

Abstract
We report a detailed investigation into the application of visible light-mediated photocatalysis to a challenging bond construction in a complex pharmaceutical target. The optimized reaction allowed the direct coupling of N-methylmorpholine with an unfunctionalized pyridazine in good yield and selectivity, and with high purity of the product isolated via crystallization. The reaction also facilitated the expedient synthesis of a range of analogues via the use of other commercially available N-methyl substituted tertiary amines, and therefore it represents an attractive tool for medicinal chemistry. Furthermore, a number of other interesting photoredox reactions were discovered during the course of this investigation, such as a formal methylation reaction via C–N bond cleavage, functionalization of C–H bonds alpha to amides, and a visible light-mediated iminium ion reduction.
Introduction
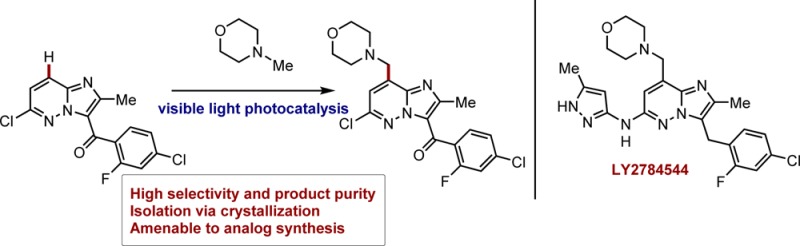
LY2784544 has been identified as a selective inhibitor of JAK2-V617F and has undergone clinical trials for the treatment of several myeloproliferative disorders (Scheme 1).1 A significant challenge associated with the formation of the functionalized imidazopyridazine is the introduction of the benzylic morpholine unit. A first-generation synthesis employed a silver-catalyzed Minisci radical alkylation2 with N-phthaloylglycine (Scheme 1). This method suffered from poor selectivity for the desired C-8 position, as well as forming significant quantities of a bis-alkylation product. At large scale, the Minisci chemistry proved challenging due to the insolubility of the products and the thick mixtures that were formed. Additionally, from a route design perspective, formation of the morpholine in this fashion was undesirable due to the use of a protecting group.
Scheme 1. Previous Process Routes to LY2784544.

In order to address these concerns, efforts were directed toward introduction of the desired morpholine in a single step.3 A second-generation synthesis, which employed a vanadium-catalyzed addition of N-methylmorpholine-N-oxide (NMO) was developed that displayed greater scalability, improved yield, and product quality. This method ultimately delivered >1 metric tonne of imidiazopyridizine 2 in a highly reproducible manner. While the exact mechanistic pathway for this reaction has not been elucidated, one possibility proceeds via the intermediacy of an α-amino radical. Other methods for generation of the intermediate radical were subsequently investigated, including the use of photochemistry. Initial studies found that under irradiation with a Hg-lamp, a range of sensitizers were successful in promoting the addition of N-methylmorpholine (NMM) to the pyridazine core, albeit in low conversions and with relatively high levels of impurities. The use of NMM would represent a significant advantage as it is available at greatly reduced cost and the safety4 and bulk sourcing concerns associated with anhydrous NMO would be alleviated.
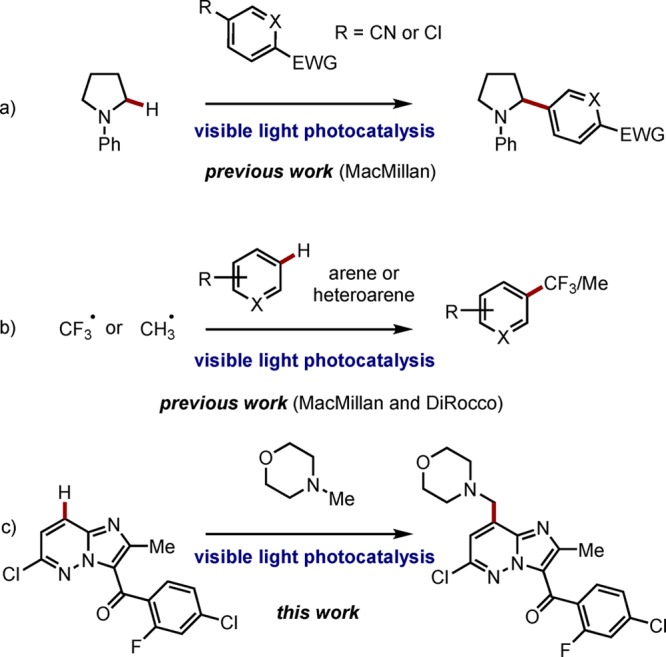
Visible light-mediated photocatalysis has developed into a defined research field with a recent increase in both the number and diversity of reported transformations.5 Within this arena the functionalization of amine containing compounds has received significant attention.6 The generation of α-amino radicals7 has been demonstrated from both unfunctionalized amines,8 and those containing adjacent activating groups such as CO2H9 or TMS.10 While most reports demonstrate the addition of α-amino radicals to activated alkenes, MacMillan8h,8i,9c (Scheme 2a) has shown the efficient arylation of simple N-aryl amines with either cyano or chloro- substituted arenes and heteroarenes. Furthermore, given the quantity of established methods for the functionalization of amine containing compounds, there remain relatively few efforts that have demonstrated the applicability of photocatalysis to a pharmaceutical or agrochemical setting, especially with regard to the formation of challenging bond constructs. Recent elegant studies by Nagib and MacMillan11 as well as DiRocco and co-workers,12 among others8c,8g,13 have identified photocatalysis as an efficient strategy for the functionalization of biologically relevant heterocycles (Scheme 2b). These reported methods appear ideally suited for the rapid generation of a collection of compounds during early stage discovery chemistry. Later stage development demands a different set of criteria, notably selectivity, efficiency, and scalability. A limited number of recent reports have demonstrated the use of photocatalysis en route to known pharmaceutical compounds,9c,14 such as the efficient 3-step synthesis of pregabalin,9b albeit in a racemic fashion and with the key step conducted on <100 mg scale.
