mutational events that lead to a critical role for type II hexokinase. J. Bioenerg. Biomembr. 1997;29:339–343. doi: 10.1023/a:1022494613613. [DOI] [PubMed] [Google Scholar]
- (71).Neary CL, Pastorino JG. Nucleocytoplasmic shuttling of hexokinase II in a cancer cell. Biochem. Biophys. Res. Commun. 2010;394:1075–1081. doi: 10.1016/j.bbrc.2010.03.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J. Exp Biol. 2003;206:2049–2057. doi: 10.1242/jeb.00241. [DOI] [PubMed] [Google Scholar]
- (73).Kim S, Kim DH, Jung WH, Koo JS. Succinate dehydrogenase expression in breast cancer. SpringerPlus. 2013;2:299. doi: 10.1186/2193-1801-2-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, Skoldberg F, Husebye ES, Eng C, Maher ER. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 2001;69:49–54. doi: 10.1086/321282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Ricketts C, Woodward ER, Killick P, Morris MR, Astuti D, Latif F, Maher ER. Germline SDHB mutations and familial renal cell carcinoma. J. Natl. Cancer Inst. 2008;100:1260–1262. doi: 10.1093/jnci/djn254. [DOI] [PubMed] [Google Scholar]
- (76).Bardella C, Pollard PJ, Tomlinson I. SDH mutations in cancer. Biochim. Biophys. Acta. 2011;1807:1432–1443. doi: 10.1016/j.bbabio.2011.07.003. [DOI] [PubMed] [Google Scholar]
- (77).Briere JJ, Favier J, Gimenez-Roqueplo AP, Rustin P. Tricarboxylic acid cycle dysfunction as a cause of human diseases and tumor formation. Am. J. Physiol.: Cell Physiol. 2006;291:C1114–C1120. doi: 10.1152/ajpcell.00216.2006. [DOI] [PubMed] [Google Scholar]
- (78).Jackson JB. Proton translocation by transhydrogenase. FEBS Lett. 2003;555:176–177. doi: 10.1016/s0014-5793(03)01123-2. [DOI] [PubMed] [Google Scholar]
- (79).Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M, Motohashi H. Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell. 2012;22:66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- (80).Phang JM, Liu W, Hancock C, Christian KJ. The proline regulatory axis and cancer. Front. Oncol. 2012;2:60. doi: 10.3389/fonc.2012.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Colombo SL, Palacios-Callender M, Frakich N, Carcamo S, Kovacs I, Tudzarova S, Moncada S. Molecular basis for the differential use of glucose and glutamine in cell proliferation as revealed by synchronized HeLa cells. Proc. Natl. Acad. Sci. U. S. A. 2011;108:21069–21074. doi: 10.1073/pnas.1117500108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Slamenova D, Kozics K, Hunakova L, Melusova M, Navarova J, Horvathova E. Comparison of biological processes induced in HepG2 cells by tert-butyl hydroperoxide (t-BHP) and hydroperoxide (H2O2): The influence of carvacrol. Mutation Res. 2013;757:15–22. doi: 10.1016/j.mrgentox.2013.03.014. [DOI] [PubMed] [Google Scholar]
- (83).Chen D, Wilkinson CR, Watt S, Penkett CJ, Toone WM, Jones N, Bahler J. Multiple pathways differentially regulate global oxidative stress responses in fission yeast. Mol. Biol. Cell. 2008;19:308–317. doi: 10.1091/mbc.E07-08-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Campbell MR, Karaca M, Adamski KN, Chorley BN, Wang X, Bell DA. Novel hematopoietic target genes in the NRF2-mediated transcriptional pathway. Oxid. Med. Cell. Longevity. 2013;2013:120305. doi: 10.1155/2013/120305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Brunner M, Schneider HC, Lill R, Neupert W. Dissection of protein translocation across the mitochondrial outer and inner membranes. Cold Spring Harbor Symp. Quant. Biol. 1995;60:619–627. doi: 10.1101/sqb.1995.060.01.066. [DOI] [PubMed] [Google Scholar]
- (86).De Mena L, Coto E, Sanchez-Ferrero E, Ribacoba R, Guisasola LM, Salvador C, Blazquez M, Alvarez V. Mutational screening of the mortalin gene (HSPA9) in Parkinson’s disease. J. Neural Transm. 2009;116:1289–1293. doi: 10.1007/s00702-009-0273-2. [DOI] [PubMed] [Google Scholar]
- (87).Wu PK, Hong SK, Veeranki S, Karkhanis M, Starenki D, Plaza JA, Park JI. A mortalin/HSPA9-mediated switch in tumor-suppressive signaling of Raf/MEK/extracellular signal-regulated kinase. Mol. Cell. Biol. 2013;33:4051–4067. doi: 10.1128/MCB.00021-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Harrison PM, Arosio P. The ferritins: molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta. 1996;1275:161–203. doi: 10.1016/0005-2728(96)00022-9. [DOI] [PubMed] [Google Scholar]
