

The Eut microcompartment is a protein-based organelle that carries out B12-dependent degradation of ethanolamine in diverse bacteria. This work characterizes a binding interaction between cobalamin (B12) and the inward-facing surface of EutL, a shell protein proposed to be involved in cofactor transport in the Eut microcompartment.
Keywords: bacterial microcompartments, EutL shell protein, Clostridium perfringens
Abstract
The EutL shell protein is a key component of the ethanolamine-utilization microcompartment, which serves to compartmentalize ethanolamine degradation in diverse bacteria. The apparent function of this shell protein is to facilitate the selective diffusion of large cofactor molecules between the cytoplasm and the lumen of the microcompartment. While EutL is implicated in molecular-transport phenomena, the details of its function, including the identity of its transport substrate, remain unknown. Here, the 2.1 Å resolution X-ray crystal structure of a EutL shell protein bound to cobalamin (vitamin B12) is presented and the potential relevance of the observed protein–ligand interaction is briefly discussed. This work represents the first structure of a bacterial microcompartment shell protein bound to a potentially relevant cofactor molecule.
1. Introduction
Bacterial microcompartments (MCPs) are giant protein complexes that function as metabolic organelles in prokaryotes (Cheng et al., 2008 ▶; Yeates et al., 2008 ▶, 2010 ▶; Kerfeld et al., 2010 ▶). These highly ordered structures consist of a thin polyhedral shell that surrounds a series of sequentially acting metabolic enzymes, thereby sequestering them from the cytosol in order to enhance metabolic flux and to retain intermediate compounds that might otherwise be lost by diffusion across the plasma membrane or become toxic in the cell at elevated concentrations (Penrod & Roth, 2006 ▶; Sampson & Bobik, 2008 ▶). The so-called BMC (bacterial microcompartment) family of proteins are the primary component of MCP shells, and they are largely responsible for the ability of these elaborate protein complexes to act as metabolic organelles in bacteria (Yeates et al., 2011 ▶, 2013 ▶). BMC shell proteins oligomerize to form hexamers, which further tile together into a two-dimensional lattice forming the flat facets of the polyhedral MCP (Kerfeld et al., 2005 ▶; Yeates et al., 2011 ▶, 2013 ▶; Fig. 1 ▶ a).
Figure 1.

The ethanolamine-utilization (Eut) microcompartment. (a) BMC-domain shell protein hexamers, such as EutL, interact along their edges, forming a thin, but tightly packed, two-dimensional layer. Flat facets combine to create a polyhedral shell assembled around a series of internal enzymes that are responsible for the initial steps of ethanolamine catabolism. The first enzymatic step of the encapsulated pathway, catalyzed by EutBC, produces a volatile intermediate (acetaldehyde; blue box) and requires a vitamin B12 (cobalamin) cofactor. The need to regenerate this cofactor implies that the shell allows the passage of molecules much larger than the substrates and products of the encapsulated enzymes (red dashed arrow). (b) EutL is a tandem BMC-domain polypeptide that forms a pseudohexameric trimer. The previous observation of both ‘open-pore’ and ‘closed-pore’ conformations of E. coli EutL indicated that this shell protein is likely to function in the gated transport of large cofactor molecules.
The MCP shell must act as a semi-permeable diffusion barrier, allowing the passage of substrates, products and cofactors, while simultaneously restricting the efflux of key metabolic intermediates. This functional complexity is exemplified by the shell of the ethanolamine-utilization (Eut) MCP (Fig. 1 ▶ a). This MCP shell must permit the inward diffusion of ethanolamine as well as the outward diffusion of ethanol (formed by EutG) and acetyl phosphate (formed by EutD and EutE), while simultaneously restricting the mass transport of the acetaldehyde intermediate (Roof & Roth, 1988 ▶; Kofoid et al., 1999 ▶; Tanaka et al., 2010 ▶; Held et al., 2013 ▶). This feat is even more impressive considering that, in addition to regulating the flux of small substrates and products, the shell of the Eut MCP must also coordinate the exchange of large cofactor molecules required by the EutBC enzyme (Tanaka et al., 2010 ▶), which are roughly an order of magnitude larger than the reaction substrates and products. The exact details of cofactor transport in the Eut MCP system are unclear, but the requirement for the regeneration of vitamin B12 (cobalamin) cofactors for internal ethanolamine-ammonia lyase (EutBC) enzymes (Toraya, 2003 ▶; Sheppard et al., 2004 ▶) appears to necessitate the transport of either ATP, or some form of the cobalamin cofactor itself, across the shell (Buan et al., 2004 ▶; Mori et al., 2004 ▶).
