

Abstract
Although Ca2+ is critically important in activity-dependent neuronal development, not much is known about the regulation of dendritic Ca2+ signals in developing neurons. Here, we used ratiometric Ca2+ imaging to investigate dendritic Ca2+ signalling in rat hippocampal pyramidal cells during the first 1–4 weeks of postnatal development. We show that active dendritic backpropagation of Nav channel-dependent action potentials (APs) evoked already large dendritic Ca2+ transients in animals aged 1 week with amplitudes of ∼150 nm, similar to the amplitudes of ∼160 nM seen in animals aged 4 weeks. Although the AP-evoked dendritic Ca2+ load increased about four times during the first 4 weeks, the peak amplitude of free Ca2+ concentration was balanced by a four-fold increase in Ca2+ buffer capacity κs (∼70 vs. ∼280). Furthermore, Ca2+ extrusion rates increased with postnatal development, leading to a slower decay time course (∼0.2 s vs. ∼0.1 s) and more effective temporal summation of Ca2+ signals in young cells. Most importantly, during prolonged theta-burst stimulation dendritic Ca2+ signals were up to three times larger in cells at 1 week than at 4 weeks of age and much larger than predicted by linear summation, which is attributable to an activity-dependent slow-down of Ca2+ extrusion. As Ca2+ influx is four-fold smaller in young cells, the larger Ca2+ signals are generated using four times less ATP consumption. Taken together, the data suggest that active backpropagations regulate dendritic Ca2+ signals during early postnatal development. Remarkably, during prolonged AP firing, Ca2+ signals are several times larger in young than in mature cells as a result of activity-dependent regulation of Ca2+ extrusion rates.
Introduction
During the first few weeks of rodent brain development, there is substantial growth of neuronal dendrites and a high rate of synaptogenesis. Neuronal activity is critically important to the development of neuronal circuits (Katz & Schatz, 1996; Hensch, 2005), and intracellular Ca2+ is an important signalling molecule contributing to the activity-dependent growth and remodelling of neuronal processes (Wong & Ghosh, 2002; Spitzer, 2006). A global rise in the intracellular Ca2+ concentration may activate nuclear protein kinases and transcription factors like CaM-kinase IV (CaMKIV) to control transcription and promote neuronal outgrowth (Deisseroth et al. 2003; Aizawa et al. 2004). However, local dendritic Ca2+ signalling may activate CaMKII to phosphorylate guanine nucleotide exchange factors (GEFs) interacting with RhoGTPases and the cytoskeleton to promote growth cone turning, as well as retraction and stabilization of new dendritic branches (Jin et al. 2005; Konur & Ghosh, 2005).
In developing rat hippocampal pyramidal cells, dendritic spines are largely absent at birth and are subsequently formed during the first few postnatal weeks in an activity-dependent manner (Fiala et al. 1998; Kirov et al. 2004). It has been shown that new dendritic filopodia and spines are generated by the release of glutamate from neighbouring presynaptic boutons activating extrasynaptic NMDA receptors on dendritic shafts (Engert & Bonhoeffer, 1999; Maletic-Savatic et al. 1999; Kwon & Sabatini, 2011). This process is Ca2+-dependent and is most probably mediated via a Ca2+-dependent adenylate cyclase activating PKA and MAP kinase (Kwon & Sabatini, 2011). In addition, CaMKII is localized to newly formed dendritic spines and may contribute to the maturation and maintenance of nascent spines (Tolias et al. 2005; Ahmed et al. 2006; Xie et al. 2007). Thus, the regulation of the intracellular Ca2+ concentration is of major importance not only to dendritic outgrowth, but also to activity-dependent synapse formation.
Activity-dependent dendritic Ca2+ transients were found to be extensively characterized in juvenile rat hippocampal pyramidal cells at 2–3 weeks after birth (Helmchen et al. 1996; Sabatini et al. 2002; Scheuss et al. 2006). In animals younger than postnatal day 12, however, action potential (AP)-induced dendritic Ca2+ transients are thought to be very small and negligible in CA1 pyramidal cells (Isomura & Kato, 1999). This appears to contradict the suggestion that Ca2+-dependent processes contribute to dendritic growth and activity-dependent synapse formation at a younger age. Therefore, we investigated dendritic Ca2+ influx, buffering and extrusion in rat CA1 pyramidal cells during the first 1–5 weeks of postnatal rat development. We have focused on backpropagating APs (bAPs), which generate δ-like Ca2+ influx, which is perfectly suited to the study of dendritic Ca2+ buffering and Ca2+ extrusion because influx and extrusion are temporally separated. We show for the first time that single Nav channel-dependent bAPs induce large dendritic Ca2+ transients already in young neurons at P7–P9 similar to mature pyramidal cells. Surprisingly, during continuous theta-burst firing, dendritic Ca2+ signals are 2- to 3-fold larger at 1 week after birth as compared to 4-week-old animals.
Methods
Ethical approval
The use and care of experimental animals (Wistar rats) in this project was approved by the Animal Advisory Committee of the Regierungspräsidium Freiburg (Germany) and the Kanton Basel (Kantonales Verterinäramt BS, Switzerland).
Slice preparation
Transverse slices of 350 μm in thickness were cut from the hippocampus of Wistar rats aged 5−37 days using a Leica VT1200 vibratome (Leica Microsystems GmbH, Wetzlar, Germany) (Geiger et al. 2002; Bischofberger et al. 2006). Slices were taken from the middle third along the dorso−ventral axes of the hippocampus. For data analysis, P7−P9 and P26−P29 refer to rats aged 1 week and 4 weeks, respectively. The animals were anaesthetized with isoflurane added to the oxygenated inspiration airflow (4–5%; Abbott GmbH & Co. KG, Ludwigshafen, Germany) and killed by decapitation in accordance with national and institutional guidelines. Slices were kept at 35°C for 30 min after slicing and then stored at room temperature in artificial cerebrospinal fluid (ACSF) containing 125 mm NaCl, 25 mm NaHCO3, 2.5 mm KCl, 1.25 mm NaH2PO4, 2 mm CaCl2, 1 mm MgCl2 and 25 mm glucose. For tissue from animals older than 2 weeks, we used a sucrose-based solution for cutting and storage containing 87 mm NaCl, 25 mm NaHCO3, 2.5 mm KCl, 1.25 mm NaH2PO4, 75 mm sucrose, 0.5 mm CaCl2, 7 mm MgCl2 and 10 mm glucose (equilibrated with 95% O2 to 5% CO2).
Electrophysiology
CA1 pyramidal neurons were visually identified using infrared differential interference contrast (IR-DIC) video microscopy. For all age groups we selected superficial pyramidal cells with the soma located close to the stratum radiatum. For electrophysiological experiments, slices were continuously superfused with ACSF at near physiological temperature (approximately 33°C). Patch pipettes (4–7 MΩ) were pulled from borosilicate glass tubing with a 2.0 mm outer diameter and 0.5 mm wall thickness (Hilgenberg GmbH, Malsfeld, Germany). The pipettes were filled with a solution containing 120–135 mm K-gluconate, 20 mm KCl, 4 mm MgCl2, 2–4 mm Na2ATP, 0.3 mm NaGTP, 2 mm Na-ascorbate, 2 mm Na2-phosphocreatine, 10 mm HEPES and 0.2% biocytin. The pH was adjusted to 7.3 by adding potassium hydroxide (KOH).
Voltage signals were measured with a MultiClamp 700 A amplifier (Molecular Devices, Inc., Palo Alto, CA, USA) in current clamp mode, filtered at 10 kHz and digitized at 20 kHz using a CED Power1401 interface (Cambridge Electronic Design, Cambridge, UK). Data acquisition and analysis were achieved using custom software (FPulse 3.33; U. Fröbe, Physiological Institute Freiburg, Freiburg, Germany) running under IGOR Pro 6.21 (WaveMetrics, Lake Oswego, OR, USA) and GraphPad Prism Version 5.0 (GraphPad Software, Inc., San Diego, CA, USA). All neurons chosen in this study generated overshooting APs during injection of depolarizing current. At 4 weeks (P26−P29), pyramidal cells had a resting membrane potential of between −75 mV and −65 mV. In rats aged 1 week (P7–P9), pyramidal cells were slightly more depolarized between −70 mV and −60 mV. The membrane potential of all neurons was adjusted to about −70 mV throughout the experiment by small depolarizing or hyperpolarizing current injection.
Cells were filled for at least 15 min in whole-cell voltage clamp with a stable series resistance of RS < 15 MΩ. Subsequently, bAPs were elicited in current clamp mode via current injections of 2 ms in duration with an amplitude in the range of 0.5–2.5 nA to reliably evoke APs. Series resistance and pipette capacitance were compensated. To examine the contribution of Nav channels to dendritic backpropagation, AP firing was blocked with 1 μm of TTX (Alomone Labs, Jerusalem, Israel). Mock APs were generated by adjusting the amplitude of 2 ms current pulses until the peak amplitude of the passive somatic voltage response was similar to the previously measured AP amplitude in young (115.9 ± 2.8 mV vs. 103.5 ± 1.4 mV, average of five APs, n = 4 cells) and mature (115.2 ± 2.7 mV vs. 104.8 ± 1.7 mV, average of five APs, n = 4 cells) pyramidal cells.
Fluorescent measurements
For ratiometric Ca2+ imaging, the Ca2+-sensitive fluorescent dye Fura-FF (100–200 μm) or Fura-2 (50–200 μm) (Invitrogen, Inc., Eugene, OR, USA) was added to the pipette solution. The excitation light source (Polychrome V; TILL Photonics GmbH, Munich, Germany) was coupled to the epifluorescent port of the microscope via fibre optics (Zeiss Examiner, equipped with a 63× NA1.0 water immersion objective; Carl Zeiss Microscopy GmbH, Jena, Germany). To minimize bleaching, the light intensity was reduced to 5–10% by neutral density filters. The filter combination for fluorescence excitation and emission comprised a beam splitter (BSP400) and an emission filter (BP430–630) from AHF Analysentechnik AG (Tübingen, Germany).