- (89).Rucker P, Torti FM, Torti SV. Role of H and L subunits in mouse ferritin. J. Biol. Chem. 1996;271:33352–33357. doi: 10.1074/jbc.271.52.33352. [DOI] [PubMed] [Google Scholar]
- (90).Theil EC. The ferritin family of iron storage proteins. Adv. Enzymol. Relat. Areas Mol. Biol. 1990;63:421–449. doi: 10.1002/9780470123096.ch7. [DOI] [PubMed] [Google Scholar]
- (91).Bogenhagen DF, Rousseau D, Burke S. The layered structure of human mitochondrial DNA nucleoids. J. Biol. Chem. 2008;283:3665–3675. doi: 10.1074/jbc.M708444200. [DOI] [PubMed] [Google Scholar]
- (92).Jansa P, Mason SW, Hoffmann-Rohrer U, Grummt I. Cloning and functional characterization of PTRF, a novel protein which induces dissociation of paused ternary transcription complexes. EMBO J. 1998;17:2855–2864. doi: 10.1093/emboj/17.10.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Curtis CD, Likhite VS, McLeod IX, Yates JR, Nardulli AM. Interaction of the tumor metastasis suppressor nonmetastatic protein 23 homologue H1 and estrogen receptor alpha alters estrogen-responsive gene expression. Cancer Res. 2007;67:10600–10607. doi: 10.1158/0008-5472.CAN-07-0055. [DOI] [PubMed] [Google Scholar]
- (94).Baltz AG, Munschauer M, Schwanhausser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, Wyler E, Bonneau R, Selbach M, Dieterich C, Landthaler M. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell. 2012;46:674–690. doi: 10.1016/j.molcel.2012.05.021. [DOI] [PubMed] [Google Scholar]
- (95).Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, Krijgsveld J, Hentze MW. Insights into RNA Biology from an Atlas of Mammalian mRNA-Binding Proteins. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- (96).Nowikovsky K, Bernardi P. LETM1 in mitochondrial cation transport. Front. Physiol. 2014;5:83. doi: 10.3389/fphys.2014.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Burhans WC, Heintz NH. The cell cycle is a redox cycle: Linking phase-specific targets to cell fate. Free Radical Biol. Med. 2009;47:1282–1293. doi: 10.1016/j.freeradbiomed.2009.05.026. [DOI] [PubMed] [Google Scholar]
- (98).Chiu J, Dawes IW. Redox control of cell proliferation. Trends Cell Biol. 2012;22:592–601. doi: 10.1016/j.tcb.2012.08.002. [DOI] [PubMed] [Google Scholar]
- (99).Sutliff RL, Hilenski LL, Amanso AM, Parastatidis I, Dikalova AE, Hansen L, Datla SR, Long JS, El-Ali AM, Joseph G, Gleason RL, Jr., Taylor WR, Hart CM, Griendling KK, Lassegue B. Polymerase delta interacting protein 2 sustains vascular structure and function. Arterioscler., Thromb., Vasc. Biol. 2013;33:2154–2161. doi: 10.1161/ATVBAHA.113.301913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Liu L, Rodriguez-Belmonte EM, Mazloum N, Xie B, Lee MY. Identification of a novel protein, PDIP38, that interacts with the p50 subunit of DNA polymerase delta and proliferating cell nuclear antigen. J. Biol. Chem. 2003;278:10041–10047. doi: 10.1074/jbc.M208694200. [DOI] [PubMed] [Google Scholar]
- (101).Maga G, Crespan E, Markkanen E, Imhof R, Furrer A, Villani G, Hubscher U, van Loon B. DNA polymerase delta-interacting protein 2 is a processivity factor for DNA polymerase lambda during 8-oxo-7,8-dihydroguanine bypass. Proc. Natl. Acad. Sci. U. S. A. 2013;110:18850–18855. doi: 10.1073/pnas.1308760110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Tissier A, Janel-Bintz R, Coulon S, Klaile E, Kannouche P, Fuchs RP, Cordonnier AM. Crosstalk between replicative and translesional DNA polymerases: PDIP38 interacts directly with Pol eta. DNA Repair. 2010;9:922–928. doi: 10.1016/j.dnarep.2010.04.010. [DOI] [PubMed] [Google Scholar]
- (103).Klaile E, Kukalev A, Obrink B, Muller MM. PDIP38 is a novel mitotic spindle-associated protein that affects spindle organization and chromosome segregation. Cell Cycle. 2008;7:3180–3186. doi: 10.4161/cc.7.20.6813. [DOI] [PubMed] [Google Scholar]
- (104).Wong A, Zhang SF, Mordue D, Wu JM, Zhang ZT, Darzynkiewicz Z, Lee EYC, Lee MYWT. PDIP38 is translocated to the spliceosomes/nuclear speckles in response to UV-induced DNA damage and is required for UV-induced alternative splicing of MDM2. Cell Cycle. 2013;12:3184–3193. doi: 10.4161/cc.26221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Dutertre M, Sanchez G, Barbier J, Corcos L, Auboeuf D. The emerging role of pre-messenger RNA splicing in stress responses: sending alternative messages and silent messengers. RNA Biol. 2011;8:740–747. doi: 10.4161/rna.8.5.16016. [DOI] [PubMed] [Google Scholar]