Scheme 2. Previous Examples and Proposed Transformation.
Considering the scarcity of industrially relevant examples and the unanswered questions of how visible light photocatalysis should best be conducted at production scale, it currently remains unclear whether it can be successfully applied in a process development setting.
With these questions in mind, we envisioned the introduction of the benzylic morpholine in LY2784544 as an ideal opportunity to test the viability of a photoredox transformation in a complex pharmaceutical setting (Scheme 2c). We were particularly drawn to the mild reaction conditions and functional group tolerance typically associated with visible light-mediated photocatalysis and furthermore were encouraged by the precedent for the efficient formation of α-amino radicals.
Results and Discussion
Proof of Concept
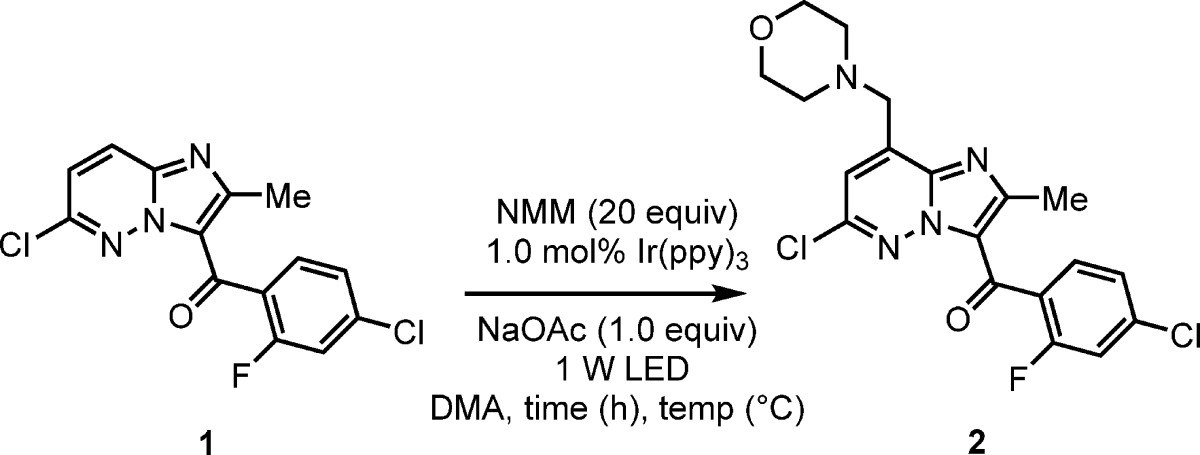
We started with initial conditions similar to those reported for the α-arylation of N-aryl amines8h but employed oxygen as the terminal oxidant by running the reactions open to air. Our light source consisted of 2 × 4 W blue LED light strips15 which raised the temperature of the reaction to 35 °C. After 7 h, using 2 mol % of Ir(ppy)3,16 with 20 equiv of NMM, all of the imidazopyridazine starting material had been consumed (as measured by HPLC). The desired exo isomer 2 (the product of addition from a speculated radical at the exocyclic CH3 position) was identified as the major product (63% area/area (a/a) HPLC) (Scheme 3). An expected major impurity, the endo regioisomer 3 (the product of addition from a speculated radical at the endocyclic CH2 group), was present in 11% (or 5.7:1 exo:endo) which is greater than that observed in the second generation route employing NMO and VO(acac)2 where the endo isomer was formed in <5%. Consistent with the previous Minisci alkylation the competitive addition of the α-amino radical to the C-7 position of the imidazopyridazine (4) was observed, albeit in greatly reduced quantity (3% vs 25%).
Scheme 3. Initial Reaction with Observed Impurities.

Further impurities unique to the photocatalysis protocol were identified as the reduction of the carbon-chlorine bond (5) (6%), which is not unexpected given the precedent for photocatalysed aryl halide bond reduction.17 More interestingly the product of formal methylation of the imidazopyridazine (6) (5%) and of the addition of DMA (7) (12%) were also present. While the crude 63% a/a of the desired product appeared high under these conditions, the reaction was inconsistent and the isolation of pure 2 via chromatography proved challenging. Furthermore, the bis-alkylation product 8, a significant impurity in the N-phthaloylglycine Minisci reaction, could not be reliably resolved using the existing HLPC method.