Studies of the BMC shell proteins present in the Eut MCP system have revealed EutL to be the conduit through which large cofactor molecules are likely to be transported. EutL is a tandem BMC-domain polypeptide, which assembles as a pseudohexameric homotrimer that maintains the overall hexagonal shape of a typical BMC shell protein oligomer (Sagermann et al., 2009 ▶; Takenoya et al., 2010 ▶; Tanaka et al., 2010 ▶; Fig. 1 ▶ b). In the case of EutL, the broken oligomeric symmetry has an important functional consequence, allowing a conformational change that opens a large central pore capable of allowing the passage of requisite cofactors (Takenoya et al., 2010 ▶; Tanaka et al., 2010 ▶; Fig. 1 ▶ b). Many questions remain with respect to understanding the presumptive cofactor-transport function of EutL. Here, we describe the X-ray crystal structure of EutL from Clostridium perfringens (cpEutL) bound to cobalamin, in an attempt to shed light on the potential transport substrates of the EutL protein pore.
2. Materials and methods
2.1. Cloning, expression and purification of recombinant protein
For preparation of the cpEutL construct, we amplified the eutL gene from C. perfringens chromosomal DNA (ATCC) and ligated the amplicon into the multiple cloning site of the pET-22b expression vector (Novagen). We used the NdeI and XhoI restriction sites, thus incorporating the noncleavable Leu-Glu-His6 affinity tag at the C-terminus of the 217-residue native sequence. In a similar manner, we also prepared a construct containing an N-terminal His6-ENLYFQG sequence, which acts as a cleavable affinity tag. Dideoxy chain-termination sequencing (Sanger et al., 1977 ▶) confirmed the recombinant DNA sequences.
We expressed recombinant protein using transformed Escherichia coli BL21 (DE3) Rosetta cells (Novagen). During the exponential phase of cell growth in selective Luria–Bertani (LB) broth, 1 mM isopropyl β-d-1-thiogalactopyranoside was added to induce protein expression for 4 h at 30°C. Cells were collected by centrifugation for 15 min at 5000g.
Purification of cpEutL protein samples for crystallization began with cell lysis by high-pressure emulsification (EmulsiFlex C3, Avestin). The lysis buffer consisted of 20 mM Tris buffer, 300 mM NaCl pH 8.0 with a protease-inhibitor additive (Sigma–Aldrich), 10 mM MgCl2, 1 mg ml−1 lysozyme and 100 units ml−1 of both DNase and RNase. We clarified the cell lysate by centrifugation at 30 000g for 30 min and then used a HisTrap nickel-affinity column (GE Healthcare) to purify cpEutL from clarified lysates, eluting the bound protein using lysis buffer supplemented with 300 mM imidazole. The eluate was finally dialyzed against a buffer consisting of 20 mM Tris, 100 mM NaCl pH 8.0. This single purification step resulted in protein of sufficient purity for crystallizing the construct bearing the (noncleavable) C-terminal His tag.
When working with the N-terminally tagged cpEutL, the following steps, in addition to the initial metal-affinity purification, were taken to remove the affinity tag and produce highly pure protein samples. Firstly, the peptide tag was removed by overnight cleavage with TEV protease (1 mg ml−1) while dialyzing the reaction mixture against buffer consisting of 20 mM Tris pH 8.0, 100 mM NaCl, 1 mM dithiothreitiol (DTT). Following overnight protease treatment, the protein sample was once again passed over a nickel-affinity column, except this time the untagged protein was collected from the column flowthrough. The protein was then subjected to two additional purification steps, first using a HiTrap Q anion-exchange column and then a Superdex 200 gel-filtration column (both from GE Healthcare). The final purification step left the protein in buffer consisting of 20 mM Tris, 100 mM NaCl pH 8.0.