- (106).Nam SY, Sabapathy K. p53 promotes cellular survival in a context-dependent manner by directly inducing the expression of haeme-oxygenase-1. Oncogene. 2011;30:4476–4486. doi: 10.1038/onc.2011.150. [DOI] [PubMed] [Google Scholar]
- (107).Shen J, Sheng X, Chang Z, Wu Q, Wang S, Xuan Z, Li D, Wu Y, Shang Y, Kong X, Yu L, Li L, Ruan K, Hu H, Huang Y, Hui L, Xie D, Wang F, Hu R. Iron Metabolism Regulates p53 Signaling through Direct Heme-p53 Interaction and Modulation of p53 Localization, Stability, and Function. Cell Rep. 2014;7:180–193. doi: 10.1016/j.celrep.2014.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Klaile E, Muller MM, Kannicht C, Otto W, Singer BB, Reutter W, Obrink B, Lucka L. The cell adhesion receptor carcinoembryonic antigen-related cell adhesion molecule 1 regulates nucleocytoplasmic trafficking of DNA polymerase delta-interacting protein 38. J. Biol. Chem. 2007;282:26629–26640. doi: 10.1074/jbc.M701807200. [DOI] [PubMed] [Google Scholar]
- (109).Lyle AN, Deshpande NN, Taniyama Y, Seidel-Rogol B, Pounkova L, Du P, Papaharalambus C, Lassegue B, Griendling KK. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ. Res. 2009;105:249–259. doi: 10.1161/CIRCRESAHA.109.193722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Cheng X, Kanki T, Fukuoh A, Ohgaki K, Takeya R, Aoki Y, Hamasaki N, Kang D. PDIP38 associates with proteins constituting the mitochondrial DNA nucleoid. J. Biochem. 2005;138:673–8. doi: 10.1093/jb/mvi169. [DOI] [PubMed] [Google Scholar]
- (111).Xie B, Li H, Wang Q, Xie SQ, Rahmeh A, Dai W, Lee MYWT. Further characterization of human DNA polymerase delta interacting protein 38. J. Biol. Chem. 2005;280:22375–22384. doi: 10.1074/jbc.M414597200. [DOI] [PubMed] [Google Scholar]
- (112).Grinchuk OV, Motakis E, Kuznetsov VA. Complex sense-antisense architecture of TNFAIP1/POLDIP2 on 17q11.2 represents a novel transcriptional structural-functional gene module involved in breast cancer progression. BMC Genomics. 2010;11(Suppl 1):S9. doi: 10.1186/1471-2164-11-S1-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Pickering AM, Staab TA, Tower J, Sieburth D, Davies KJ. A conserved role for the 20S proteasome and Nrf2 transcription factor in oxidative stress adaptation in mammals, Caenorhabditis elegans and Drosophila melanogaster. J. Exp. Biol. 2013;216:543–553. doi: 10.1242/jeb.074757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Sainsbury R, Proctor I, Rodriguez S, Loddo M, Tudzarova S, Stoeber K, Williams G. Targeting, D. N. A. replication before it starts: Cdc7 as a therapeutic target in p53 mutant Her2 and triple negative breast cancer. Breast Cancer Res. 2010;12:S15–S16. doi: 10.2353/ajpath.2010.100421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discovery. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- (116).Hagenbuchner J, Ausserlechner MJ. Mitochondria and FOXO3: breath or die. Front. Physiol. 2013;4:147. doi: 10.3389/fphys.2013.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Szeto J, Kaniuk NA, Canadien V, Nisman R, Mizushima N, Yoshimori T, Bazett-Jones DP, Brumell JH. ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy. 2006;2:189–199. doi: 10.4161/auto.2731. [DOI] [PubMed] [Google Scholar]
- (118).Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, Kimura M, Sato S, Hattori N, Komatsu M, Tanaka K, Matsuda N. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells. 2010;15:887–900. doi: 10.1111/j.1365-2443.2010.01426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6:1090–1106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Pankiv S, Lamark T, Bruun JA, Overvatn A, Bjorkoy G, Johansen T. Nucleocytoplasmic shuttling of p62/SQSTM1 and its role in recruitment of nuclear polyubiquitinated proteins to promyelocytic leukemia bodies. J. Biol. Chem. 2010;285:5941–5953. doi: 10.1074/jbc.M109.039925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- (123).Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD, Johansen T. p62/SQSTM1 Is a Target Gene for Transcription Factor NRF2 and Creates a Positive Feedback Loop by Inducing Antioxidant Response Element-driven Gene Transcription. J. Biol. Chem. 2010;285:22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279–296. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Lamark T, Johansen T. Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int. J. Cell Biol. 2012;2012:736905. doi: 10.1155/2012/736905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Sakurai S, Kitano K, Yamaguchi H, Hamada K, Okada K, Fukuda K, Uchida M, Ohtsuka E, Morioka H, Hakoshima T. Structural basis for recruitment of human flap endonuclease 1 to PCNA. EMBO J. 2005;24:683–693. doi: 10.1038/sj.emboj.7600519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Subramani S, Malhotra V. Non-autophagic roles of autophagy-related proteins. EMBO Rep. 2013;14:143–151. doi: 10.1038/embor.2012.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Moscat J, Diaz-Meco MT. p62: a versatile multitasker takes on cancer. Trends Biochem. Sci. 2012;37:230–236. doi: 10.1016/j.tibs.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Al-Mehdi AB, Pastukh VM, Swiger BM, Reed DJ, Patel MR, Bardwell GC, Pastukh VV, Alexeyev MF, Gillespie MN. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci. Signaling. 2012;5:ra47. doi: 10.1126/scisignal.2002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Twig G, Shirihai OS. The Interplay Between Mitochondrial Dynamics and Mitophagy. Antioxid. Redox Signaling. 2011;14:1939–1951. doi: 10.1089/ars.2010.3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Bezprozvanny I, Hiesinger PR. The synaptic maintenance problem: membrane recycling, Ca2+ homeostasis and late onset degeneration. Mol. Neurodegener. 2013;8:23. doi: 10.1186/1750-1326-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Kotiadis VN, Duchen MR, Osellame LD. Mitochondrial quality control and communications with the nucleus are important in maintaining mitochondrial function and cell health. Biochim. Biophys. Acta. 2014;1840:1254–1265. doi: 10.1016/j.bbagen.2013.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Chini EN, Chini CCS, Nin V, Escande C. Deleted in breast cancer-1 (DBC-1) in the interface between metabolism, aging and cancer. Biosci. Rep. 2013;33:637–643. doi: 10.1042/BSR20130062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Joshi P, Quach OL, Giguere SS, Cristea IM. A Functional Proteomics Perspective of DBC1 as a Regulator of Transcription. J. Proteomics Bioinf. 2013;(Suppl2):002. [PMC free article] [PubMed] [Google Scholar]
- (135).Close P, East P, Dirac-Svejstrup AB, Hartmann H, Heron M, Maslen S, Chariot A, Soding J, Skehel M, Svejstrup JQ. DBIRD complex integrates alternative mRNA splicing with RNA polymerase II transcript elongation. Nature. 2012;484:386–389. doi: 10.1038/nature10925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Trauernicht AM, Kim SJ, Kim NH, Boyer TG. Modulation of estrogen receptor alpha protein level and survival function by DBC-1. Mol. Endocrinol. 2007;21:1526–1536. doi: 10.1210/me.2007-0064. [DOI] [PubMed] [Google Scholar]
- (137).Koyama S, Wada-Hiraike O, Nakagawa S, Tanikawa M, Hiraike H, Miyamoto Y, Sone K, Oda K, Fukuhara H, Nakagawa K, Kato S, Yano T, Taketani Y. Repression of estrogen receptor beta function by putative tumor suppressor DBC1. Biochem. Biophys. Res. Commun. 2010;392:357–362. doi: 10.1016/j.bbrc.2010.01.025. [DOI] [PubMed] [Google Scholar]
- (138).Sundararajan R, Chen G, Mukherjee C, White E. Caspase-dependent processing activates the proapoptotic activity of deleted in breast cancer-1 during tumor necrosis factor-alpha-mediated death signaling. Oncogene. 2005;24:4908–4920. doi: 10.1038/sj.onc.1208681. [DOI] [PubMed] [Google Scholar]
- (139).Chen JQ, Cammarata PR, Baines CP, Yager JD. Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochim. Biophys. Acta. 2009;1793:1540–1570. doi: 10.1016/j.bbamcr.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Ngo JK, Pomatto LC, Davies KJ. Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease, and aging. Redox Biol. 2013;1:258–264. doi: 10.1016/j.redox.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Suzuki CK, Rep M, vanDijl JM, Suda K, Grivell LA, Schatz G. ATP-dependent proteases that also chaperone protein biogenesis. Trends Biochem. Sci. 1997;22:118–123. doi: 10.1016/s0968-0004(97)01020-7. [DOI] [PubMed] [Google Scholar]