In order to quantify the reaction products with more certainty, an analytical UPLC (ultra performance liquid chromatography) method was developed with both a photodiode array (PDA) and mass detection (QDa) analyzer, which proved pivotal for the further optimization of this reaction. These analytical systems allowed the analysis of a reaction mixture to a higher level of sensitivity in greatly reduced time. At this juncture, we decided to control the reaction temperature via the use of a jacketed beaker connected to a recirculating chiller. Previously, the temperature of the reaction had been influenced by the light-source, a feature that can be often overlooked. Running the reaction at 22 °C and using a 1 W LED strip, while monitoring with the updated analytical methods, the reaction was found to be complete (<5% 1 remained) in 16 h. The desired exo isomer 2, was again the main component in the reaction mixture (65% a/a) with a calculated UPLC yield of 57% and 59% respectively, for the two quantitation methods (UPLC-PDA and UPLC-QDa) (Table 1 entry 1).18 Under these conditions the exo:endo ratio was 5.5:1, while the other quantifiable impurities were individually controlled to <5%.19 Through improved chromatography conditions, 2 could be isolated in 43% yield. This agreement between the two analytical yields and the isolated material provided us with confidence in our analytical method going forward.
Table 1. Effect of Temperature, Oxygen, and Scale.
| entry | T (°C) | t (h) | yield of 2a | yield of 1a | exo:endo ratioa |
|---|---|---|---|---|---|
| 1 | 22 | 16 | 57 | <5 | 5.5:1 |
| 2 | 5 | 8 | 53 | <5 | 5.1:1 |
| 3b | 22 | 16 | 24 | 42 | 4.3:1 |
| 4c | 5 | 12.5 | 6 | 80 | 3.3:1 |
| 5d | 5 | 12.5 | 22 | 44 | 2.7:1 |
| 6e | 5 | 12.5 | 33 | 29 | 4.6:1 |
| 7f | 5 | 13 | 40 | 26 | 3.5:1 |
| 8g | 5 | 13 | 40 | 23 | 3.3:1 |
Calculated by UPLC (PDA).
Reaction performed at a 0.50 mmol scale.
Reaction deoxygenated via freeze–pump–thaw.
Reaction performed under a balloon of oxygen.
Reaction performed under a balloon of air.
Reaction sparged continuously with air (0.25 mmol scale).
Reaction sparged continuously with air (0.50 mmol scale).
Reaction Optimization
We next sought to utilize the jacketed beaker system by exploring the reaction at lower temperature. At 5 °C a similar yield and exo:endo ratio could be obtained in only 8 h, potentially due to higher oxygen solubility at this temperature (Table 1, entry 2).20 Further evidence of the important role of oxygen was observed when the reaction was run at twice the scale, resulting in a significant decrease in yield (entry 3). This is attributed to a decrease of relative surface area of solution exposed to air.21 Further probing the effect of oxygen we found a deoxygenated reaction gave minimal product (<10%) (entry 4). Conducting the reaction under an atmosphere of oxygen gave both a reduction in the quantity of 2 and in the exo:endo ratio (entry 5). Furthermore, a control reaction utilizing a balloon of air equally gave reduced yield and exo:endo ratio (entry 6). In an effort to address the issue of scalability, air was continuously sparged through the reaction leading to identical reactions at different scales, but with significantly decreased exo:endo ratio and with increased reaction time (entries 7 and 8). The poor scalability of this batch reaction was not considered a critical factor at this juncture. Any scale up would likely be more efficiently conducted via continuous processing;22 therefore, optimization was continued at the current scale.
The reaction also showed a high degree of sensitivity to the light source. In the absence of LED light, or Ir(ppy)3, less than <5% 2 was observed. Varying the total LED power, independently of temperature, showed that increasing LED power had a detrimental effect on selectivity (Figure 1).23 Most strikingly, after 4 h with 9 W LED irradiation <10% 1 remained; however, the exo product 2 was only observed in 23% yield. The remaining material balance (by UPLC) contained elevated amounts of previously identified side products (5 5%, 4 7%) but also comprised a large quantity of uncharacterized material that made quantitation of other known impurities such as 6 and 7 difficult (Figure 1 right).
Figure 1.
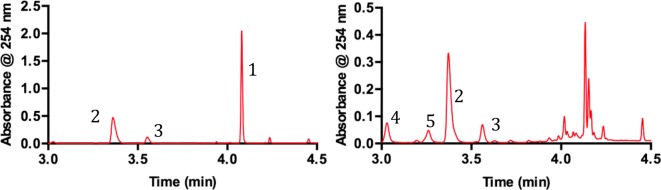
UPLC chromatograms showing reaction profile at 4 h using a 1 W LED (left) and a 9 W LED (right).