2.2. Crystallization
Prior to crystallization experiments, we concentrated the protein samples to approximately 20 mg ml−1 in 20 mM Tris pH 8.0, 100 mM NaCl using Amicon Ultra concentrators (Millipore). We carried out initial crystallization screening of the C-terminally tagged protein by the vapor-diffusion method in 96-well hanging-drop format using a Mosquito robot and several commercial screening kits, including the JCSG+ Suite (Qiagen), Crystal Screen and Crystal Screen 2 (Hampton Research), and the Wizard I and II screens (Emerald Bio). Initial crystallization trials were performed in the absence of the vitamin B12 ligand. We identified initial crystal hits of the tetragonal crystal form in condition B4 of the JCSG+ Suite, which consisted of 0.1 M HEPES buffer pH 7.5, 10%(w/v) PEG 8000, 8% ethylene glycol. These conditions were further optimized by grid screening in 24-well hanging-drop format (VDX plates, Hampton Research). The highest quality crystals (as judged by diffraction) were approximately 0.5–1 mm in length and grew from 0.1 M HEPES buffer pH 7.0, 5% PEG 8000, 8% ethylene glycol (Figs. 2 ▶ a and 2 ▶ b). The crystals of B12-bound cpEutL used for structure determination were obtained by overnight soaking of the native crystals in mother liquor with 10 mM hydroxocobalamin (Fig. 2 ▶ c). Alternatively, cobalamin-bound protein crystals could be obtained by using a fine needle to add a few small crystals of solid hydroxocobalamin to crystallization drops containing pre-formed protein crystals. It was evident from their color that the added cofactor had entered the cpEutL crystals (Figs. 2 ▶ c and 2 ▶ d).
Figure 2.

Crystals of cpEutL. Native crystals of cpEutL (a) show birefringence when viewed through a polarizer (b). High-quality crystals of ligand-bound cpEutL could be obtained by soaking pre-formed protein crystals in mother liquor containing the hydroxocobalamin ligand (a form of vitamin B12) (c) or by adding small crystals of pure hydroxocobalamin to crystallized protein (d). Additionally, crystals of cpEutL bound to hydroxocobalamin were obtained by co-crystallization (e), (f). These crystals appeared to be hexagonal in nature, but did not diffract well enough for structure determination.
In addition to the cobalamin-bound crystals obtained by adding the cofactor to pre-formed crystals of cpEutL (described above), we also grew crystals of cpEutL in the presence of cofactor. These crystals were obtained using a cpEutL sample from which the N-terminal affinity tag had been removed by protease treatment in order to prevent spurious binding of the cofactor. These crystallization trials were performed with varying concentrations of hydroxocobalamin (1–10 mM). Both systematic and sparse-matrix crystallization screening yielded numerous co-crystals from solutions containing cpEutL and hydroxocobalamin. All of these crystals appeared with hexagonal morphologies, commonly displaying facets that met at 120° angles (Figs. 2 ▶ e and 2 ▶ f), and contained cofactor as judged by their deep color. However, despite their well defined morphology, the crystals obtained in this manner universally suffered from lattice-translocation disorders and severe diffraction anisotropy, as evident from their diffraction patterns (Supplementary Fig. S11). The severity of the disorder prevented the collection of useful diffraction data from the co-crystallized complex.
2.3. X-ray data collection and processing
Prior to X-ray data collection, we harvested the crystals and cryoprotected them using 50% mother liquor with 2 M trimethylamine N-oxide (Mueller-Dieckmann et al., 2011 ▶) before flash-cooling in liquid nitrogen. The cryoprotectant solution did not contain cobalamin ligand.
We collected single-crystal X-ray diffraction data on beamline 24-ID-C at the Advanced Photon Source. This beamline was equipped with a PILATUS 6M-F detector and an MD2 microdiffractometer. The experimental apparatus, as well as the dimensions of the cpEutL crystals, allowed us to collect X-ray diffraction data using a vector-scan method, thereby spreading the X-ray dose throughout the length of the crystal. We maintained the crystals at a cryogenic temperature (−173°C) throughout the course of data collection.
