

Abstract
Induction of several forms of synaptic plasticity, a cellular basis for learning and memory, depends on the activation of Ca2+/calmodulin (CaM)-dependent protein kinase II (CaMKII). CaMKII acts as a holoenzyme consisting of α and β subunits (α- and βCaMKII). However, it remains elusive how the subunit composition of a CaMKII holoenzyme affects its activation and hence synaptic plasticity. We addressed this issue by focusing on long-term potentiation (LTP) at inhibitory synapses on cerebellar Purkinje neurons (PNs) (called rebound potentiation, RP). The contribution of each subunit to RP was examined by selective knock-down or overexpression of that subunit. Electrophysiological recording from a rat cultured PN demonstrated that βCaMKII is essential for RP induction, whereas αCaMKII suppresses it. Thus, RP was negatively regulated due to the greater relative abundance of αCaMKII compared to βCaMKII, suggesting a critical role of CaMKII subunit composition in RP. The higher affinity of βCaMKII to Ca2+/CaM compared with αCaMKII was responsible for the predominant role in RP induction. Live-cell imaging of CaMKII activity based on the Förster resonance energy transfer (FRET) technique revealed that βCaMKII enrichment enhances the total CaMKII activation upon a transient conditioning depolarization. Taken together, these findings clarified that α- and βCaMKII oppositely regulate CaMKII activation, controlling the induction of inhibitory synaptic plasticity in a PN, which might contribute to the adaptive information processing of the cerebellar cortex.
Introduction
Several forms of synaptic plasticity depend on the activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII), which functions as a holoenzyme consisting of two distinct types of subunit, α- and βCaMKII. When two neighbouring subunits in a holoenzyme are activated by Ca2+/CaM, autophosphorylation occurs at Thr286 (αCaMKII) or Thr287 (βCaMKII), rendering the subunit Ca2+/CaM-independently active (Miller & Kennedy, 1986). Mutant mice with replacement of Thr286 of αCaMKII by Ala exhibited impairment of both long-term potentiation (LTP) at hippocampal excitatory synapses and spatial learning (Giese et al. 1998). Thus, the positive feedback activation of CaMKII through Thr286/287 autophosphorylation has been thought to be important for synaptic plasticity and learning (Lisman et al. 2002).
In contrast to the pivotal roles of αCaMKII, which is abundantly expressed in excitatory pyramidal cells in the hippocampus and cerebral cortex, the roles of βCaMKII in synaptic plasticity remain elusive. βCaMKII specifically associates with cytoskeletal F-actin (Shen et al. 1998; O'Leary et al. 2006), and involvement of βCaMKII in synaptic plasticity through F-actin association was demonstrated (Borgesius et al. 2011). In addition, βCaMKII was reported to have higher affinity for Ca2+/CaM (Kd: ∼10 nm) compared with αCaMKII (Kd: ∼100 nm) (Meyer et al. 1992; Brocke et al. 1999), although the impact of this difference on synaptic regulation remains unknown. Considering these differences between α- and βCaMKII, we hypothesized that the subunit composition of the CaMKII holoenzyme might affect its activation and/or localization, leading to modulation of synaptic plasticity. To examine this issue, we focused on CaMKII-dependent LTP at inhibitory synapses on a cerebellar Purkinje neuron (PN), in which both subunits are expressed. The relative expression level of βCaMKII compared to αCaMKII is higher in the cerebellum (β:α, about 4:1) than in the forebrain (1:2–1:3) (McGuinness et al. 1985; Walaas et al. 1988).
At GABAergic synapses from inhibitory interneurons (basket and stellate neurons) onto a PN, postsynaptic depolarization such as that caused by climbing fibre inputs induces LTP of the γ-aminobutyric acid type A receptor (GABAAR)-mediated transmission for longer than 30 min (Kano et al. 1992; Hirano & Kawaguchi, 2014). This inhibitory synaptic LTP induced by heterosynaptic excitatory inputs is called rebound potentiation (RP). Distinct from NMDA receptor-dependent LTP at excitatory synapses, RP induction depends on the Ca2+ influx through voltage-gated Ca2+ channels and the resultant activation of CaMKII (Kano et al. 1996). CaMKII was shown to directly phosphorylate GABAARs augmenting the channel currents (Houston & Smart, 2006; Houston et al. 2008), although the roles of the direct phosphorylation in RP remain elusive. In addition, CaMKII modulates GABAAR-associated protein (GABARAP) which binds to GABAARs and microtubules, playing a critical role in the establishment and maintenance of RP (Kawaguchi & Hirano, 2007). Transgenic mice in which the association between GABARAP and GABAARs is disturbed exhibit impaired RP induction and suppression of a form of motor learning (Tanaka et al. 2013). Similarly to hippocampal LTP (Strack et al. 1997; Lisman et al. 2002,2012), the induction of RP by CaMKII is controlled by protein phosphatases (PPs) including PP-1 and calcineurin, which is regulated by cAMP-dependent protein kinase (protein kinase A) through dopamine and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32) (Kawaguchi & Hirano, 2002; Kitagawa et al. 2009). Thus, signalling mechanisms regulating RP induction and its critical role in cerebellar adaptive information processing have been clarified. However, it remains unknown whether α- and βCaMKII contribute equally or differently to RP, and how RP is affected by alterations of the subunit composition. In order to address these issues, we performed whole-cell patch clamp recording and FRET imaging on cultured PNs in which the expression levels of α- and βCaMKII were altered by RNA interference (RNAi) or overexpression. Electrophysiological experiments showed that αCaMKII negatively regulates RP, while βCaMKII is necessary for RP induction. FRET imaging showed that CaMKII activity is augmented by the higher affinity of βCaMKII for Ca2+/CaM, which contributes to RP establishment.
Methods
Ethical approval
Experimental procedures were performed in accordance with the principles of UK regulations, the policies on the use of animals and humans in neuroscience research in the USA, and the guidelines regarding care and use of animals for experimental procedures of Kyoto University, and were approved by the local committee for handling experimental animals in the Graduate School of Science, Kyoto University.
Cell culture
Primary cultures of cerebellar neurons were prepared from newborn Wistar rats of both sexes by a method similar to that in a previous study (Kawaguchi & Hirano, 2013). Briefly, rat pups were decapitated on the day of birth (P0), and cerebella were dissected, followed by treatment with 0.1% trypsin. Cells were then dissociated by trituration and cultured in a defined medium. All experiments with living PNs were performed 3 weeks after preparation of the culture.
DNA construction
cDNAs of mouse α- and βCaMKII were cloned by PCR of a cDNA template obtained by reverse transcription of mRNAs prepared from a mouse cerebellar culture. cDNAs of α- and βCaMKII were inserted into pCAGplay expression vector (Kawaguchi & Hirano, 2006). For α- and βCaMKII short hairpin RNA interference (shRNAi) constructs, the following sequences were used as target sequences in accord with previous studies (Okamoto et al. 2007): αCaMKII, 5′-CCACTACCTTATCTTCGAT-3′; βCaMKII, 5′-GAGTATGCAGCTAAGATCA-3′. A negative control shRNAi for βCaMKII was mutated in six nucleotides (underlined) as follows, 5′-GACTCTGGAGTAAAGACCA-3′. The shRNAs were expressed with a plasmid-based expression vector, pSuper (Oligoengine, Seattle, WA, USA). An RNAi-resistant form of βCaMKII was constructed by introducing silent mutations in the corresponding cDNA sequence as follows (underlined), 5′-GAATACGCCGCGAAAATAA-3′. Venus-αCaMKII (Vα) and Venus-βCaMKII (Vβ) were constructed by ligating Venus (a variant of yellow fluorescent protein) cDNA to the N-termini of α- and βCaMKII cDNAs, respectively. FRET probes Venus-αCaMKII-ECFP (enhanced cyan fluorescent protein) (VαC), Venus-βCaMKII-ECFP (VβC) and ECFP-αCaMKII-Venus (CαV) were constructed by tandem ligation of cDNAs for αCaMKII or βCaMKII, Venus and ECFP, in pCAGplay expression vector. Because fusion of βCaMKII to CFP at the C-terminal region resulted in cleavage around the junction, the C-terminal region (amino acids 344–479) of αCaMKII was substituted for the corresponding region (amino acids 408–594) of βCaMKII. The mutant probes were produced by PCR-mediated introduction of mutation. An expression plasmid (50 ng μl−1) encoding α- or βCaMKII, or shRNA for α- or βCaMKII, was transfected together with an expression plasmid encoding enhanced green fluorescent protein (EGFP) or red fluorescent protein (RFP). An expression plasmid encoding a FRET probe was transfected by itself or with expression plasmid for α- or βCaMKII. DNA transfection in a PN was performed by microinjection into the nucleus of a PN through a sharp glass pipette. The immunocytochemical, electrophysiological, Ca2+ imaging or FRET experiments were performed 1–2 days after injection. HEK293T or HEK293 cells were transfected with Vα, Vβ, wild-type α- or βCaMKII using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
Electrophysiology
The methods for electrophysiological experiments were similar to those in earlier studies (Kawaguchi et al. 2011). Briefly, whole-cell patch-clamp recording from a cultured PN grown in culture for 3 weeks was performed with an amplifier (EPC10, HEKA Electronics, Lambrecht, Germany) in an external solution containing (in mm): 145 NaCl, 5 KOH, 2 CaCl2, 1 MgCl2, 10 Hepes, and 10 glucose (pH 7.3) at room temperature (20–24°C). The solution also contained 6-cyano-7-nitroquinoxaline-2,3-dione disodium (CNQX, 10 μm, Tocris Bioscience, Bristol, UK), tetrodotoxin (TTX, 1 μm, Wako, Osaka, Japan, or Tocris), and SCH50911 (10 μm, Tocris) to inhibit glutamatergic EPSCs, action potentials, and GABABR activation, respectively. Actin polymerization inhibitor latrunculin A (LatA, 2 μm, Calbiochem, San Diego, CA, USA) and protein phosphatase (PP) inhibitor nodularin (2 nm, Calbiochem) were added to the external solution in some experiments. AIPII (1 μm, Calbiochem) was added to the internal solution. PNs were visually identified by their large cell body and thick dendrites. A patch pipette used to record from a PN was filled with an internal solution (pH 7.3, adjusted with CsOH) containing (in mm): 150 CsCl, 10.5 CsOH, 0.5 EGTA, 10 Hepes, 2 Mg-ATP, and 0.2 Na-GTP. The membrane potential of a PN was held at −70 mV. Only recordings with input resistance of more than 100 MΩ and series resistance of less than 25 MΩ were accepted. Series resistance and input resistance were monitored every 2 min and experiments were terminated when a change of more than 20% was detected. The method for iontophoretic application of GABA was similar to that in earlier studies (Kawaguchi & Hirano, 2000,2002,2006,2007). A glass pipette containing 10 mm GABA (pH 7.2) was aimed at a proximal or secondary dendrite (∼40 μm distance from the soma), and 20 ms positive voltage pulses were applied every 20 s. Estimation of GABA diffusion from the tip of the GABA-containing pipette was performed using an outside-out membrane patch excised from a PN soma, and it was suggested that more than 90% of the whole-cell GABA-induced current occurred at the dendritic plasma membrane (not shown). To minimize the voltage-clamp error, the amplitude of the GABA response at the beginning of each experiment was set at around 200 pA. In the mIPSCs analysis, events larger than 5 pA with appropriate time courses were selected visually, and amplitudes were measured with MiniAnalysis software (Synaptosoft, Inc., Decatur, GA, USA). The mean amplitude was calculated from 200–400 events.