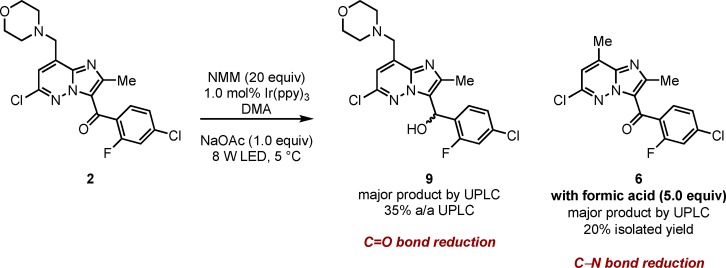
While the exo product 2 is stable to the reaction conditions under 1 W LED irradiation, increasing the power to 8 W and resubjection to the reaction conditions gave ketone reduction (9) as the major product (35%) among a complex mixture also containing 6 (7%) and dechlorinated 2 (7%). The addition of 5 equiv of formic acid provided a cleaner reaction profile leading to methylation product 6, isolated in 20% yield (Scheme 4). This strongly suggests that 6 is derived from 2 and presumably is formed via C–N bond reduction. No significant decomposition of 2 was observed in the absence of Ir(ppy)3 or NMM, and was greatly reduced in the absence of oxygen (72% 2 remained after 8 h) suggesting this potentially useful24 transformation is a photocataylsed process.25
Scheme 4. Stability of 2 to the Reaction Conditions.
Given the limited solubility of NaOAc in DMA a range of both organic and inorganic bases were screened.24 The initial choice of 1 equiv of NaOAc proved optimal with other bases delivering reduced yield, and surprisingly, reduced exo:endo ratio (Table 2, entries 1–6). The exo:endo ratio also varied with the amount of NMM. Using 60 equiv a 1.2:1 exo:endo ratio was obtained (entry 7), lowering the equivalents below 20 displayed a clear trend for increased exo:endo ratio with decreasing quantities of NMM, albeit at the expense of 2 (entries 8–10). This is potentially rationalized by competition between radical addition of the α-amino radical and C–H abstraction from a molecule of NMM. The primary α-amino radical could abstract the weaker secondary C–H bond to give the secondary α-amino radical that then may add to the imidazopyridazine core to ultimately generate the endo isomer 3.26
Table 2. Effect of Base and Equivalents of NMM.
| entry | base | NMM (equiv) | yield of 2a | yield of 1a | exo:endo ratioa |
|---|---|---|---|---|---|
| 1 | NaOAc | 20 | 53 | <5 | 5.1:1 |
| 2 | None | 20 | 41 | 17 | 3.8:1 |
| 3 | K2HPO4 | 20 | 36 | 30 | 3.2:1 |
| 4 | 2,6-lutidine | 20 | 33 | 30 | 2.9:1 |
| 5 | Imidazole | 20 | 22 | 48 | 2.1:1 |
| 6 | DBU | 20 | <5 | >95 | N/A |
| 7b | none | 60 | 33c | <5 | 1.2:1d |
| 8e | NaAOc | 15 | 49 | 17 | 6.1:1 |
| 9e | NaAOc | 10 | 39 | 26 | 6.9:1 |
| 10e | NaAOc | 5 | 35 | 38 | 9.6:1 |
Reactions performed at a 0.231 mmol scale, at 5 °C with 1.0 mol % Ir(ppy)3, 1.0 equiv of base, 20 equiv of NMM for 8 h unless otherwise stated. Calculated by UPLC (PDA).
Reaction performed at a 0.774 mmol scale, at 35 °C with 2.0 mol % Ir(ppy)3, for 12 h.
Isolated yield
Calculated via 1H NMR analysis of the crude reaction mixture.
Reaction time of 10 h.
Seeking to lower the cost burden associated with the use of iridium, we screened a wide range of other common photocatalysts.24 All ruthenium complexes tested under the standard reaction conditions returned only starting material (Table 3, entries 1–2). These photocatalysts would likely operate via reductive quenching of the excited state by NMM and their consistent failure to promote the reaction may indicate a general problem when applying reductive quenching to this particular system. More promisingly, 5 mol % of the organic photocatalyst Eosin Y provided a 22% yield of 2 after 15 h, but with a reduced exo:endo ratio of 3.7:1 (entry 3). Other iridium-based photocatalysts were capable of promoting the reaction but in greatly reduced yield (11–21% yield) and with lower exo:endo selectivity (entries 4–6). The unique efficiency of Ir(ppy)3 with regard to yield and exo:endo selectivity is intriguing, especially given the similar reduction potentials of Ir(Fppy)3 (E1/2IV/III* = −1.48 V and E1/2IV/III = +1.28 V vs SCE)27 to Ir(ppy)3 (E1/2IV/III* = −1.73 V and E1/2IV/III = +0.77 vs SCE).28
Table 3. Variation of Photocatalyst.
| entry | photocataylst | yield of 2a | yield of 1a | exo:endo ratioa |
|---|---|---|---|---|
| 1b | [Ru(bpy)3]Cl2 | <5 | 86 | N/A |
| 2b | [Ru(bpz)3](PF6)2 | <5 | 86 | N/A |
| 3c | Eosin Y (5 mol %) | 22 | 48 | 3.7:1 |
| 4 | Ir(Fppy)3 | 21 | 48 | 3.6:1 |
| 5 | [Ir(py)2(dtbbpy)]PF6 | 11 | 71 | 2.3:1 |
| 6 | [Ir{(dF(CF3)ppy)2(dtbbpy)}]PF6 | 17 | 58 | 2.9:1 |
Reactions performed at a 0.231 mmol scale, at 5 °C with 1.0 mol % photocatalyst, 1.0 equiv of base, 20 equiv of NMM for 8 h unless otherwise stated. Calculated by UPLC (PDA).