Fluorescence was measured with a cooled frame-transfer CCD camera system (Sensicam; TILL Photonics GmbH). Images were obtained with 8 × 32 binning at a 100 Hz repetition rate using TillVision 4.0. Fluorescence was measured in dendritic regions of interest (ROIs; typical size 5 × 10 μm) along the apical dendrite of the CA1 pyramidal cells at a distance of ∼25 μm (proximal) or ∼100 μm (stratum radiatum) from the centre of the soma. The fluorescence signals were corrected for background fluorescence, which was measured in neighbouring ROIs shifted by approximately 10−15 μm with respect to the original ROI, avoiding the presence of dendritic processes (Normann et al. 2000; Aponte et al. 2008).
The background fluorescence signal often contained some stray light from dendritic fluorescence. As a result, an AP-dependent reduction of the background fluorescence up to ∼5% was observed, indicating a scattering of Fura emission into the background ROI. Therefore, we developed a method to subtract this AP-dependent Fura fluorescence from background ROI signals. The stray light factor f was estimated by calculating the ratio of the peak amplitude of the AP-induced reduction of fluorescence in the background ROI and the dendritic ROI. As the fluorescence of our ROIs is the sum of a Ca2+-dependent Fura signal F and a Ca2+-independent background signal BG, we assume:
| (1) |
| (2) |
Eliminating F den from (1) and (2) resulted in an estimate of the Fura-independent background signal BG:
| (3) |
which no longer showed any AP-induced reduction in fluorescence intensity. These Ca2+-independent BG fluorescence traces were subtracted from the ROI den fluorescence signals according to eqn (1) to finally obtain the dendritic Fura fluorescence F den.
Calibration of ratiometric Ca2+ measurements
To convert the fluorescence signals into Ca2+ concentrations, we used the isosbestic ratioing method as described previously (Normann et al. 2000; Aponte et al. 2008). Briefly, the AP-induced fluorescence change was recorded at an excitation wavelength of 380 nm for a duration of 2–8 s. The isosbestic fluorescence was measured immediately before and after these sweeps for about 100–150 ms, respectively, using an excitation wavelength of 357 nm (the Ca2+-insensitive wavelength in our experimental conditions). To obtain the dendritic Fura fluorescence at the excitation wavelength of 380 nm (F380), the Ca2+-independent BG fluorescence traces (eqn (3)) were low pass-filtered using a binomial filter (∼20–40 points) and subtracted from the ROIden fluorescence signals according to eqn (1). For the isosbestic excitation wavelength, eqn (2) was subtracted from eqn (1) to calculate the background-corrected F357. Finally, F357 was linearly extrapolated from the beginning to the end of the sweep.
The ratio of the background corrected fluorescence signals R = F357/F380 was then calculated and converted into the Ca2+ concentration using the equation (Grynkiewicz et al. 1985):
| (4) |
with Keff = Kd (Rmax/Rmin); where Kd is the dissociation constant, Rmin the ratio in Ca2+-free solution and Rmax the ratio when Fura is completely saturated with Ca2+. Rmin and Rmax were determined with internal solutions containing either 10 mm EGTA or 10 mm CaCl2. Using these solutions, we obtained ratio values of Rmin = 0.44 ± 0.01 (n = 5) and Rmax = 3.26 ± 0.04 (n = 5) for Fura-2. The corresponding values for Fura-FF were estimated as Rmin = 0.40 ± 0.01 (n = 4) and Rmax = 3.20 ± 0.05 (n = 6). The dissociation constant was assumed to be Kd = 286 nm and Kd = 4.18 μm for Fura-2 and Fura-FF, respectively (Helmchen et al. 1996; Aponte et al. 2008). The resting Ca2+ concentration obtained with 100 μm Fura-2 and the standard internal solution was on average 53 ± 7 nm (n = 39), which is similar to previously reported values for CA1 pyramidal neurons (Helmchen et al. 1996; Normann et al. 2000).
Data analysis and a single-compartment model
Ca2+ traces were analysed using custom programs running under Igor 6. To optimize signal-to-noise ratio, 10 to 20 sweeps were averaged and low pass-filtered using a three-point binomial filter. The exponential decay of the Ca2+ concentration was either fitted with a monoexponential function, or after theta-burst stimulation (TBS), with the sum of two exponentials, and the amplitude-weighted time constant was calculated (Aponte et al. 2008). To analyse dendritic Ca2+ buffering, a single-compartment model was used assuming that influx and equilibration with intracellular Ca2+ buffers are fast compared with Ca2+ extrusion mechanisms as previously described (Helmchen et al. 1996; Aponte et al. 2008).
Briefly, the fraction of Ca2+ which binds to endogenous Ca2+ buffers (S) during a short AP-evoked Ca2+ influx can be quantified by the Ca2+ binding ratio defined as:
| (5) |
where Δ[SCa] represents the increase in buffer-bound Ca2+ and Δ[Ca2+]i the increase in the free Ca2+ concentration (Neher & Augustine, 1992). Similarly the Ca2+ binding ratio of fura (at a certain concentration B) can be expressed as κB Δ[BCa] / Δ[Ca2+]i. Assuming that the intracellular Ca2+ concentration within the dendritic compartment of the recorded ROI is homogeneously elevated during an AP from a resting level [Ca2+]1 to the peak level [Ca2+]2, this binding ratio can be calculated from the law of mass action as:
| (6) |
with Bt and Kd representing the total concentration and the Ca2+ dissociation constant of fura, respectively (Neher & Augustine, 1992). As already mentioned, we assumed that Ca2+ ions enter a dendritic compartment with the volume V and rapidly bind and distribute between the different buffers. The total amount of Ca2+ ions (Ca2+tot) will partially increase the Ca2+-bound fraction of fura (Δ[BCa]), the Ca2+-bound fraction of the endogenous Ca2+ buffers (Δ[SCa]), and will also appear as an increase in the free Ca2+ concentration (Δ[Ca2+]i). This relationship can be expressed as:
| (7) |
with 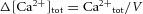 , representing the total increase of the intracellular Ca2+ concentration. Therefore, the increase in the intracellular Ca2+ concentration is linearly related to the Ca2+ binding ratio of fura-2:
, representing the total increase of the intracellular Ca2+ concentration. Therefore, the increase in the intracellular Ca2+ concentration is linearly related to the Ca2+ binding ratio of fura-2:
| (8) |
After the rapid increase, the Ca2+ transient exponentially decays back to resting Ca2+ concentration according to the equation:
| (9) |
with the peak amplitude A = Δ[Ca2+]tot/(1 + κB + κS) and the decay time constant τ = (1 + κB + κS)/γ, which is dependent on Ca2+ pumps and transporters, characterized by a single lumped extrusion rate γ. The endogenous Ca2+ binding ratio κS was estimated by variation of κB via different concentrations of the exogenous Ca2+ buffers Fura-2 and Fura-FF. The inverse amplitude of the Ca2+ transients was plotted against κB to obtain κS as well as the total Ca2+ load Δ[Ca2+]tot by linear extrapolation from the slope (1/Δ[Ca2+]tot) and the x-axes intercept (1+κS) according to:
| (10) |
In the buffer competition experiments (Fig.3), the peak amplitude of single AP-evoked transients was estimated by extrapolating the exponential time course to the mid-point of the rising phase A = Ameasured exp(Δt/τ). Similarly, the decay time constant τ was plotted against κB to obtain the Ca2+ extrusion rate γ by linear extrapolation according to:
Figure 3. Small Ca2+ buffer capacity and slow extrusion rate in young pyramidal cells.
A, action potential (AP)-evoked Ca2+ transients in cells from rats aged 1 week (left) and 4 weeks (right) measured with Fura-FF and different concentrations of Fura 2 (single AP, black traces). Red and blue lines represent monoexponential functions fitted to the measured data. B, inverse peak amplitudes plotted vs. exogenous calcium buffer capacity κB at 1 week (n = 23). C, open circles represent measured monoexponential decay time constants plotted vs. exogenous calcium buffer capacity κB. The red lines in B and C show a linear regression through the data points. D, inverse amplitudes plotted vs. exogenous calcium buffer capacity κB in cells from rats aged 3−4 weeks (n = 28). E, open circles represent measured monoexponential decay time constants for transients plotted vs. exogenous calcium buffer capacity κB. The blue lines in D and E show a linear regression through the data points.
| (11) |
Modelling of non-linear dendritic Ca2+ signalling
In order to simulate dendritic Ca2+ accumulation, we calculated the linear summation of single AP-evoked Ca2+ transients using the amplitude A1 and the decay time constant τ1 measured with Fura FF for a single AP. For single bursts of n APs with an interspike interval Δt, the peak amplitude was calculated either numerically as the linear sum of single transients:
| (12) |
or analytically via geometric progression as:
| (13) |
similar as previously described (Regehr & Tank, 1994; Helmchen et al. 1996).
During theta-burst stimulation (TBSn), brief bursts of n APs (3–10 APs at 100 Hz) were repeated 10 times at a rate of 5 Hz. To simulate Ca2+ accumulation during TBS firing, we first calculated the integral of the Ca2+ signals as a linear summation of single AP-evoked transients from eqn (9):
| (14) |
As a second approach we modelled TBS more precisely, taking into account that the Ca2+ influx, and therefore the slope of rise of the individual burst-evoked Ca2+ transients, decreased during TBS. This was represented by an experimentally obtained normalized rise-time factor ri ≤ 1. Furthermore, the time-dependent slow-down of the Ca2+ decay time constant was calculated as an exponential transition from single burst decay τ1 towards the decay time constant measured after the last burst τ10. Thus, the integral of the Ca2+ signal can be expressed as:
 |
(15) |
with Δt = 0.2 s (5 Hz TBS) and TTBS representing the time constant of the slow-down of the Ca2+ extrusion rate measured after the individual burst during the TBS paradigm.