Immunocytochemistry
Cultured neurons were fixed with 4% paraformaldehyde in phosphate buffered saline, permeabilized with 0.5% Tween 20, then blocked with 2% skimmed milk, and finally labelled with primary and secondary antibodies. The following antibodies were used: rabbit polyclonal antibody (pAb) against calbindin (1:1000, Chemicon, Temecula, CA, USA), rabbit pAb against GABAA receptor α1 subunit (1:1000, Alomone Labs, Jerusalem, Israel), rabbit pAb against vesicular GABA transporter (VGAT, 1:1000, Synaptic Systems, Goettingen, Germany), rabbit pAb against glutamate receptor δ (GluD, 1:1000, Millipore, Darmstadt, Germany), mouse monoclonal antibody (mAb) against αCaMKII (1:1000, Chemicon), mouse mAb against βCaMKII (1:1000 or 1:10,000, Zymed Laboratories, Inc., South San Francisco, CA, USA), mouse mAb against calbindin (1:1000, Swant), chick pAb against GFP (1: 1000, Chemicon), Alexa 568-conjugated pAb against mouse IgG (1:400, Molecular Probes, Eugene, OR, USA), and Alexa 488-conjugated pAbs against rabbit or chick IgG (1:400, Molecular Probes). Alexa 568-conjugated phalloidin (1:50, Molecular Probes) was used to stain F-actin. Fluorescence images were recorded with a confocal laser microscope (FV1000 imaging system, Olympus, Japan), and analysed using IP lab software (Solution Systems, Funahashi, Japan) or ImageJ software (NIH, Bethesda, MA, USA). Averaged fluorescence signals for α- or βCaMKII in a soma, or proximal or distal dendrites of transfected PNs in the area positive for EGFP were compared. Some quantifications of α- and βCaMKII signals were performed in a blind condition, i.e. the transfection, experiment, data recording and analysis were performed without knowing the identity of the transfected cDNAs. Similar results were obtained between blind and non-blind experiments, and pooled data are presented. To estimate the expression ratio of α- and βCaMKII in a PN, fluorescence signals for each endogenous subunit were normalized as follows. Each subunit tagged with Venus was exogenously expressed in cells from the human embryonic kidney cell line HEK293T, in which endogenous CaMKII expression levels are marginal compared with the overexpressed amounts, and cells were stained with anti-GFP antibody and also with antibody against either α- or βCaMKII. Then, the relative staining levels of anti-α- or -βCaMKII antibody to anti-GFP antibody was quantified, and finally that of anti-αCaMKII to anti-βCaMKII antibody was calculated.
Biochemical assay and western blotting
Cultured neurons or HEK293T cells transfected with Venus-tagged α- or βCaMKII were lysed in sample loading buffer (50 mm Tris–HCl, 4% SDS, 20% glycerol, 10% 2-mercaptoethanol, 0.001% bromophenol blue) and boiled for 5 min. To evaluate the efficiency of RNAi, the subunit-encoding expression vector for either α- or βCaMKII was transfected to HEK293 cells together with RNAi plasmid encoding shRNA for the respective CaMKII. To assay Ca2+/CaM activation of wild-type or mutant CaMKII subunits, they were expressed in HEK293T cells and collected and purified in an assay buffer (40 mm Hepes, 0.1 mm EGTA, 5 mm magnesium acetate, 0.01% Tween 20, and 1 mm DTT, pH 8.0), as described in previous studies (Takao et al. 2005). Each sample was treated with different concentrations of CaM (0, 10, 20, 50, 100, 200 nm) for 10 s at 20°C in the presence of 1 mm Ca2+ and 100 μm ATP. Then, reactions were terminated by adding the sample buffer. Samples were separated by 10% SDS–PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes, followed by incubation with primary antibody (mouse mAb against GFP (1:1000, Nacalai Tesque, Kyoto, Japan), αCaMKII (1:10,000, Chemicon), βCaMKII (1:1000, Zymed), or β-actin (1:2000, Sigma, St. Louis, MO, USA), or rabbit pAb against Thr286/287-phosphorylated CaMKII, 1: 1000, Promega, Madison, WI, USA), and HRP-conjugated secondary antibody (goat pAb against mouse or rabbit IgG, 1:1000, Molecular Probes). Signals were detected with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, Waltham, MA, USA). Images were acquired using LAS 3000 (Fuji Film, Tokyo, Japan), and analysed using ImageJ (NIH). Apparent EC50 values for the CaM-triggered autophosphorylation of wild-type and mutant CaMKII subunits relative to 200 nm CaM were estimated by fitting dose–response curves to the Hill equation assuming a Hill coefficient of 1.
Ca2+ imaging
[Ca2+]i was measured with a Ca2+ imaging system (Aquacosmos, Hamamatsu Photonics, Japan) mounted on an upright microscope (BX50WI, Olympus) using Fura-4F (50 μm, Invitrogen). Fura-4F was loaded into a PN through a patch pipette, and excited alternately at 340 and 380 nm for 120 ms. Each fluorescence image was recorded at 2 Hz, and the fluorescence ratio (the fluorescence excited at 340 nm divided by that at 380 nm) was calculated. Transfected PNs were identified by co-expression of RFP. The background signal arising from slight excitation of RFP by UV light was subtracted. Based on in vitro calibration, the fluorescence ratio was converted to absolute [Ca2+]i as follows: [Ca2+]i = 590 × (ratio − Rmin)0.82/(Rmax − ratio)0.82. Rmax and Rmin are the maximum and minimum ratio values determined by in vivo calibration, respectively. Rmax was measured by applying 5 μm ionomycin to PNs in the presence of 2 mm Ca2+ in the extracellular solution. Rmin was obtained by applying 15 mm Cs-BAPTA into a PN in 0 Ca2+ extracellular solution. Because of the background fluorescence of RFP, both Rmax and Rmin were measured for both non-transfected PNs and RFP-expressing PNs (non-transfected, Rmax = 3.39, Rmin = 0.44; RFP-expressing, Rmax = 4.41, Rmin = 0.29).
FRET imaging
FRET images were obtained from a PN transfected with VαC, VβC, CαV, or mutated probes using an upright microscope BX61WI equipped with an FV1000 fluorescence imaging system (Olympus). FRET probes were excited by a 405 or 440 nm laser, and the emission between 460 and 500 nm (for ECFP) and that between 515 and 615 nm (for Venus) were spectroscopically separated and recorded by two photomultipliers. Time-lapse imaging of FRET change was performed from a whole-cell patch-clamped PN, and each image was obtained every 10 s. Fluorescence intensities of ECFP and Venus in a shaft of a proximal dendrite of a PN were measured, and the fluorescence ratio (ECFP/Venus) was calculated. Fluorescence spectrums of FRET probes were obtained by systematically changing the wavelength of image acquisition spectroscopically (10 nm width, 5 nm step) under the illumination of a 405 nm laser. The bleaching of Venus was performed by illumination at maximum laser intensity of 515 nm.
Statistics
Data are presented as mean ± SEM. Statistical significance was assessed by Student's t test or one-way ANOVA followed by post hoc Dunnett T3 test.
Results
Expression of α- and βCaMKII in a PN
We first confirmed that CaMKII plays a critical role in RP induction. RP was monitored by whole-cell recording of current responses to GABA iontophoretically applied to a proximal or secondary dendrite of a rat cultured PN. As shown in Fig.1A, conditioning depolarization (0 mV for 500 ms, 0.5 Hz, 5 times) induced RP, i.e. the amplitude of GABA-induced current was increased for longer than 30 min (154 ± 9% of basal, at 30 min). This RP induction was suppressed by inhibition of CaMKII activity using a specific inhibitor AIPII (1 μm) (100 ± 14%, P < 0.05 compared with control). Thus, in accordance with previous studies (Kano et al. 1996; Kawaguchi & Hirano, 2002), CaMKII activity is essential for RP induction.
Figure 1. α- and βCaMKII in a PN.
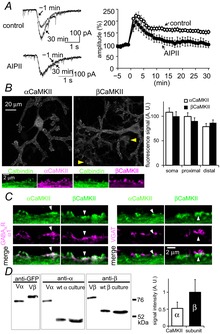
A, representative traces and time courses of GABA current amplitudes before and after the conditioning depolarization in the absence (control) or presence of AIPII (1 μm). n = 5 for each. B, immunofluorescence images of endogenous α- or βCaMKII in PNs. The area of a PN was defined as the calbindin-positive area (not shown). Yellow arrowheads indicate βCaMKII expression in neurons other than a PN, which were calbindin-negative. Bottom, high-resolution images of each CaMKII subunit (magenta) and calbindin (green, a PN marker). Right, fluorescence intensities of anti-αCaMKII and anti-βCaMKII signal in soma, proximal and distal dendrites. Staining efficiency of each antibody is calibrated, and the signal is normalized to that of βCaMKII in soma. n = 20 cells for each. C, representative immunofluorescence images of GABAAR α1 or VGAT, and α- or βCaMKII. D, western blotting of Venus-tagged CaMKII subunits (Vα and Vβ) and wild-type CaMKII subunits (wt α and wt β) prepared from HEK293T cells, and endogenous α- and βCaMKII prepared from a cerebellar culture (n = 3 cultures). Quantified signals for α- and βCaMKII after calibration of staining efficiency of each antibody are shown. A.U., arbitrary units.