Reaction time of 10 h.
Reaction time of 15 h.
Screening a range of solvents typically employed in visible light photocatalysis revealed DMF as comparable to DMA at room temperature (53%, 5.3:1 exo:endo, 16 h). DMSO provided an increased exo:endo ratio but significantly lower conversion and yield (25% 1, 38% 2, 6.5:1 exo:endo, 16 h). Increasing the reaction time to 24 h provided 50% yield of 2, with 13% 1 remaining, but further increase in reaction time did not improve this yield. MeCN did not efficiently solubilize 1 at 5 °C and at 20 °C provided only 12% of the desired product 2. Concentration and analysis of the MeCN reaction mixture revealed the presence of NMO,29 the morpholine source in the corresponding VO(acac)2 chemistry. A subsequent control reaction showed that NMO is not an effective starting material in this process. Interestingly the use of DMPU gave 45% of the solvent addition products 10 and 11. Excluding the NMM from the reaction provided clean conversion to a 1:5 ratio of exo:endo with a combined 53% yield of the inseparable products (Scheme 5). Presumably this process occurs via C–H abstraction of the solvent from the HO2• radical, generated following oxygen quenching of the Ir(ppy)3 excited state and protonation, and could provide a method for heterocyclic functionalization in its own right.30
Scheme 5. Use of DMPU as the Solvent in the Absence of NMM.

Given the high exo:endo ratio observed for the NMO/VO(acac)2 system run in EtOH, we next screened a range of polar protic solvent additives.24 While the addition of EtOH or MeOH had limited effect on both the yield and exo:endo ratio, the use of water (10:1 DMA:H2O) gave considerably improved exo:endo selectivity (12:1) at the expense of reaction time, requiring 24 h to achieve 55% yield with ∼10% 1 remaining. The increased reaction time is possibly due to the lower solubility of Ir(ppy)3 in the solvent mixture, whereas the cause of increased in exo:endo remains to be elucidated.
Further optimization of the DMA:H2O conditions by reducing the reaction temperature to 0 °C and using only 0.5 mol % of Ir(ppy)3 resulted in a precipitation of 2 during the course of the reaction. While this served to both ease isolation and prevent the product from further reaction, premature precipitation at moderate levels of conversion effectively blocked light penetration and stalled the reaction. The solids could be filtered and the filtrate further progressed; however, this method proved impractical and gave material with reduced purity (80–90% a/a UPLC) and yield (45–49%). Attempts to increase the scale of the reaction while employing these conditions, again proved problematic. The scale of the reaction was increased by a factor of 4 (300 mg, 0.92 mmol 1) and the surface area by two, in an effort to mitigate the previously observed rate decrease upon scale up. At this scale the premature precipitation of 2 was a more significant issue, and the reaction time increased to 72 h. Even after this duration only a 37% isolated yield of 2 was obtained in 80% purity. Returning to the standard scale, and diluting the reaction to 0.09 M retained 2 in solution for an increased duration, allowing greater conversion (approximately 75% of 2, 12:1 exo:endo). Under the fully optimized process, significant quantities of 2 precipitated after 32 h. Addition of a solution of 3:1 H2O:EtOH followed by filtration, provided 56% isolated yield with a 90% purity (a/a UPLC) (Scheme 6). The purity of this material could be further improved to >98% with simple extraction into a solution of 1 N HCl and reisolation via basification to pH 12.
Scheme 6. Optimized Reaction Conditions.

The final optimized reaction is superior to the initial first generation Minisci route with regard to selectivity for 2, step efficiency, and yield. Furthermore, replacement of AgNO3 and TFA with 0.5 mol % Ir(ppy)3 is an attractive prospect from a waste management perspective, although the limited scalability and cost concerns surrounding the use of iridium represent significant issues to be addressed. Ultimately, any procedure on scale employing photocatalysis would likely need to be conducted via continuous processing; this offers an exciting opportunity to address these current limitations and research in this direction is underway.
Reaction Generality
Application to the Synthesis of Analogues of 2
Despite the challenges that were encountered in attempts to scale the methodology, the demonstration of efficiency and selectivity prompted us to explore the reaction as a tool for discovery chemistry. Both the previous routes are not ideal for the synthesis of analogues, limited by high step count or the availability of the requisite amine-N-oxide. Testing a range of simple, commercially available tertiary amines we were rapidly able to build a small collection of imidazopyridizine analogues that would be difficult to prepare by other means (Scheme 7). The isolated yields were moderate (20–56%), yet synthetically useful, and the scope of the tertiary amine component encompassed aliphatic, benzylic, and aromatic N-methyl tertiary amines. In certain cases (such as 12) the reaction appeared to stall and increasing both the catalyst loading and the equivalents of amine had limited effect. Radical addition was selective through the N-methyl group when employing N,N-dimethylglycine methyl ester or N,N-dimethybenzylamine, possibly due to the increased nucleophilicity of a primary radical when compared to the secondary radical.31
Scheme 7. Scope of the Amine Component.