Statistics
Two-tailed unpaired signalling was used to calculate P-values of normally distributed data (peak amplitudes of calcium transients); Wilcoxon−Mann−Whitney tests were used otherwise. Normality was tested using the D'Agostino and Pearson's omnibus normality test implemented in GraphPad Prism (Version 5.0).
Results
Active dendritic backpropagation in young CA1 pyramidal cells
To study the postnatal development of AP-induced dendritic Ca2+ transients in CA1 pyramidal cells, we performed ratiometric Ca2+ imaging in acute hippocampal brain slices from 5- to 37-day-old rats. Brief current injections (2 ms) evoked large overshooting APs in all recorded neurons consistent with previous reports (Costa et al. 1991). Using the low-affinity Ca2+-sensitive dye Fura-FF, we found large AP-evoked dendritic Ca2+ transients in the pyramidal cells from all age groups including P7–P9 (Fig.1). A brief burst of five APs evoked Ca2+ transients in apical dendrites of young pyramidal cells with an amplitude of 363 ± 27 nm (n = 10) at a distance of 25 μm and 438 ± 40 nm (n = 9) at 100 μm from the soma (Fig.1A and B), with no significant deviation from normal distribution (P = 0.98 and P = 0.20). In cells from rats aged 4 weeks, dendritic Ca2+ transients were slightly larger at proximal locations (551 ± 34 nm, 25 μm, n = 11), but had amplitudes similar to those of young cells at 100 μm from soma (467 ± 33 nm, n = 12) (P = 0.58) (Fig.1E and F), which were also normally distributed (P = 0.97 and P = 0.54, respectively). These data suggest that growing dendrites of developing CA1 pyramidal cells can generate large dendritic Ca2+ signals already 1 week after birth that are similar to those in mature neurons.
Figure 1. Active dendritic action potential (AP) backpropagation in young CA1 pyramidal cells.
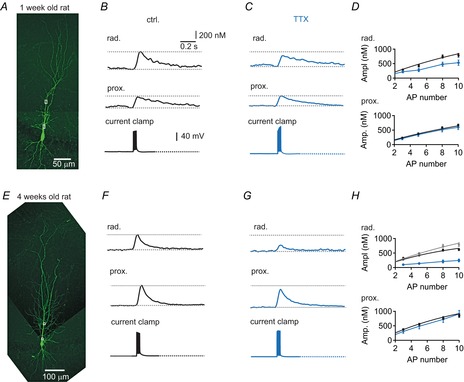
A, biocytin-filled young pyramidal neuron (P7) with recorded regions of interest (ROIs) indicated by white boxes. B, AP-evoked dendritic Ca2+ transient (five APs at 100 Hz) measured in stratum radiatum and proximal apical dendrite from the cell in A (top), and corresponding current clamp recordings of evoked APs (bottom). C, traces for the same cell as in A and B after bath-applying 1 μm TTX. D, average amplitudes for cells from rats aged 1 week in control (n = 10, black) and in 1 μm TTX (n = 4, blue) in stratum radiatum (top), and average amplitudes in control (black) and in 1 μm TTX in proximal dendrites (bottom). E, biocytin-filled mature pyramidal neuron (P28). F, AP-evoked dendritic Ca2+ transient (five APs at 100 Hz) measured in stratum radiatum and proximal apical dendrite from the cell in E (top), and corresponding current clamp recordings of evoked APs (bottom). G, example traces for the cell in E after bath-applying 1 μm TTX in saline. H, average amplitudes for cells from rats aged 4 weeks in control (n = 12, black) and in 1 μm TTX (n = 4, blue) in stratum radiatum (top), with average amplitudes for cells from animals aged 1 week shown for comparison in grey (taken from D top, black), and proximal dendrites at 4 weeks (bottom). Continuous black lines in panel D and H represent linear Ca2+ accumulation [eqn (13), see text].
To analyse the contribution of voltage-gated sodium (Nav) channels to dendritic backpropagation, we bath-applied 1 μm TTX and generated brief bursts of three to 10 mock APs with an amplitude similar to that in Nav channel-dependent APs in young and mature pyramidal cells (Spruston et al. 1995; Bischofberger & Jonas, 1997). Whereas the mock APs generated similar Ca2+ transients in proximal dendrites in young (Fig.1C and D, blue) and mature (Fig.1G and H, blue) pyramidal cells, the amplitude in the middle of stratum radiatum (∼100 μm) was significantly reduced to ∼60% (5–10 APs, n = 4; P < 0.05) and ∼30% (5–10 APs, n = 4; P < 0.001) of control at 1 week and 4 weeks, respectively. This indicates that, in the absence of TTX, APs actively propagate into the dendritic tree of CA1 pyramidal cells starting already at early developmental stages around 1 week after birth.
When plotting the amplitude of backpropagation-induced dendritic Ca2+ transients vs. the age of animals (Fig.2A and C), we found that dendritic Ca2+ transients rapidly increased after P5. An exponential function fitted to values recorded at a distance of 100 μm from the soma indicated that the amplitude was already 81% at P8 and 99% at P14 relative to that in animals aged 4 weeks. Similarly, the decay time course of Ca2+ transients was slower in young animals, but rapidly decreased to 175% and 107% at P8 and P14, respectively (Fig.2B and D). These data show that APs actively backpropagate into apical dendrites of immature CA1 pyramidal cells, generating large Ca2+ transients with mature-like amplitude at 1 week and mature-like decay at 2 weeks after birth.
Figure 2. Rapid postnatal development of dendritic Ca2+ transients.
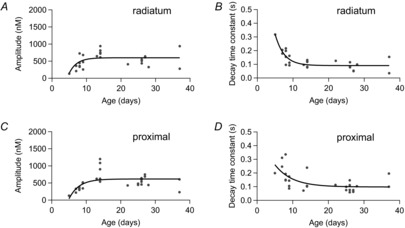
A, filled circles represent measured amplitudes in stratum radiatum (100 μm from soma) for brief bursts of five action potentials (APs) at 100 Hz vs. age of the rats. B, corresponding measured monoexponential decay time constants for Ca2+ transients in A. C, amplitudes of Ca2+ transients in proximal dendrites (25 μm from soma) for brief bursts of five APs at 100 Hz vs. age of the rats. D, corresponding monoexponential decay time constants for Ca2+ transients in C. Data points in A−D were fitted with an exponential function represented by black traces (n = 28).
Postnatal development of Ca2+ buffering and extrusion
How can immature neurons generate such large dendritic Ca2+ signals at only 1 week after birth? The large amplitudes in young animals may be generated by a high ‘mature-like’ density of voltage-gated Ca2+ channels or a smaller Ca2+ influx combined with reduced dendritic Ca2+ buffering in young cells. To understand the underlying mechanisms, we compared dendritic Ca2+ influx and buffering in animals aged 1 week and 3–4 weeks using Fura-FF and different concentrations of Fura-2 to differentially compete with endogenous dendritic Ca2+ buffers (Helmchen et al. 1996). As shown in Fig.3, the application of 100 μm Fura-2 substantially reduced the amplitude of Ca2+ transients in young and mature cells evoked by single bAPs in comparison with Fura-FF (Fig.3A). However, the effect of Fura-2 was larger at 1 week than at 4 weeks after birth (Fig.3A, dotted lines). To estimate the total Ca2+ influx Δ[Ca2+]tot and the endogenous Ca2+ binding ratio κS, we plotted the inverse amplitude against the exogenous binding ratio introduced by Fura-2 (Fig.3B and D). Consistent with a linear compartment model, we were able to fit the data from young cells by linear regression, which revealed Δ[Ca2+]tot = 10 ± 2 μm and an endogenous binding ratio κS = 69 ± 52 from the slope and x-axes intercept, respectively (r2 = 0.60) [eqn (10), Table1]. By contrast, in mature cells the Ca2+ load (Δ[Ca2+]tot = 44 ± 5 μm, r2 = 0.74) as well as the endogenous binding ratio (κS = 276 ± 45) were about four times larger. As a result, the amplitude of single AP-evoked Ca2+ transients revealed by the inverse y-axes intercept was similar in young (A0 = 145 nm) and mature cells (A0 = 160 nm). The extrapolated amplitudes were also similar to the amplitudes measured directly with the low-affinity dye Fura-FF in animals aged 1 week (A = 108 ± 20 nm, n = 6) and 4 weeks (A = 103 ± 12 nm, n = 7), which did not differ significantly between the two age groups (P = 0.80). These values are smaller than the amplitudes obtained from buffer competition experiments, probably as a result of the non-zero buffer capacity of Fura-FF (κB ≈ 36). These data indicate that there is a large increase in dendritic Ca2+ influx during the first 4 weeks of postnatal development. However, this seems to be compensated by a similar increase in endogenous buffer capacity. As a result the amplitude of AP-evoked dendritic Ca2+ signals is very similar throughout postnatal development (Fig.2).
Table 1.
Ca2+ dynamics in apical dendrites of developing CA1 pyramidal cells
| Animal age |
|||
|---|---|---|---|
| 1 week | 2 weeks | 3−4 weeks | |
| A0 | 145 nm | 150 nm * | 160 nm |
| [Ca2+]tot | 10 μm | 25 μm* | 44 μm |
| κs (from A(1) | 69 | 168* | 276 |
| τdecay | 0.17 s+ | 0.09 s* | 0.09 s+ |
| ΔIntegral per spike | 15–30 nm s+ | ∼15 nm s* | ∼10 nm s+ |
Taken from Helmchen et al. (1996).