Next, localization of CaMKII in PNs was examined by immunofluorescent staining using a specific antibody against α- or βCaMKII antibody. Both α- and βCaMKII were observed in dendritic shafts and around the plasma membrane (Fig.1B). Co-staining of GluD2 which is enriched at dendritic spines in a PN showed that CaMKII signals were found not only around spines but also GluD-negative membranous regions in dendritic shafts (not shown). To estimate the localization of CaMKII at inhibitory synapses, we stained either CaMKII subunit together with GABAAR α1 subunit or vesicular GABA transporter (VGAT; a marker for GABAergic presynaptic terminals). Some α- and βCaMKII signals were co-localized with GABAAR α1 signals (Fig.1C left, arrowheads), and were also found adjacent to VGAT signals (Fig.1C right, arrowheads). Thus, both α- and βCaMKII were located not only at excitatory postsynaptic spines but also around GABAergic postsynaptic membrane.
We next estimated the relative abundance of α- and βCaMKII in a PN. Toward this aim, we expressed Venus (a variant of GFP)-tagged α- or βCaMKII (Vα or Vβ, respectively) in HEK293T cells and stained them with specific antibodies against α- or βCaMKII and GFP. Then, the fluorescence intensity of the α- or βCaMKII signal was compared with that of Venus, and the staining efficiency of anti-α- or βCaMKII antibody relative to that of anti-GFP antibody was estimated. In our staining conditions, anti-βCaMKII antibody was 1.05 times more efficient than anti-αCaMKII antibody. Based on this calibration, the amount of αCaMKII was estimated to be 1.10 ± 0.08 times that of βCaMKII in a proximal dendrite of a PN (soma, 1.08 ± 0.09 times; distal dendrite, 0.91 ± 0.08 times) (Fig.1B). We also quantified relative amounts of α- and βCaMKII in a cerebellar culture by western blotting (Fig.1D). Based on the relative efficiency of anti-αCaMKII antibody compared to anti-βCaMKII antibody (1.55 times) estimated with a method similar to that used for immunocytochemistry, the amount of αCaMKII was 0.52 ± 0.19 times that of βCaMKII in cultured cerebellar neurons (Fig.1D). The βCaMKII abundance in cerebellar culture was about half of a previous estimation in slices (McGuinness et al. 1985), probably due to a reduced relative number of granule cells in culture. αCaMKII expression is limited to PNs in the cerebellum, whereas βCaMKII is expressed in not only PNs but also granule neurons and inhibitory interneurons (see Fig.1B; Walaas et al. 1988). Thus, the relative abundance of β- to αCaMKII in a PN should be lower than the estimated value by western blotting. Together, these results suggest that α- and βCaMKII are expressed almost equally in a PN.
βCaMKII is essential for RP induction
To examine whether α- and βCaMKII contribute to RP equally or differently, we first knocked down the expression of each subunit in a cultured PN using RNAi. Subunit-specific RNAi target sequences for α- and βCaMKII used in previous studies (Okamoto et al. 2007) were expressed using a plasmid-based short hairpin RNA (shRNA) expression vector. In accordance with previous studies (Okamoto et al. 2007), shRNA transfection reduced the amount of α- or βCaMKII exogenously expressed in HEK293 cells to 14 ± 1% or 37 ± 14%, respectively (Fig.2A and B). Also in a PN, αCaMKII-targeting RNAi plasmid (α KD (knock-down)) significantly decreased the fluorescence signal intensity of αCaMKII (Fig.2C and D; 64 ± 6% of control, P < 0.005 compared to that in control PNs transfected with null plasmid, Dunnett T3 test), but not that of βCaMKII (95 ± 9%, P = 0.93). Conversely, the shRNA against βCaMKII (β KD) reduced the signal of βCaMKII but not αCaMKII (Fig.2C and D; αCaMKII, 90 ± 4%, P = 0.15; βCaMKII, 62 ± 4%, P < 0.001). The reduction of either CaMKII subunit by corresponding shRNA was observed similarly in a soma, proximal and distal dendrites irrespective of membranous or cytoplasmic regions. Knock-down of either CaMKII subunit caused no apparent changes in CaMKII localization, the number of spines, or morphology of dendrites of PNs (not shown).
Figure 2. Critical role of βCaMKII in RP.
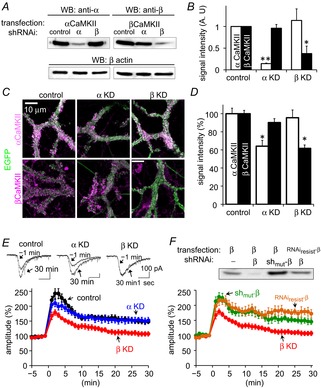
A and B, western blotting (A) and its quantification (B) of α- (left in A) and βCaMKII (right in A) exogenously expressed in HEK293 cells in which RNAi expression vector without (control) or with shRNA for α- or βCaMKII was expressed. For negative control, blotting against β-actin is also shown. **P < 0.005; *P < 0.05. n = 3 cultures. C, representative immunofluorescence images of α- or βCaMKII (magenta) and EGFP (green) in PNs transfected with shRNA expression vectors. D, quantification of fluorescence signal intensity for α- or βCaMKII. For αCaMKII, n = 69 (control), 39 (α KD), and 53 (β KD) cells. For βCaMKII, n = 73 (control), 74 (α KD), and 65 (β KD) cells. *P < 0.001, Dunnett T3 test. E, representative traces and time courses of GABA-induced current amplitudes before and after the conditioning depolarization in a PN transfected with RNAi expression vector without (control) or with shRNA for α- (α KD) or βCaMKII (β KD). n = 5 for each. F, western blotting of βCaMKII exogenously expressed in HEK293T cells (top) and time courses of RP in a PN (bottom). shRNA for βCaMKII without (β KD) or with the RNAi-resistant form of βCaMKII (RNAiresist-β), or mutated shRNA (shmut-β) was transfected to HEK293 cells or to a PN. RP time course for β KD is the same as that in E, and presented for comparison. n = 5 for each.
Then, we examined how the reduction of each subunit affected RP. In a control PN transfected with an RNAi plasmid lacking the target sequence for either CaMKII subunit, conditioning depolarization pulses induced RP (Fig.2E; 148 ± 6% of basal at 30 min). In spite of the requirement of CaMKII in RP induction (as shown in Fig.1A), knock-down of αCaMKII did not affect RP induction (Fig.2E; 152 ± 10%, P = 0.94, compared to control vector, Dunnett T3 test). In contrast, reduction of βCaMKII expression suppressed long-lasting potentiation of the GABA response (Fig.2E; 107 ± 8%, P < 0.005), although the potentiation soon after the depolarization remained. When a PN was transfected with a negative control shRNA for βCaMKII in which six nucleotides of the target sequence were mutated, RP induction was not affected (145 ± 9%, P > 0.8, Fig.2F). Importantly, the RP induction impaired by βCaMKII shRNA was rescued by additional transfection of an RNAi-resistant form of βCaMKII (172 ± 14%, P < 0.01 compared to β KD, Fig.2F). Thus, RP induction seemed to preferentially depend on βCaMKII but not on αCaMKII. Neither knock-down of αCaMKII nor of βCaMKII significantly affected basal inhibitory synaptic transmission as estimated by miniature inhibitory postsynaptic current (mIPSC) (amplitude, control, 100 ± 4%; α KD, 111 ± 8%; β KD, 90 ± 11%; P = 0.18, one-way ANOVA; frequency, control, 100 ± 8%; α KD, 99 ± 6%; β KD, 105 ± 11%; P = 0.85).
The amplitude of [Ca2+]i increase caused by the conditioning depolarization was measured by [Ca2+]i imaging using fluorescent Ca2+ indicator Fura-4F (50 μm) loaded to a PN through a glass patch pipette for longer than 5 min. The fluorescence ratio (F340/F380) in a proximal dendrite of a PN during the conditioning depolarization was not affected by knock-down of either subunit (control, 3.52 ± 0.51; α KD, 3.46 ± 0.50; β KD, 3.30 ± 0.41; P = 0.94, one-way ANOVA). These ratios corresponded to about 1.7, 1.6 and 1.3 μm [Ca2+]i, respectively. Taken together, these results suggest that βCaMKII preferentially contributes to RP induction.
Overexpression of αCaMKII suppressed RP
To further examine the respective role of each CaMKII subunit, we next examined the effect of increase in each subunit on RP by whole-cell recording from a PN transfected with either subunit. Transfection of a plasmid encoding α- or βCaMKII (α or β OE (overexpression)) increased the fluorescence signal intensity of the corresponding subunit in a PN (Fig.3A and B; αCaMKII, 884 ± 42% of EGFP-transfected control cells, P < 0.001 compared to control PNs, Dunnett T3 test; βCaMKII, 555 ± 29%, P < 0.001). Transfection of either subunit slightly, but not significantly, decreased the amount of the other subunit, probably through some unknown compensatory mechanism (βCaMKII in αCaMKII-overexpressing PNs, 84 ± 4%, P = 0.89; αCaMKII in βCaMKII-overexpressing PNs, 86 ± 6%, P = 0.96). In a control PN transfected with EGFP alone, conditioning depolarization induced RP (Fig.3C; 150 ± 6% of basal, at 30 min). Surprisingly, RP induction was impaired by overexpression of αCaMKII, but not by that of βCaMKII (Fig.3C; EGFP, 150 ± 6%, α OE, 111 ± 8%, P < 0.001; β OE, 153 ± 6%, P = 0.98). Overexpression of α-or βCaMKII did not affect the amplitude or the frequency of mIPSCs (amplitude, EGFP, 100 ± 5%; α OE, 105 ± 8%; β OE, 106 ± 10%; P = 0.84, one-way ANOVA; frequency, EGFP, 100 ± 9%; α OE, 84 ± 4%; β OE, 83 ± 7%; P = 0.15), or the [Ca2+]i increase caused by the conditioning stimulation (F340/F380: RFP, 3.51 ± 0.30; α OE, 3.56 ± 0.37; β OE, 3.61 ± 0.14, P = 0.97, corresponding to about 1.7, 1.8 and 1.9 μm [Ca2+]i, respectively). According to our simulation of Ca2+-caused CaMKII activation using a simple model, total CaMKII activity primarily depends on βCaMKII with higher CaM affinity, and is weakened by an aberrant increase of αCaMKII due to disturbance of efficient positive-feedback autophosphorylation at Thr287 of βCaMKII in a holoenzyme (authors’ unpublished observations). Thus, the RP impairment by αCaMKII overexpression might be ascribed to insufficient activation of βCaMKII activation. Taken together, these results indicated that αCaMKII might negatively regulate RP in contrast to a positive role of βCaMKII.