Tetramethylethylenediamine (TMEDA) proved a particularly efficient amine in terms of both reaction time and regioisomeric ratio (r.r) (14, 7 h, >10:1 r.r). Postulating that this may be due to an efficient intramolecular deprotonation of the amine radical cation through a 5-membered transition state, we tested tetramethylhexanediamine. This amine proceeded with an equally high regioisomeric ratio (r.r > 10:1) but with decreased yield and increased reaction time (15, 18 h, 24%) suggesting the improved selectivity is more likely the kinetic effect of an increased number of N-methyl groups. In the case of N,N-dimethylethylamine 1.0 mol % Ir(ppy)3 was employed to increase the reaction rate to compete productively with evaporation of the amine (bp 36–38 °C), a problem not encountered with the other amines. The configurational stability of a stereogenic center alpha to nitrogen was also tested under the reaction conditions, with minimal erosion of enantiomeric ratio (e.r) observed (21). This implies that radical formation at this position is disfavored or the resultant radical cannot efficiently re-enter the catalytic cycle via H atom abstraction. N-methylpyrrolidine, N-ethylmorpholine and triethylamine failed to give any discernible product, while 1-methypiperizine gave a complex mixture, presumably due to the presence of a secondary amine. Intriguingly, given their reactivity in related systems employing electron deficient alkenes or dicyanobenzene, diphenylmethylamine and N-phenylmorpholine returned only starting material.8f,8h
Routes to LY2784544 via Alternative Heterocyclic Cores
Attempts were then made to assess the viability of the reaction to a limited selection of other imidazopyridazines that are potential intermediates in the synthesis of LY2784544. Imidazopyridazine 22 (Scheme 8) with an ethyl ester at C-3 has been extensively studied as an alternative precursor for the NMO/VO(acac)2 chemistry, although it showed initially poor exo:endo selectivity (3:1) when compared to 1 (not shown).32 Similarly, poor exo:endo selectivity was observed in this visible light-mediated system giving a 1.1:1 exo:endo ratio and only moderate isolated yield (36% 22, 19% 24, not shown). Attempting to improve the exo:endo ratio via the use of the 10:1 DMA:H2O solvent conditions employed previously led to no improvement in selectivity (Scheme 8). Interestingly under these conditions the formation of methylated imidazopyridazine 25, analogous to the formal methylation product 7, could also be observed (Scheme 8).
Scheme 8. Variation of the Heterocyclic Core.

Furthermore, while no reaction was observed on the parent heterocyclic core 28 without substitution at the C-3 position, the benzylic imidazopyridiazine 26 gave a low isolated 14% yield of the exo product 27. The next step in the synthesis of LY2784544 after 2 is the carbonyl reduction giving 27, a transformation that required significant investigation.33 The formation of the key benzylic morpholine unit from 26 would therefore offer the attractive prospect of eliminating the carbonyl reduction sequence. Additionally the reactivity of 26, albeit moderate, suggested the intriguing possibility of extending this reaction to less activated heterocycles.
To this end, a selection of pyridazines that have previously been shown to undergo Minisci-type radical addition of N-protected amino acid derived radicals were initially tested.34 Unfortunately, no significant addition products could be detected other than in the case of 3,6-dichloropyridazine 29, where a mixture of products consistent with radical addition were detected but proved difficult to isolate.24 Other simple heterocyclic scaffolds representative of common fragments found in pharmaceutical and agrochemical compounds were subsequently investigated (Scheme 8). In all cases the majority of the starting material was recovered and no significant formation of radical addition products could be detected. The NMO/VO(acac)2 chemistry also displays limited substrate scope for the heterocyclic portion, raising questions over the seemingly unique reactivity of the substituted imidazopyridazine and the mechanistic pathways of both these reactions. While the current methodology appears limited with regards to the generality of the heterocyclic portion, the ease and simplicity of the reaction set up, combined with the availability of N-methyl tertiary amine coupling partners renders this an attractive reaction to test on existing compound libraries.
Mechanistic Discussion
Given the demonstrated sensitivity of this reaction to a range of factors such as oxygen, light source, base, and photocatalyst, it is a strong possibility that multiple reaction mechanisms may be operational. One simplistic mechanistic pathway is depicted in Figure 2 whereby the Ir(ppy)3 undergoes initial photoexcitation to a long-lived excited state that can undergo bimolecular quenching with oxygen to generate Ir(IV) and the O2– radical. The resultant Ir(IV) (E1/2IV/III = +0.77 vs SCE) may then oxidize NMM35 to the radical cation and return to the ground state. The primary α-amino radical 30 is then generated via deprotonation of the radical cation before addition to imidazopyridazine 1 and rearomatization to yield benzylic morpholine 2.36 Stern–Volmer analysis,37 showed that imidazopyridazine 1 is a quencher of the excited state Ir(ppy)3 in the presence of oxygen,38 and therefore cannot exclude mechanisms proceeding from this initial event.39 We probed the possibility of radical propagation contributing to the mechanistic complexity by conducting a lights-on-lights-off experiment.24 Minimal reaction was observed during the periods of dark suggesting a long-lived radical propagation is not an major pathway under these conditions.40 Additionally the exo:endo selectivity observed is opposed to that expected if the intermediate α-amino radical was formed via C–H abstraction.
