

Abstract
Resurgent Na+ current results from a distinctive form of Na+ channel gating, originally identified in cerebellar Purkinje neurons. In these neurons, the tetrodotoxin-sensitive voltage-gated Na+ channels responsible for action potential firing have specialized mechanisms that reduce the likelihood that they accumulate in fast inactivated states, thereby shortening refractory periods and permitting rapid, repetitive, and/or burst firing. Under voltage clamp, step depolarizations evoke transient Na+ currents that rapidly activate and quickly decay, and step repolarizations elicit slower channel reopening, or a ‘resurgent’ current. The generation of resurgent current depends on a factor in the Na+ channel complex, probably a subunit such as NaVβ4 (Scn4b), which blocks open Na+ channels at positive voltages, competing with the fast inactivation gate, and unblocks at negative voltages, permitting recovery from an open channel block along with a flow of current. Following its initial discovery, resurgent Na+ current has been found in nearly 20 types of neurons. Emerging research suggests that resurgent current is preferentially increased in a variety of clinical conditions associated with altered cellular excitability. Here we review the biophysical, molecular and structural mechanisms of resurgent current and their relation to the normal functions of excitable cells as well as pathophysiology.
Introduction
In all excitable cells, voltage-gated Na+ channels are closed (deactivated) at negative potentials, open (activate) upon depolarization, and then rapidly become non-conducting (inactivated, but see below), decreasing current flow by about 99% within a few milliseconds (Hodgkin & Huxley, 1952a). In voltage clamp, the brief macroscopic Na+ current evoked by a step depolarization is therefore often referred to as a ‘transient Na+ current’, while the proportionately tiny, residual Na+ current that lasts throughout the step is called ‘persistent Na+ current’. In many cells, relief of inactivation occurs only at strongly negative potentials, without additional current flow (Hodgkin & Huxley, 1952c; Kuo & Bean, 1994). In cells such as cerebellar Purkinje neurons, however, non-conducting Na+ channels have the distinctive characteristic of reopening in response to repolarization after steps to positive potentials. The current that flows through channels that reopen in response to negative voltage changes, after the decay of macroscopic transient current, is called ‘resurgent Na+ current’ (Fig.1A; Raman & Bean, 1997).
Figure 1. Transient, resurgent and persistent Na+ currents.
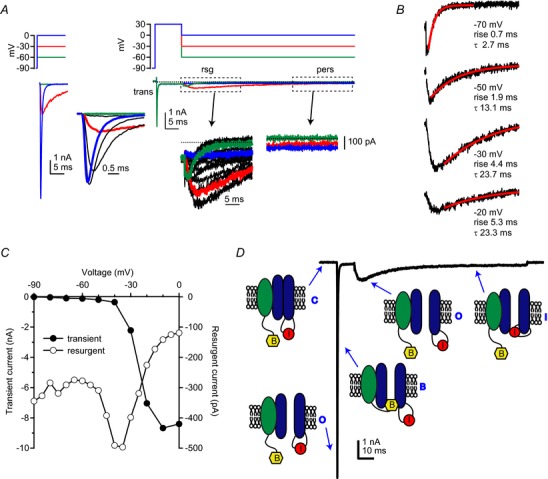
A, TTX-sensitive Na+ currents in a voltage-clamped Purkinje neuron. Left, a family of transient currents evoked by step depolarizations from −90 mV to −60 (green), −30 (red), 0 (blue) mV. Right, in a different cell, transient current evoked by a 10 ms step to +30 mV followed by resurgent and persistent currents elicited by repolarization to the same voltages. Recordings were made at room temperature, in normal Na+ gradients (150 mm external, 10 mm internal). Recordings were repeated in 900 nm TTX to block all current and subtractions gave the illustrated TTX-sensitive current. Insets, currents at higher gain or faster sweep speed, with currents at more voltages shown for transient (Δ10 mV) and resurgent (Δ5 mV) components. In physiological Na+ concentration gradients, resurgent current can be maximized by step depolarizations to +30 mV for 5–50 ms (depending on the decay time of transient current; slower decay requires a longer step) followed by repolarization to −30 to −40 mV; thus this voltage protocol probably maximizes the resurgent current amplitude. B, resurgent current kinetics and amplitude vary with repolarization potential. Currents from (A) are shown from the repolarization step only. The time of peak relative to the time of the repolarization step (rise) is prolonged at less negative repolarizations, reflecting less efficient displacement of the blocker. The single exponential decay time constant (τ) also increases with less negative steps, owing to less concerted transitions into non-conducting states as well as the slower transition of open channels into inactivated, rather than closed states. C, current–voltage relation of transient and resurgent Na+ currents from (A). Maximal Na+ conductance was 120 nS in the cell from which transient currents are shown and 45 nS in the cell from which resurgent currents are shown. Resurgent current scale is 20× the transient current scale. D, schematic of the primary channel states occupied during different phases of transient and resurgent current. The α subunit is blue with a red inactivation gate ‘I’, and the blocking subunit (possibly NaVβ4) is green with a yellow blocking domain ‘B’. Upon depolarization, channels make transitions from closed (‘C’) to open (‘O’) states, eliciting a large transient current. This current decays as channels become blocked (‘B’). Upon repolarization, channels briefly revisit open states before making transitions into fast inactivated states (‘I’). Traces from Aman & Raman (2010). pers, persistent (current); rsg, resurgent (current); trans, transient (current); TTX, tetrodotoxin.
Properties and distribution of resurgent current
Although resurgent current flows through the same channels as transient and persistent current, it exhibits different voltage dependence and kinetics. Resurgent current is distinct from persistent current because it is dynamically gating; at any given potential, it has a rising phase, a peak and a falling phase. With a prolonged repolarizing step, the resurgent current decays to a persistent (steady-state) current. It can be useful to subtract this persistent component when measuring resurgent current amplitudes. Besides being activated by negative rather than positive voltage steps, resurgent current activates and decays more slowly than transient current. For instance, in Purkinje neurons at room temperature, transient current evoked by a step depolarization to −30 mV activates within 500 μs and decays in about 1 ms, while resurgent current evoked by a step repolarization to −30 mV rises in 3–5 ms and has a decay constant near 20 ms (Fig.1B). The slow rise of resurgent current distinguishes it from tail current, which rises instantaneously, flowing through the few channels that remain open throughout the depolarizing step. Tail currents also scale directly with driving force, increasing in amplitude at more negative potentials (Hodgkin & Huxley, 1952b), whereas resurgent currents have a non-monotonic voltage dependence on repolarization. Resurgent current is generally not detectable until the membrane potential is repolarized below 0 mV. With progressively more negative steps, the peak current increases until about −30 mV, and then decreases, remaining measurable to at least −90 mV (Fig.1C). At its maximum near −30 mV, peak resurgent current is about 5–10% of the amplitude of transient current measured at 0 mV (Aman & Raman, 2007; Lewis & Raman, 2011). The mechanistic bases for these properties are further discussed below.
Historically, resurgent current was considered a quirk of Purkinje neurons. Since its identification, however, resurgent current has been found in 19 other classes of cells throughout the nervous system (Table1). The cerebellum and brainstem contain several cell types in which every neuron has resurgent current; in the globus pallidus, parts of the hippocampus and the dorsal root ganglia (DRG), only subsets of certain neuronal classes express the current. Conversely, resurgent current is consistently absent from CA3 pyramidal neurons of the hippocampus, some spinal neurons and neurons of the superior cervical ganglion (Raman & Bean, 1997; Pan & Beam, 1999; Han et al. 2012).
Table 1.