We performed indexing, integration and scaling of the X-ray data using XDS and XSCALE, and then converted the intensities to structure-factor amplitudes with XDSCONV (Kabsch, 2010 ▶). Diffraction data were analyzed with phenix.xtriage to check for crystal pathologies (Zwart et al., 2005 ▶; Adams et al., 2010 ▶). A resolution cutoff (2.1 Å) was taken where the redundancy-independent merging R value (R r.i.m.) rose to a value of 1, which was justified by the high value of CC1/2 (0.93) at that resolution. Further information regarding data collection and processing is presented in Table 1 ▶.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell. R factors and correlation coefficients are calculated with riding H atoms present in the model.
| Wavelength () | 0.9795 |
| Resolution range () | 83.272.10 (2.152.10) |
| Unit-cell parameters (, ) | a = b = 88.23, c = 252.01, = = = 90 |
| Space group | P43212 |
| Unique reflections | 59035 |
| Multiplicity | 12.5 |
| Completeness (%) | 100 (100) |
| Mean I/(I) | 8.6 (2.0) |
| Wilson B factor (2) | 47.1 |
| Mosaicity () | 0.1 |
| R merge | 0.067 (0.993) |
| R r.i.m. † | 0.072 (1.006) |
| R p.i.m. ‡ | 0.021 (0.276) |
| CC1/2 § | 0.999 (0.934) |
| CC*§ | 1.000 (0.983) |
| CCwork § | 0.948 (0.917) |
| CCfree § | 0.947 (0.902) |
| R work ¶ | 0.1845 |
| R free ¶ | 0.2104 |
| No. of test reflections | 3023 |
| No. of refined atoms | 5098 |
| Protein residues | 650 |
| Ligand atoms | 92 |
| Ligand occupancy | 0.95 |
| Solvent molecules | 102 |
| Average B factor (2) | |
| Protein | 60.7 |
| Ligand | 97.9 |
| R.m.s.d.bonds () | 0.003 |
| R.m.s.d.angles () | 0.731 |
| Ramachandran plot | |
| Favored | 98.4 |
| Allowed | 1.6 |
| Outliers | 0.0 |
| MolProbity clashscore†† | 0.7 |
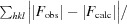
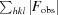
2.4. Structure determination
As a starting point for determining the structure of cpEutL bound to cobalamin, we performed rigid-body refinement of a previously determined, ligand-free cpEutL structure (PDB entry 4edi, M. C. Thompson, D. Cascio, C. S. Crowley, J. S. Kopstein & T. O. Yeates, unpublished work) against our X-ray data using phenix.refine (Adams et al., 2010 ▶; Afonine et al., 2012 ▶). Our crystals were nearly isomorphous with the crystals used to determine this ligand-free structure of the protein, and we were careful to preserve the free set of reflections used in refinement of the ligand-free structure.
Next, using electron-density maps calculated after initial rigid-body refinement, we rebuilt the missing or incorrect parts of the structure and then performed initial atomic refinement with simulated annealing to remove residual model bias. Additional iterative steps of manual model rebuilding and atomic refinement were performed, during which the cobalamin molecule became evident in the electron-density maps. The ligand was added to the model and successive refinement with TLS parameters, a riding hydrogen model, occupancy refinement of the ligand and automatic weight optimization led to a final, high-quality model. Further information regarding model building and refinement is presented in Table 1 ▶. The final model coordinates were deposited in the Protein Data Bank (PDB; Bernstein et al., 1977 ▶) under accession code 4u6i.
All model building was performed using Coot (v.0.6.2; Emsley et al., 2010 ▶) and refinement steps were performed with phenix.refine (v.1.8.4-dev1555) within the PHENIX suite (Adams et al., 2010 ▶; Afonine et al., 2012 ▶). Restraints for the cobalamin ligand were based on the small-molecule structure of adenosylcobalamin (Mebs et al., 2009 ▶), as provided by Dr Oliver Smart (https://www.globalphasing.com/buster/wiki/index.cgi?B12Dictionary). Additional metal-coordination restraints were determined by phenix.ready_set. Omit maps were calculated by removing the coordinates of the ligand and His32 and performing automated refinement with phenix.refine (Afonine et al., 2012 ▶). Real-space correlation coefficients (RSCCs) were calculated using phenix.get_cc_mtz_pdb (Adams et al., 2010 ▶).