- (142).Liu T, Lu B, Lee I, Ondrovicova G, Kutejova E, Suzuki CK. DNA and RNA binding by the mitochondrial Lon protease is regulated by nucleotide and protein substrate. J. Biol. Chem. 2004;279:13902–13910. doi: 10.1074/jbc.M309642200. [DOI] [PubMed] [Google Scholar]
- (143).Matsushima Y, Goto Y, Kaguni LS. Mitochondrial Lon protease regulates mitochondrial DNA copy number and transcription by selective degradation of mitochondrial transcription factor A (TFAM) Proc. Natl. Acad. Sci. U. S. A. 2010;107:18410–18415. doi: 10.1073/pnas.1008924107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337:587–590. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Lo SC, Hannink M. PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Exp. Cell Res. 2008;314:1789–1803. doi: 10.1016/j.yexcr.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Fernandez-Real JM, Manco M. Effects of iron overload on chronic metabolic diseases. Lancet. 2013;2(6):513–526. doi: 10.1016/S2213-8587(13)70174-8. [DOI] [PubMed] [Google Scholar]
- (147).Chtourou Y, Fetoui H, Gdoura R. Protective Effects of Naringenin on Iron-Overload-Induced Cerebral Cortex Neurotoxicity Correlated with Oxidative Stress. Biol. Trace Elem. Res. 2014;158(3):376–383. doi: 10.1007/s12011-014-9948-0. [DOI] [PubMed] [Google Scholar]
- (148).Lamy PJ, Durigova A, Jacot W. Iron homeostasis and anemia markers in early breast cancer: Iron and breast cancer. Clin. Chim. Acta. 2014;434:34–40. doi: 10.1016/j.cca.2014.04.011. [DOI] [PubMed] [Google Scholar]
- (149).Marques O, da Silva BM, Porto G, Lopes C. Iron homeostasis in breast cancer. Cancer Lett. 2014;347:1–14. doi: 10.1016/j.canlet.2014.01.029. [DOI] [PubMed] [Google Scholar]
- (150).Hamara K, Bielecka-Kowalska A, Przybylowska-Sygut K, Sygut A, Dziki A, Szemraj J. Alterations in expression profile of iron-related genes in colorectal cancer. Mol. Biol. Rep. 2013;40:5573–5585. doi: 10.1007/s11033-013-2659-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Buss JL, Greene BT, Turner J, Torti FM, Torti SV. Iron chelators in cancer chemotherapy. Curr. Top. Med. Chem. 2004;4:1623–1635. doi: 10.2174/1568026043387269. [DOI] [PubMed] [Google Scholar]
- (152).Anderson GJ, Vulpe CD. Mammalian iron transport. Cell. Mol. Life Sci. 2009;66:3241–3261. doi: 10.1007/s00018-009-0051-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Lawen A, Lane DJ. Mammalian iron homeostasis in health and disease: uptake, storage, transport, and molecular mechanisms of action. Antioxid. Redox Signaling. 2013;18:2473–2507. doi: 10.1089/ars.2011.4271. [DOI] [PubMed] [Google Scholar]
- (154).Diab HI, Kane PM. Loss of Vacuolar H+-ATPase (V-ATPase) Activity in Yeast Generates an Iron Deprivation Signal That Is Moderated by Induction of the Peroxiredoxin TSA2. J. Biol. Chem. 2013;288:11366–11377. doi: 10.1074/jbc.M112.419259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Cyert MS, Philpott CC. Regulation of cation balance in Saccharomyces cerevisiae. Genetics. 2013;193:677–713. doi: 10.1534/genetics.112.147207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Hamza I, Dailey HA. One ring to rule them all: trafficking of heme and heme synthesis intermediates in the metazoans. Biochim. Biophys. Acta. 2012;1823:1617–1632. doi: 10.1016/j.bbamcr.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Braschi E, Goyon V, Zunino R, Mohanty A, Xu LQ, McBride HM. Vps35 Mediates Vesicle Transport between the Mitochondria and Peroxisomes. Curr. Biol. 2010;20:1310–1315. doi: 10.1016/j.cub.2010.05.066. [DOI] [PubMed] [Google Scholar]
- (158).Mohanty A, McBride HM. Emerging roles of mitochondria in the evolution, biogenesis, and function of peroxisomes. Front. Physiol. 2013;4:268. doi: 10.3389/fphys.2013.00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Arnoult D, Rismanchi N, Grodet A, Roberts RG, Seeburg DP, Estaquier J, Sheng M, Blackstone C. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr. Biol. 2005;15:2112–2118. doi: 10.1016/j.cub.2005.10.041. [DOI] [PubMed] [Google Scholar]