Distribution of resurgent Na+ current, NaVβ4 expression and firing properties
| Cell type with resurgent current | NaVβ4 | Spike pattern | References |
|---|---|---|---|
| Cerebellar Purkinje cells | Yes | Spontaneous; repetitive; bursts* | Raman & Bean, 1997; Yu et al. 2003, Buffington & Rasband, 2013 |
| Subthalamic nuclei | Yes | Spontaneous; repetitive; bursts | Do & Bean, 2003; Buffington & Rasband, 2013 |
| Cerebellar nuclei | Yes | Spontaneous; repetitive; bursts | Raman et al. 2000; Yu et al. 2003; Afshari et al. 2004 |
| Cerebellar granule cells | Yes | Repetitive; bursts | D'Angelo et al. 2001; Chadderton et al. 2004; Afshari et al. 2004; Magistretti et al. 2006; Bant & Raman, 2010 |
| Cerebellar unipolar brush cells | Probably† | Spontaneous; repetitive; bursts | Yu et al. 2003; Afshari et al. 2004; Russo et al. 2007 |
| Large dorsal root ganglion cells | Yes | Repetitive | Abdulla & Smith, 2001; Yu et al. 2003; Cummins et al. 2005 |
| Mesencephalic trigeminal neurons | Yes | Repetitive; bursts | Yu et al. 2003; Enomoto et al. 2006; Buffington & Rasband, 2013 |
| Medial nucleus of the trapezoid body | Yes | Repetitive | Wang et al. 1998; Leão et al. 2006; Buffington & Rasband, 2013 |
| Medial vestibular nuclei: GABAergic | Yes | Spontaneous; repetitive | Gittis & du Lac, 2008; Kodama et al. 2012 |
| Medial vestibular nuclei: Non-GABA | No | Spontaneous; repetitive | Gittis & du Lac, 2008; Kodama et al. 2012 |
| Cartwheel cells (cochlear nucleus) | Probably† | Spontaneous; repetitive; bursts | Raman & Trussell unpublished; Yu et al. 2003; Kim & Trussell, 2007 |
| Globus pallidus (some cells) | Some | Spontaneous; repetitive | Yu et al. 2003; Mercer et al. 2007 |
| Layer II pyramidal cells of perirhinal cortex, area 35 (most cells) | Probably not‡ | Repetitive (most cells) | Yu et al. 2003; Castelli et al. 2007a |
| Hippocampal dentate gyrus (60%) | No | Other | Yu et al. 2003; Castelli et al. 2007b |
| Ventral CA1 pyramidal cells (35%) | Low | Other | Yu et al. 2003; Castelli et al. 2007b; Buffington & Rasband, 2013 |
| Calyx of Held (bushy cells of ventral cochlear nucleus) | Probably† | Repetitive | Yu et al. 2003; Kim et al. 2010 |
| 10% small dorsal root ganglia cells | Yes | Repetitive | Abdulla & Smith, 2001; Yu et al. 2003; Jarecki et al. 2010 |
| Substantia nigra pars reticulata (GABAergic) | Yes | Spontaneous; repetitive | Yu et al. 2003; Ding et al. 2011 |
| Substantia nigra pars compacta (dopaminergic) – small currents | Some | Spontaneous; repetitive | Yu et al. 2003; Ding et al. 2011 |
| Medial entorhinal cortex | Possibly | Repetitive with accommodation | Allen Brain Atlas; Hargus et al. 2011; Nigro et al. 2012 |
Spontaneous, fires regularly without synaptic input; repetitive, fires with little accommodation with repeated or sustained depolarizing input; bursts, fire bursts of action potentials ± depolarizing input; other, none of the above;
based on high expression in brainstem and cerebellum;
based on low expression in this layer of cortex.
Open channel block as a mechanism for resurgent Na+ current
The observation of Na+ currents apparently gated by both positive and negative voltage steps raises questions of how such currents arise. Analysing the currents can reveal specific channel states, probably corresponding to one or more physical conformations of ion channel proteins (Fig.1D). Non-conducting channels that are ready or ‘available’ to open in response to depolarization are defined as being in ‘closed’ states, ‘C’, and conducting channels are in ‘open’ states, ‘O’. Non-conducting channels that are unavailable to open – usually because of ongoing or previous depolarization – are in ‘inactivated’ states, ‘I’. Most voltage-gated Na+ channels recover from inactivation directly (I→C), without reopening and passing measurable current (Kuo & Bean, 1994). In cells with resurgent current, however, the non-conducting states favoured at positive voltages evidently make transitions directly to open states even at moderately negative potentials (just below –10 mV). These non-conducting states must therefore be distinct from classical inactivated states.
Early studies of Na+ currents in squid axons demonstrated that open channels can be reversibly blocked or plugged by exogenous compounds applied to the intracellular face of the membrane. Such compounds, including N-methyl-strychnine, pancuronium ion and thiazine dyes, obstruct channels opened by depolarization, but cannot pass through the pore (Yeh & Narahashi, 1977; Cahalan & Almers, 1979; Armstrong & Croop, 1982); the pentapeptide KIFMK works similarly in expressed Na+ channels (Tang et al. 1996). Repolarization of channels blocked by these agents generates currents with a slow rising phase, termed ‘hooked tail currents’ (Yeh & Narahashi, 1977; see also Armstrong, 1971). The ‘hook’ arises as the blocking agent slowly dissociates from its binding site at negative potentials, after which open channels again pass current before changing conformation into more stable non-conducting states. Binding of the blocking agent therefore appears voltage-dependent. Moreover, the voltage-sensitivity arises because unbinding is facilitated by inwardly permeating ions that repel and displace the blocker (Armstrong, 1971; Tang et al. 1996). The similarity of hooked tails to resurgent current suggests that cells with resurgent current contain an endogenous factor that functions as an open channel blocking particle, which can drive open channels into a ‘blocked’ state, ‘B’. Thus, most voltage-gated Na+ channels of Purkinje cells open with depolarization (C→O) but then, rather than inactivating, they become blocked by an open channel blocking particle (O→B). Upon repolarization, blocked channels reopen as the blocker unbinds (B→O), producing resurgent current. Open channels either inactivate (>∼−50 mV, O→I) or deactivate into closed states (<∼-50 mV, O→C). The various channel conformations and state transitions have been quantitatively elaborated in a 13-state kinetic scheme, comprising five closed states, six inactivated states, one open state and one blocked state, which describes Purkinje Na+ currents recorded in 50 mm extracellular Na+ (Raman & Bean, 2001). Here, we qualitatively summarize the constraints on the model, which future simulations that improve on the original must retain.
Most importantly, a channel cannot be both blocked and inactivated. Despite being mutually exclusive, these states differ in both onset and stability. Inactivation – specifically, fast inactivation owing to binding of the fast inactivation gate (see below) – unlike block, does not require open channels, and inactivation is an absorbing state, whereas blocker binding is rendered unstable by permeating Na+. In fact, O→B is more rapid than O→I at all potentials (Raman & Bean, 2001), such that a depolarization to any voltage, even below 0 mV, initially favours open channel block. The higher the driving force on Na+, the more rapidly the blocker will be displaced – within a few milliseconds at −30 mV but over hundreds of milliseconds at +60 mV – producing an apparent voltage dependence of open channel block (Afshari et al. 2004; Aman & Raman, 2007). Indeed, in reverse concentration gradients, when extracellular Na+ is low, the blocker remains bound at negative potentials and resurgent current does not flow (Aman & Raman, 2010). In a fixed Na+ gradient, the rate of the key transition B→O depends on the affinity of the blocker for the channel; a higher affinity will yield slower dissociation, prolonging the rise of resurgent current. It will likewise lengthen the decay time by desynchronizing the O→I transition. At moderately negative potentials, channels may re-open and re-block repeatedly, delaying inactivation and yielding burst-like single channel openings that increase the amplitude and duration of resurgent current (Raman & Bean, 1997; Aman & Raman, 2010). The O→I transition rate will also contribute to current amplitude and decay time, with slower transitions producing larger and longer resurgent currents. At more negative voltages, the O→C rate begins to dominate, leading to a more rapid curtailment of resurgent current at progressively more negative potentials (Raman & Bean, 2001; Lewis & Raman, 2011).
Transient, resurgent and persistent current thus represent different components of voltage-clamped Na+ current produced by a single voltage-gated Na+ channel. Strictly speaking, because all current flows through a common open state, current can only be identified as belonging to one of the three categories in a voltage-clamped cell, in which channels have been pre-equilibrated into primarily closed or blocked states. In a non-voltage-clamped cell, in which voltage is continually changing and channels make transitions among all states, reference to transient, persistent or resurgent current is rendered ambiguous. For convenience, however, resurgent current can be defined as the current arising as channels unblock (B→O), as distinguished from transient current that arises as channels open from available states (C→O).
Physiological roles of resurgent current
Classical Hodgkin–Huxley kinetics (Hodgkin & Huxley, 1952d) cannot effectively model Na+ channels with resurgent current, because the activation and inactivation gates are not independent. Instead, the onset of block is contingent on channels being open and not inactivated; conversely, a blocked channel can neither inactivate nor deactivate (see Armstrong, 1971). Nevertheless, the physiological effects of resurgent current can be predicted largely by analysing channel gating. The central point is that, upon repolarization, displacement of the blocker is much faster than recovery from fast inactivation (Raman & Bean, 2001; Aman & Raman, 2007, 2010). The time course of restoration of Na+ channel availability determines the duration of refractory periods (Hodgkin & Huxley, 1952c), as well as the number of action potentials that can be fired repetitively before cumulative inactivation precludes further spiking (Colbert et al. 1997; Engel & Jonas, 2005). Thus, by preventing fast inactivation at positive potentials but dissociating rapidly at negative potentials, the blocking particle creates a cycle of opening, block and unblock that is permissive for rapid, repetitive action potential firing. Simulations, as well as experimental work modulating resurgent current (see below), confirm that repetitive firing in Purkinje cells depends upon restoration of Na+ channel availability, primarily through recovery from an open blocked state (Khaliq et al. 2003).