Values obtained from Fura-FF data.
We further analysed the decay time course of the Ca2+ transients, which could be well fitted by a monoexponential function independent of cell age. The decay time constants measured with Fura-FF were significantly slower in rats aged 1 week (τ = 170 ± 9 ms, n = 6 cells) than in those aged 4 weeks (τ = 93 ± 5 ms, n = 7 cells) (Fig.3A, Fig.4E). The Ca2+ transients measured with different concentrations of Fura-2 showed a markedly slower decay time course compared to Fura-FF, which is consistent with the linear compartment model (Fig.3). Similarly to the amplitude analysis, we plotted the decay time constant τ against κB for the different ages (Fig.3C and E). Linear regression revealed a substantially smaller extrusion rate of γ = 337 ± 35 s−1 at 1 week (r2 = 0.81) vs. γ = 1444 ± 263 s−1 at 4 weeks (r2 = 0.54) [eqn (11)]. This four- to five-fold difference in Ca2+ extrusion translates into a smaller difference in decay time course (0.17 s vs. 0.09 s), most probably because of the different Ca2+ binding ratios in young (κS = 70) and mature (κS = 280) cells.
Figure 4. Slower decay of Ca2+ transients in young cells during brief bursts of APs.
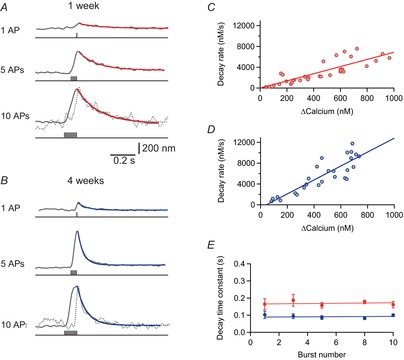
A, Ca2+ transients in a pyramidal cell dendrite (100 μm from soma) from a rat aged 1 week evoked with different numbers of action potentials (APs) at 100 Hz measured with Fura-FF (200 μm). Decay was fitted monoexponentially in red. The dotted line represents the measured Ca2+ trace obtained with one AP, scaled to the amplitude size of 10 APs. B, example traces from a cell aged 4 weeks. The dotted line represents the measured Ca2+ trace obtained with one AP, scaled to the amplitude size of 10 APs. C, decay rate vs. peak amplitude in cells from rats aged 1 week. Linear regression through the data points is shown by a red line (r = 0.84 ± 0.10, n = 6 cells). D, decay rate vs. peak amplitude in cells from rats aged 4 weeks. Linear regression through the data points is shown by a blue line (r = 0.87 ± 0.10, n = 7 cells). E, decay time constants after brief bursts vs. number of APs in the burst. The red and blue lines are linear fits through the data points for cells from rats aged 1 and 4 weeks, respectively.
Taken together, the buffer competition data show that the Ca2+ influx in CA1 pyramidal cell dendrites is four times larger at 4 weeks than it is at 1 week of age (40 μm vs. 10 μm). This seems to be balanced by a four-fold upregulation of the endogenous Ca2+ buffer capacity (280 vs. 70), and a five-fold increase in Ca2+ extrusion rate, resulting in Ca2+ transients with similar amplitudes in young and mature pyramidal cells.
Effective linear summation of Ca2+ transients during brief bursts of APs
To investigate the activity-dependent summation of dendritic Ca2+ transients during repetitive AP firing, we first compared the decay of Ca2+ transients after single APs with the decay after brief bursts (100 Hz) of three, five, eight and 10 APs using Fura-FF (Fig.4). As already shown in Fig.2, the decay is slower in younger cells and gradually speeds up with postnatal development. The decay time course did not change between different stimulation paradigms (Fig.4A and B) and linear regression analysis shows that decay of Ca2+ transients evoked by one to 10 APs is not significantly different (Fig.4E) (1 week, n = 13 cells: P = 0.88; 4 weeks, n = 13 cells: P = 0.76). Next, we plotted the initial decay after the peak against the peak calcium concentration. The initial decay could be well fitted by linear regression (r = 0.84 ± 0.10 for 1 week; r = 0.87 ± 0.10 for 4 weeks), indicating that the Ca2+ extrusion rate γ is independent of Ca2+ concentration within the investigated range up to ∼1 μm. Therefore, we modelled the peak amplitude of brief bursts as a linear superposition of single APs using the average amplitude and decay time course of single AP-evoked Ca2+ transients (Fig.5A−C). The numerical simulations [red and blue line; eqn (13)] could nicely fit the measured data (grey). The peak amplitude (789 ± 64 nm vs. 619 ± 35 nm for 10 APs; P = 0.028) was slightly larger in young cells than in mature cells as a result of the slower decay time course and thus a more effective temporal summation.
Figure 5. Effective temporal summation of dendritic Ca2+ transients during brief bursts of action potentials (APs).
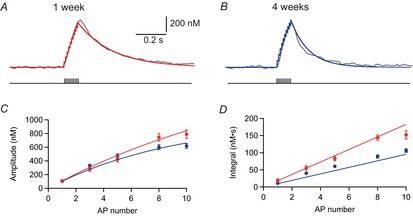
A, B, Ca2+ transients evoked by 10 APs at 100 Hz in a young (A, P7) and mature (B, P28) pyramidal cell with simulated Ca2+ concentrations, modelling linear superposition of single AP-evoked transients in red and blue, respectively. C, peak amplitudes of Ca2+ transients evoked by brief bursts in young (red) and mature (blue) cells plotted vs. the AP number. Continuous lines show the simulated amplitude predicted by linear superposition. D, integral of the Ca2+ transients plotted vs. the burst AP number. Continuous lines represent the modelled values. Data points in C and D represent means of n = 6−13 cells (young) and n = 7−13 cells (mature).
Similarly, we measured the total integral of dendritic Ca2+ signals evoked by brief bursts of bAPs (Fig.5D), which was significantly larger in young (152 ± 12 nm s for 10 APs, n = 13) than in mature (106 ± 5 nm s for 10 APs, n = 13) cells (P = 0.005). We further calculated the integral of Ca2+ transients as the linear sum of the integral of single APs (young: 18.7 ± 1.0 nm s vs. mature: 10.9 ± 0.8 nm s, n = 6 each), which again nicely reproduced the measured burst data (Fig.5D, continuous line). Similar data were obtained at proximal dendritic locations exemplified by the linear superposition of calcium transients [Fig.1D and H, continuous black lines; eqn (13)], which nicely reproduced the measured data. Taken together, the data show the linear summation of dendritic Ca2+ signals during brief bursts in both young and mature cells, and, secondly, a more effective temporal summation in the young neurons generated by the slower decay time course of dendritic Ca2+ signals.
Activity-dependent spike broadening during burst firing
Previously, it was shown that the AP waveform in CA1 pyramidal cells is developmentally regulated (Costa et al. 1991). A potentially slower time course of bAPs in young pyramidal cells may contribute to the large dendritic Ca2+ signals. Therefore, we analysed somatic APs at different developmental stages, which should be largely identical to the voltage waveform in the proximal dendrites (∼25 μm). A short burst of 10 APs was evoked with brief somatic current injections in young and mature neurons (Fig.6). The amplitude of the first AP was significantly smaller in at 1 week than at 4 weeks (94.1 ± 2.3 mV vs. 111.9 ± 2.8 mV, n = 12 each; P < 0.001). Similarly, the maximal slope of rise was smaller in the younger neurons (246 ± 22 V s−1) than in mature pyramidal cells (450 ± 22 V s−1, n = 12 each) (P < 0.0001), indicating a two-times lower expression density of voltage-gated Nav channels. By contrast, AP duration at half maximal amplitude and the maximal rate of decay were relatively similar at 1 and 4 weeks [half-duration: 1.08 ± 0.08 ms vs. 1.00 ± 0.03 ms, n = 12 each (P = 0.84); slope of decay: 92 ± 4 V s−1 vs. 102 ± 5 V s−1, n = 12 each (P = 0.10)]. This suggests that APs are slightly smaller at 1 week compared with 4 weeks, at least at the soma and proximal dendritic locations.
Figure 6. Activity-dependent spike broadening during brief bursts.
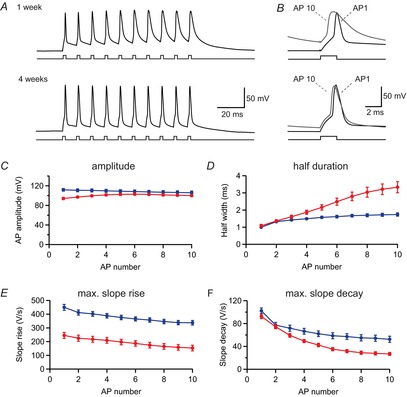
A, a brief burst of 10 action potentials (APs) was evoked in a young (P9, upper trace) and a mature-like (P27, lower trace) pyramidal cell by brief 2 ms current injections at 100 Hz. B, the first and last APs from the recordings in A were plotted at an expanded time scale to show the pronounced AP broadening in the young cell (upper trace). C, peak amplitude was measured for all APs during the burst and plotted against spike number in young (red, n = 12) and mature (blue, n = 12) cells. D−F, similarly, the half-duration (D), the maximal slope of rise (E) and the maximal slope of decay (F) were plotted for the same cells as in C.