Figure 3. αCaMKII negatively regulates RP.
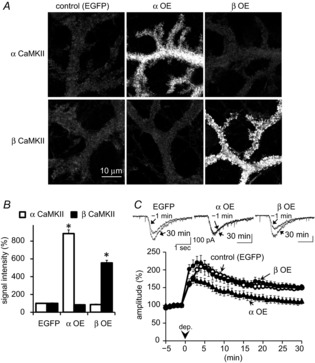
A, immunofluorescence images of α- or βCaMKII in a PN in which either subunit was overexpressed. To avoid saturation of signal, these images were taken with low detector sensitivity. B, relative immunofluorescence signal intensities of α- and βCaMKII in PN dendrites transfected with EGFP and α- or βCaMKII. For αCaMKII, n = 58 (EGFP), 52 (α OE), and 46 (β OE) cells; for βCaMKII, n = 49 (EGFP), 62 (α OE), and 56 (β OE) cells. *P < 0.001, Dunnett T3 test. C, representative traces and time courses of GABA-induced current amplitudes before and after the conditioning depolarization in a PN with overexpression of α- or βCaMKII. n = 5 for each.
Impact of CaMKII subunit composition on RP induction
Positive and negative regulations of RP by β- and αCaMKII as described above suggested that the subunit composition of a CaMKII holoenzyme might be important for RP induction. To test this idea, the relative abundance of α- and βCaMKII subunits changed by knock-down or overexpression of either subunit was restored by additional manipulation of the other subunit. Additional βCaMKII overexpression rescued RP from the impairment by αCaMKII overexpression (Fig.4A; 146 ± 7% of basal, P < 0.05, compared with α OE alone). Co-transfection of α- and βCaMKII increased the total amount of CaMKII to about 5 times (537 ± 38% for αCaMKII, n = 38 cells, 502 ± 30% for βCaMKII, n = 45 cells; both P < 0.001 compared with control). Furthermore, the RP suppression by βCaMKII knock-down was also rescued by additional knock-down of αCaMKII (Fig.4B; 152 ± 3% of basal, P < 0.005, compared with β KD alone). Our model simulation of CaMKII activation also suggested that recovery of subunit composition by reducing αCaMKII or by increasing βCaMKII leads to stronger activation of βCaMKII which is critical for RP induction (authors’ unpublished observations). Co-expression of shRNAs for α- and βCaMKII together reduced the total amount of CaMKII to about 60% (63 ± 5% for αCaMKII, n = 53 cells, 56 ± 3% for βCaMKII, n = 46 cells; both P < 0.001 compared with control). Thus, the subunit composition, rather than the absolute amount of CaMKII, is critical for RP induction.
Figure 4. CaMKII subunit composition is critical for RP.
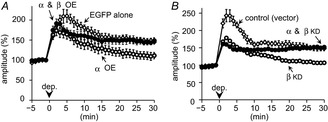
A, rescue of RP in a PN with overexpression of both α- and βCaMKII. Time course of RP with αCaMKII overexpression alone and that with EGFP alone (same as Fig.3C) are also presented for comparison. n = 5 for each. B, rescue of RP in a PN with knock-down of both α- and βCaMKII. Time course of RP with βCaMKII knock-down and that with control RNAi vector (same as Fig.2E) are also presented for comparison. n = 5 for each.
PP mediates RP suppression by αCaMKII overexpression or βCaMKII reduction
The CaMKII activity is balanced by the protein phosphatase (PP) activity, which controls RP induction (Kawaguchi & Hirano, 2002). To test whether PP is involved in the RP impairment caused by αCaMKII overexpression, a PP inhibitor, nodularin (2 nm; Honkanen et al. 1991), was added to the external solution. Application of nodularin by itself did not affect RP induction (Fig.5A, 150 ± 9% of basal at 30 min, P = 0.75). In the presence of nodularin, RP was rescued from the suppression by βCaMKII knock-down (Fig.5B; 139 ± 10%, P < 0.05) or by αCaMKII overexpression (Fig.5C; 156 ± 6%, P < 0.005). Nodularin affected neither the amplitude nor the frequency of mIPSCs (amplitude, 94 ± 4%, P > 0.05, compared with the control; frequency, 93 ± 9%, P > 0.05; n = 11 cells), nor the [Ca2+]i increase (F340/F380, 2.58 ± 0.30 corresponding to about 1.7 μm, P > 0.05 compared with the control; n = 7 cells). These results suggested that the RP impairment caused by aberrant subunit composition of a CaMKII holoenzyme is due to insufficient CaMKII activity relative to the PP activity.
Figure 5. Involvement of PP in RP impairment by αCaMKII-dominant subunit composition.
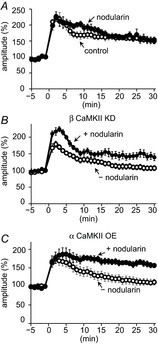
Time course of GABA current amplitudes before and after the conditioning depolarization in the absence or presence of nodularin (2 nm) in a PN without (A) or with transfection with shRNA for βCaMKII (B) or with αCaMKII (C).
βCaMKII contributes to RP via high Ca2+/CaM affinity but not association with F-actin
Compared with αCaMKII, βCaMKII has about 10 times higher affinity for Ca2+/CaM, suggesting that βCaMKII could be activated even by smaller Ca2+ increase. In addition, βCaMKII has a site for association with filamentous actin (F-actin), and reciprocal regulation of βCaMKII and F-actin has been suggested (Okamoto et al. 2009), which has been shown to be involved in plasticity at excitatory synapses (Borgesius et al. 2011). To address how βCaMKII preferentially contributes to RP, we examined which of high CaM affinity or association with actin cytoskeleton is critical for RP. For this aim, we used two mutant βCaMKII subunits: one is a deletion mutant of βCaMKII lacking F-actin association, and the other is a chimera of α- and βCaMKII which can bind to F-actin but has CaM affinity similar to αCaMKII. Previous studies showed that the variable region of βCaMKII is critical for association with F-actin, and the catalytic and CaM binding regions are responsible for higher CaM affinity (O'Leary et al. 2006; Deguchi et al. 2008). Accordingly, deletion of a segment (V1) in the variable region abolished the co-localization of βCaMKII and F-actin stained with Alexa568-conjugated phalloidin in HEK293T cells (Fig.6A and B). Biochemical assay for CaMKII subunit autophosphorylation at Thr286/287 demonstrated that the F-actin association-deficient mutant βΔV1 was activated by tens of nanomolar Ca2+/CaM (apparent EC50 = 29 nm) similar to wild-type βCaMKII (apparent EC50 = 33 nm) (Fig.6C). Thus, βΔV1 has high affinity for Ca2+/CaM similar to βCaMKII in line with previous studies (Deguchi et al. 2008). When expressed together with αCaMKII, the βΔV1 mutant rescued RP induction from the impairment by αCaMKII overexpression (Fig.6D, 153 ± 12%, P < 0.05, compared with α OE). Thus, association with F-actin seemed to be dispensable for the contribution of βCaMKII to RP induction.
Figure 6. High CaM affinity of βCaMKII, but not F-actin association, is critical for RP.
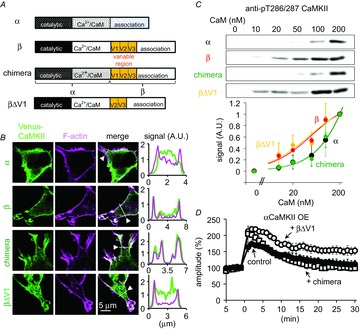
A, domain structures of α- and βCaMKII, chimera of α- and βCaMKII, and βCaMKII deletion mutant βΔV1. B, localization of αCaMKII, βCaMKII, chimera, and βΔV1 (green) and F-actin (magenta) in HEK293T cells. The distribution patterns along a line indicated in each panel were plotted. C, western blotting of T286 or T287-autophosphorylated wild-type or mutant CaMKII subunits. Each subunit was treated with different concentrations of Ca2+/CaM. Bottom, dose–response curves for CaM-triggered T286/287 autophosphorylation of CaMKII subunits. n = 3 trials for α, β and chimera; n = 4 for βΔV1. D, time courses of GABA-induced current amplitudes before and after the conditioning depolarization in a PN with overexpression of αCaMKII together with chimera or βΔV1. n = 5 for each.
Previously, it was reported that a chimera CaMKII, composed of the catalytic and CaM binding domains of αCaMKII and the variable and association domains of βCaMKII, has low CaM affinity similar to αCaMKII (Deguchi et al. 2008). Our biochemical assay also showed that the chimera CaMKII required hundreds of nanomolar CaM for effective activation (apparent EC50 = 89 nm) similar to αCaMKII (apparent EC50 = 90 nm) (Fig.6C). On the other hand, the chimera CaMKII expressed in HEK cells exhibited intensive co-localization with F-actin (Fig.6B), in accord with previous studies suggesting association of βCaMKII with F-actin through the variable region (O'Leary et al. 2006; Deguchi et al. 2008). When the chimera was expressed together with αCaMKII in a PN, RP was not induced (Fig.6D, 99 ± 8%, P = 0.3, compared with α OE). Thus, a mutant CaMKII which has F-actin binding ability and lower CaM affinity could not abolish the suppressive effect of αCaMKII overexpression, supporting the idea that the higher CaM affinity of βCaMKII is critical. Taken together, these results indicated that the preferential contribution of βCaMKII to RP depends not on association with F-actin but on high affinity for Ca2+/CaM.