Indeed, many neurons with resurgent current share a capacity for repetitive spiking: rapid spontaneous firing, continuous regular firing during steady depolarization or burst firing (Table1). The spiking pattern of any given cell, however, depends on its full complement of channels (Fig.2); resurgent current facilitates but does not guarantee rapid, spontaneous, or repetitive firing, much as hyperpolarization-activated cation current (Ih) produces pacemaking in some cells but not others (McCormick & Pape, 1990; Magee, 1998). In Purkinje neurons, which fire action potentials 50–100 spikes s–1 spontaneously and up to ∼250 spikes s–1 with depolarization (Khaliq & Raman, 2005; Monsivais et al. 2005), the effect of resurgent current is shaped by extremely large, high-threshold, rapidly deactivating K+ currents (Raman & Bean, 1999; Khaliq et al. 2003), carried mostly by KV3 channels (Akemann & Knöpfel, 2006) and BK channels (Benton et al. 2013). These K+ channels are closed at interspike potentials, permitting a high input resistance against which even small Na+ currents can depolarize effectively. The K+ currents activate powerfully with depolarization, however, truncating Purkinje action potentials so that they are among the briefest in the nervous system (Carter & Bean, 2009). Na+ current flows on the downstroke of action potentials, from unblock of blocked channels as well as from tail currents through open channels (Raman & Bean, 1997; Khaliq et al. 2003; Carter & Bean, 2009). Importantly, the K+ currents are big enough to counteract this depolarization and terminate the action potential, but deactivate quickly enough to prevent a deep afterhyperpolarization that could preclude another action potential from initiating spontaneously (Raman & Bean, 1999; Khaliq et al. 2003). Shifting the balance of channel availability to favour Na+ channels, e.g. via hyperpolarization, can change simple spikes to bursts resembling complex spikes (Raman & Bean, 1997). Firing rates, however, are set mostly by K+ currents; in the medial vestibular nucleus, for example, a strong but short-lasting repolarization is provided by KV3 currents in fast-spiking neurons and by BK currents in more slowly spiking GABAergic neurons (Gittis et al. 2010). In fact, introducing an open Na+ channel blocker into neurons with small K+ currents that lack a native blocker can actually impede rather than favour repetitive firing (Bant et al. 2013).
Figure 2. Firing patterns of different neurons, with and without an open channel Na+ blocker.
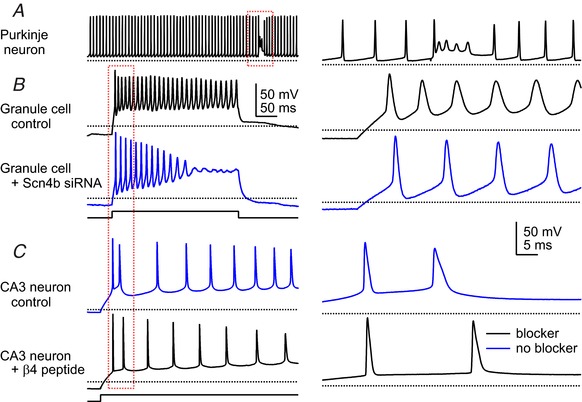
A, spontaneous firing in a Purkinje cell in a cerebellar slice, with an evoked complex spike (within box). This cell has an endogenous blocking protein. The brevity of spikes results largely from repolarization by high-voltage-activated K+ currents. B, action potentials evoked by current injection in two different cultured granule cells, transfected with non-targeted siRNA (control) or siRNA targeted against Scn4b to reduce native NaVβ4 expression. The primary difference is in the ability of the cells to fire throughout the step. C, action potentials evoked by current injection in two different CA3 pyramidal cells in a hippocampal slice, either without a blocker (control) or with the β4 peptide added intracellularly. With peptide, the firing rate does not always increase but the spike waveform consistently narrows and threshold is reduced. Left scale bars apply to all left records, right scale bars to right records. Black dotted line, –60 mV. Boxes on left indicate regions expanded on right. All recordings are from mouse. Traces from Khaliq and Raman (2005, Purkinje), Bant and Raman (2010, granule) and Bant et al. (2013, CA3).
Molecular basis for resurgent Na+ current
The first hint toward the molecular basis of resurgent current came from studies of mice lacking the neuronal voltage-gated Na+ channel α subunit NaV1.6 (Scn8a) (Burgess et al. 1995). In NaV1.6-null Purkinje cells, transient current amplitudes fall by only ∼30%, consistent with expression of NaV1.1 (Scn1a) and NaV1.2 (Scn2a). In contrast, resurgent current decreases by ∼90% (50 mm external Na+; Raman et al. 1997). A similarly drastic preferential reduction of resurgent current is seen in NaV1.6-null neurons from the DRG (large sensory neurons), mesencephalic trigeminal ganglia and hippocampal CA1 region (Cummins et al. 2005; Enomoto et al. 2007; Royeck et al. 2008), demonstrating the necessity of NaV1.6 for normal resurgent current in these cell types. Surprisingly, in NaV1.1-null mice, transient and resurgent current in Purkinje cells decrease in parallel (Kalume et al. 2007), raising the possibility that NaV1.1 somehow regulates NaV1.6 expression in these cells.
Considerable evidence suggests, however, that NaV1.6 is neither solely responsible, nor even an obligate factor, for resurgent current. First, in CA3 pyramidal neurons and large spinal motoneurons, as well as in heterologous expression systems, expressing NaV1.6 does not generate resurgent current, suggesting that NaV1.6 lacks an intrinsic blocking domain (Raman & Bean, 1997; Smith et al. 1998; Pan & Beam, 1999). Second, even in Purkinje neurons of NaV1.6-null mice, resurgent current can be restored by peptide toxins that slow the rate of fast inactivation (Grieco & Raman, 2004). Third, heterologously expressing α subunits of skeletal muscle (NaV1.4), cardiac muscle (NaV1.5) or peripheral neurons (NaV1.7) in DRG neurons that contain an endogenous blocking protein can also produce resurgent currents (Jarecki et al. 2010). Fourth, in the subthalamic nuclei, cerebellar nuclei, globus pallidus and cerebellar granule layer, loss of NaV1.6 only mildly to moderately reduces resurgent current (Do & Bean, 2004; Aman & Raman, 2007; Mercer et al. 2007; Osorio et al. 2010), suggesting that NaV1.1 and NaV1.2 normally carry resurgent current in these cells. Interestingly, NaV1.6 independent, tetrodotoxin-sensitive resurgent current in cerebellar granule cells as well as tetrodotoxin-resistant NaV1.8-mediated resurgent currents in DRG cells both have remarkably slow decay times of ∼50 ms and ∼800 ms respectively, hinting at effects of the α subunit on resurgent current kinetics (Bant & Raman, 2010; Tan et al. 2014). Thus, the native blocking particle, unlike the fast inactivation gate, is not part of NaV1.6 or any other α subunit. Because it is retained in excised patches and appears subject to proteolysis and dephosphorylation (see below), the blocker is probably a protein within the Na+ channel complex (Grieco et al. 2002). It can interact with several α subunits, although blocking efficiency apparently varies across cells.
At present, the only identified endogenous open channel blocker is NaVβ4 (Scn4b) (Grieco et al. 2005; Bant & Raman, 2010). Like other known Na+ channel β subunits (Scn1b, Scn2b, Scn3b), this ∼200 amino acid subunit has one transmembrane domain, an extracellular domain with an immunoglobulin-like fold, and a short cytoplasmic C-terminus (Yu et al. 2003, Gilchrist et al. 2013). Like NaVβ2, NaVβ4 associates covalently with α subunits (Yu et al. 2003; Buffington & Rasband, 2013; Fig.3A). A distinguishing feature of NaVβ4, however, is that it contains a nine-amino-acid insert in the C-terminus (residues 158–166). In mice, the amino acids immediately after the transmembrane segment, which include the insert, have the sequence KKLITFILKKTREK (residues 154–167). Based on its loose resemblance to the KIFMK peptide, Grieco et al. (2005) tested whether a peptide with this sequence (the ‘β4 peptide’) could block Na+ channels. In inside–out patches from Purkinje neurons, the native blocking particle was first destroyed by brief exposure of inside–out patches to trypsin or alkaline phosphatase, which enlarged and slowed transient currents and removed resurgent currents. Next, the β4 peptide was applied to the intracellular face of the patches. It blocked the channel upon depolarization and unblocked upon repolarization, shortening transient currents and restoring resurgent-like currents. Importantly, the kinetics of peptide-dependent and native transient and resurgent currents were indistinguishable. The β4 peptide and native blocking particle therefore must have similar affinities for Purkinje Na+ channels. Thus, NaVβ4 emerged as a good candidate for an endogenous open channel blocker.
Figure 3. .