3. Results and discussion
3.1. Ligand soaking yields high-quality crystals of cpEutL bound to cobalamin
We were able to obtain crystals of cpEutL bound to cobalamin by co-crystallization and by crystal soaking (Figs. 2 ▶ c–2 ▶ f). When cpEutL was co-crystallized with the ligand, broad screening of crystallization conditions gave many initial hits, virtually all of which demonstrated a flat, plate-like habit and a distinct pink hue characteristic of cobalamin compounds (Figs. 2 ▶ e and 2 ▶ f). Exposure to X-rays showed that the plate-like crystals universally suffered from lattice-translocation disorder (Supplementary Fig. S1), which prevented structure determination. Fortunately, we could also obtain high-quality crystals of cpEutL bound to cobalamin by soaking the compound into native crystals. The soaking procedure resulted in dark pink or red crystals (Figs. 2 ▶ c and 2 ▶ d), which remained pink even after washing and back-soaking the crystals overnight in mother liquor without the ligand.
3.2. Structural overview of B12-bound cpEutL
The cpEutL crystals obtained by soaking in hydroxocobalamin diffracted to a resolution of 2.1 Å (Fig. 3 ▶ a). These crystals belong to the tetragonal space group P43212, with a single cpEutL homotrimer in each asymmetric unit (Fig. 3 ▶ b). The trimer in the asymmetric unit of the crystallographic model derived from these data contains 650 amino-acid residues, one bound cobalamin ligand and 102 ordered water molecules (Fig. 3 ▶ c). Interestingly, there were a number of relatively strong electron-density features in both 2mF o − DF c and mF o − DF c maps that could not be adequately modeled, possibly resulting from bulky hydroxocobalamin molecules becoming trapped in random orientations within the solvent channels of the crystal. Despite the presence of some uninterpretable electron-density features, our final model of cpEutL bound to cobalamin fits well to the observed diffraction data and the model also has excellent protein stereochemistry (Table 1 ▶).
Figure 3.

Overview of cpEutL–B12 structure determination. (a) A representative diffraction image obtained from the X-ray experiment. The red ring represents the 2.1 Å resolution cutoff applied during data reduction. (b) The tetragonal unit cell of the cpEutL crystal contains a single trimer in the asymmetric unit. In the diagrams of the unit cell, each asymmetric unit is colored differently. (c) The structure of cpEutL bound to the hydroxocobalamin ligand (yellow spheres) shows the ligand protruding from the luminal face of the trimer.
The crystal structure of cpEutL bound to cobalamin represents the previously characterized ‘closed-pore’ conformation of EutL (Sagermann et al., 2009 ▶; Takenoya et al., 2010 ▶; Tanaka et al., 2010 ▶). In our cobalamin-bound structure, as in other ‘closed-pore’ structures of EutL, the protein adopts a tandem BMC-domain fold and oligomerizes to form a pseudohexameric trimer (Fig. 3 ▶ c). In the observed conformation, three symmetry-related β3–β4 loops are tightly packed against one another at the center of the trimer, resulting in a completely occluded central pore (Fig. 3 ▶ c).
3.3. Details of the cpEutL–cobalamin interaction
The novel feature in our structure is the presence of the vitamin B12 (cobalamin) ligand. Electron-density maps calculated early in the model-building and refinement process showed clear, positive difference density features (17σ in mF o − DF c maps) that matched the shape of the corrin ring of the cobalamin molecule. Placement of cobalamin into the density positions the imidazole ring of His32 as an axial ligand to the Co atom of the cobalamin. Further atomic refinement and phase improvement resulted in electron-density maps in which more of the ligand, including the dimethylbenzimidazole (DMB) tail, was visible (Figs. 4 ▶ a and 4 ▶ b). At the end of the atomic refinement process, the placement of the cobalamin ligand in the electron density was verified by inspection of an omit map in which the ligand molecule and the His32 side chain were excluded from atomic refinement and subsequent phase calculations (Fig. 4 ▶ c), and also by calculating the RSCC between the modeled ligand atoms and the corresponding electron density, which was 0.877. Additionally, the ligand geometry was judged to be acceptable based on statistics reported by the PDB structure validation server.