To further confirm the idea, we examined whether F-actin by itself was involved in RP. When cultured PNs were treated for 30 min with an actin polymerization inhibitor latrunculin A (LatA, 2 μm), the F-actin signal markedly decreased in both proximal and distal dendrites (Fig.7A; 26 ± 1% of control, P < 0.001, Student's t test), accompanied by the change of the spine morphology to a filopodia-like thin structure (Fig.7B). However, the total number of spines and filopodia did not change (from 8.1 ± 0.6 (10 μm)–1 to 7.5 ± 0.4 (10 μm)–1, 10 cells for each). Indeed, excitatory postsynaptic density stained with GluD antibody still remained in small protrusions, although the number of GluD puncta tended to be reduced slightly (77 ± 9% of control, P > 0.09) (Fig.7B). Whereas the LatA treatment did not affect the total signal intensity of either α- or βCaMKII in a proximal dendrite (αCaMKII, 103 ± 6% of control, P = 0.60; βCaMKII, 101 ± 4%, P = 0.89), the membranous localization of each CaMKII subunit was changed to relatively homogeneous localization in the cytoplasm (Fig.7C and D). Thus, the membranous localization of CaMKII is controlled by actin cytoskeleton. Even after the distribution of CaMKII was disturbed by F-actin destruction, RP was induced by the conditioning depolarization (Fig.7E, 147 ± 4% at 30 min, P = 0.54 compared with control, Student's t test). Thus, the enriched localization of CaMKII around the plasma membrane seems not to be required for RP induction, probably due to its abundant expression in a PN. We did not detect significant LatA-induced alteration of the mIPSC amplitude or frequency (amplitude, 83 ± 5%, P > 0.05; frequency, 121 ± 8%, P > 0.05), or of the [Ca2+]i increase caused by the conditioning depolarization (F340/F380, control, 2.77 ± 0.33; LatA, 2.58 ± 0.30, P = 0.89, corresponding to about 1.7 and 1.3 μm [Ca2+]i, respectively). Thus, F-actin does not affect the basal inhibitory synaptic transmission or the Ca2+ increase during RP induction. Taken together, these results were consistent with the idea that the predominant role of βCaMKII in RP induction does not depend on association with F-actin.
Figure 7. F-actin destruction altering CaMKII distribution does not affect RP.
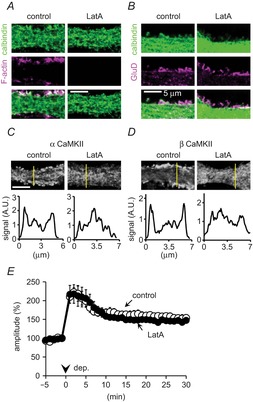
A, B, C and D, representative fluorescence images of F-actin (A, magenta), GluD (a spine marker of PNs, B), αCaMKII (C) or βCaMKII (D), and calbindin (green, a PN marker, A and B) before and after LatA treatment (2 μm) for 30 min. Line plots (along the yellow line indicated in each image) of signal intensity for α- and βCaMKII are shown in C and D. E, time courses of GABA-induced current amplitudes before and after the conditioning depolarization in a PN with or without LatA treatment. n = 5 for each. The control time course is the same as that shown in Fig.1A (shown for comparison).
Enhanced CaMKII activity by βCaMKII
The above results suggested that the βCaMKII dominant subunit composition is critical for RP induction due to its higher affinity with Ca2+/CaM. Then, does βCaMKII dominance lead to higher CaMKII activity in a PN? To address this issue, we attempted to monitor the CaMKII activity using a FRET technique. A FRET probe Camuiα was previously developed by tandem fusion of Venus, αCaMKII and CFP, and has been used to image the CaMKII activity in hippocampal neurons (Takao et al. 2005; Lee et al. 2009). The conformational change of Camuiα caused by Ca2+/CaM binding or by autophosphorylation at Thr286 was shown to reduce the fluorescent energy transfer from CFP to Venus. Therefore, the CaMKII activity can be estimated by measuring the ratio of CFP to Venus. We constructed a probe (here we call it VαC for distinction from other probes used below), which is essentially similar to Camuiα except for linker sequences (several amino acids) derived from restriction enzyme sequences between αCaMKII and fluorophores (Fig.8A). Even if two fluorophores were fused, VαC showed CaM-dependent activation similar to αCaMKII (Fig.8B, apparent EC50 = 105 nm). VαC distributed throughout somatodendritic regions in a PN as endogenous αCaMKII, and emitted fluorescence with intensity peaks for ECFP (∼470 nm) and for Venus (∼520 nm) upon illumination by a 405 nm laser similarly to Camuiα, indicating that FRET occurred in the basal condition (Fig.8C and D). Indeed, bleaching of Venus by 515 nm laser illumination resulted in an increase of the fluorescence of ECFP. Camuiα was shown to be incorporated into a holoenzyme (Takao et al. 2005), so that it can be used for detection of the CaMKII activity change through subunit interactions.
Figure 8. FRET monitoring of CaMKII activation in a PN.
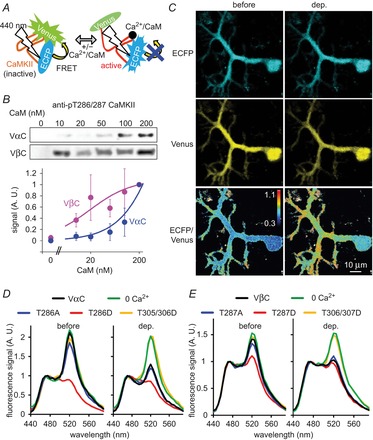
A, illustration of alteration in FRET between ECFP and Venus in a CaMKII FRET probe reflecting its conformational change by activation. B, western blotting of T286- and T287-autophosphorylated VαC and VβC probes upon treatment with different concentrations of Ca2+/CaM, respectively. Bottom, dose–response curves for CaM-triggered T286/287 autophosphorylation of VαC (n = 3) and VβC (n = 4). C, representative images of ECFP, Venus, and ECFP/Venus ratio (shown as pseudo colour) for VαC before and during the conditioning depolarization in a PN. D and E, fluorescence emission spectrums of VαC and its mutants (D) or of VβC and its mutants (E) excited by a 405 nm laser before and during the conditioning depolarization. The fluorescence intensities were normalized by the peak around 470 nm.
The conditioning depolarization for RP induction in a VαC-expressing PN under whole-cell recording markedly decreased the fluorescence peak for Venus, and consequently increased the ECFP/Venus ratio (Fig.8C and D). Removal of Ca2+ from the external solution abolished the depolarization-induced decrease of Venus signal (Fig.8D, 0 Ca2+), suggesting that the FRET change reflects the Ca2+-induced conformational change of VαC. Indeed, replacement of amino acids Thr305/306 of VαC by aspartate, which suppresses the Ca2+/CaM association, abolished the FRET change induced by depolarization (Fig.8D, T305/306D). Clear occurrence of FRET and its Ca2+-dependent decrease were also observed with another FRET probe in which positions of ECFP and Venus at the N- and C-termini were exchanged (not shown). These results suggest that the Ca2+-dependent CaMKII activation accompanied by the conformational change can be detected by the change of FRET between Venus and ECFP in each probe molecule. It should be noted that slight inter-molecular FRET was detected between Vα and αC, although depolarization-induced change of the signal did not take place (not shown). This suggests that fluorophore-tagged CaMKII can form a multimer causing inter-molecular FRET, but the FRET change observed upon Ca2+ increase predominantly reflects conformational change of each subunit.
Next, to evaluate the effects of Thr286 autophosphorylation on FRET, we also constructed mutant probes in which Thr286 of αCaMKII in VαC was replaced by alanine or aspartate, named Vα(T286A)C or Vα(T286D)C, respectively. Vα(T286A)C showed a similar emission spectrum to VαC before and soon after the depolarization (Fig.8D). On the other hand, Vα(T286D)C, which is expected to take an autonomously active conformation, showed much lower FRET in the basal condition, and the conditioning depolarization did not change it (Fig.8D). Together, these results suggest that VαC detected the conformational changes of αCaMKII caused by Ca2+/CaM association upon the [Ca2+]i increase and by autophosphorylation at Thr286, in accord with previous studies (Takao et al. 2005).
To measure the time course of CaMKII activation after RP induction, we performed time-lapse imaging with the VαC probe (Fig.9A and B). When depolarization pulses were given to a PN through a whole-cell patch pipette, the signal intensity of Venus decreased and then returned to the basal level within several minutes (Fig.9B). As a result, the ECFP/Venus signal ratio increased immediately after the depolarization (136 ± 4% of basal, at 30 s), and then returned to the basal level in several minutes (Fig.9A, B and C; 103 ± 2% at 6 min, 98 ± 2% at 18 min). Abolishment of autophosphorylation by mutation at Thr286 of the probe did not affect the time course of the FRET signal (Fig.9C; 97 ± 2% at 18 min, P > 0.05, compared to VαC at 18 min, Student's t test). Thus, the autonomous activity relying on Thr286 autophosphorylation was marginal, if any, and the CaMKII activation seemed not to last for a long time in a PN expressing VαC. To examine whether βCaMKII with higher affinity for Ca2+/CaM augments the CaMKII activation or not, we also constructed a βCaMKII-based probe VβC. The VβC probe showed CaM-dependent activation similar to βCaMKII (see Fig.8B, apparent EC50 = 17 nm), and exhibited a FRET change during Ca2+ increase similar to VαC (see Fig.8E). As shown in Fig.9D, E and F, VβC exhibited sustained increase of CFP/Venus ratio (i.e. FRET decrease) upon the conditioning depolarization (110 ± 2% at 18 min, P < 0.01). This sustained FRET change was abolished by mutation of Thr287 of the probe into alanine (100 ± 2% at 18 min, P < 0.01 compared with VβC), suggesting that the sustained FRET change of VβC was mediated by autophosphorylation of that residue. Thus, these results indicated that βCaMKII is activated more persistently than αCaMKII in a PN.
Figure 9. βCaMKII is more strongly activated than αCaMKII in a PN.