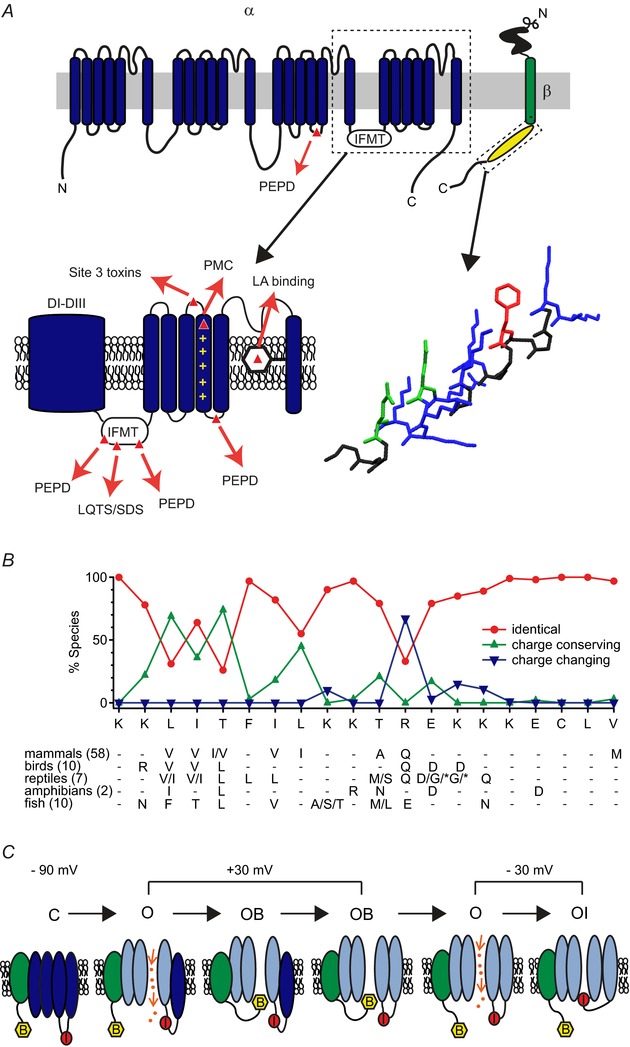
A, schematic of the topology of Na+ channel α (upper left) and β (upper right) subunits. Boxed regions are shown in more detail in lower panels. Mutations in the α subunit that affect resurgent current are highlighted in red, and correspond to the human diseases paroxysmal extreme pain disorder (V1299F, T1464I, M1627K in NaV1.7), LQTS/SDS (F1468L in NaV1.5) and PMC (R1448P in NaV1.4) (Jarecki et al. 2010; Theile et al. 2011). The site 3 toxin binding site and a residue implicated in both LA and β4 peptide binding (F1760K in NaV1.5) are also highlighted (Wang et al. 2006; Hanck & Sheets, 2007). Lower right, the β4 peptide rendered as an idealized α-helix in DeepView. Charged residues are blue (positive) and green (negative); the phenylalanine that contributes to stable block is red. B, evolutionary conservation and divergence among species of the region of NaVβ4 corresponding to the β4 peptide. Sequences from 86 vertebrate species were compared to the mouse β4 sequence with each residue assigned as identical, charge conserving, or charge changing. Charged residues and a phenylalanine residue are particularly highly conserved. Below, list of substitutions at each residue for each class. C, schematic illustrating how voltage sensor movement influences the susceptibility of a channel to block. At rest, all four domains (blue ellipses) are in an inward position and the channel is non-conducting (‘C’). Upon depolarization, the voltage sensors of DI–DIII deploy, opening the channel and allowing current to flow (‘O’). Deployment of the voltage sensor of DIV is delayed relative to the other domains. The blocker can bind (‘OB’) before the DIV sensor deploys. Subsequent outward movement of DIV, which usually favours fast inactivation, stabilizes the bound blocker. Upon repolarization, inwardly permeating Na+ ions knock off the blocker, returning the channel to a conducting state. With prolonged time at a moderately negative potential (e.g. −30 mV), where block is unstable, channels eventually make transitions into fast inactivated states (‘OI’). IFMT, isoleucine, phenylalanine, methionine, and threonine (motif); LA, local anaesthetic; LQTS, long QT syndrome; PEPD, paroxysmal extreme pain disorder; PMC, paramyotonia congenita.
Indeed, when expression of NaVβ4 is reduced by siRNA in cultured cerebellar granule cells, resurgent current is reduced proportionately to the degree of knockdown, and repetitive firing is compromised. (Note that these cells do not fire spontaneously.) The β4 peptide restored both resurgent current and sustained repetitive firing, supporting the idea that both siRNA-induced changes result from the loss of an open channel blocking action by the subunit. Strikingly, NaVβ4 knockdown also negatively shifted steady-state inactivation curves of transient currents, and the β4 peptide restored the curves to normal, more depolarized values, illustrating the intimate relation between block – or the blocking sequence itself – and fast inactivation (Bant & Raman, 2010). The β4 peptide can also induce a resurgent-like current in neurons lacking endogenous blocking proteins, including CA3 pyramidal cells (Grieco et al. 2005) and HEK cells expressing NaV1.1, 1.4, 1.5, 1.6 and 1.7 (Wang et al. 2006; Aman et al. 2009; Theile et al. 2011), demonstrating generalizability of an open channel block by this peptide and its utility for studying resurgent current.
Despite strong evidence that NaVβ4 is a functional blocking protein in some cells, however, it has been impossible to reconstitute resurgent current in heterologous systems of non-excitable cells. Expressing full-length NaVβ4 with NaV1.6, NaV1.1 or NaV1.7 in HEK cells produces channels without resurgent current (Chen et al. 2008; Aman et al. 2009; Theile et al. 2011), revealing that the subunit does not function as a blocking protein under all conditions. Possibly, other proteins or modifications to NaVβ4 are required for the subunit to act as a blocker or for the α subunit to be receptive to block by the protein. The lack of reconstitution may also indicate peculiarities of the expression system, however. Na+ channel gating properties in HEK cells are greatly altered from those in excitable cells, with activation curves of transient currents shifted 20–30 mV depolarized (Aman & Raman, 2007; Aman et al. 2009; Fu et al. 2011). Such changes may influence the efficacy of an open channel block; even β4 peptide-induced block is unstable in HEK cells, requiring extremely large depolarizations (+60 mV) for binding and only weak repolarization (−10 mV) for unbinding (Aman et al. 2009).
Reconstitution notwithstanding, many neurons with resurgent current indeed express NaVβ4, and some cells without resurgent current (CA3 pyramidal neurons) lack NaVβ4 (Table1). Moreover, much like NaV1.6 (Caldwell et al. 2000; Royeck et al. 2008), NaVβ4 expression is enriched in axons and nodes of Ranvier (Buffington & Rasband, 2013), positioning it to participate in spike initiation in several cell types. In fact, in neurons from the perirhinal cortex, resurgent current appears restricted to the initial segment (Castelli et al. 2007a). In many spontaneously firing neurons, which usually have high densities of somatic Na+ channels, cell bodies have resurgent current, however (Raman & Bean, 1997; Do & Bean, 2003; Afshari et al. 2004). Even pharmacologically silencing Na+ channels in Purkinje initial segments or first nodes of Ranvier does not disrupt action potential generation (Khaliq & Raman, 2006), suggesting a broader subcellular distribution of NaVβ4 in some neurons. Moreover, the roles of NaVβ4 may be quite diverse; it appears involved in neurite outgrowth and is specifically downregulated in mouse models of Huntington's disease (Ohyama et al. 2006).
Not all cells with resurgent current express detectable NaVβ4, however; two salient examples are GABAergic neurons of the medial vestibular nucleus (Gittis & du Lac, 2008; Kodama et al. 2012) and hippocampal dentate granule neurons, in which just over half the cells have resurgent current (Yu et al. 2003; Castelli et al. 2007b). Such observations raise the possibility that additional blocking proteins remain to be discovered. If they exist, these blocking proteins may share structural features with NaVβ4 that permit them to interact with a binding site (or sites) in the permeation pathway, and these features are probably evolutionarily conserved. To date, resurgent current has been recorded in Purkinje neurons from rat, mouse, electric fish and chick (Raman & Bean, 1997; Raman et al. 1997; de Ruiter et al. 2006; Lewis & Raman, 2011). Across species, the putative blocking sequence of NaVβ4 is highly conserved at some sites and diverges at others; in 50 vertebrates, the sequence is K+ΦΦxFΦΦK+xxxKK−CLV, where Φ, + and – indicate neutral, positive and negative residues, and x is a non-conservative substitution. Four other species have either an F6L or a K10S substitution (Fig.3B). Despite the variability, β4 peptides with mouse, human, chick, frog and cow sequences all generate resurgent current when introduced into mouse CA3 neurons, suggesting that the most relevant residues for the open channel block include the aromatic phenylalanine and the positively charged residues (Lewis & Raman, 2011). Indeed, altering the properties or position of F6 or the charge or side chain branching pattern of K10/K11 greatly changes the affinity of the blocker. Such mutational studies also suggest that the channel-bound peptide may assume a helical conformation, with the phenylalanine stabilizing the open channel block at all potentials and the unbranched alkylammonium lysine side chains favouring binding at positive voltages while allowing unbinding with repolarization (Grieco et al. 2005; Lewis & Raman, 2011). Thus, the FΦΦK+ motif may be a guide toward finding additional open channel blocking subunits.
