

This article describes the preliminary crystallographic data of a complex of Feline infectious peritonitis virus main protease with its inhibitor N3.
Keywords: Feline infectious peritonitis virus, main protease, N3 inhibitor
Abstract
Feline infectious peritonitis virus (FIPV) causes a lethal systemic granulomatous disease in wild and domestic cats around the world. Currently, no effective vaccines or drugs have been developed against it. As a member of the genus Alphacoronavirus, FIPV encodes two polyprotein precursors required for genome replication and transcription. Each polyprotein undergoes extensive proteolytic processing, resulting in functional subunits. This process is mainly mediated by its genome-encoded main protease, which is an attractive target for antiviral drug design. In this study, the main protease of FIPV in complex with a Michael acceptor-type inhibitor was crystallized. The complex crystals diffracted to 2.5 Å resolution and belonged to space group I422, with unit-cell parameters a = 112.3, b = 112.3, c = 102.1 Å. There is one molecule per asymmetric unit.
1. Introduction
Feline coronaviruses (FCoVs), which belong to the genus Alphacoronavirus of the subfamily Coronavirinae, are the leading pathogen of Felidae around the world (Horzinek & Osterhaus, 1979 ▶). FCoVs have two antigenically distinct serotypes: type I and type II (Balint, Farsang, Szeredi et al., 2014 ▶). Both serotypes occur in two pathotypes: Feline enteric coronavirus (FECV) and Feline infectious peritonitis virus (FIPV). FECV mainly replicates in the lower portion of the intestinal tract, spreads by the faecal–oral route, and presents clinically as mild or inapparent enteritis (Pedersen et al., 1981 ▶; Herrewegh et al., 1997 ▶; Balint, Farsang, Zadori et al., 2014 ▶). On the other hand, FIPV efficiently replicates in macrophages/monocytes, leading to feline infectious peritonitis (FIP), a highly lethal systemic granulomatous disease of wild and domestic cats (Addie et al., 2009 ▶; Pedersen, 2009 ▶; Balint, Farsang, Szeredi et al., 2014 ▶; Balint, Farsang, Zadori et al., 2014 ▶; Kipar & Meli, 2014 ▶). FIP, as an immune-mediated disease, features the antibody-dependent enhancement (ADE) phenomenon (Weiss & Scott, 1981 ▶; Vennema et al., 1990 ▶; Tirado & Yoon, 2003 ▶). The virus-specific antibodies induced upon infection by FIPV in cats do not protect them, but enhance the infection by FIPV and thus accelerate the disease symptoms (Weiss & Scott, 1981 ▶; Vennema et al., 1990 ▶; Tirado & Yoon, 2003 ▶; Takano et al., 2008 ▶). Hence, there has been little progress in the development of clinical vaccines against FIPV, although a variety of methods have been tried, such as avirulent vaccines, attenuated live FIPV vaccines and recombinant vaccines (Pedersen, 2009 ▶).
Like other coronaviruses, FIPV contains a single-stranded positive-sense polyadenylated RNA genome that encodes two large polyproteins (pp1a and pp1ab) which need to be processed into 16 nonstructural proteins (nsp1–16) for genome replication (Dye & Siddell, 2005 ▶). This process is mediated by two virus-encoded proteinases. Nsp5, also named main protease (Mpro), is responsible for 11 out of 15 cleavage sites, thus playing a pivotal role in this digestion process and being indispensable for viral replication (Dye & Siddell, 2005 ▶). The critical role of nsp5 in virus replication makes it an ideal target for anti-FIPV drug design (Yang et al., 2005 ▶; Anand et al., 2003 ▶). To date, several crystal structures of other coronavirus main proteases have been solved (Anand et al., 2002 ▶, 2003 ▶; Yang et al., 2003 ▶, 2005 ▶; Hilgenfeld, 2014 ▶). Based on the structural analysis of these main proteases, the idea of designing wide-spectrum inhibitors against CoVs has been proposed. In this study, we report the crystallization and preliminary crystallographic study of FIPV main protease in complex with a designed Michael acceptor inhibitor named N3 (Supplementary Fig. S11).