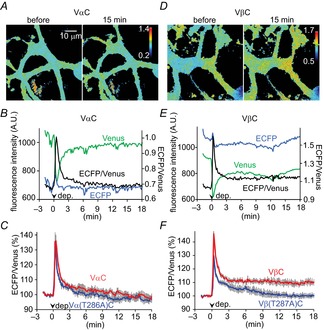
A and B, representative images (A) and time courses of fluorescence signal of ECFP, Venus and ECFP/Venus (B) of VαC before and after the conditioning depolarization. C, averaged time courses of ECFP/Venus before and after the conditioning depolarization in a PN expressing VαC or Vα(T286A)C. n = 7 for each. D and E, representative images (D) and time courses of fluorescence signal of ECFP, Venus and ECFP/Venus (E) of VβC before and after the conditioning depolarization. F, averaged time courses of ECFP/Venus before and after the conditioning depolarization in a PN expressing VβC or Vβ(T287A)C. n = 6 for each.
We hypothesized that VαC expression might have made the total CaMKII subunit composition α-dominant, resulting in a weakening of the total activation. To test this possibility, we restored the subunit composition by expressing βCaMKII in a PN together with VαC. Additional expression of βCaMKII markedly weakened the increase of the ECFP/Venus signal of VαC soon after the depolarization (Fig.10A, 105 ± 1% of basal, at 30 s), probably due to the decrease of available Ca2+/CaM by competition between VαC and overexpressed βCaMKII. However, remarkably, the FRET ratio remained high until the end of the experiment depending on autophosphorylation at the Thr286 residue of VαC (Fig.10A; VαC and βCaMKII, 106 ± 1% at 18 min; Vα(T286A)C and βCaMKII, 100 ± 1%, P < 0.01, t test). In contrast, additional expression of αCaMKII did not sustain the FRET change (Fig.10B; VαC and αCaMKII, 97 ± 2% at 18 min; Vα(T286A)C and αCaMKII, 99 ± 3%; P = 0.7, t test). As shown in Fig.10C and D, the ability of βCaMKII to sustain the FRET change of VαC was abolished by mutation of K43R (kinase activity-dead, 100 ± 4%, P = 0.47) or T305/306D (suppressed CaM binding, 100 ± 5%, P = 0.49), indicating that the activity of βCaMKII contributes to prolonging the VαC FRET change. βCaMKII-mediated prolongation of FRET change was also observed with the CαV probe (Fig.10D; CαV and βCaMKII, 105 ± 1% at 18 min, P < 0.05, CαV and αCaMKII, 97 ± 2%, P = 0.97, compared to CαV alone, Dunnett T3 test). Thus, if enough βCaMKII is expressed relative to αCaMKII, CaMKII activation seems to be prolonged based on autophosphorylation at Thr286/287 by overcoming the counteraction by PPs. Consistently, when PP activity was inhibited by an inhibitor nodularin (nod), PN expressing VαC alone, but not Vα(T286A)C, exhibited sustained FRET change for longer than 18 min (Fig.10C and D; VαC + nod, 109 ± 2% of basal, at 18 min; Vα(T286A)C + nod, 99 ± 2%, P < 0.005, t test). Taking these results together, the β-subunit dominant composition of CaMKII determines whether or not its activation is persistent or transient.
Sustained CaMKII activity depending on βCaMKII.
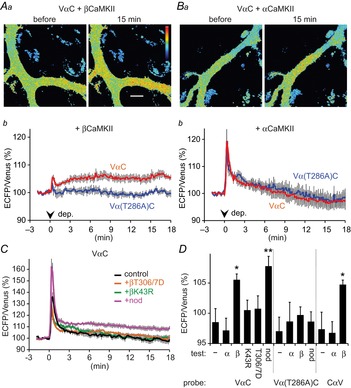
A and B, representative ratio images (Aa and Ba) and time courses (Ab and Bb) of ECFP/Venus of VαC before and after the conditioning depolarization in PNs with additional transfection of βCaMKII (A) or αCaMKII (B). In Ab and Bb, the results obtained for Vα(T286A)C are also shown. n = 7 for each. C, time courses of the ECFP/Venus of VαC without (control) or with additional expression of mutant βCaMKII (T306/7D and K43R) or with nodularin (nod). n = 7 (control and nod) and 5 (T306/7D and K43R). D, ECFP/Venus ratio at 18 min of VαC, Vα(T286A)C, or CαV without (–) or with additional expression of αCaMKII, βCaMKII, mutant βCaMKII (T306/7D or K43R) or with nodularin (nod). n = 7 except for K43R or T306/7D (n = 5). **P < 0.01; *P < 0.05.
Discussion
Using patch clamp recording and FRET imaging on cultured PNs in which relative expression levels of CaMKII subunits were modulated, the present study revealed contrasting roles of α- and βCaMKII in RP induction. Electrophysiological experiments demonstrated that knock-down of βCaMKII, but not that of αCaMKII, impaired RP induction. On the other hand, overexpression of αCaMKII, but not that of βCaMKII, suppressed RP. Thus, βCaMKII is essential for RP induction, whereas αCaMKII negatively regulates it, indicating importance of CaMKII subunit composition in RP induction. Distinct roles of α- and βCaMKII in RP were not mediated by specific association of βCaMKII with F-actin, but by their different affinities for Ca2+/CaM. FRET imaging showed that predominance of βCaMKII in a holoenzyme prolongs the total CaMKII activity. Taking all these results together, we conclude that the subunit composition of CaMKII is critical for its activation and hence for RP.
Opposite roles of α- and βCaMKII in synaptic plasticity
The present results demonstrated an essential role of βCaMKII and a suppressive role of αCaMKII in RP at inhibitory synapses on a PN. This negative control of plasticity induction by αCaMKII contrasts with a pivotal role of αCaMKII in NMDA receptor-dependent LTP at hippocampal synapses. αCaMKII knockout mice and mutant mice with amino acid replacement of Thr286 by Ala show deficits in hippocampal LTP at excitatory synapses and in spatial learning (Silva et al. 1992; Giese et al. 1998). Importantly, αCaMKII has a site that associates with NMDA receptor subunit NR2B (Bayer et al. 2006), and this binding sustains activation of αCaMKII (Bayer et al. 2001), thereby contributing to LTP (Barria & Malinow, 2005; Lisman et al. 2012). Thus, the NMDA receptor might not only mediate the [Ca2+]i increase but also compensate for low Ca2+/CaM affinity of αCaMKII, thereby contributing to effective CaMKII activation and hence LTP induction. NMDA receptor activation also induces LTP at inhibitory synapses in the hippocampus through αCaMKII-mediated increased transport of GABAARs toward the plasma membrane (Marsden et al. 2010).
In contrast to hippocampal pyramidal neurons, a cerebellar PN has few NMDA receptors until 4 weeks after birth (Llano et al. 1991; Piochon et al. 2007), and this might be responsible for the lack of a clear αCaMKII contribution to RP induction in cultured PNs. Thus, at inhibitory synapses on a PN, αCaMKII might contribute to stable transmission by restricting excessive synaptic plasticity. It should be noted that our results do not necessarily rule out a possibility that complete loss of αCaMKII impairs RP through a large reduction in the amount of total CaMKII. According to our simulation model, abundant αCaMKII such as that caused by overexpression seems to compete with endogenous βCaMKII for a limited amount of Ca2+/CaM during the [Ca2+]i increase, resulting in dispersed CaM binding to CaMKII subunits located in different holoenzymes and hence weakened Thr286/287 autophosphorylation (authors’ unpublished observations). On the other hand, reduction of αCaMKII such as that caused by knock-down tends to augment the βCaMKII activation by reduced competition for Ca2+/CaM, but the augmented βCaMKII activation does not necessarily lead to facilitation of the extent of RP (see Fig.2), probably due to saturated activation of RP-inducing signalling cascades downstream of CaMKII (Kitagawa et al. 2009). αCaMKII is also involved in long-term depression (LTD) at excitatory parallel fibre synapses on PNs, and its ablation impairs adaptation of an ocular reflex in mice (Hansel et al. 2006). Coordinated regulation of both RP and LTD through the CaMKII activity has been suggested (Kawaguchi & Hirano, 2013). Furthermore, CaMKII is also implicated in another form of plasticity in a PN, depolarization-induced depression of inhibitory transmission (DDI), through altering the intracellular chloride concentration (Satoh et al. 2013). Thus, CaMKII-mediated regulation of multiple forms of plasticity at both excitatory and inhibitory synapses on a PN seems to be critical for adaptive information processing in the cerebellar cortex.
In clear contrast to a negative contribution of αCaMKII to RP, we demonstrated a positive role of βCaMKII. Information about the involvement of βCaMKII in synaptic plasticity has been limited. A recent study using βCaMKII knockout mice showed that association of βCaMKII with F-actin is critical for hippocampal LTP through controlling the distribution of αCaMKII (Borgesius et al. 2011). In addition, it was recently shown that preferential association of βCaMKII to Arc (activity-regulated cytoskeleton-associated protein), which also associates with actin, is implicated in synaptic plasticity regulation (Okuno et al. 2012). We observed a tendency for CaMKII to accumulate at membranous regions in PNs, including inhibitory postsynaptic membrane, probably through F-actin association (see Fig.7), which might partly contribute to RP induction by ensuring its higher expression level at synapses. However, F-actin destruction by LatA had little effect on RP. Rather, higher Ca2+/CaM affinity of βCaMKII was critical for RP induction. Using FRET imaging, we demonstrated that greater relative abundance of βCaMKII in a PN strengthened the total CaMKII activity. The facilitating effect of βCaMKII on the total CaMKII activity is likely to contribute to its preferential role in RP.
As far as we are aware, opposite regulation of synaptic plasticity by α- and βCaMKII is a novel mechanism for dynamically controlling synaptic strength. Very recently, using α- or βCaMKII knockout mice different contributions of both subunits to LTP of inhibitory synaptic transmission to a PN were reported (Gao et al. 2014). Consistently to our observation, ablation of βCaMKII was shown to impair LTP depending on phosphatase activities. On the other hand, in contrast to our data, knockout of αCaMKII also resulted in impaired LTP which could not be rescued by the phosphatase inhibition. It should be noted that LTP monitored in Gao et al. was a mixture of two forms of CaMKII-dependent plasticity, RP (augmented GABAAR response) and DDI (reduced inhibition by chloride concentration change) (Satoh et al. 2013), which might be responsible for the discrepancy about the roles of αCaMKII between the two studies. Interestingly, the CaMKII subunit composition changes depending on neuronal activity and on developmental stages (Wu et al. 1998; Bayer et al. 1999; Ouyang et al. 1999; Thiagarajan et al. 2002; Fink et al. 2003). Expression of βCaMKII was shown to precede that of αCaMKII in a developing rodent brain (Bayer et al. 1999), which might underlie facilitated synaptic plasticity and learning ability in young animals (Diana et al. 1995). Moreover, the pharmacological manipulation of activities of cultured hippocampal neurons was shown to change the expression ratio of α- and βCaMKII, causing altered synaptic strength in a direction to compensate for the manipulated neuronal activity (Thiagarajan et al. 2002). Thus, in the future it will be interesting to examine whether activity- or development-dependent alteration of CaMKII subunit composition affects synaptic plasticity in the cerebellum and also in other brain regions.