Clues about the blocker binding site come from studies of local anaesthetics. The β4 peptide cannot block NaV1.5 channels in which a phenylalanine residue in the local anaesthetic binding site has been mutated (Ragsdale et al. 1994; Wang et al. 2006). Likewise, briefly exposing inside–out Purkinje cell patches to chymotrypsin, which preferentially cleaves aromatic residues, not only removes native open channel block, but also makes channels insensitive to blockade by the local anaesthetic derivative QX-314 (Grieco et al. 2005). Additionally, the native Purkinje open channel blocker and the β4 peptide each protect Na+ channels from inhibition by lidocaine (Bant et al. 2013). Together, these data support the idea that native open channel blockers, and probably NaVβ4, may be endogenous ligands for the local anaesthetic binding site. As in the case of local anaesthetics (Ragsdale et al. 1994), binding may involve interactions of the conserved aromatic rings of the α subunit and the blocking protein, in a manner that permits the charged lysine side chains to obstruct the permeation pathway.
Structural determinants of the open channel block
Ultimately, a full mechanistic description of resurgent current requires a structural understanding of the Na+ channel complex and its gating properties. Much is known about the physical counterparts of the four main states, C, O, I and B. Voltage-gated Na+ channels comprise four domains (DI, DII, DIII, DIV), which form a central pore that opens and closes in response to movements of the voltage-sensing regions (which include the fourth transmembrane segment, S4) of each domain (Stühmer et al. 1989). At strongly negative voltages, the voltage sensors of all domains are in the resting (inward) position, holding the channel closed. With depolarization, the sensors move, each with distinct voltage dependence and kinetics, and the voltage sensor of DIV deploys less readily than those of DI–III. The open state is favoured only after the voltage sensors of DI, DII and DIII all have moved to the activated (outward) position (Cha et al. 1999; Chanda & Bezanilla, 2002). The DIV voltage sensor need not activate for channel opening, but its deployment is required for stable binding of the fast inactivation gate, thought to be the intracellular linker between DIII and DIV (Chahine et al. 1994; Eaholtz et al. 1994; Sheets et al. 1999; Capes et al. 2013).
A central question is how an open channel blocker from a separate subunit binds the Na+ channel more rapidly than a highly localized intrinsic inactivation gate. As mentioned above, when toxins or mutations restrict DIV voltage sensor movement, binding of the fast inactivation gate is slowed (Hanck & Sheets, 2007). Even with DIVS4 in the resting position, however, native or exogenous blocking proteins can readily bind the open channel. Thus, the longer the gap between DI–DIII voltage sensor movements (opening) and DIVS4 movement (inactivation), the more effective an open channel blocker may be at binding during the interval (Grieco & Raman, 2004; Lewis & Raman, 2013). Notably, DIVS4 can deploy after activated channels have been blocked and stabilizes binding of the blocking protein, preventing inactivation even when its onset would otherwise be likely (Lewis & Raman, 2013). Upon repolarization, resurgent current flows at voltages that favour the maintained deployment of DI–DIII (i.e., an open pore) as well as blocker unbinding (Fig.3C).
Ultimately, therefore, the amplitude of resurgent current depends on several factors (Raman & Bean, 2001; Aman & Raman, 2007, 2010; Lewis & Raman, 2011, 2013). First, the number of channels that previously have undergone open channel block. The greater the proportion of channels that bind a blocker upon opening by depolarization, the larger the ratio of resurgent to transient current will be. Second, the binding affinity of the blocker, which can have opposing effects; tighter binding increases the proportion of blocked channels at positive potentials, which can increase resurgent current amplitudes upon repolarization. A higher affinity also slows the current rise time, however, which desynchronizes reopening, reducing peaks, while prolonging current flow. Third, the duration of the step preceding repolarization. With long depolarizations, permeating Na+ eventually unblocks channels, permitting channels to inactivate stably; thus, longer depolarizations elicit smaller resurgent currents upon repolarization. Fourth, the repolarization potential; more negative voltages generate higher driving forces on Na+, increasing both the ease of displacement of bound blocking proteins as well as the synchrony of reopening, and thereby raising resurgent current amplitudes. The effect of hyperpolarization is counterbalanced, however, by the fact that less negative voltages permit repeated channel reopening before inactivation, enlarging as well as prolonging resurgent currents. Consequently, resurgent current tends to be maximal near −30 mV, but this value may change with alterations of Na+ gradients or voltage sensing by any domain. Fifth, the transition rates of the voltage sensors and fast inactivation gate. Upon depolarization, faster DIVS4 deployment limits the open channel block by favouring fast inactivation, setting the stage for small resurgent currents. Upon repolarization, faster DI–DIII deactivation rates or faster binding of the inactivation gate will curtail peak resurgent current as well as speed its decay.
It is worth noting that recovery from any form of inactivation while channels remain activated will generate currents resembling resurgent current. Such effects have been produced pharmacologically by toxins that either shift the voltage dependence of voltage-sensing domains so that DIS4, DIIS4 and/or DIIIS4 activate more negatively or destabilize DIVS4 deployment so that channels recover at more depolarized voltages (Schiavon et al. 2006, 2012). Under these conditions, after depolarization-evoked opening and fast inactivation, subsequent repolarization permits DIVS4 to deactivate while other domains remain temporarily activated, leading to a repolarization-dependent flow of current. Such current is mechanistically distinct from open channel block-dependent resurgent current. It demonstrates the generalizable principle, however, that recovery through open states can be simply mimicked by non-conducting states, entered at positive voltages, becoming unstable at negative potentials at which some channels either remain activated or deactivate more slowly than the inactivation gate unbinds. It may also provide a means by which some cells can make a resurgent-like current without a blocking protein, although no such instances have yet been described. Such cells would be typified by large window currents and slow channel closure. In these cases, the observed resurgent-like current might be relatively insensitive to Na+ gradients.
Resurgent current and disease
These biophysical considerations may be relevant to research suggesting that the resurgent current is altered in several disease states (Fig.3A), including paroxysmal extreme pain disorder, long QT syndrome, paramyotonia congenita (Jarecki et al. 2010), epilepsy (Hargus et al. 2011, 2013) and chemotherapy-induced neuropathy (Sittl et al. 2012). Increases in inflammatory mediators can also increase resurgent currents through NaV1.8 (Tan et al. 2014). Some of these conditions result from mutations in α subunits that slow the rate of fast inactivation: In paroxysmal extreme pain disorder and long QT, the fast inactivation gate of NaV1.7 or NaV1.5 is directly disrupted; in paramyotonia congenita, DIVS4 of NaV1.4 is mutated. When channels with these mutations are transfected into neurons that contain a native blocking particle, they generate resurgent currents that are much larger than in wild-type channels; in fact, transfection of wild-type NaV1.4 makes no resurgent current (Jarecki et al. 2010).
In rat models of status epilepticus, resurgent current is increased in layer II stellate neurons of the medial entorhinal cortex, correlating with increased NaV1.6 expression (Hargus et al. 2013). The transient current activation curve shifts slightly negative and inactivation curve slightly positive, increasing the window in which resurgent current can flow. NaV1.6 also appears implicated in cooling-induced neuropathy induced by the anticancer drug oxaliplatin; treatment slows inactivation of NaV1.6 in pain-sensing DRG neurons (Sittl et al. 2012). The slowing of fast inactivation associated with nearly all these conditions is expected to alter firing patterns by prolonging transient currents, increasing persistent current and enlarging resurgent current, thus reducing accumulation of channels in non-conducting states. In non-voltage-clamped cells, such changes will increase subthreshold Na+ current, probably generating the increases in action potential firing rate or duration that underlie the disorders (Jarecki et al. 2010). If so, amelioration of the conditions might be achieved by reducing Na+ currents, most simply by stabilizing inactivation. Indeed, β4 peptide-induced resurgent current in expression systems can be decreased by carbamazepine, riluzole or anandamide (Theile & Cummins, 2011). Such drugs are expected to be effective in reducing rapid or excessive firing in cells with or without a native blocking particle. Another approach, to target only those cells with resurgent current, might be to stabilize native open channel blockers with compounds that increase blocker channel affinity, making them less easily displaced.