2. Materials and methods
2.1. Macromolecule production
The coding sequence for FIPV main protease was synthesized and was cloned into the vector pGEX-6P-1 using the BamHI and XhoI restriction sites (Table 1 ▶). The recombinant plasmid was verified by sequencing and then transformed into Escherichia coli strain BL21 (DE3) for protein expression. Cultures were grown in LB medium containing 0.1 mg ml−1 ampicillin at 310 K until the optical density at 600 nm reached 0.6. Isopropyl β-d-1-thiogalactopyranoside was then added to a final concentration of 0.5 mM and the cultures were induced to express FIPV main protease at 289 K for 16 h. Thereafter, centrifugation was used to harvest the cells and the bacterial pellets were resuspended in PBS (140 mM NaCl, 10 mM Na2HPO4, 2.7 mM KCl, 1.8 mM KH2PO4 pH 7.3) supplemented with 1 mM dithiothreitol (DTT) and 10% glycerol. After sonication at 277 K, the bacterial lysate was centrifuged at 12 000g for 50 min at 277 K and the precipitate was discarded. The supernatant was loaded onto a disposable column containing glutathione Sepharose 4B affinity resin (Pharmacia) to purify the GST-tagged FIPV main protease. The fusion protein was then subjected to on-column cleavage using commercial PreScission protease (Pharmacia) at 277 K for 18 h. The protease was added to a final concentration of 0.25 mg ml−1 for proteolysis in PBS. Five additional residues (GPLGS) were left at the N-terminus of FIPV main protease. The resulting protein of interest was further purified by anion-exchange chromatography using a HiTrap Q column (GE Healthcare) with a linear gradient from 25 to 250 mM NaCl in 20 mM Tris–HCl pH 8.0, 10% glycerol, 1 mM DTT and reached more than 90% purity by SDS–PAGE analysis (Fig. 1 ▶ a).
Table 1. Macromolecule-production information.
| Source organism | FIPV strain 791146 (GenBank accession No. AF326575) |
| DNA source | Synthetic plasmid |
| Forward primer† | CGGGATCCAGCGGTCTGCGTAAAATG |
| Reverse primer‡ | CCGCTCGAGTTACTGCAGATTAACGCCATAC |
| Cloning vector | pGEX-6P-1 |
| Expression vector | pGEX-6P-1 |
| Expression host | E. coli BL21 (DE3) |
| Complete amino-acid sequence of the construct produced | GPLGSSGLRKMAQPSGVVEPCIVRVAYGNNVLNGLWLGDEVICPRHVIASDTSRVINYENELSSVRLHNFSIAKNNAFLGVVSAKYKGVNLVLKVNQVNPNTPEHKFKSVRPGESFNILACYEGCPGSVYGVNMRSQGTIKGSFIAGTCGSVGYVLENGTLYFVYMHHLELGNGSHVGSNLEGEMYGGYEDQPSMQLEGTNVMSSDNVVAFLYAALINGERWFVTNTSMTLESYNAWAKTNSFTEIVSTDAFNMLAAKTGYSVEKLLECIVRLNKGFGGRTILSYGSLCDEFTPTEVIRQMYGVNLQ |
Underlined sequence: BamHI site.
Underlined sequence: XhoI site.
Figure 1.

Purification and crystallization of FIPV main protease. (a) SDS–PAGE analysis of purified FIPV main protease. The molecular masses of the marker and FIPV main protease are indicated in kDa. (b) Typical complex crystals of FIPV main protease with inhibitor N3 grown by the hanging-drop method. These crystals, with typical dimensions of 0.03 × 0.03 × 0.03 mm, were used for subsequent diffraction and data collection.
2.2. Crystallization
The purified protein was immediately supplemented with 10% DMSO and concentrated to 1 mg ml−1. The previously reported inhibitor N3 (Yang et al., 2005 ▶), dissolved in 100% DMSO to a final concentration of 10 mM as a stock, was added to the purified protein to give a molar ratio of between 3:1 and 5:1. After mixing at 4°C for 4 h, the protein complex was centrifuged at 12 000g for 10 min and exchanged into a buffer consisting of 10 mM HEPES pH 7.5, 150 mM NaCl, 1 mM DTT using Thermo iCON concentrators. The final protein was concentrated to 8 mg ml−1 for crystallization. In the initial stage, commercial screening kits, including Crystal Screen, Crystal Screen 2, PEG/Ion and Index (Hampton Research, Laguna Niguel, California, USA), were used to screen for preliminary crystallization conditions for FIPV main protease with the inhibitor N3. Crystallization trials were set up in 16-well crystallization plates at 291 K using the hanging-drop vapour-diffusion method. Crystallization drops were carefully set up by mixing 1.0 µl protein solution with 1.0 µl reservoir solution and were then left to equilibrate against 200 µl reservoir solution. Initial crystals of FIPV main protease in complex with N3 inhibitor were obtained after 24 h under condition No. 45 from Crystal Screen, which consists of 0.2 M zinc acetate dihydrate, 0.1 M sodium cacodylate trihydrate pH 6.5, 18%(w/v) polyethylene glycol 8000. The optimized condition consisted of 0.2 M zinc acetate dihydrate, 0.1 M sodium cacodylate trihydrate pH 6.5, 14%(w/v) polyethylene glycol 8000 (Fig. 1 ▶ b). The crystallization information is summarized in Table 2 ▶.