Sustained CaMKII activation and synaptic plasticity
Bistable switch-like activation of CaMKII through autophosphorylation at Thr286/287 has attracted much attention as a mechanism for molecular memory or synaptic tag (Lisman et al. 2002; Okamoto et al. 2009; Lucchesi et al. 2011). Our FRET imaging demonstrated that a relatively high abundance of βCaMKII sustains the total CaMKII activity in a manner dependent on positive-feedback autophosphorylation at Thr286/287. In contrast, increase of αCaMKII did not sustain the total CaMKII activity probably due to its low affinity for Ca2+/CaM. Thus, the sustainability of CaMKII activation is controlled by the balance of expression levels of α- and βCaMKII. It remains controversial whether sustained CaMKII activation takes place and plays an essential role in synaptic plasticity. Previous biochemical and FRET imaging studies showed that CaMKII activation lasts from 10 min to hours in response to transient bath application of NMDA or LTP-inducing electrical stimulation in the hippocampus (Fukunaga et al. 1993; Ouyang et al. 1997; Takao et al. 2005; Marsden et al. 2010). In contrast, a recent study using two-photon fluorescence lifetime FRET imaging showed that the CaMKII activity attenuates within 1 min after LTP-inducing glutamate uncaging stimulation (Lee et al. 2009). Prolongation of the total CaMKII activation by the βCaMKII subunit is likely to be more evident in PNs because of their more abundant expression of βCaMKII (α:β, 1:1) compared with pyramidal neurons in the hippocampus (3:1).
The duration of CaMKII activation required for RP remains unclear. Here, our FRET imaging showed that CaMKII activation persists longer than 15 min if the CaMKII subunit composition is β subunit-dominant. Previous immunocytochemical studies showed that the sustained autophosphorylation of CaMKII at Thr286/287 takes place for longer than 2 h upon transient high K+ treatment of cultured PNs (Kitagawa et al. 2009). Direct application of pre-activated (Thr286-phosphorylated) αCaMKII was shown to induce RP by itself (Kano et al. 1996). Considering the direct potentiating effect of GABAAR phosphorylation by CaMKII (Houston & Smart, 2006; Houston et al. 2008), sustained CaMKII activity is a potential mechanism for expression and maintenance of RP. However, KN62 treatment after RP induction was previously shown to have little effect on the once-established RP in a slice preparation (Kano et al. 1996). Consistently, our preliminary data suggested that RP was not cancelled by CaMKII inhibition from 5 min after the induction (data not shown). Thus, long-term CaMKII activation may not be essential for maintenance of RP, as is the case for hippocampal LTP at excitatory synapses (Murakoshi et al. 2011). Rather than direct augmentation of GABAAR function by CaMKII-mediated phosphorylation, RP maintenance might depend on the association of GABAAR with GABARAP modulated by CaMKII (Kawaguchi & Hirano, 2007). GABARAP, which binds to microtubules, is thought to positively regulate GABAAR function through facilitated transport of GABAARs towards the plasma membrane and/or through direct modulation of channel kinetics.
To summarize, our results highlighted contrasting roles of α- and βCaMKII in the activation of total CaMKII and the resultant induction of inhibitory synaptic plasticity, which might contribute to the fine-tuning of information processing in the cerebellar cortex.
Acknowledgments
We thank Drs T. Sakaba, Y. Tagawa and E. Nakajima for their critical reading of the manuscript and helpful comments.
Glossary
- αC
CFP-tagged αCaMKII
- CaM
calmodulin
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- DARPP-32
dopamine and cAMP-regulated phosphoprotein of 32 kDa
- ECFP
enhanced cyan fluorescent protein
- EGFP
enhanced green fluorescent protein
- FRET
Förster resonance energy transfer
- GABAAR
γ-aminobutyric acid type A receptor
- GABARAP
GABAAR-associated protein
- GluD
glutamate receptor δ
- KD
knock-down
- LatA
latrunculin A
- LTD
long-term depression
- LTP
long-term potentiation
- OE
overexpression
- PP
protein phosphatase
- PN
Purkinje neuron
- RFP
red fluorescent protein
- RNAi
RNA interference
- RP
rebound potentiation
- shRNA
short hairpin RNA
- Vα
Venus-tagged αCaMKII
- Vβ
Venus-tagged βCaMKII
- VGAT
vesicular GABA transporter
Key points
At inhibitory synapses on a cerebellar Purkinje neuron, depolarization-caused Ca2+ increase induces long-term potentiation (known as rebound potentiation, RP).
We show that the subunit composition of a Ca2+/calmodulin-dependent protein kinase II (CaMKII) holoenzyme, consisting of α- and βCaMKII, is critical for RP induction.
βCaMKII plays an essential role in RP induction depending on the high affinity for Ca2+/CaM but not on the F-actin-binding property, whereas αCaMKII negatively regulates it.
βCaMKII enrichment prolongs total CaMKII activation depending on Thr286/287 autophosphorylation.
α- and βCaMKII expression ratio might play critical roles in regulation of synaptic functions in the central nervous system.
Additional information
Competing interests
The authors declare that they have no competing financial interests.
Author contributions
N.N. and S.K. designed and performed experiments; N.N., T.H. and S.K. interpreted data and wrote the manuscript. All authors approved the final version. This work was mainly performed in the Graduate School of Science, Kyoto University.
Funding
This work was supported by grants from MEXT, Japan, to S.K., T.H. and N.N. (for JSPS Fellows), from the Naito Foundation and the Takeda Science Foundation to S.K., from the Uehara Memorial Foundation and Takeda Science Foundation to T.H., by the Global COE programme A06 of Kyoto University, and by a Grant for Excellent Graduate Schools to the Division of Biological Science, Graduate School of Science, Kyoto University, from MEXT, Japan.
References
- Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. &. [DOI] [PubMed] [Google Scholar]
- Bayer K-U, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–805. doi: 10.1038/35081080. &. [DOI] [PubMed] [Google Scholar]
- Bayer KU, LeBel É, McDonald GL, O'Leary H, Schulman H, De Koninck P. Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J Neurosci. 2006;26:1164–1174. doi: 10.1523/JNEUROSCI.3116-05.2006. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer K-U, Löhler J, Schulman H, Harbers K. Developmental expression of the CaM kinase II isoforms: ubiquitous γ- and δ-CaM kinase II are the early isoforms and most abundant in the developing nervous system. Brain Res Mol Brain Res. 1999;70:147–154. doi: 10.1016/s0169-328x(99)00131-x. &. [DOI] [PubMed] [Google Scholar]
- Borgesius NZ, van Woerden GM, Buitendijk GHS, Keijzer N, Jaarsma D, Hoogenraad CC, Elgersma Y. βCaMKII plays a nonenzymatic role in hippocampal synaptic plasticity and learning by targeting αCaMKII to synapses. J Neurosci. 2011;31:10141–10148. doi: 10.1523/JNEUROSCI.5105-10.2011. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocke L, Chiang LW, Wagner PD, Schulman H. Functional implications of the subunit composition of neuronal CaM kinase II. J Biol Chem. 1999;274:22713–22722. doi: 10.1074/jbc.274.32.22713. &. [DOI] [PubMed] [Google Scholar]
- Deguchi A, Hata M, Uhara T, Yamauchi T. Domain structure responsible for the different properties between αand β Ca2+/calmodulin-dependent protein kinase II analyzed by their chimera enzymes. Brain Res. 2008;1238:1–11. doi: 10.1016/j.brainres.2008.07.070. &. [DOI] [PubMed] [Google Scholar]
- Diana G, Domenici MR, Scotti de Carolis A, Loizzo A, Sagratella S. Reduced hippocampal CA1 Ca2+-induced long-term potentiation is associated with age-dependent impairment of spatial learning. Brain Res. 1995;686:107–110. doi: 10.1016/0006-8993(95)00440-2. &. [DOI] [PubMed] [Google Scholar]
- Fink CC, Bayer K-U, Myers JW, Ferrell JE, Jr, Schulman H, Meyer T. Selective regulation of neurite extension and synapse formation by the β but not the α isoform of CaMKII. Neuron. 2003;39:283–297. doi: 10.1016/s0896-6273(03)00428-8. &. [DOI] [PubMed] [Google Scholar]
- Fukunaga K, Stoppini L, Miyamoto E, Muller D. Long-term potentiation is associated with an increased activity of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1993;268:7863–7867. &. [PubMed] [Google Scholar]
- Gao Z, van Woerden GM, Elgersma Y, De Zeeuw CI, Hoebeek FE. Distinct roles of α- and βCaMKII in controlling long-term potentiation of GABAA-receptor mediated transmission in murine Purkinje cells. Front Cell Neurosci. 2014;8:16. doi: 10.3389/fncel.2014.00016. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the αcalcium-calmodulin kinase II in LTP and learning. Science. 1998;279:870–873. doi: 10.1126/science.279.5352.870. &. [DOI] [PubMed] [Google Scholar]
- Hansel C, de Jeu M, Belmeguenai A, Houtman SH, Buitendijk GHS, Andreev D, De Zeeuw CI, Elgersma Y. αCaMKII is essential for cerebellar LTD and motor learning. Neuron. 2006;51:835–843. doi: 10.1016/j.neuron.2006.08.013. &. [DOI] [PubMed] [Google Scholar]
- Hirano T, Kawaguchi S. Regulation and functional roles of rebound potentiation at cerebellar stellate cell – Purkinje cell synapse. Front Cell Neurosci. 2014;8:42. doi: 10.3389/fncel.2014.00042. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honkanen RE, Dukelow M, Zwiller J, Moore RE, Khatra BS, Boynton AL. Cyanobacterial nodularin is a potent inhibitor of type 1 and type 2A protein phosphatases. Mol Pharmacol. 1991;40:577–583. &. [PubMed] [Google Scholar]