Conclusion
Voltage-gated Na+ channels that underlie action potentials in all excitable cells – in central and peripheral neurons and in cardiac and skeletal muscle – have the capacity to produce resurgent as well as transient and persistent current. Whether resurgent current is normally present depends on two main factors: first, the expression of an open channel blocking protein within the Na+ channel complex, a role that can be assumed by NaVβ4, though not necessarily uniquely, and, second, the gating properties of the specific Na+ channel complex, particularly the movement of the DIV voltage sensor relative to channel opening. The cycle of open channel block and unblock prevents channels from entering absorbing inactivated states, thereby keeping Na+ channel availability high enough for repetitive and/or burst firing. Modulating either Na+ channel gating or the blocking particle itself can alter this cycle, as well as the firing properties that it facilitates, which may have significant consequences for both normal physiology and disease.
Acknowledgments
We thank Drs. Zayd Khaliq, Teresa Aman and Jason Bant for the recordings reproduced in the Figures and for helpful comments on the manuscript. Supported by NIH grant R01-NS39395 (IMR).
Biography
Indira Raman is a Professor in the Department of Neurobiology at Northwestern University, where she holds the Bill and Gayle Cook Chair in Biology. She completed her PhD in Neuroscience at the University of Wisconsin-Madison and postdoctoral training at the Vollum Institute and Harvard Medical School. She and her postdoctoral advisor Bruce Bean discovered resurgent sodium current in 1996. Her research is in the areas of ion channel biophysics, synaptic transmission and cerebellar physiology. Amanda Lewis completed her PhD in Biological Sciences at Northwestern University in the laboratory of Indira Raman. She is currently a postdoctoral associate in the Ion Channel Research Unit at Duke University, where she is investigating the mechanism of activation of mechanosensitive ion channels.

Additional information
Author contributions
AHL and IMR wrote the manuscript and made figures together. Both authors approved the final version for publication.
References
- Abdulla FA, Smith PA. Axotomy- and autotomy-induced changes in the excitability of rat dorsal root ganglion neurons. J Neurophysiol. 2001;85(2):630–643. doi: 10.1152/jn.2001.85.2.630. &. [DOI] [PubMed] [Google Scholar]
- Afshari FS, Ptak K, Khaliq ZM, Grieco TM, Slater NT, McCrimmon DR, Raman IM. Resurgent Na currents in four classes of neurons of the cerebellum. J Neurophysiol. 2004;92:2831–2843. doi: 10.1152/jn.00261.2004. &. [DOI] [PubMed] [Google Scholar]
- Akemann W, Knopfel T. Interaction of Kv3 potassium channels and resurgent sodium current influences the rate of spontaneous firing of Purkinje neurons. J Neurosci. 2006;26:4602–4612. doi: 10.1523/JNEUROSCI.5204-05.2006. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aman TK, Raman IM. Subunit dependence of Na channel slow inactivation and open channel block in cerebellar neurons. Biophys J. 2007;92:1938–1951. doi: 10.1529/biophysj.106.093500. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aman TK, Raman IM. Inwardly permeating Na ions generate the voltage dependence of resurgent Na current in cerebellar Purkinje neurons. J Neurosci. 2010;30:5629–5634. doi: 10.1523/JNEUROSCI.0376-10.2010. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aman TK, Grieco-Calub TM, Chen C, Rusconi R, Slat EA, Isom LL, Raman IM. Regulation of persistent Na current by interactions between beta subunits of voltage-gated Na channels. J Neurosci. 2009;29:2027–2042. doi: 10.1523/JNEUROSCI.4531-08.2009. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J Gen Physiol. 1971;58:413–437. doi: 10.1085/jgp.58.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM, Croop RS. Simulation of Na channel inactivation by thiazine dyes. J Gen Physiol. 1982;80:641–662. doi: 10.1085/jgp.80.5.641. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bant JS, Raman IM. Control of transient, resurgent, and persistent current by open-channel block by Na channel beta4 in cultured cerebellar granule neurons. Proc Natl Acad Sci U S A. 2010;107:12357–12362. doi: 10.1073/pnas.1005633107. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bant JS, Aman TK, Raman IM. Antagonism of lidocaine inhibition by open-channel blockers that generate resurgent Na current. J Neurosci. 2013;33:4976–4987. doi: 10.1523/JNEUROSCI.3026-12.2013. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton MD, Lewis AH, Bant JS, Raman IM. Iberiotoxin-sensitive and -insensitive BK currents in Purkinje neuron somata. J Neurophysiol. 2013;109:2528–2541. doi: 10.1152/jn.00127.2012. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffington SA, Rasband MN. Na+ channel-dependent recruitment of Navbeta4 to axon initial segments and nodes of Ranvier. J Neurosci. 2013;33:6191–6202. doi: 10.1523/JNEUROSCI.4051-12.2013. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess DL, Kohrman DC, Galt J, Plummer NW, Jones JM, Spear B, Meisler MH. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant ‘motor endplate disease. Nat Genet. 1995;10:461–465. doi: 10.1038/ng0895-461. , & ’. [DOI] [PubMed] [Google Scholar]
- Cahalan MD, Almers W. Block of sodium conductance and gating current in squid giant axons poisoned with quaternary strychnine. Biophys J. 1979;27:57–73. doi: 10.1016/S0006-3495(79)85202-9. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell JH, Schaller KL, Lasher RS, Peles E, &Levinson SR. Sodium channel NaV1.6 is localized at nodes of Ranvier, dendrites, and synapses. Proc Natl Acad Sci U S A. 2000;97(10):5616–5620. doi: 10.1073/pnas.090034797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capes DL, Goldschen-Ohm MP, Arcisio-Miranda M, Bezanilla F, Chanda B. Domain IV voltage-sensor movement is both sufficient and rate limiting for fast inactivation in sodium channels. J Gen Physiol. 2013;142:101–112. doi: 10.1085/jgp.201310998. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter BC, Bean BP. Sodium entry during action potentials of mammalian neurons: incomplete inactivation and reduced metabolic efficiency in fast-spiking neurons. Neuron. 2009;64:898–909. doi: 10.1016/j.neuron.2009.12.011. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli L, Nigro MJ, Magistretti J. Analysis of resurgent sodium-current expression in rat parahippocampal cortices and hippocampal formation. Brain Res. 2007a;1163:44–55. doi: 10.1016/j.brainres.2007.05.065. &. [DOI] [PubMed] [Google Scholar]
- Castelli L, Biella G, Toselli M, Magistretti J. Resurgent Na+ current in pyramidal neurones of rat perirhinal cortex: axonal location of channels and contribution to depolarizing drive during repetitive firing. J Physiol. 2007b;582:1179–1193. doi: 10.1113/jphysiol.2007.135350. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha A, Ruben PC, George AL, Jr, Fujimoto E, Bezanilla F. Voltage sensors in domains III and IV, but not I and II, are immobilized by Na+ channel fast inactivation. Neuron. 1999;22:73–87. doi: 10.1016/s0896-6273(00)80680-7. &. [DOI] [PubMed] [Google Scholar]
- Chadderton P, Margrie TW, Häusser M. Integration of quanta in cerebellar granule cells during sensory processing. Nature. 2004;428(6985):856–860. doi: 10.1038/nature02442. &. [DOI] [PubMed] [Google Scholar]
- Chahine M, George AL, Jr, Zhou M, Ji S, Sun W, Barchi RL, Horn R. Na channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 1994;12:281–294. doi: 10.1016/0896-6273(94)90271-2. &. [DOI] [PubMed] [Google Scholar]
- Chanda B, Bezanilla F. Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J Gen Physiol. 2002;120:629–645. doi: 10.1085/jgp.20028679. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yu FH, Sharp EM, Beacham D, Scheuer T, Catterall WA. Functional properties and differential neuromodulation of NaV1.6 channels. Mol Cell Neurosci. 2008;38:607–615. doi: 10.1016/j.mcn.2008.05.009. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbert CM, Magee JC, Hoffman DA, Johnston D. Slow recovery from inactivation of Na+ channels underlies the activity-dependent attenuation of dendritic action potentials in hippocampal CA1 pyramidal neurons. J Neurosci. 1997;17(17):6512–6521. doi: 10.1523/JNEUROSCI.17-17-06512.1997. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Dib-Hajj SD, Herzog RI, Waxman SG. NaV1.6 channels generate resurgent sodium currents in spinal sensory neurons. FEBS Lett. 2005;579:2166–2170. doi: 10.1016/j.febslet.2005.03.009. &. [DOI] [PubMed] [Google Scholar]