During burst firing of 10 APs, however, substantial changes developed in young as well as in mature neurons. In mature pyramidal cells there was a slow-down of the maximal rate of AP repolarization to 51 ± 5% of control (n = 12) (Fig.6F, blue), leading to significant broadening of the half-duration of the 10th AP to 1.74 ± 0.09 ms (n = 12; P < 0.001) (Fig.6D, blue), probably attributable to K-channel inactivation (Kim et al. 2005). Remarkably, this activity-dependent K-channel inactivation was much more pronounced in young neurons, showing a reduced rate of decay to 29 ± 2% (n = 12) (Fig.6F, red) and a broadening of the half-duration to 3.33 ± 0.32 ms (n = 12) (Fig.6D, red) after 10 APs, significantly larger than in mature cells (P < 0.001). This may be generated by a relatively higher density of transiently inactivating K-currents in young CA1 pyramidal cells (Klee et al. 1995). As voltage-gated Ca2+ influx is proportional to the AP half-duration, the activity-dependent spike broadening would indicate that the Ca2+ influx per spike increases during burst firing in both young and mature cells, leading to a supralinear increase of Ca2+ transients with increasing AP number. By contrast, we observed a rather linear increment of the integral of the Ca2+ transients with AP number in both young and mature neurons (Fig.5). This would suggest that somatic spike broadening is counterbalanced with dendritic Ca2+ channel inactivation (Magee & Johnston, 1995; Kavalali et al. 1997) and/or a decrease in backpropagation efficiency with increasing AP number (Spruston et al. 1995). As linear summation of Ca2+ transients was also observed at proximal dendritic locations close to the soma (Fig.1D and H), Ca2+ channel inactivation contributes significantly. As AP broadening is nearly twice as pronounced in the younger cells (AP half-duration: 3.33 ms vs. 1.75 ms), Ca2+ channel inactivation may be more pronounced in the younger neurons, as suggested previously (Thompson & Wong 1991).
Taken together, APs in young pyramidal cells show on average a smaller amplitude and a longer half-duration than mature cells. Furthermore, Ca2+ influx during AP firing is probably a complex interplay between changes in AP waveform and Ca2+ channel inactivation, which, however, results in an overall relatively constant Ca2+ influx per spike during brief bursts in both young and mature neurons.
Supralinear summation of dendritic Ca2+ signals in young CA1 pyramidal cells during prolonged AP firing
During spatial exploration in vivo, CA1 pyramidal cells fire brief bursts of APs repeated at theta frequency (5–8 Hz) (Buzsaki & Draguhn, 2004; Wills et al. 2010). Thus we analysed the shape of APs during theta-burst firing with bursts of three to 10 APs repeated at 5 Hz. As shown in Fig.7, the shape of the last AP burst after 10 repetitions (burst 10, grey) was largely identical to the first theta-cycle (burst 1, black) of the sequence in both young and mature cells (Fig.7A and B). The mean peak amplitude of the 10 APs was calculated for each burst and plotted against burst number (Fig.7C), which revealed that the amplitudes do not change significantly between theta cycles. Further, the mean maximal slope of rise, the maximal slope of decay and the mean half-duration were relatively similar (Fig.7D−F).
Figure 7. Small changes of action potential (AP) waveform during repetitive theta-burst firing.
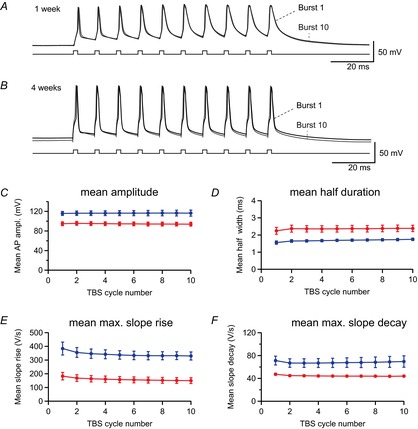
A, the first (black) and 10th (grey) bursts recorded during a 5 Hz theta-burst stimulation (TBS) are superimposed for a young pyramidal cell (P9). B, similarly, the first (black) and 10th (grey) bursts are superimposed for a mature cell (P28). C−F, the mean amplitude (C), mean half-duration (D), mean maximal slope of rise (E) and mean maximal slope of decay (F) were calculated from all 10 APs for each burst and plotted against burst number in animals aged 1 week (red, n = 4) and 4 weeks (blue, n = 6).
We further investigated Ca2+ signalling during TBS paradigms with three to 10 APs per burst corresponding to an average AP frequency of 15–50 Hz (Fig.8). Surprisingly, the maximal peak amplitude of the Ca2+ signals during theta-burst firing with 10 APs per burst (TBS10) was about 2.5 times larger in cells from young animals (1449 ± 236 nm, n = 4) than in those from 4-week-old animals (594 ± 46 nm, n = 6) (P = 0.002) (Fig.8A and B). Furthermore, the young cells showed a roughly three times larger integral [2690 ± 463 nm s (n = 4) vs. 894 ± 126 nm s (n = 6); P = 0.0095] (Fig.8C) and a 2.5-fold larger mean plateau value [1000 ± 170 nm (n = 4) vs. 387 ± 47 nm (n = 6); P = 0.0095] than mature cells.
Figure 8. Supralinear summation of dendritic Ca2+ transients in young cells during theta-burst firing.
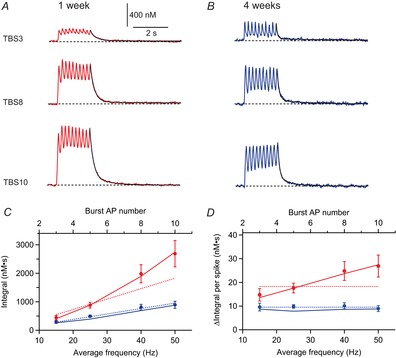
A, B, Ca2+ transients evoked by theta-burst stimulation of backpropagating action potentials (bAPs) with three, eight or 10 APs evoked at 100 Hz repeated at a rate of 5 Hz, from a young (A, P7) and a mature (B, P28) pyramidal cell. The decay time course was fitted with a biexponential function (black traces). C, integral of the Ca2+ transients measured in pyramidal cells at 1 week (red, n = 4) and at 4 weeks (blue, n = 6) plotted vs. the average AP frequency. Dotted lines are the modelled values predicted by linear superposition [eqn (14)]. D, the relative contribution of single spikes to the total theta-burst stimulation (TBS) integral was plotted vs. the average AP frequency (same cells as in C). Dotted lines represent the theoretically predicted contribution per spike [A1×τ1; cf. eqns (12), (13)] for linear superposition. continuous lines in C and D show simulated values for the Integral (C) and Integral per spike (D) predicted by a more realistic model considering the activity-dependent reduction in Ca2+ influx and slow-down of extrusion [eqn (15)] during TBS firing.
We calculated the Ca2+ accumulation as a linear superposition of the previously measured single burst transients [eqn (14)]. The simulation shows a linear increase of the TBS-evoked Ca2+ signal integral with increasing number of APs (Fig.8C, dotted lines), in contradiction with the measured TBS data in young cells, which show a non-linear behaviour. For mature cells, however, the linear prediction was similar to the measured data (blue lines). Furthermore, we calculated the average increment of the TBS integral per spike (Δ Integral per spike), which should be equal to A1*τ1 according to eqn (14), and independent of the AP frequency (Fig.8D, dotted lines). Again, this nicely fitted the data from pyramidal cells from rats aged 4 weeks (Δ Integral per spike ≈ 10 nm s), but substantially deviated from the measured data in young neurons (Δ Integral per spike ≈ 13–27 nm s). Thus, the data from mature cells would be consistent with the linear summation of dendritic Ca2+ transients as described previously for apical dendrites in CA1 pyramidal cells (Helmchen et al. 1996) (Table1), which may provide a linear ‘calcium code’ for firing rate (Johnston, 1996). However, in the young neurons, the data strongly deviate from the linear summation regime during continuous AP firing.
Activity-dependent slow-down of Ca2+ extrusion during theta-burst firing
What might be the reason for this activity-dependent supralinear increase in Ca2+ concentration in young pyramidal cells at 1 week after birth? It could be mediated via an activity-dependent increase in Ca2+ influx and/or Ca2+-induced Ca2+ release (CICR), or buffer saturation, or activity-dependent reduction in Ca2+ extrusion (Llano et al. 1994; Scheuss et al. 2006). To distinguish between these possibilities, the relative change of Ca2+ influx and extrusion was analysed during TBS firing (Fig.9). Measuring the rising slope of the Ca2+ concentration during each burst of APs revealed a reduction of Ca2+ influx for TBS10 during the second burst of APs to 70 ± 5% and 64 ± 4% of control in young (n = 4) and mature (n = 6) cells, respectively (Fig.9C). From the third burst onwards, however, the influx was relatively constant, approaching ∼50% in young and mature cells at the 10th burst. This argues against an activity-dependent increase in Ca2+ influx, CICR or buffer saturation. Similar results were obtained for proximal dendrites showing a reduction of Ca2+ influx at the 10th burst of 50% and 85% in young and mature cells, respectively. As the somatic AP waveform is relatively similar for the different theta cycles, the reduction of Ca2+ influx may reflect incomplete recovery from Ca2+ channel inactivation between theta cycles, at least in proximal dendrites.
Figure 9. Activity-dependent slow-down of extrusion during theta-burst firing.
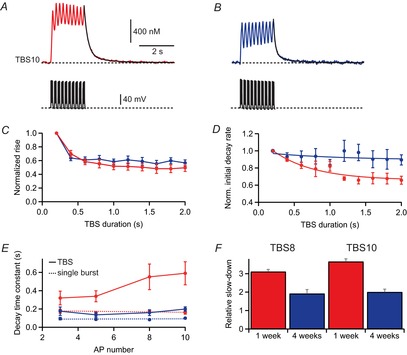
A, B, Ca2+ transients evoked by backpropagating action potentials (bAPs) during theta-burst stimulation (TBS) 10 in a young (P7) and a mature (P28) pyramidal cell (top). Corresponding APs measured in current clamp (bottom). C, rising slope of the Ca2+ concentration during each burst in TBS was normalized to the first burst and plotted against the theta cycle in young (n = 4, red) and mature (n = 6, blue) pyramidal cells. D, the initial decay rate after each burst was divided by the peak amplitude, normalized to decay of the first burst and plotted against the theta cycle. The solid lines represent an exponential fit through the measured data points to estimate TTBS [eqn (15)]. E, amplitude-weighted decay time constants after the end of theta-burst firing. Dotted lines represent the linear regression lines obtained from the decay of brief bursts (Fig.4E). F, the ratio of amplitude-weighted TBS decay time constant divided by the average brief burst decay time constant (from Fig.4) for TBS8 and TBS10 (eight or 10 APs at 100 Hz repeated 10 times at 5 Hz).