Figure 4.

Crystallographic evidence of the cpEutL–B12 interaction. Placement of the hydroxocobalamin ligand (yellow sticks) in the model is justified by 2mF o − DF c and mF o − DF c electron-density maps (a), (b) and also by an omit map (c). The electron density supports a binding mode in which His32 (light blue sticks) ligates the Co atom (violet) at the center of the corrin ring. The gray mesh represents a 2mF o − DF c map contoured at 1.0σ, and the green/red mesh represents an mF o − DF c difference map (or similar type of omit map) contoured at 3.0σ.
In our structure, cobalamin is ligated to His32 through a His–Co coordination bond (Fig. 4 ▶ b) and is positioned so that it protrudes from the luminal face of the cpEutL pseudohexamer (Fig. 3 ▶ c). Only one ligand molecule is present per trimer (at approximately 95% occupancy as estimated by automated refinement) within the crystal, even though the cpEutL trimer presents three chemically identical binding sites. This is likely to be due to occlusion of the other two potential binding sites by crystal packing interactions that were pre-formed prior to soaking the ligand into the crystal. The position of the bound ligand molecule also suggests why co-crystallization results in lattice-translocation disorders; the bound cobalamin molecules would likely interfere with ordered stacking of two-dimensional molecular sheets in a hexagonally layered cpEutL crystal.
In the observed binding orientation, the cobalamin ligand makes little contact with the protein aside from the His–Co interaction (Fig. 4 ▶ b), presumably giving the ligand some degree of orientational freedom, which may explain several idiosyncrasies of the structure determination. Firstly, the disordered nature of the ligand is reflected in our electron-density maps. Specifically, the map features corresponding to cobalamin, while unambiguous, are not especially well resolved (Figs. 4 ▶ a and ▶ b), which might result from displacement of the ligand in one unit cell relative to another. Secondly, the intrinsic flexibility of this large ligand could explain the relatively high average B factors for the ligand atoms relative to the protein molecules (97.9 versus 60.7 Å2). Additionally, the refined value for the ligand occupancy was 0.95. It is worth noting, however, that in the present case, in which the ligand is somewhat disordered relative to the associated protein molecule, it is very difficult to differentiate between disorder (high B factors) and low occupancy as the cause of reduced scattering contribution from the ligand molecule. Finally, the optimal TLS parameterization for atomic refinement (judged by minimization of R work, R free ▶ and the R free–R work gap) was one in which the three protein chains of the trimer and the cobalamin ligand were all treated as separate TLS groups. The need to treat the ligand as a separate TLS group is consistent with its proposed flexibility and limited atomic interactions with the protein. Interestingly, the average TLS contribution to the total B factors for the ligand atoms is large relative to the protein atoms (42.9 versus 22.6 Å2).
3.4. Biological implications and open questions
While several features of the cpEutL–cobalamin co-crystal structure are consistent with expected features of protein–cobalamin interactions, there are also some puzzling aspects of the observed binding mode. Other cobalamin ligation motifs similar to the one observed in our structure, involving a histidine and an acidic residue (Glu30 in cpEutL), have been seen before in other cobalamin-bound protein structures (Drennan et al., 1994 ▶; Wuerges et al., 2007 ▶; Koutmos et al., 2009 ▶). Often in these motifs the imidazole group of a histidine side chain coordinates the cobalamin by displacing the axial DMB ligand to the Co atom. In our structure of cobalamin bound to cpEutL, the histidine side chain instead appears to displace the hydroxyl group of hydroxocobalamin, creating a complex in which both the histidine and the axial DMB group are simultaneously ligated to the Co atom at the center of the corrin ring (Fig. 4 ▶ b), similar to the ligation arrangement observed in the bovine transcobalamin protein (Wuerges et al., 2007 ▶). Another point of uncertainty is that the apparent ligation motif (sequential amino acids 30EPH32) that seems to be important for cobalamin binding is not well conserved across EutL homologs, which is not what would normally be expected for a functionally relevant amino-acid sequence. In the structure reported here, the ligand molecule is protruding from the luminal face of the pseudohexamer (Fig. 3 ▶ c), and it may be that the observed binding site is an intermediate binding position that is occupied during the transfer of cobalamin molecules to or from ethanolamine-ammonia lyase enzymes (EutBC) inside the microcompartment, but this idea is speculative at present. Indeed, more work will be required to validate the physiological relevance of the observed protein–ligand interaction and place it into a meaningful biochemical context.