Table 2. Crystallization.
| Method | Hanging-drop vapour diffusion |
| Plate type | 16-well crystallization plate |
| Temperature (K) | 291 |
| Protein concentration (mgml1) | 8 |
| Buffer composition of protein solution | 10mM HEPES pH 7.5, 150mM NaCl, 1mM DTT |
| Composition of reservoir solution | 0.2M zinc acetate dihydrate, 0.1M sodium cacodylate trihydrate pH 6.5, 14%(w/v) polyethylene glycol 8000 |
| Volume and ratio of drop | 1.0l + 1.0l |
| Volume of reservoir (l) | 200 |
2.3. X-ray data collection and processing
The crystals were cryoprotected in a solution consisting of 0.2 M zinc acetate dihydrate, 0.1 M sodium cacodylate trihydrate pH 6.5, 14%(w/v) polyethylene glycol 8000, 20% glycerol and were then mounted in a nylon loop and flash-cooled in a nitrogen stream at 100 K. Data were collected using an ADSC Q315r detector on beamline BL17U of the Shanghai Synchrotron Radiation Facility (SSRF) at a wavelength of 0.97923 Å. A complete data set was collected from a single crystal which diffracted to 2.5 Å resolution (Fig. 2 ▶). All intensity data were indexed, integrated and scaled with the HKL-2000 package (Otwinowski & Minor, 1997 ▶). The related data-collection and processing statistics are summarized in Table 3 ▶.
Figure 2.

A typical diffraction pattern of an FIPV main protease complex crystal collected on beamline BL17U of the Shanghai Synchrotron Radiation Facility (SSRF). The edge of the frame is at 1.76 Å resolution. The box shows diffraction spots in the outer resolution shell.
Table 3. Data-collection and processing statistics.
Values in parentheses are for the outer shell.
| Diffraction source | Beamline BL17U, SSRF |
| Wavelength () | 0.97923 |
| Temperature (K) | 100 |
| Detector | ADSC Q315r |
| Crystal-to-detector distance (mm) | 250 |
| Rotation range per image () | 1 |
| Total rotation range () | 95 |
| Space group | I422 |
| Unit-cell parameters (, ) | a = b = 112.3, c = 102.1, = = = 90 |
| Mosaicity () | 0.83 |
| Resolution range () | 50.02.45 (2.492.45) |
| Total No. of reflections | 89406 (4704) |
| No. of unique reflections | 12082 (619) |
| Completeness (%) | 97.8 (99.4) |
| Multiplicity | 7.4 (7.6) |
| I/(I) | 44.1 (8.6) |
| R merge † (%) | 7.0 (44.1) |
| R meas ‡ (%) | 7.5 (47.3) |
R
merge = 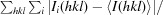
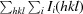 , where Ii(hkl) is the intensity of the ith observation of reflection hkl and I(hkl) is the average intensity.
, where Ii(hkl) is the intensity of the ith observation of reflection hkl and I(hkl) is the average intensity.
R meas is calculated by multiplying the R merge value by the factor [N/(N 1)]1/2.
3. Results and discussion
FIP, which is mainly caused by FIPV, is a lethal systemic granulomatous disease of cats around the world (Addie et al., 2009 ▶; Pedersen, 2009 ▶; Balint, Farsang, Szeredi et al., 2014 ▶; Balint, Farsang, Zadori et al., 2014 ▶; Kipar & Meli, 2014 ▶). However, upon virus infection the induced antibodies further enhance the infection instead of neutralizing the virus. This is the so-called ‘antibody-dependent enhancement’ (Weiss & Scott, 1981 ▶; Vennema et al., 1990 ▶; Tirado & Yoon, 2003 ▶). Thus, no clinical vaccines against FIPV have been successfully developed to date. For this reason, the FIPV main protease, which is indispensable for virus replication, is an alternative target for antiviral therapy and has been subjected to crystallographic studies.