Similarly, we also tried to estimate the extrusion rate after each burst by normalizing the initial decay slope to the peak amplitude. This analysis revealed a slight reduction in initial extrusion rate in mature pyramidal cells to about 87 ± 6% after the 10th cycle relative to the first cycle (n = 6) (Fig.9D). Remarkably, this effect was more pronounced in the young neurons, showing a reduction of the initial decay slope to 66 ± 6% after the last burst of the TBS (n = 4; P = 0.038 compared with mature cells). The gradual slow-down could be fitted with a monoexponential function and developed with a time constant of TTBS = 1.0 s in young and TTBS = 0.58 s in mature cells (Fig.9D).
By contrast with the single burst, the decay of the Ca2+ concentration after TBS showed a pronounced biexponential time course (Fig.9A and B). Fitting of the decay after TBS revealed a substantially larger slow-down in young cells (Fig.9E and F, red) than in mature cells (blue). The amplitude-weighted time constant after TBS10 was three times slower in young (0.59 ± 0.13 s, n = 4) than in mature (0.20 ± 0.02 s, n = 6) cells (P = 0.0095). This slow-down of decay after TBS was more pronounced with increasing number of APs per burst (Fig.9E), indicating that this phenomenon might be dependent on the intracellular accumulation of Ca2+ or Na+ ions. The slow-down of Ca2+ extrusion after TBS10 relative to single bursts was 365 ± 15% (n = 4) in young cells and thus significantly larger than the relative slow-down in mature cells (200 ± 14%, n = 6) using the same stimulation pattern (Fig.9F). Similar results were obtained at proximal dendritic locations with a slow-down of Ca2+ extrusion after TBS10 relative to single bursts to 420 ± 160% of control in young cells (TBS10: 0.67 ± 0.22 s, n = 4 vs. one AP: 0.16 ± 0.03 s, n = 7) and to 200 ± 30% in mature pyramidal cells (TBS10: 0.22 ± 0.02 s, n = 6 vs. one AP: 0.11 ± 0.02 s, n = 7), indicating that the strong activity-dependent slow-down of extrusion in young neurons is present throughout the apical dendrite. For comparison, the membrane potential after TBS decays back to resting potential much faster with monoexponential time constants of 13.3 ± 4.4 ms and 17.8 ± 3.2 ms (Fig.7) in young and mature cells, respectively. These results show that dendritic Ca2+ influx is similarly reduced in both young and mature cells during prolonged theta-burst activity. However, the decay time course, and hence the extrusion rate, after TBS is substantially slower in cells from young rats than in those from 4-week-old rats.
We further tried to implement the reduction in Ca2+ influx and the activity-dependent slow-down of extrusion to model the Ca2+ transients during theta-burst firing. Therefore, we assumed that the slow time course of extrusion developed with the time constant TTBS obtained for the slow-down of the initial decay rate during TBS [Fig.9D, eqn (15)], measured at each age group, respectively. As shown in Fig.8C and D, the simulations (solid lines) nicely fit the measured data. This analysis shows that the pronounced activity-dependent slow-down of Ca2+ extrusion in young pyramidal cells is sufficient to explain the remarkable supralinear increase in the dendritic Ca2+ concentration and the total integral of the Ca2+ signal during theta-burst firing.
Discussion
The analysis of AP-evoked dendritic Ca2+ transients in rat CA1 pyramidal cells during the first 4 weeks of postnatal development revealed several unexpected results. The first surprising finding was that AP-induced dendritic Ca2+ transients rapidly increase after P5 and achieve an amplitude of ∼145 nm per AP already 1 week after birth (P7–P9) similar to the amplitude measured in cells from 4-week-old rats (∼160 nm per AP). Secondly, we were able to show that these similar amplitudes are generated by the balanced upregulation of both the total dendritic calcium load per AP (from ∼10 μm to ∼40 μm) and the endogenous Ca2+ buffer capacity (from ∼70 to ∼280) (Table1). Furthermore, the Ca2+ extrusion after the APs was about five times slower in cells at 1 week than at 4 weeks, resulting in a slower decay in young (τ = 0.17 s) than in mature (τ = 0.09 s) cells and a more effective temporal summation during brief bursts of APs. Finally, during continuous theta-burst firing dendritic Ca2+ concentration was up to three-fold larger 1 week after birth than in 4-week-old animals. We show that this reflects an activity-dependent slow-down of Ca2+ extrusion and a resulting supralinear temporal summation of AP-evoked Ca2+ signals specifically in young pyramidal cells.
Postnatal development of Ca2+ influx and buffering
The amplitude of our AP-evoked dendritic Ca2+ transients (∼150 nm) and the decay time course of τ ≈ 0.1 s in cells from animals aged 2−4 weeks are consistent with several previously published results (Helmchen et al. 1996; Maravall et al. 2000). However, the data obtained with younger animals (P7−P9 pyramidal cells) contrast strongly with findings in previous publications showing calcium transients per AP that are three to four times smaller at 1 week compared with postnatal week 3 [Isomura & Kato, 1999 (Fig.1)].
What are the reasons for this discrepancy? Isomura & Kato (1999) used a concentration of 1 mm Fura-2 in addition to 0.2 mm EGTA. Assuming a resting Ca2+ concentration close to zero, this will correspond to an additional buffer capacity of κB = 4000 [eqn (6)]. According to our analysis, the endogenous buffer capacity in young and mature pyramidal cells is 70 and 280, respectively. Therefore, the addition of 1 mm Fura-2 plus 0.2 mm EGTA will completely overwrite the endogenous Ca2+ buffers. Consequently, the change in fluorescence reports a relative difference in total Ca2+ influx, which is four times smaller in young than in mature pyramidal cells. This nicely explains the difference between our data and previously reported data for cells in animals aged 1 week. Actually, the relative fluorescence change obtained by Isomura & Kato (1999) for P8 animals (ΔF380/F360 ≈ 0.03) can be converted into a ratio value of R = 1.03*Rmin, assuming that resting [Ca2+] is close to zero with 1.2 mm Fura2/EGTA. Using our calibration values, this would correspond to an amplitude of 10.3 nm per four APs. Using the buffer capacity we obtained for animals aged 1 week (κS = 70) and assuming κB = 4000, this translates into an unperturbed amplitude of 150 nm per AP, which is very similar to our new data. Therefore, the data of the different studies converge to a matched upregulation of total dendritic Ca2+ influx and buffering to obtain large AP-induced dendritic Ca2+ transients already at 1 week after birth and onwards (Table1).
In fine dendrites and spines of CA1 pyramidal cells, however, the scenario seems to differ. Buffer competition experiments in single dendritic spines using two-photon imaging revealed a much smaller buffer capacity of κS = 20 (Sabatini et al. 2002, P14−P20). As a consequence Ca2+ transients in spines show 10 times larger (A = 1.5 μm) and 10-fold faster AP-evoked Ca2+ signals (τ = 12 ms) than transients in larger apical dendrites. Furthermore, the free Ca2+ concentration in spines can reach huge amplitudes during synaptic transmission and follow the time course of NMDA-induced Ca2+ influx with high fidelity, which is of major importance for activity-dependent synaptic plasticity (Magee & Johnston, 1997; Sabatini et al. 2002; Dan & Poo, 2006; Hao & Oertner, 2012).
Activity-dependent slow-down of Ca2+ extrusion
Ca2+ extrusion was four to five times slower in young pyramidal cells at 1 week than in neurons from 4-week-old rats. This is consistent with several previous reports about the strong postnatal upregulation of all the major Ca2+ transport systems including the plasma membrane Ca2+ ATPase (PMCA), the sodium–calcium exchanger (NCX) and the smooth endoplasmatic Ca2+ ATPase (Usachev et al. 2001; Jensen et al. 2004; Kip et al. 2006; Lee et al. 2007; Stocca et al. 2008). However, our results substantially extend previous findings on the expression density of transporters by showing that this translates into only a relatively small increase in the decay time course of AP-evoked Ca2+ transients in developing CA1 pyramidal cell dendrites (0.17 s vs. 0.09 s), which is a direct consequence of the reduced buffer capacity. Overall this leads to a slightly but significantly larger temporal summation of Ca2+ transients during brief burst firing in young neurons.
During prolonged theta-burst firing, however, the behaviour changes dramatically. First of all, the Ca2+ influx during the individual bursts decreases in both young and mature cells to about 50% of control (Fig.9). As we have seen a similar decrement of Ca2+ influx during TBS firing at distances of 100 μm and 25 μm from the soma, this reduction may reflect an activity-dependent inhibition of dendritic voltage-gated calcium channels (Magee & Johnston, 1995; Kavalali et al. 1997; Budde et al. 2002). Secondly, Ca2+ extrusion slows down during TBS firing in both young and mature cells. Interestingly, the decay after TBS is three times slower in young neurons (τ ≈ 0.6 s) than it is in mature cells (τ ≈ 0.2 s). As a consequence there was a supralinear summation of the Ca2+ transients during TBS with an up to three-fold larger peak amplitude and integral of Ca2+ signals in young than in mature pyramidal cell dendrites (Fig.8). This is remarkable because total Ca2+ influx per AP is about four times smaller in young cells. Therefore, the young neurons can generate three-fold larger calcium signals during continuous TBS firing using four times less ATP relative to mature cells.