4. Conclusion
A major shortcoming in our knowledge of MCP function is our insufficient understanding of molecular-transport phenomena through the semi-permeable protein shell that separates the MCP interior from the cytoplasm. In terms of understanding how large cofactors traverse the shell, much of our knowledge has been derived from studying the EutL shell protein. Despite much progress, we are still unaware of the identity of the molecule that passes through the central pore of EutL when it is in the open conformation. The chemistry of the Eut MCP, however, suggests that it is either ATP or some form of cobalamin, but uncertainty about the subcellular localization of some Eut enzymes leaves ambiguity. Our co-crystal structure of cpEutL bound to cobalamin suggests that cobalamin compounds might bind to EutL and that these compounds could be the transport substrates of the open EutL pore. In our structure, cobalamin is ligated to His32, which displaces the hydroxyl group from hydroxocobalamin, forming an axial His–Co coordination bond opposite the axial Co–DMB coordination bond. In this orientation the ligand is positioned so that it protrudes from the luminal face of the EutL pseudohexamer. The structure presented here provides the basis for a detailed biochemical analysis of the interaction between EutL shell proteins and cobalamin compounds, and for an investigation of the role of this interaction in MCP function.
Supplementary Material
PDB reference: EutL bound to cobalamin, 4u6i
. DOI: 10.1107/S2053230X1402158X/wd5239sup1.pdf
Diffraction images from crystals showing disorder
Acknowledgments
The authors wish to thank Drs Michael Sawaya and Duilio Cascio for helpful suggestions regarding X-ray structure determination, as well as Jason Navarro and Michael Collazo for their technical assistance with crystallization screening. This work was supported by NIH grant R01AI081146 (TOY and TAB) and by the BER program of the DOE Office of Science. X-ray data collection was supported by DOE grant DE-FC02-02ER63421 and the NE-CAT beamlines of the Advanced Photon Source, which are supported by National Institutes of Health grant RR-15301(NCRR). We thank the NE-CAT beamline staff for their technical assistance. Use of the Advanced Photon Source is supported by the DOE, Office of Basic Energy Sciences under Contract DE-AC02-06CH11357.
Footnotes
Supporting information has been deposited in the IUCr electronic archive (Reference: WD5239).
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Afonine, P. V., Grosse-Kunstleve, R. W., Echols, N., Headd, J. J., Moriarty, N. W., Mustyakimov, M., Terwilliger, T. C., Urzhumtsev, A., Zwart, P. H. & Adams, P. D. (2012). Acta Cryst. D68, 352–367. [DOI] [PMC free article] [PubMed]
- Bernstein, F. C., Koetzle, T. F., Williams, G. J. B., Meyer, E. F. Jr, Brice, M. D., Rodgers, J. R., Kennard, O., Shimanouchi, T. & Tasumi, M. (1977). J. Mol. Biol. 112, 535–542. [DOI] [PubMed]
- Buan, N. R., Suh, S.-J. & Escalante-Semerena, J. C. (2004). J. Bacteriol. 186, 5708–5714. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Cheng, S., Liu, Y., Crowley, C. S., Yeates, T. O. & Bobik, T. A. (2008). Bioessays, 30, 1084–1095. [DOI] [PMC free article] [PubMed]