Figure 2.
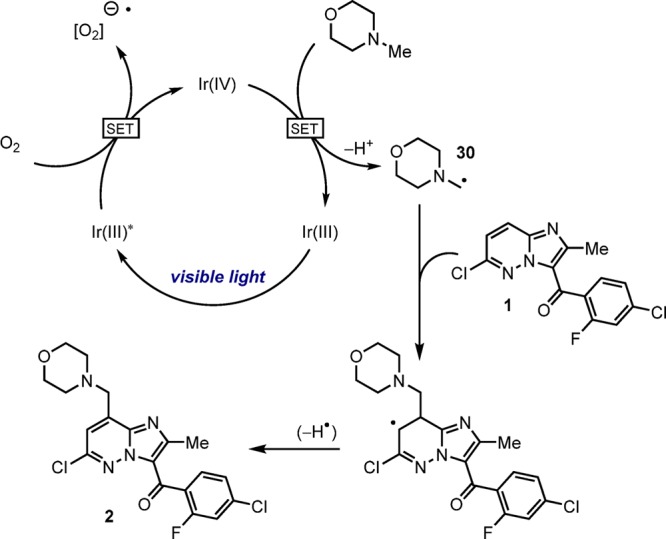
Possible mechanistic pathway.
A potential mechanism, which we deem unlikely, is the generation of an iminium species and Friedel–Crafts type addition of the imidazopyridazine core. The use of preformed iminium species 31 in the absence of photocatalyst gave no observable coupling product. However, the use of iminium 31 under photocatalytic conditions in the absence of oxygen provided 2 with high selectivity, albeit in low yield (13%, >20:1 exo:endo, Scheme 9). This may demonstrate the possibility of a redox neutral process by which reduction of the iminium by the excited state of Ir(ppy)3 provides the α-amino radical 30. Alternatively direct reduction of the imidazopyridazine followed by addition to the iminium provides another mechanistic possibility that cannot be ruled out.
Scheme 9. Use of the Preformed Iminium 31.

Conclusion
In conclusion, we have successfully developed a visible light-mediated photochemical transformation to address a challenging bond construction en route to the JAK2 inhibitor LY2784544. The introduction of this benzylic morpholine unit occurs in moderate yield, with good selectivity and product purity and is competitive with previously reported routes in these respects. Importantly, NMM is utilized directly, which has considerable benefit when compared to the NMM surrogates employed in the first and second generation syntheses. Furthermore, the methodology was amenable to a range of simple, commercially available tertiary amines and delivered a range of analogues in synthetically useful yields. While the developed batch conditions are not currently scalable, the potential application to continuous processing is an attractive method to allow the further development of this step, and efforts in this direction are ongoing. During the course of this investigation a number of interesting photoredox transformations have been observed, such as formal methylation via C–N bond cleavage, functionalization of C–H bonds alpha to amides, and iminium ion reduction. Overall, we believe that this initial demonstration warrants the wider consideration of visible light-mediated photocatalysis as a tool in process development as well as the potential application of this specific transformation to the diversification of pharmaceutical and agrochemical scaffolds.
Experimental Section
General Methods
Chemicals and solvents (DMA, DMPU) were used as received. 1 was prepared according to the literature.41 Ir(ppy)3 was either purchased from Sigma or prepared via the known methods. Solvents (MeCN, DMF, DMSO) were obtained from a solvent purification system. Reactions that were monitored by TLC were visualized by a dual short wave/long wave UV lamp and stained with an ethanolic solution of potassium permanganate. Column flash chromatography was performed using 230–400 mesh silica gel or via automated column chromatography. Yields refer to chromatographically and spectroscopically pure compounds, unless otherwise noted. LED lights were purchased from Creative Lighting Solutions (http://www.creativelightings.com). Jacketed beakers were obtained from Chemglass and connected to a recirculating heater chiller with Sil 180 silicone oil as the coolant.
1H, 13C and 19F NMR spectra were recorded using an internal deuterium lock on 400, 500, or a 700 MHz spectrometer. All signals are reported in ppm with the internal reference of the specified solvent. J couplings are reported in Hz. Data are presented as follows: integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad, app = apparent, dd = doublet of doublet, dt = doublet of triplet, etc.) and coupling constant (J/Hz).