In mature cells the slow-down of extrusion is less pronounced (∼200%). In combination with the ∼50% decrease in Ca2+ influx, this apparently leads to a near linear increase in the average calcium concentration with increasing AP frequency, as has been reported for apical dendrites in pyramidal cells aged at P14 (Helmchen et al. 1996). Together with our data, this indicates that supralinear Ca2+ signalling in apical dendrites of CA1 pyramidal cells is restricted to animals younger than 2 weeks. By contrast, Scheuss et al. (2006) found an activity-dependent slow-down of Ca2+ extrusion in small dendrites and spines of P16−P20 rats, generating supralinear Ca2+ transients during prolonged AP firing. Probably the difference with apical dendrites at this developmental stage is related to the larger accumulation of Ca2+ and Na+ ions in fine dendrites and spines, which was shown to be important to the activity-dependent reduction of Ca2+ extrusion in these small structures (Scheuss et al. 2006).
Functional significance
In CA1 pyramidal cells, spine synapses start to form after birth and density increases rapidly to reach adult-like patterns after 3 weeks of postnatal development (Fiala et al. 1998; Kirov et al. 2004). The formation of new dendritic spines can be triggered by the release of glutamate from neighbouring presynaptic boutons, activating extrasynaptic NMDA receptors on dendritic shafts (Engert & Bonhoeffer, 1999; Maletic-Savatic et al. 1999; Kwon & Sabatini, 2011). Furthermore, this process is Ca2+-dependent and restricted to a certain critical time window which lasts from birth to about the second postnatal week (Kwon & Sabatini, 2011). The induction threshold for glutamate-induced spine formation is very low at the end of the first postnatal week (P8–P12), increases at 2 weeks (P14–P15) and seems to be very high at 3 weeks after birth (Kwon & Sabatini, 2011). Similarly, activation of Ca2+-permeable nicotinic ACh receptors can facilitate spine formation at P7 but not at P15 or later (Lozada et al. 2012). Dendritic Ca2+ buffering properties would nicely support this critical window behaviour because buffer capacity is lowest at P7 (∼70), increases towards P14 (∼170) (Helmchen et al. 1996) and reaches ∼280 at 4 weeks of age (Table1).
Furthermore, synapse formation will be supported by hippocampal theta-gamma oscillations, which start to emerge around P7 (Lahtinen et al. 2007; Mohns et al. 2008). Activity-dependent slow-down of Ca2+ extrusion generates large Ca2+ signals via supralinear temporal summation in young dendrites during prolonged firing, providing a large dynamic range of the intracellular Ca2+ signals to control Ca2+-dependent growth and synapse formation. This will help to effectively shape network connectivity in the developing brain in an activity-dependent manner (Brockmann et al. 2011). As a consequence of rapid network formation, hippocampal pyramidal cells show spatially selective place field firing at only about 2 weeks after birth (Wills et al. 2010; Langston et al. 2010).
After postnatal development, dendritic plasticity and synapse formation strongly decrease (Trachtenberg et al. 2002; Wilbrecht et al. 2010; Romand et al. 2011). Mature pyramidal cells may support stable network connectivity by reducing Ca2+-dependent plasticity in larger dendrites via the upregulation of dendritic Ca2+ buffer capacity. Only dendritic spines and fine dendritic branches may show low Ca2+ buffering in adulthood, facilitating local Ca2+-dependent plasticity during learning in the adult nervous system (Holtmaat & Svoboda, 2009).
We have shown the developmental regulation of dendritic Ca2+ influx, buffering and extrusion providing up to three-fold larger Ca2+ signals during theta-burst firing in young neurons in comparison with events in mature hippocampal pyramidal cells. These age-specific and activity-dependent Ca2+ signals are very likely instrumental in shaping Ca2+-dependent plasticity and synapse formation during brain development.
Acknowledgments
We thank Jan Schulz for helpful comments on the manuscript and Selma Becherer for histochemical stainings and technical assistance.
Glossary
- ACSF
artificial cerebrospinal fluid
- AP
action potential
- bAP
backpropagating action potential
- CICR
Ca2+-induced Ca2+ release
- ROI
region of interest
- TBS
theta-burst stimulation
Key points
Because intracellular Ca2+ is important for activity-dependent growth and development, we studied action potential (AP)-evoked dendritic Ca2+ influx and Ca2+ buffering in developing rat CA1 pyramidal cells during the first 1–4 weeks after birth.
We show for the first time that active dendritic backpropagation of APs generates large Ca2+ transients in pyramidal cell dendrites even after the first postnatal week.
The amplitude of Ca2+ transients at 1 week is similar to that at 4 weeks because a four-fold upregulation of calcium influx per AP is balanced by a similar increase in endogenous Ca2+ buffer capacity during this period.
The calcium extrusion after APs was about five times slower at 1 week than at 4 weeks, resulting in a slower decay in young cells and a more effective temporal summation during brief bursts of APs.
During continuous theta-burst firing, dendritic calcium concentration was up to three-fold larger in animals aged 1 week than in those aged 4 weeks, generated via an activity-dependent slow-down of Ca2+ extrusion, which may allow Ca2+-dependent control of growth and development with a large dynamic range.
Additional information
Competing interests
None declared.
Author contributions
J.P. performed the experiments and data analysis. J.B. conceived and designed the research and wrote the paper. Both authors read and approved the final version of the paper.
Funding
The work was supported by the Swiss National Science Foundation (SNSF) (31003A_153276) and by Deutsche Forschungsgemeinschaft (GRK 843).
References
- Ahmed R, Zha XM, Green SH, Dailey ME. Synaptic activity and F-actin coordinately regulate CaMKIIalpha localization to dendritic postsynaptic sites in developing hippocampal slices. Mol Cell Neurosci. 2006;31:37–51. doi: 10.1016/j.mcn.2005.08.020. [DOI] [PubMed] [Google Scholar]
- Aizawa H, Hu SC, Bobb K, Balakrishnan K, Ince G, Gurevich I, Cowan M, Ghosh A. Dendrite development regulated by CREST, a calcium-regulated transcriptional activator. Science. 2004;303:197–202. doi: 10.1126/science.1089845. &. [DOI] [PubMed] [Google Scholar]
- Aponte Y, Bischofberger J, Jonas P. Efficient Ca2+ buffering in fast spiking basket cells of rat hippocampus. J Physiol. 2008;586:2061–2075. doi: 10.1113/jphysiol.2007.147298. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischofberger J, Engel D, Li L, Geiger JRP, Jonas P. Patch clamp recording from mossy fiber terminals in hippocampal slices. Nature Protocols. 2006;1:2075–2081. doi: 10.1038/nprot.2006.312. &. [DOI] [PubMed] [Google Scholar]
- Bischofberger J, Jonas P. Action potential propagation into the presynaptic dendrites of rat mitral cells. J Physiol. 1997;504:359–365. doi: 10.1111/j.1469-7793.1997.359be.x. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockmann MD, Pöschel B, Cichon N, Hanganu-Opatz IL. Coupled oscillations mediate directed interactions between prefrontal cortex and hippocampus of the neonatal rat. Neuron. 2011;71:332–347. doi: 10.1016/j.neuron.2011.05.041. &. [DOI] [PubMed] [Google Scholar]
- Budde T, Meuth S, Pape HC. Calcium-dependent inactivation of neuronal calcium channels. Nat Rev Neurosci. 2002;3:873–883. doi: 10.1038/nrn959. &. [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Draguhn A. Neuronal oscillations in cortical networks. Science. 2004;304:1926–1929. doi: 10.1126/science.1099745. &. [DOI] [PubMed] [Google Scholar]
- Costa PF, Ribeiro MA, Santos AI. Afterpotential characteristics and firing patterns in maturing rat hippocampal CA1 neurones in in vitro slices. Brain Res Dev Brain Res. 1991;62:263–272. doi: 10.1016/0165-3806(91)90174-h. &. [DOI] [PubMed] [Google Scholar]
- Dan Y, Poo MM. Spike timing-dependent plasticity, from synapse to perception. Physiol Rev. 2006;86:1033–1048. doi: 10.1152/physrev.00030.2005. &. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Mermelstein PG, Xia H, Tsien RW. Signaling from synapse to nucleus: the logic behind the mechanisms. Curr Opin Neurobiol. 2003;13:354–365. doi: 10.1016/s0959-4388(03)00076-x. &. [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. &. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Feinberg M, Popov V, Harris KM. Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J Neurosci. 1998;18:8900–8911. doi: 10.1523/JNEUROSCI.18-21-08900.1998. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger JRP, Bischofberger J, Vida I, Fröbe U, Pfitzinger S, Weber HJ, Haverkampf K, Jonas P. Patch-clamp recording in brain slices with improved slicer technology. Pflugers Arch. 2002;443:491–501. doi: 10.1007/s00424-001-0735-3. &. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. &. [PubMed] [Google Scholar]
- Häusser M, Spruston N, Stuart GJ. Diversity and dynamics of dendritic signaling. Science. 2000;290:739–744. doi: 10.1126/science.290.5492.739. &. [DOI] [PubMed] [Google Scholar]
- Hao J, Oertner TG. Depolarization gates spine calcium transients and spike-timing-dependent potentiation. Curr Opin Neurobiol. 2012;22:509–515. doi: 10.1016/j.conb.2011.10.004. &. [DOI] [PubMed] [Google Scholar]