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Mol. Biol. 4, 269–275. [DOI] [PubMed]
- Drennan, C. L., Matthews, R. G. & Ludwig, M. L. (1994). Curr. Opin. Struct. Biol. 4, 919–929. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Held, M., Quin, M. B. & Schmidt-Dannert, C. (2013). J. Mol. Microbiol. Biotechnol. 23, 308–320. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Karplus, P. A. & Diederichs, K. (2012). Science, 336, 1030–1033. [DOI] [PMC free article] [PubMed]
- Kerfeld, C. A., Heinhorst, S. & Cannon, G. C. (2010). Annu. Rev. Microbiol. 64, 391–408. [DOI] [PubMed]
- Kerfeld, C. A., Sawaya, M. R., Tanaka, S., Nguyen, C. V., Phillips, M., Beeby, M. & Yeates, T. O. (2005). Science, 309, 936–938. [DOI] [PubMed]
- Kofoid, E., Rappleye, C., Stojiljkovic, I. & Roth, J. (1999). J. Bacteriol. 181, 5317–5329. [DOI] [PMC free article] [PubMed]
- Koutmos, M., Datta, S., Pattridge, K. A., Smith, J. L. & Matthews, R. G. (2009). Proc. Natl Acad. Sci. USA, 106, 18527–18532. [DOI] [PMC free article] [PubMed]
- Mebs, S., Henn, J., Dittrich, B., Paulmann, C. & Luger, P. (2009). J. Phys. Chem. A, 113, 8366–8378. [DOI] [PubMed]
- Mori, K., Bando, R., Hieda, N. & Toraya, T. (2004). J. Bacteriol. 186, 6845–6854. [DOI] [PMC free article] [PubMed]
- Mueller-Dieckmann, C., Kauffmann, B. & Weiss, M. S. (2011). J. Appl. Cryst. 44, 433–436.
- Penrod, J. T. & Roth, J. R. (2006). J. Bacteriol. 188, 2865–2874. [DOI] [PMC free article] [PubMed]
- Roof, D. M. & Roth, J. R. (1988). J. Bacteriol. 170, 3855–3863. [DOI] [PMC free article] [PubMed]
- Sagermann, M., Ohtaki, A. & Nikolakakis, K. (2009). Proc. Natl Acad. Sci. USA, 106, 8883–8887. [DOI] [PMC free article] [PubMed]
- Sampson, E. M. & Bobik, T. A. (2008). J. Bacteriol. 190, 2966–2971. [DOI] [PMC free article] [PubMed]
- Sanger, F., Nicklen, S. & Coulson, A. R. (1977). Proc. Natl Acad. Sci. USA, 74, 5463–5467. [DOI] [PMC free article] [PubMed]
- Sheppard, D. E., Penrod, J. T., Bobik, T., Kofoid, E. & Roth, J. R. (2004). J. Bacteriol. 186, 7635–7644. [DOI] [PMC free article] [PubMed]
- Takenoya, M., Nikolakakis, K. & Sagermann, M. (2010). J. Bacteriol. 192, 6056–6063. [DOI] [PMC free article] [PubMed]
- Tanaka, S., Sawaya, M. R. & Yeates, T. O. (2010). Science, 327, 81–84. [DOI] [PubMed]
- Toraya, T. (2003). Chem. Rev. 103, 2095–2127. [DOI] [PubMed]
- Weiss, M. S. (2001). J. Appl. Cryst. 34, 130–135.
- Wuerges, J., Geremia, S., Fedosov, S. N. & Randaccio, L. (2007). IUBMB Life, 59, 722–729. [DOI] [PubMed]
- Yeates, T. O., Crowley, C. S. & Tanaka, S. (2010). Annu. Rev. Biophys. 39, 185–205. [DOI] [PMC free article] [PubMed]
- Yeates, T. O., Jorda, J. & Bobik, T. A. (2013). J. Mol. Microbiol. Biotechnol. 23, 290–299. [DOI] [PMC free article] [PubMed]
- Yeates, T. O., Kerfeld, C. A., Heinhorst, S., Cannon, G. C. & Shively, J. M. (2008). Nature Rev. Microbiol. 6, 681–691. [DOI] [PubMed]
- Yeates, T. O., Thompson, M. C. & Bobik, T. A. (2011). Curr. Opin. Struct. Biol. 21, 223–231. [DOI] [PMC free article] [PubMed]
- Zwart, P. H., Grosse-Kunstleve, R. W. & Adams, P. D. (2005). CCP4 Newsl. Protein Crystallogr. 43, contribution 7.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: EutL bound to cobalamin, 4u6i
. DOI: 10.1107/S2053230X1402158X/wd5239sup1.pdf
Diffraction images from crystals showing disorder