- Houston CM, Hosie AM, Smart TG. Distinct regulation of β2 and β3 subunit-containing cerebellar synaptic GABAA receptors by calcium/calmodulin-dependent protein kinase II. J Neurosci. 2008;28:7574–7584. doi: 10.1523/JNEUROSCI.5531-07.2008. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston CM, Smart TG. CaMK-II modulation of GABAA receptors expressed in HEK293, NG108-15 and rat cerebellar granule neurons. Eur J Neurosci. 2006;24:2504–2514. doi: 10.1111/j.1460-9568.2006.05145.x. &. [DOI] [PubMed] [Google Scholar]
- Kano M, Kano M, Fukunaga K, Konnerth A. Ca2+-induced rebound potentiation of γ-aminobutyric acid-mediated currents requires activation of Ca2+/calmodulin-dependent kinase II. Proc Natl Acad Sci USA. 1996;93:13351–13356. doi: 10.1073/pnas.93.23.13351. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Rexhausen U, Dreessen J, Konnerth A. Synaptic excitation produces a long-lasting rebound potentiation of inhibitory synaptic signals in cerebellar Purkinje cells. Nature. 1992;356:601–604. doi: 10.1038/356601a0. &. [DOI] [PubMed] [Google Scholar]
- Kawaguchi S, Hirano T. Suppression of inhibitory synaptic potentiation by presynaptic activity through postsynaptic GABAB receptors in a Purkinje neuron. Neuron. 2000;27:339–347. doi: 10.1016/s0896-6273(00)00041-6. &. [DOI] [PubMed] [Google Scholar]
- Kawaguchi S, Hirano T. Signaling cascade regulating long-term potentiation of GABAA receptor responsiveness in cerebellar Purkinje neurons. J Neurosci. 2002;22:3969–3976. doi: 10.1523/JNEUROSCI.22-10-03969.2002. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi S, Hirano T. Integrin α3β1 suppresses long-term potentiation at inhibitory synapses on the cerebellar Purkinje neuron. Mol Cell Neurosci. 2006;31:416–426. doi: 10.1016/j.mcn.2005.10.012. &. [DOI] [PubMed] [Google Scholar]
- Kawaguchi S, Hirano T. Sustained structural change of GABAA receptor-associated protein underlies long-term potentiation at inhibitory synapses on a cerebellar Purkinje neuron. J Neurosci. 2007;27:6788–6799. doi: 10.1523/JNEUROSCI.1981-07.2007. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi S, Hirano T. Gating of long-term depression by Ca2+/calmodulin-dependent protein kinase II through enhanced cGMP signalling in cerebellar Purkinje cells. J Physiol. 2013;591:1707–1730. doi: 10.1113/jphysiol.2012.245787. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi S, Nagasaki N, Hirano T. Dynamic impact of temporal context of Ca2+ signals on inhibitory synaptic plasticity. Sci Rep. 2011;1:143. doi: 10.1038/srep00143. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa Y, Hirano T, Kawaguchi S. Prediction and validation of a mechanism to control the threshold for inhibitory synaptic plasticity. Mol Syst Biol. 2009;5:280. doi: 10.1038/msb.2009.39. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-JR, Escobedo-Lozoya Y, Szatmari EM, Yasuda R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature. 2009;458:299–304. doi: 10.1038/nature07842. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. &. [DOI] [PubMed] [Google Scholar]
- Lisman J, Yasuda R, Raghavachari S. Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci. 2012;13:169–182. doi: 10.1038/nrn3192. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano I, Marty A, Armstrong CM, Konnerth A. Synaptic- and agonist-induced excitatory currents of Purkinje cells in rat cerebellar slices. J Physiol. 1991;434:183–213. doi: 10.1113/jphysiol.1991.sp018465. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchesi W, Mizuno K, Giese KP. Novel insights into CaMKII function and regulation during memory formation. Brain Res Bull. 2011;85:2–8. doi: 10.1016/j.brainresbull.2010.10.009. &. [DOI] [PubMed] [Google Scholar]
- McGuinness TL, Lai Y, Greengard P. Ca2+/calmodulin-dependent protein kinase II. Isozymic forms from rat forebrain and cerebellum. J Biol Chem. 1985;260:1696–1704. &. [PubMed] [Google Scholar]
- Marsden KC, Shemesh A, Bayer KU, Carroll RC. Selective translocation of Ca2+/calmodulin protein kinase IIα (CaMKIIα). to inhibitory synapses. Proc Natl Acad Sci USA. 2010;107:20559–20564. doi: 10.1073/pnas.1010346107. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer T, Hanson PI, Stryer L, Schulman H. Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science. 1992;256:1199–1202. doi: 10.1126/science.256.5060.1199. &. [DOI] [PubMed] [Google Scholar]
- Miller SG, Kennedy MB. Regulation of brain type II Ca2+/calmodulin-dependent protein kinase by autophosphorylation: a Ca2+-triggered molecular switch. Cell. 1986;44:861–870. doi: 10.1016/0092-8674(86)90008-5. &. [DOI] [PubMed] [Google Scholar]
- Murakoshi H, Wang H, Yasuda R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature. 2011;472:100–104. doi: 10.1038/nature09823. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Bosch M, Hayashi Y. The roles of CaMKII and F-actin in the structural plasticity of dendritic spines: a potential molecular identity of a synaptic tag. Physiology. 2009;24:357–366. doi: 10.1152/physiol.00029.2009. &. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Narayanan R, Lee SH, Murata K, Hayashi Y. The role of CaMKII as an F-actin-bundling protein crucial for maintenance of dendritic spine structure. Proc Natl Acad Sci USA. 2007;104:6418–6423. doi: 10.1073/pnas.0701656104. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno H, Akashi K, Ishii Y, Yagishita-Kyo N, Suzuki K, Nonaka M, Kawashima T, Fujii H, Takemoto-Kimura S, Abe M, Natsume R, Chowdhury S, Sakimura K, Worley PF, Bito H. Inverse synaptic tagging of inactive synapses via dynamic interaction of Arc/Arg3.1 with CaMKIIβ. Cell. 2012;149:886–898. doi: 10.1016/j.cell.2012.02.062. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary H, Lasda E, Bayer KU. CaMKIIβ association with the actin cytoskeleton is regulated by alternative splicing. Mol Biol Cell. 2006;17:4656–4665. doi: 10.1091/mbc.E06-03-0252. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Y, Kantor D, Harris KM, Schuman EM, Kennedy MB. Visualization of the distribution of autophosphorylated calcium/calmodulin-dependent protein kinase II after tetanic stimulation in the CA1 area of the hippocampus. J Neurosci. 1997;17:5416–5427. doi: 10.1523/JNEUROSCI.17-14-05416.1997. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Y, Rosenstein A, Kreiman G, Schuman EM, Kennedy MB. Tetanic stimulation leads to increased accumulation of Ca2+/calmodulin-dependent protein kinase II via dendritic protein synthesis in hippocampal neurons. J Neurosci. 1999;19:7823–7833. doi: 10.1523/JNEUROSCI.19-18-07823.1999. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piochon C, Irinopoulou T, Brusciano D, Bailly Y, Mariani J, Levenes C. NMDA receptor contribution to the climbing fiber response in the adult mouse Purkinje cell. J Neurosci. 2007;27:10797–10809. doi: 10.1523/JNEUROSCI.2422-07.2007. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh H, Qu L, Suzuki H, Saitow F. Depolarization-induced depression of inhibitory transmission in cerebellar Purkinje cells. Physiol Rep. 2013;1:e00061. doi: 10.1002/phy2.61. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Teruel MN, Subramanian K, Meyer T. CaMKIIβ functions as an F-actin targeting module that localizes CaMKIIα/β heterooligomers to dendritic spines. Neuron. 1998;21:593–606. doi: 10.1016/s0896-6273(00)80569-3. &. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Paylor R, Wehner JM, Tonegawa S. Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257:206–211. doi: 10.1126/science.1321493. &. [DOI] [PubMed] [Google Scholar]
- Strack S, Barban MA, Wadzinski BE, Colbran RJ. Differential inactivation of postsynaptic density-associated and soluble Ca2+/calmodulin-dependent protein kinase II by protein phosphatases 1 and 2A. J Neurochem. 1997;68:2119–2128. doi: 10.1046/j.1471-4159.1997.68052119.x. &. [DOI] [PubMed] [Google Scholar]
- Takao K, Okamoto K-I, Nakagawa T, Neve RL, Nagai T, Miyawaki A, Hashikawa T, Kobayashi S, Hayashi Y. Visualization of synaptic Ca2+/calmodulin-dependent protein kinase II activity in living neurons. J Neurosci. 2005;25:3107–3112. doi: 10.1523/JNEUROSCI.0085-05.2005. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Kawaguchi SY, Shioi G, Hirano T. Long-term potentiation of inhibitory synaptic transmission onto cerebellar Purkinje neurons contributes to adaptation of vestibulo-ocular reflex. J Neurosci. 2013;33:17209–17220. doi: 10.1523/JNEUROSCI.0793-13.2013. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajan TC, Piedras-Renteria ES, Tsien RW. α- and βCaMKII: inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron. 2002;36:1103–1114. doi: 10.1016/s0896-6273(02)01049-8. &. [DOI] [PubMed] [Google Scholar]
- Walaas SI, Lai Y, Gorelick FS, DeCamilli P, Moretti M, Greengard P. Cell-specific localization of the α-subunit of calcium/calmodulin-dependent protein kinase II in Purkinje cells in rodent cerebellum. Mol Brain Res. 1988;4:233–242. doi: 10.1016/0169-328x(88)90029-0. &. [DOI] [PubMed] [Google Scholar]
- Wu L, Wells D, Tay J, Mendis D, Abbott M-A, Barnitt A, Quinlan E, Heynen A, Fallon JR, Richter JD. CPEB-mediated cytoplasmic polyadenylation and the regulation of experience-dependent translation of α-CaMKII mRNA at synapses. Neuron. 1998;21:1129–1139. doi: 10.1016/s0896-6273(00)80630-3. &. [DOI] [PubMed] [Google Scholar]