- (160).Lyamzaev KG, Nepryakhina OK, Saprunova VB, Bakeeva LE, Pletjushkina OY, Chernyak BV, Skulachev VP. Novel mechanism of elimination of malfunctioning mitochondria (mitoptosis): formation of mitoptotic bodies and extrusion of mitochondrial material from the cell. Biochim. Biophys. Acta. 2008;1777:817–825. doi: 10.1016/j.bbabio.2008.03.027. [DOI] [PubMed] [Google Scholar]
- (161).Daum B, Walter A, Horst A, Osiewacz HD, Kuhlbrandt W. Age-dependent dissociation of ATP synthase dimers and loss of inner-membrane cristae in mitochondria. Proc. Natl. Acad. Sci. U. S. A. 2013;110:15301–15306. doi: 10.1073/pnas.1305462110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Habersetzer J, Larrieu I, Priault M, Salin B, Rossignol R, Brethes D, Paumard P. Human F1F0 ATP synthase, mitochondrial ultrastructure and OXPHOS impairment: a (super-)complex matter? PLoS One. 2013;8:e75429. doi: 10.1371/journal.pone.0075429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Bernardi P. The mitochondrial permeability transition pore: a mystery solved? Front. Physiol. 2013;4:95. doi: 10.3389/fphys.2013.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabo I, Lippe G, Bernardi P. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. U. S. A. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Greco T, Shafer J, Fiskum G. Sulforaphane inhibits mitochondrial permeability transition and oxidative stress. Free Radical Biol. Med. 2011;51:2164–2171. doi: 10.1016/j.freeradbiomed.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Greco T, Fiskum G. Brain mitochondria from rats treated with sulforaphane are resistant to redox-regulated permeability transition. J. Bioenerg. Biomembr. 2010;42:491–497. doi: 10.1007/s10863-010-9312-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Xu JM, Hao ZM, Gou XB, Tian W, Jin YL, Cui SX, Guo J, Sun YJ, Wang Y, Xu ZL. Imaging of reactive oxygen species burst from mitochondria using laser scanning confocal microscopy. Microsc. Res. Tech. 2013;76:612–617. doi: 10.1002/jemt.22207. [DOI] [PubMed] [Google Scholar]
- (168).Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Wang W, Mattson MP, Kao JP, Lakatta EG, Sheu SS, Ouyang K, Chen J, Dirksen RT, Cheng H. Superoxide flashes in single mitochondria. Cell. 2008;134:279–290. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Wang Y, Nartiss Y, Steipe B, McQuibban GA, Kim PK. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy. 2012;8:1462–1476. doi: 10.4161/auto.21211. [DOI] [PubMed] [Google Scholar]
- (170).Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, McBride HM. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 2012;22:135–141. doi: 10.1016/j.cub.2011.11.057. [DOI] [PubMed] [Google Scholar]
- (171).Kimura I, Nakayama Y, Konishi M, Terasawa K, Ohta M, Itoh N, Fujimoto M. Functions of MAPR (Membrane-Associated Progesterone Receptor) Family Members As Heme/Steroid-Binding Proteins. Curr. Protein Pept. Sci. 2012;13:687–696. doi: 10.2174/138920312804142110. [DOI] [PubMed] [Google Scholar]
- (172).Bali N, Morgan TE, Finch CE. Pgrmc1: new roles in the microglial mediation of progesterone-antagonism of estradiol-dependent neurite sprouting and in microglial activation. Front. Neurosci. 2013;7:157. doi: 10.3389/fnins.2013.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Petersen SL, Intlekofer KA, Moura-Conlon PJ, Brewer DN, Del Pino Sans J, Lopez JA. Novel progesterone receptors: neural localization and possible functions. Front. Neurosci. 2013;7:164. doi: 10.3389/fnins.2013.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Xu JB, Zeng CB, Chu WH, Pan FH, Rothfuss JM, Zhang FJ, Tu ZD, Zhou D, Zeng DX, Vangveravong S, Johnston F, Spitzer D, Chang KC, Hotchkiss RS, Hawkins WG, Wheeler KT, Mach RH. Identification of the PGRMC1 protein complex as the putative sigma-2 receptor binding site. Nat. Commun. 2011;2:380. doi: 10.1038/ncomms1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Ahmed IS, Rohe HJ, Twist KE, Craven RJ. Pgrmc1 (progesterone receptor membrane component 1) associates with epidermal growth factor receptor and regulates erlotinib sensitivity. J. Biol. Chem. 2010;285:24775–24782. doi: 10.1074/jbc.M110.134585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Bali N, Arimoto JM, Morgan TE, Finch CE. Progesterone antagonism of neurite outgrowth depends on microglial activation via Pgrmc1/S2R. Endocrinology. 