- Helmchen F, Imoto K, Sakmann B. Ca2+ buffering and action potential-evoked Ca2+ signaling in dendrites of pyramidal neurons. Biophys J. 1996;70:1069–1081. doi: 10.1016/S0006-3495(96)79653-4. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch TK. Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 2005;6:877–888. doi: 10.1038/nrn1787. [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. &. [DOI] [PubMed] [Google Scholar]
- Isomura Y, Kato N. Action potential-induced dendritic calcium dynamics correlated with synaptic plasticity in developing hippocampal pyramidal cells. J Neurophysiol. 1999;82:1993–1999. doi: 10.1152/jn.1999.82.4.1993. &. [DOI] [PubMed] [Google Scholar]
- Jensen TP, Buckby LE, Empson RM. Expression of plasma membrane Ca2+ ATPase family members and associated synaptic proteins in acute and cultured organotypic hippocampal slices from rat. Brain Res Dev Brain Res. 2004;152:129–136. doi: 10.1016/j.devbrainres.2004.06.004. &. [DOI] [PubMed] [Google Scholar]
- Jin M, Guan CB, Jiang YA, Chen G, Zhao CT, Cui K, Song YQ, Wu CP, Poo MM, Yuan XB. Ca2+-dependent regulation of rho GTPases triggers turning of nerve growth cones. J Neurosci. 2005;25:2338–2347. doi: 10.1523/JNEUROSCI.4889-04.2005. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston D. The calcium code. Biophys J. 1996;70:1095. doi: 10.1016/S0006-3495(96)79674-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. &. [DOI] [PubMed] [Google Scholar]
- Kaiser KM, Zilberter Y, Sakmann B. Back-propagating action potentials mediate calcium signalling in dendrites of bitufted interneurons in layer 2/3 of rat somatosensory cortex. J Physiol. 2001;535:17–31. doi: 10.1111/j.1469-7793.2001.t01-1-00017.x. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET, Zhuo M, Bito H, Tsien RW. Dendritic Ca2+ channels characterized by recordings from isolated hippocampal dendritic segments. Neuron. 1997;18:651–663. doi: 10.1016/s0896-6273(00)80305-0. &. [DOI] [PubMed] [Google Scholar]
- Kim J, Wei DS, Hoffman DA. Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. J Physiol. 2005;569:41–57. doi: 10.1113/jphysiol.2005.095042. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kip SN, Gray NW, Burette A, Canbay A, Weinberg RJ, Strehler EE. Changes in the expression of plasma membrane calcium extrusion systems during the maturation of hippocampal neurons. Hippocampus. 2006;16:20–34. doi: 10.1002/hipo.20129. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirov SA, Goddard CA, Harris KM. Age-dependence in the homeostatic upregulation of hippocampal dendritic spine number during blocked synaptic transmission. Neuropharmacology. 2004;47:640–648. doi: 10.1016/j.neuropharm.2004.07.039. &. [DOI] [PubMed] [Google Scholar]
- Klee R, Ficker E, Heinemann U. Comparison of voltage-dependent potassium currents in rat pyramidal neurons acutely isolated from hippocampal regions CA1 and CA3. J Neurophysiol. 1995;74:1982–1995. doi: 10.1152/jn.1995.74.5.1982. &. [DOI] [PubMed] [Google Scholar]
- Konur S, Ghosh A. Calcium signaling and the control of dendritic development. Neuron. 2005;46:401–405. doi: 10.1016/j.neuron.2005.04.022. &. [DOI] [PubMed] [Google Scholar]
- Kwon HB, Sabatini BL. Glutamate induces de novo growth of functional spines in developing cortex. Nature. 2011;474:100–104. doi: 10.1038/nature09986. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston RF, Ainge JA, Couey JJ, Canto CB, Bjerknes TL, Witter MP, Moser EI, Moser MB. Development of the spatial representation system in the rat. Science. 2010;328:1576–1580. doi: 10.1126/science.1188210. &. [DOI] [PubMed] [Google Scholar]
- Lahtinen H, Palva JM, Sumanen S, Voipio J, Kaila K, Taira T. Postnatal development of rat hippocampal gamma rhythm in vivo. J Neurophysiol. 2002;88:1469–1474. doi: 10.1152/jn.2002.88.3.1469. &. [DOI] [PubMed] [Google Scholar]
- Lee SH, Park KH, Ho WK, Lee SH. Postnatal developmental changes in Ca2+ homeostasis in supraoptic magnocellular neurons. Cell Calcium. 2007;41:441–450. doi: 10.1016/j.ceca.2006.08.003. &. [DOI] [PubMed] [Google Scholar]
- Llano I, DiPolo R, Marty A. Calcium-induced calcium release in cerebellar Purkinje cells. Neuron. 1994;12:663–673. doi: 10.1016/0896-6273(94)90221-6. &. [DOI] [PubMed] [Google Scholar]
- Lozada AF, Wang X, Gounko NV, Massey KA, Duan J, Liu Z, Berg DK. Induction of dendritic spines by β2-containing nicotinic receptors. J Neurosci. 2012;32:8391–8400. doi: 10.1523/JNEUROSCI.6247-11.2012. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Johnston D. Characterization of single voltage-gated Na+ and Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons. J Physiol. 1995;487:67–90. doi: 10.1113/jphysiol.1995.sp020862. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. &. [DOI] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. &. [DOI] [PubMed] [Google Scholar]
- Maravall M, Mainen ZF, Sabatini BL, Svoboda K. Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys J. 2000;78:2655–2667. doi: 10.1016/S0006-3495(00)76809-3. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohns EJ, Blumberg MS. Synchronous bursts of neuronal activity in the developing hippocampus: modulation by active sleep and association with emerging gamma and theta rhythms. J Neurosci. 2008;28:10134–10144. doi: 10.1523/JNEUROSCI.1967-08.2008. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normann C, Peckys D, Schulze CH, Walden J, Jonas P, Bischofberger J. Associative long-term depression in the hippocampus is dependent on postsynaptic N-type Ca2+ channels. J Neurosci. 2000;20:8290–8297. doi: 10.1523/JNEUROSCI.20-22-08290.2000. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Augustine GJ. Calcium gradients and buffers in bovine chromaffin cells. J Physiol. 1992;450:273–301. doi: 10.1113/jphysiol.1992.sp019127. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Tank DW. Dendritic calcium dynamics. Curr Opin Neurobiol. 1994;4:373–382. doi: 10.1016/0959-4388(94)90099-x. &. [DOI] [PubMed] [Google Scholar]
- Romand S, Wang Y, Toledo-Rodriguez M, Markram H. Morphological development of thick-tufted layer V pyramidal cells in the rat somatosensory cortex. Front Neuroanat. 2011;5:5. doi: 10.3389/fnana.2011.00005. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Oertner TG, Svoboda K. The life cycle of Ca2+ ions in dendritic spines. Neuron. 2002;33:439–452. doi: 10.1016/s0896-6273(02)00573-1. &. [DOI] [PubMed] [Google Scholar]
- Scheuss V, Yasuda R, Sobczyk A, Svoboda K. Nonlinear [Ca2+] signaling in dendrites and spines caused by activity-dependent depression of Ca2+ extrusion. J Neurosci. 2006;26:8183–8194. doi: 10.1523/JNEUROSCI.1962-06.2006. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer NC. Electrical activity in early neuronal development. Nature. 2006;444:707–712. doi: 10.1038/nature05300. [DOI] [PubMed] [Google Scholar]
- Spruston N, Schiller Y, Stuart G, Sakmann B. Activity-dependent action potential invasion and calcium influx into hippocampal CA1 dendrites. Science. 1995;268:297–300. doi: 10.1126/science.7716524. &. [DOI] [PubMed] [Google Scholar]
- Stocca G, Schmidt-Hieber C, Bischofberger J. Differential dendritic Ca2+ signalling in young and mature hippocampal granule cells. J Physiol. 2008;586:3795–3811. doi: 10.1113/jphysiol.2008.155739. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SM, Wong RK. Development of calcium current subtypes in isolated rat hippocampal pyramidal cells. J Physiol. 1991;439:671–689. doi: 10.1113/jphysiol.1991.sp018687. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolias KF, Bikoff JB, Burette A, Paradis S, Harrar D, Tavazoie S, Weinberg RJ, Greenberg ME. The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron. 2005;45:525–538. doi: 10.1016/j.neuron.2005.01.024. &. [DOI] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 2002;420:788–794. doi: 10.1038/nature01273. &. [DOI] [PubMed] [Google Scholar]
- Usachev YM, Toutenhoofd SL, Goellner GM, Strehler EE, Thayer SA. Differentiation induces upregulation of plasma membrane Ca2+-ATPase and concomitant increase in Ca2+ efflux in human neuroblastoma cell line IMR-32. J Neurochem. 2001;76:1756–1765. doi: 10.1046/j.1471-4159.2001.00169.x. &. [DOI] [PubMed] [Google Scholar]
- Vizard TN, O'Keeffe GW, Gutierrez H, Kos CH, Riccardi D, Davies AM. Regulation of axonal and dendritic growth by the extracellular calcium-sensing receptor. Nat Neurosci. 2008;11:285–291. doi: 10.1038/nn2044. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilbrecht L, Holtmaat A, Wright N, Fox K, Svoboda K. Structural plasticity underlies experience-dependent functional plasticity of cortical circuits. J Neurosci. 2010;30:4927–4932. doi: 10.1523/JNEUROSCI.6403-09.2010. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills TJ, Cacucci F, Burgess N, O'Keefe J. Development of the hippocampal cognitive map in preweanling rats. Science. 2010;328:1573–1576. doi: 10.1126/science.1188224. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RO, Ghosh A. Activity-dependent regulation of dendritic growth and patterning. Nat Rev Neurosci. 2002;3:803–812. doi: 10.1038/nrn941. &. [DOI] [PubMed] [Google Scholar]
- Xie Z, Srivastava DP, Photowala H, Kai L, Cahill ME, Woolfrey KM, Shum CY, Surmeier DJ, Penzes P. Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron. 2007;56:640–656. doi: 10.1016/j.neuron.2007.10.005. &. [DOI] [PMC free article] [PubMed] [Google Scholar]