FIPV main protease was expressed as a GST-tagged protein, digested using commercial PreScission protease (Pharmacia) and was then purified using anion-exchange chromatography on a HiTrap Q column. The final protein used for crystallization trials reached greater than 90% purity as monitored by SDS–PAGE. Crystals could be obtained from condition No. 45 of Crystal Screen. The optimized crystals diffracted to a highest resolution of 2.5 Å using 0.2 M zinc acetate dihydrate, 0.1 M sodium cacodylate trihydrate pH 6.5, 14%(w/v) polyethylene glycol 8000, 20% glycerol as a cryoprotectant. The crystal belonged to space group I422, with unit-cell parameters a = 112.3, b = 112.3, c = 102.1 Å. Based on the molecular weight of the monomer, the Matthews coefficient (Matthews, 1968 ▶) was calculated to be 2.43 Å3 Da−1 and the solvent content was 49.5%, assuming the presence of one molecule per asymmetric unit. Further structural and biochemical analysis of FIPV main protease in complex with the Michael acceptor N3 will lead to better design and optimization of antivirals against FIP.
Supplementary Material
Chemical structure of the compound N3.. DOI: 10.1107/S2053230X14022390/no5064sup1.pdf
Acknowledgments
We would like to thank Zuokun Lu for his help with data collection on beamline BL17U of the Shanghai Synchrotron Radiation Facility (SSRF). This work was supported by the National Natural Science Foundation of China (31300150), the Specialized Research Fund for the Doctoral Program of Higher Education of China (20130032120090) and Tianjin Municipal Natural Science Foundation (General Program: 13JCYBJC42500).
Footnotes
Supporting information has been deposited in the IUCr electronic archive (Reference: NO5064).
References
- Addie, D. et al. (2009). J. Feline Med. Surg. 11, 594–604. [DOI] [PMC free article] [PubMed]
- Anand, K., Palm, G. J., Mesters, J. R., Siddell, S. G., Ziebuhr, J. & Hilgenfeld, R. (2002). EMBO J. 21, 3213–3224. [DOI] [PMC free article] [PubMed]
- Anand, K., Ziebuhr, J., Wadhwani, P., Mesters, J. R. & Hilgenfeld, R. (2003). Science, 300, 1763–1767. [DOI] [PubMed]
- Balint, A., Farsang, A., Szeredi, L., Zadori, Z. & Belak, S. (2014). Vet. Microbiol. 169, 154–162. [DOI] [PMC free article] [PubMed]
- Balint, A., Farsang, A., Zadori, Z. & Belak, S. (2014). PLoS One, 9, e88758. [DOI] [PMC free article] [PubMed]
- Dye, C. & Siddell, S. G. (2005). J. Gen. Virol. 86, 2249–2253. [DOI] [PMC free article] [PubMed]
- Herrewegh, A. A., Mähler, M., Hedrich, H. J., Haagmans, B. L., Egberink, H. F., Horzinek, M. C., Rottier, P. J. & de Groot, R. J. (1997). Virology, 234, 349–363. [DOI] [PMC free article] [PubMed]
- Hilgenfeld, R. (2014). FEBS J. 281, 4085–4096. [DOI] [PMC free article] [PubMed]
- Horzinek, M. C. & Osterhaus, A. D. (1979). Am. J. Vet. Res. 40, 1487–1492. [PubMed]
- Kipar, A. & Meli, M. L. (2014). Vet. Pathol. 51, 505–526. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Pedersen, N. C. (2009). J. Feline Med. Surg. 11, 225–258. [DOI] [PMC free article] [PubMed]
- Pedersen, N. C., Boyle, J. F., Floyd, K., Fudge, A. & Barker, J. (1981). Am. J. Vet. Res. 42, 368–377. [PubMed]
- Takano, T., Kawakami, C., Yamada, S., Satoh, R. & Hohdatsu, T. (2008). J. Vet. Med. Sci. 70, 1315–1321. [DOI] [PubMed]
- Tirado, S. M. C. & Yoon, K.-J. (2003). Viral Immunol. 16, 69–86. [DOI] [PubMed]
- Vennema, H., de Groot, R. J., Harbour, D. A., Dalderup, M., Gruffydd-Jones, T., Horzinek, M. C. & Spaan, W. J. (1990). J. Virol. 64, 1407–1409. [DOI] [PMC free article] [PubMed]
- Weiss, R. C. & Scott, F. W. (1981). Comp. Immunol. Microbiol. Infect. Dis. 4, 175–189. [DOI] [PMC free article] [PubMed]
- Yang, H. et al. (2005). PLoS Biol. 3, e324. [DOI] [PMC free article] [PubMed]
- Yang, H., Yang, M., Ding, Y., Liu, Y., Lou, Z., Zhou, Z., Sun, L., Mo, L., Ye, S., Pang, H., Gao, G. F., Anand, K., Bartlam, M., Hilgenfeld, R. & Rao, Z. (2003). Proc. Natl Acad. Sci. USA, 100, 13190–13195. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Chemical structure of the compound N3.. DOI: 10.1107/S2053230X14022390/no5064sup1.pdf