2013;154:2468–2480. doi: 10.1210/en.2012-2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Peluso JJ, Liu X, Gawkowska A, Lodde V, Wu CA. Progesterone inhibits apoptosis in part by PGRMC1-regulated gene expression. Mol. Cell. Endocrinol. 2010;320:153–161. doi: 10.1016/j.mce.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Peluso JJ, DeCerbo J, Lodde V. Evidence for a genomic mechanism of action for progesterone receptor membrane component-1. Steroids. 2012;77:1007–1012. doi: 10.1016/j.steroids.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (179).Mir SU, Schwarze SR, Jin L, Zhang J, Friend W, Miriyala S, St Clair D, Craven RJ. Progesterone receptor membrane component 1/Sigma-2 receptor associates with MAP1LC3B and promotes autophagy. Autophagy. 2013;9:1566–1578. doi: 10.4161/auto.25889. [DOI] [PubMed] [Google Scholar]
- (180).Thomas P, Pang YF, Dong J. Enhancement of Cell Surface Expression and Receptor Functions of Membrane Progestin Receptor alpha (mPR alpha) by Progesterone Receptor Membrane Component 1 (PGRMC1): Evidence for a Role of PGRMC1 as an Adaptor Protein for Steroid Receptors. Endocrinology. 2014;155:1107–1119. doi: 10.1210/en.2013-1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (181).Chen WS, Chen PL, Li JP, Lind AC, Lu DS. Lipid synthesis and processing proteins ABHD5, PGRMC1 and squalene synthase can serve as novel immunohistochemical markers for sebaceous neoplasms and differentiate sebaceous carcinoma from sebaceoma and basal cell carcinoma with clear cell features. J. Cutaneous Pathol. 2013;40:631–638. doi: 10.1111/cup.12147. [DOI] [PubMed] [Google Scholar]
- (182).Mir SU, Ahmed IS, Arnold S, Craven RJ. Elevated progesterone receptor membrane component 1/sigma-2 receptor levels in lung tumors and plasma from lung cancer patients. Int. J. Cancer. 2012;131:E1–E9. doi: 10.1002/ijc.26432. [DOI] [PubMed] [Google Scholar]
- (183).Cahill MA. Progesterone receptor membrane component 1: an integrative review. J. Steroid Biochem. Mol. Biol. 2007;105:16–36. doi: 10.1016/j.jsbmb.2007.02.002. [DOI] [PubMed] [Google Scholar]
- (184).Peluso JJ, Yuan A, Liu X, Lodde V. Plasminogen activator inhibitor 1 RNA-binding protein interacts with progesterone receptor membrane component 1 to regulate progesterone’s ability to maintain the viability of spontaneously immortalized granulosa cells and rat granulosa cells. Biol. Reprod. 2013;88:20. doi: 10.1095/biolreprod.112.103036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (185).Luciano AM, Lodde V, Franciosi F, Ceciliani F, Peluso JJ. Progesterone receptor membrane component 1 expression and putative function in bovine oocyte maturation, fertilization, and early embryonic development. Reproduction. 2010;140:663–72. doi: 10.1530/REP-10-0218. [DOI] [PubMed] [Google Scholar]
- (186).Van Buul JD, Fernandez-Borja M, Anthony EC, Hordijk PL. Expression and localization of NOX2 and NOX4 in primary human endothelial cells. Antioxid. Redox Signaling. 2005;7:308–317. doi: 10.1089/ars.2005.7.308. [DOI] [PubMed] [Google Scholar]
- (187).Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler., Thromb., Vasc. Biol. 2004;24:677–683. doi: 10.1161/01.ATV.0000112024.13727.2c. [DOI] [PubMed] [Google Scholar]
- (188).Diaz B, Courtneidge SA. Redox signaling at invasive microdomains in cancer cells. Free Radical Biol. Med. 2012;52:247–256. doi: 10.1016/j.freeradbiomed.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).Diaz B, Shani G, Pass I, Anderson D, Quintavalle M, Courtneidge SA. Tks5-Dependent, Nox-Mediated Generation of Reactive Oxygen Species Is Necessary for Invadopodia Formation. Sci. Signaling. 2009;2:ra53. doi: 10.1126/scisignal.2000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ. Res. 2010;106:1253–1264. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).Block K, Gorin Y, Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. U. S. A. 2009;106:14385–14390. doi: 10.1073/pnas.0906805106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (192).Graham KA, Kulawiec M, Owens KM, Li XR, Desouki MM, Chandra D, Singh KK. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol. Ther. 2010;10:223–231. doi: 10.4161/cbt.10.3.12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Matsushima S, Kuroda J, Ago T, Zhai P, Park JY, Xie LH, Tian B, Sadoshima J. Increased oxidative stress in the nucleus caused by Nox4 mediates oxidation of HDAC4 and cardiac hypertrophy. Circ. Res. 2013;112:651–663. doi: 10.1161/CIRCRESAHA.112.279760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Spencer NY, Yan Z, Boudreau RL, Zhang Y, Luo M, Li Q, Tian X, Shah AM, Davisson RL, Davidson B, Banfi B, Engelhardt JF. Control of hepatic nuclear superoxide production by glucose 6-phosphate dehydrogenase and NADPH oxidase-4. J. Biol. Chem. 2011;286:8977–8987. doi: 10.1074/jbc.M110.193821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Sciarretta S, Zhai P, Shao D, Zablocki D, Nagarajan N, Terada LS, Volpe M, Sadoshima J. Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2alpha/activating transcription factor 4 pathway. Circ. Res. 2013;113:1253–1264. doi: 10.1161/CIRCRESAHA.113.301787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).Kartha GK, Moshal KS, Sen U, Joshua IG, Tyagi N, Steed MM, Tyagi SC. Renal mitochondrial damage and protein modification in type-2 diabetes. Acta Diabetol. 2008;45:75–81. doi: 10.1007/s00592-008-0025-z. [DOI] [PubMed] [Google Scholar]
- (197).Barnes JL, Gorin Y. Myofibroblast differentiation during fibrosis: role of NAD(P)H oxidases. Kidney Int. 2011;79:944–956. doi: 10.1038/ki.2010.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discovery. 2011;10:453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Beyenbach KW, Wieczorek H. The V-type H+ ATPase: molecular structure and function, physiological roles and regulation. J. Exp. Biol. 2006;209:577–589. doi: 10.1242/jeb.02014. [DOI] [PubMed] [Google Scholar]
- (200).Milgrom E, Diab H, Middleton F, Kane PM. Loss of vacuolar proton-translocating ATPase activity in yeast results in chronic oxidative stress. J. Biol. Chem. 2007;282:7125–7136. doi: 10.1074/jbc.M608293200. [DOI] [PubMed] [Google Scholar]
- (201).Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005;12:1161–1208. doi: 10.2174/0929867053764635. [DOI] [PubMed] [Google Scholar]
- (202).Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, Hansen M, Moscat J, Diaz-Meco MT. p62 Is a Key Regulator of Nutrient Sensing in the mTORC1 Pathway. Mol. Cell. 2011;44:134–146. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Zoncu R, Bar-Peled L, Efeyan A, Wang SY, Sancak Y, Sabatini DM. mTORC1 Senses Lysosomal Amino Acids Through an Inside-Out Mechanism That Requires the Vacuolar H+-ATPase. Science. 2011;334:678–683. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).O’Callaghan KM, Ayllon V, O’Keeffe J, Wang YR, Cox OT, Loughran G, Forgac M, O’Connor R. Heme-binding Protein HRG-1 Is Induced by Insulin-like Growth Factor I and Associates with the Vacuolar H+-ATPase to Control Endosomal pH and Receptor Trafficking. J. Biol. Chem. 2010;285:381–391. doi: 10.1074/jbc.M109.063248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Brown D, Paunescu TG, Breton S, Marshansky V. Regulation of the V-ATPase in kidney epithelial cells: dual role in acid-base homeostasis and vesicle trafficking. J. Exp Biol. 2009;212:1762–1772. doi: 10.1242/jeb.028803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (206).Gu F, Gruenberg J. ARF1 regulates pH-dependent COP functions in the early endocytic pathway. J. Biol. Chem. 2000;275:8154–8160. doi: 10.1074/jbc.275.11.8154. [DOI] [PubMed] [Google Scholar]
- (207).Mangieri LR, Mader BJ, Thomas CE, Taylor CA, Luker AM, Tse TE, Huisingh C, Shacka JJ. ATP6V0C Knockdown in Neuroblastoma Cells Alters Autophagy-Lysosome Pathway Function and Metabolism of Proteins that Accumulate in Neurodegenerative Disease. PLoS One. 2014;9:e93257. doi: 10.1371/journal.pone.0093257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Krop M, Lu XF, Danser AHJ, Meima ME. The (pro)renin receptor. A decade of research: what have we learned? Pfluegers Arch. 2013;465:87–97. doi: 10.1007/s00424-012-1105-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Chiabrando D, Vinchi F, Fiorito V, Mercurio S, Tolosano E. Heme in pathophysiology: a matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014;5:61. doi: 10.3389/fphar.2014.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (210).Huatori J, Helenius A. Endosome maturation. EMBO J. 2011;30:3481–500. doi: 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.