- D'Angelo E, Nieus T, Maffei A, Armano S, Rossi P, Taglietti V, Fontana A, Naldi G. Theta-frequency bursting and resonance in cerebellar granule cells: experimental evidence and modelling of a slow K+-dependent mechanism. J Neurosci. 2001;21(3):759–770. doi: 10.1523/JNEUROSCI.21-03-00759.2001. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Wei W, Zhou FM. Molecular and functional differences in voltage-activated sodium currents between GABA projection neurons and dopamine neurons in the substantia nigra. J Neurophysiol. 2011;106:3019–3034. doi: 10.1152/jn.00305.2011. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do MT, Bean BP. Subthreshold sodium currents and pacemaking of subthalamic neurons: modulation by slow inactivation. Neuron. 2003;39:109–120. doi: 10.1016/s0896-6273(03)00360-x. &. [DOI] [PubMed] [Google Scholar]
- Do MT, Bean BP. Sodium currents in subthalamic nucleus neurons from NaV1.6-null mice. J Neurophysiol. 2004;92:726–733. doi: 10.1152/jn.00186.2004. &. [DOI] [PubMed] [Google Scholar]
- Eaholtz G, Scheuer T, Catterall WA. Restoration of inactivation and block of open sodium channels by an inactivation gate peptide. Neuron. 1994;12:1041–1048. doi: 10.1016/0896-6273(94)90312-3. &. [DOI] [PubMed] [Google Scholar]
- Engel D, Jonas P. Presynaptic action potential amplification by voltage-gated Na+ channels in hippocampal mossy fiber boutons. Neuron. 2005;45(3):405–417. doi: 10.1016/j.neuron.2004.12.048. &. [DOI] [PubMed] [Google Scholar]
- Enomoto A, Han JM, Hsiao CF, Wu N, Chandler SH. Participation of sodium currents in burst generation and control of membrane excitability in mesencephalic trigeminal neurons. J Neurosci. 2006;26:3412–3422. doi: 10.1523/JNEUROSCI.5274-05.2006. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto A, Han JM, Hsiao CF, Chandler SH. Sodium currents in mesencephalic trigeminal neurons from NaV1.6 null mice. J Neurophysiol. 2007;98(2):710–719. doi: 10.1152/jn.00292.2007. &. [DOI] [PubMed] [Google Scholar]
- Fu Y, Struyk A, Markin V, Cannon S. Gating behaviour of sodium currents in adult mouse muscle recorded with an improved two-electrode voltage clamp. J Physiol. 2011;589(Pt 3):525–546. doi: 10.1113/jphysiol.2010.199430. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist J, Das S, Van Petegem F, Bosmans F. Crystallographic insights into sodium-channel modulation by the β4 subunit. Proc Natl Acad Sci U S A. 2013;110(51):E5016–E5024. doi: 10.1073/pnas.1314557110. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittis AH, du Lac S. Similar properties of transient, persistent, and resurgent Na currents in GABAergic and non-GABAergic vestibular nucleus neurons. J Neurophysiol. 2008;99:2060–2065. doi: 10.1152/jn.01389.2007. &. [DOI] [PubMed] [Google Scholar]
- Gittis AH, Moghadam SH, du Lac S. Mechanisms of sustained high firing rates in two classes of vestibular nucleus neurons: differential contributions of resurgent Na, Kv3, and BK currents. J Neurophysiol. 2010;104(3):1625–1634. doi: 10.1152/jn.00378.2010. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieco TM, Raman IM. Production of resurgent current in NaV1.6-null Purkinje neurons by slowing sodium channel inactivation with beta-pompilidotoxin. J Neurosci. 2004;24:35–42. doi: 10.1523/JNEUROSCI.3807-03.2004. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieco TM, Afshari FS, Raman IM. A role for phosphorylation in the maintenance of resurgent sodium current in cerebellar Purkinje neurons. J Neurosci. 2002;22:3100–3107. doi: 10.1523/JNEUROSCI.22-08-03100.2002. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieco TM, Malhotra JD, Chen C, Isom LL, Raman IM. Open-channel block by the cytoplasmic tail of sodium channel b4 as a mechanism for resurgent sodium current. Neuron. 2005;45:233–244. doi: 10.1016/j.neuron.2004.12.035. &. [DOI] [PubMed] [Google Scholar]
- Han CI, Hoeijmakers JG, Liu S, Gerrits MM, te Morsche RH, Lauria G, Dib-Hajj SD, Drenth JP, Faber CG, Merkies IS, Waxman SG. Functional profiles of SCN9A variants in dorsal root ganglion neurons and superior cervical ganglion neurons correlate with autonomic symptoms in small fibre neuropathy. Brain. 2012;135(Pt 9):2613–2628. doi: 10.1093/brain/aws187. &. [DOI] [PubMed] [Google Scholar]
- Hanck DA, Sheets MF. Site-3 toxins and cardiac sodium channels. Toxicon. 2007;49(2):181–193. doi: 10.1016/j.toxicon.2006.09.017. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargus NJ, Merrick EC, Nigam A, Kalmar CL, Baheti AR, Bertram EH, 3rd, Patel MK. Temporal lobe epilepsy induces intrinsic alterations in Na channel gating in layer II medial entorhinal cortex neurons. Neurobiol Dis. 2011;41(2):361–376. doi: 10.1016/j.nbd.2010.10.004. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargus NJ, Nigam A, Bertram EH, 3rd, Patel MK. Evidence for a role of Nav1.6 in facilitating increases in neuronal hyperexcitability during epileptogenesis. J Neurophysiol. 2013;110(5):1144–1157. doi: 10.1152/jn.00383.2013. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. The dual effect of membrane potential on sodium conductance in the giant axon of Loligo. J Physiol. 1952a;116:497–506. doi: 10.1113/jphysiol.1952.sp004719. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952b;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. The components of membrane conductance in the giant axon of Loligo. J Physiol. 1952c;116:473–496. doi: 10.1113/jphysiol.1952.sp004718. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. Currents carried by sodium and potassium ions through the membrane of the giant axon of Loligo. J Physiol. 1952d;116:449–472. doi: 10.1113/jphysiol.1952.sp004717. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarecki BW, Piekarz AD, Jackson JO, 2nd, Cummins TR. Human voltage-gated sodium channel mutations that cause inherited neuronal and muscle channelopathies increase resurgent sodium currents. J Clin Invest. 2010;120:369–378. doi: 10.1172/JCI40801. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalume F, Yu FH, Westenbroek RE, Scheuer T, Catterall WA. Reduced sodium current in Purkinje neurons from Nav1.1 mutant mice: implications for ataxia in severe myoclonic epilepsy in infancy. J Neurosci. 2007;27:11065–11074. doi: 10.1523/JNEUROSCI.2162-07.2007. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliq ZM, Raman IM. Axonal propagation of simple and complex spikes in cerebellar Purkinje neurons. J Neurosci. 2005;25(2):454–463. doi: 10.1523/JNEUROSCI.3045-04.2005. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliq ZM, Raman IM. Relative contributions of axonal and somatic Na channels to action potential initiation in cerebellar Purkinje neurons. J Neurosci. 2006;26(7):1935–1944. doi: 10.1523/JNEUROSCI.4664-05.2006. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliq ZM, Gouwens NW, Raman IM. The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: An experimental and modelling study. J Neurosci. 2003;23:4899–4912. doi: 10.1523/JNEUROSCI.23-12-04899.2003. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Kushmerick C, von Gersdorff H. Presynaptic resurgent Na+ currents sculpt the action potential waveform and increase firing reliability at a CNS nerve terminal. J Neurosci. 2010;30:15479–15490. doi: 10.1523/JNEUROSCI.3982-10.2010. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Trussell LO. Ion channels generating complex spikes in cartwheel cells of the dorsal cochlear nucleus. J Neurophysiol. 2007;97(2):1705–1725. doi: 10.1152/jn.00536.2006. &. [DOI] [PubMed] [Google Scholar]
- Kodama T, Guerrero S, Shin M, Moghadam S, Faulstich M, du Lac S. Neuronal classification and marker gene identification via single-cell expression profiling of brainstem vestibular neurons subserving cerebellar learning. J Neurosci. 2012;32:7819–7831. doi: 10.1523/JNEUROSCI.0543-12.2012. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CC, Bean BP. Na+ channels must deactivate to recover from inactivation. Neuron. 1994;12(4):819–829. doi: 10.1016/0896-6273(94)90335-2. &. [DOI] [PubMed] [Google Scholar]
- Leão RN1, Naves MM, Leão KE, Walmsley B. Altered sodium currents in auditory neurons of congenitally deaf mice. Eur J Neurosci. 2006;24(4):1137–1146. doi: 10.1111/j.1460-9568.2006.04982.x. &. [DOI] [PubMed] [Google Scholar]
- Lewis AH, Raman IM. Cross-species conservation of open-channel block by Na channel beta4 peptides reveals structural features required for resurgent Na current. J Neurosci. 2011;31:11527–11536. doi: 10.1523/JNEUROSCI.1428-11.2011. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis AH, Raman IM. Interactions among DIV voltage-sensor movement, fast inactivation, and resurgent Na current induced by the NaVbeta4 open-channel blocking peptide. J Gen Physiol. 2013;142:191–206. doi: 10.1085/jgp.201310984. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC. Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci. 1998;18(19):7613–7624. doi: 10.1523/JNEUROSCI.18-19-07613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti J, Castelli L, Forti L, D'Angelo E. Kinetic and functional analysis of transient, persistent and resurgent sodium currents in rat cerebellar granule cells in situ: an electrophysiological and modelling study. J Physiol. 2006;573(Pt 1):83–106. doi: 10.1113/jphysiol.2006.106682. , &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Pape HC. Properties of a hyperpolarization-activated cation current and its role in rhythmic oscillation in thalamic relay neurones. J Physiol. 1990;431:291–318. doi: 10.1113/jphysiol.1990.sp018331. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer JN, Chan CS, Tkatch T, Held J, Surmeier DJ. NaV1.6 sodium channels are critical to pacemaking and fast spiking in globus pallidus neurons. J Neurosci. 2007;27:13552–13566. doi: 10.1523/JNEUROSCI.3430-07.2007. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsivais P, Clark BA, Roth A, Häusser M. Determinants of action potential propagation in cerebellar Purkinje cell axons. J Neurosci. 2005;25(2):464–472. doi: 10.1523/JNEUROSCI.3871-04.2005. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigro MJ, Quattrocolo G, Magistretti J. Distinct developmental patterns in the expression of transient, persistent, and resurgent Na+ currents in entorhinal cortex layer-II neurons. Brain Res. 2012;1463:30–41. doi: 10.1016/j.brainres.2012.04.049. &. [DOI] [PubMed] [Google Scholar]
- Osorio N, Cathala L, Meisler MH, Crest M, Magistretti J, Delmas P. Persistent NaV1.6 current at axon initial segments tunes spike timing of cerebellar granule cells. J Physiol. 2010;588(Pt 4):651–670. doi: 10.1113/jphysiol.2010.183798. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyama F, Miyazaki H, Sakamoto N, Becquet C, Machida Y, Kaneko K, Uchikawa C, Suzuki T, Kurosawa M, Ikeda T, Tamaoka A, Sakurai T, Nukina N. Sodium channel beta4 subunit: down-regulation and possible involvement in neuritic degeneration in Huntington's disease transgenic mice. J Neurochem. 2006;98(2):518–529. doi: 10.1111/j.1471-4159.2006.03893.x. &. [DOI] [PubMed] [Google Scholar]
- Pan F, Beam KG. The absence of resurgent sodium current in mouse spinal neurons. Brain Res. 1999;849(1–2):162–168. doi: 10.1016/s0006-8993(99)02060-0. &. [DOI] [PubMed] [Google Scholar]
- Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+ channels by local anaesthetics. Science. 1994;265(5179):1724–1728. doi: 10.1126/science.8085162. &. [DOI] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci. 1997;17:4517–4526. doi: 10.1523/JNEUROSCI.17-12-04517.1997. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci. 1999;19:1663–1674. doi: 10.1523/JNEUROSCI.19-05-01663.1999. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Inactivation and recovery of sodium currents in cerebellar Purkinje neurons: evidence for two mechanisms. Biophys J. 2001;80:729–737. doi: 10.1016/S0006-3495(01)76052-3. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Sprunger LK, Meisler MH, Bean BP. Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron. 1997;19:881–891. doi: 10.1016/s0896-6273(00)80969-1. &. [DOI] [PubMed] [Google Scholar]
- Raman IM, Gustafson AE, Padgett D. Ionic currents and spontaneous firing in neurons isolated from the cerebellar nuclei. J Neurosci. 2000;20(24):9004–9016. doi: 10.1523/JNEUROSCI.20-24-09004.2000. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royeck M, Horstmann MT, Remy S, Reitze M, Yaari Y, Beck H. Role of axonal NaV1.6 sodium channels in action potential initiation of CA1 pyramidal neurons. J Neurophysiol. 2008;100(4):2361–2380. doi: 10.1152/jn.90332.2008. &. [DOI] [PubMed] [Google Scholar]
- de Ruiter MM, De Zeeuw CI, Hansel C. Voltage-gated sodium channels in cerebellar Purkinje cells of mormyrid fish. J Neurophysiol. 2006;96(1):378–390. doi: 10.1152/jn.00906.2005. &. [DOI] [PubMed] [Google Scholar]
- Russo MJ, Mugnaini E, Martina M. Intrinsic properties and mechanisms of spontaneous firing in mouse cerebellar unipolar brush cells. J Physiol. 2007;581(2):709–724. doi: 10.1113/jphysiol.2007.129106. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavon E, Sacco T, Cassulini RR, Gurrola G, Tempia F, Possani LD, Wanke E. Resurgent current and voltage sensor trapping enhanced activation by a beta-scorpion toxin solely in NaV1.6 channel. Significance in mice Purkinje neurons. J Biol Chem. 2006;281(29):20326–20337. doi: 10.1074/jbc.M600565200. &. [DOI] [PubMed] [Google Scholar]
- Schiavon E, Pedraza-Escalona M, Gurrola GB, Olamendi-Portugal T, Corzo G, Wanke E, Possani LD. Negative-shift activation, current reduction and resurgent currents induced by β-toxins from Centruroides scorpions in sodium channels. Toxicon. 2012;59(2):283–293. doi: 10.1016/j.toxicon.2011.12.003. &. [DOI] [PubMed] [Google Scholar]
- Sheets MF, Kyle JW, Kallen RG, Hanck DA. The Na channel voltage sensor associated with inactivation is localized to the external charged residues of domain IV, S4. Biophys J. 1999;77(2):747–757. doi: 10.1016/S0006-3495(99)76929-8. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sittl R, Lampert A, Huth T, Schuy ET, Link AS, Fleckenstein J, Alzheimer C, Grafe P, Carr RW. Anticancer drug oxaliplatin induces acute cooling-aggravated neuropathy via sodium channel subtype NaV1.6-resurgent and persistent current. Proc Natl Acad Sci U S A. 2012;109:6704–6709. doi: 10.1073/pnas.1118058109. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MR, Smith RD, Plummer NW, Meisler MH, Goldin AL. Functional analysis of the mouse Scn8a sodium channel. J Neurosci. 1998;18(16):6093–6102. doi: 10.1523/JNEUROSCI.18-16-06093.1998. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stühmer W, Conti F, Suzuki H, Wang XD, Noda M, Yahagi N, Kubo H, Numa S. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339:597–603. doi: 10.1038/339597a0. &. [DOI] [PubMed] [Google Scholar]
- Tan ZY, Piekarz AD, Priest BT, Knopp KL, Krajewski JL, McDermott JS, Nisenbaum ES, Cummins TR. Tetrodotoxin-resistant sodium channels in sensory neurons generate slow resurgent currents that are enhanced by inflammatory mediators. J Neurosci. 2014;34(21):7190–7197. doi: 10.1523/JNEUROSCI.5011-13.2014. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L, Kallen RG, Horn R. Role of an S4-S5 linker in sodium channel inactivation probed by mutagenesis and a peptide blocker. J Gen Physiol. 1996;108:89–104. doi: 10.1085/jgp.108.2.89. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theile JW, Cummins TR. Inhibition of Navbeta4 peptide-mediated resurgent sodium currents in Nav1.7 channels by carbamazepine, riluzole, and anandamide. Mol Pharmacol. 2011;80:724–734. doi: 10.1124/mol.111.072751. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theile JW, Jarecki BW, Piekarz AD, Cummins TR. Nav1.7 mutations associated with paroxysmal extreme pain disorder, but not erythromelalgia, enhance Navbeta4 peptide-mediated resurgent sodium currents. J Physiol. 2011;589:597–608. doi: 10.1113/jphysiol.2010.200915. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GK, Edrich T, Wang SY. Time-dependent block and resurgent tail currents induced by mouse b4154–167 peptide in cardiac Na+ channels. J Gen Physiol. 2006;127:277–289. doi: 10.1085/jgp.200509399. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LY, Gan L, Forsythe ID, Kaczmarek LK. Contribution of the Kv3.1 potassium channel to high-frequency firing in mouse auditory neurones. J Physiol. 1998;509:183–194. doi: 10.1111/j.1469-7793.1998.183bo.x. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh JZ, Narahashi T. Kinetic analysis of pancuronium interaction with sodium channels in squid axon membranes. J Gen Physiol. 1977;69:293–323. doi: 10.1085/jgp.69.3.293. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FH, Westenbroek RE, Silos-Santiago I, McCormick KA, Lawson D, Ge P, Ferriera H, Lilly J, DiStefano PS, Catterall WA, Scheuer T, Curtis R. Sodium channel b4, a new disulfide-linked auxiliary subunit with similarity to b2. J Neurosci. 2003;23:7577–7585. doi: 10.1523/JNEUROSCI.23-20-07577.2003. &. [DOI] [PMC free article] [PubMed] [Google Scholar]