Infrared spectra were recorded on a FT-IR fitted with an ATR accessory. Absorptions are given in wavenumbers (cm–1). High resolution mass spectra were obtained on a HRMS-TOF spectrometer. Photocatalyst quenching was conducted on a fluorimeter and the values represent an average of 3 samples. UPLC analysis was conducted on a UPLC with a BEH C18 column (1.7 μm 2.1 × 50 mm). Chiral HPLC analysis was conducted on a HPLC with Chiral Pak OD-H column (4.6 × 250 mm) at 254 nm at 22 °C.
Initial Reaction (Scheme 3)
To a 25 mL round bottom flask (RBF) open to air was added 1 (125 mg, 0.386 mmol, 1.0 equiv), Ir(ppy)3 (5.00 mg, 0.008 mml, 0.02 equiv), sodium acetate (32.0 mg, 390 mmol, 1.0 equiv), DMA (2.5 mL) and N-methylmorpholine (0.850 mL, 7.71 mmol, 20 equiv) and the yellow heterogeneous solution was irradiated with 2 × 4 W LED strips placed in a circular loop around the flask. Aluminum foil was placed over the lights around the flask to contain the light (see Figure S1, Supporting Information). After 1 h the internal temperature of the flask had stabilized at 35 °C. The reaction was monitored via HPLC.
(4-Chloro-2-fluorophenyl)(2-methylimidazo[1,2-b]pyridazin-3-yl)methanone 5
νmax (ATR)/cm–1 3048, 2361, 1603, 1569, 1502, 1479, 1411, 1318, 1268, 1197, 914, 887, 806, 761 and 611; 1H NMR (500 MHz, CDCl3) δH 8.26 (1H, dd, J 4.5, 1.7), 7.95 (1 H, dd, J 9.1, 1.7), 7.63 (1H, dd, J 8.2, 7.5), 7.29 (1H, dd, J 8.4, 2.0), 7.19 (1H, dd, J 9.1, 4.5), 7.10 (1H, dd, J 9.9, 1.9) and 2.68 (3 H, s); 13C NMR (126 MHz, CDCl3) δc 180.3, 160.4 (d, J 255.4), 153.0, 143.0, 140.3, 138.9 (d, J 10.4), 131.4 (d, J 3.7), 127.2 (d, J 14.0), 125.3 (d, J 3.5), 125.2, 119.9, 116.6 (d, J 25.5) and 16.45; 19F NMR (471 MHz, CDCl3) δF −112.5 (t, J 8.5); m/z (ESI+) HRMS [M + H] C14H10ClFN3O+ found 290.0488, calcd. 290.0491.
6-Chloro-2,8-dimethylimidazo[1,2-b]pyridazin-3-yl)(4-chloro-2-fluorophenyl)methanone 6
νmax (ATR)/cm–1 2927, 1630, 1601, 1554, 1504, 1485, 1396, 1286, 1217, 1074, 919, 822, and 623; 1H NMR (500 MHz, CDCl3) δH 7.61 (1H, t, J 7.9), 7.28 (1H, dd, J 8.3, 1.8), 7.11 (1H, dd, J 10.0, 1.8), 7.01 (1H, d, J 1.0), 2.68 (3H, s) and 2.66 (1H, d, J 1.0); 13C NMR (101 MHz, CDCl3) δc 180.3, 160.5 (d, J 255), 152.1, 146.7, 139.7, 139.1 (d, J 10.5), 138.6, 131.41 (d, J 3.7), 126.7 (d, J 13.7), 125.8, 125.3 (d, J 3.2), 120.9, 116.5 (d, J 25.7), 16.8 and 16.40; 19F NMR (471 MHz, CDCl3) δF −112.4 (t, J 8.8); m/z (ESI+) HRMS [M + H] C15H10Cl2FN3O+ found 338.0263, calcd. 338.0259.
N-((6-Chloro-3-(4-chloro-2-fluorobenzoyl)-2-methylimidazo[1,2-b]pyridazin-8-yl)methyl)-N-methylacetamide 7
νmax (ATR)/cm–1 2925, 1649, 1630, 1601, 1554, 1504, 1396, 1286, 1074, 919, 822 and 617; 1H NMR (500 MHz, CDCl3) δH (as a mixture of rotamers confirmed by VT NMR) 7.61–7.58 (1H, m), 7.30–7.26 (1H, m), 7.21–7.09 (1H, m), 7.01 (1H, s, major rotamer), 6.92 (1H, s, minor rotamer), 4.95 (2H, s), 3.18 (3H, s, major rotamer), 3.05 (3H, s, minor rotamer), 2.67 (3H, s), 2.21 (3H, s, major rotamer) and 2.14 (3H, s, minor rotamer); 13C NMR (126 MHz, CDCl3) δc data for major rotamer 180.0, 171.4, 160.4 (1d, J 255.4), 152.2, 147.0, 139.1 (d, J 10.7), 138.2, 137.2, 131.3 (1d, J 3.6), 126.5 (d, J 13.6), 125.7, 125.2 (d, J 3.4), 119.1, 116.4 (d, J 25.6), 46.2, 37.4, 21.6, and 16.2; 19F NMR (471 MHz, CDCl3) δF −112.4 (t, J 8.6); m/z (ESI+) HRMS [M + H] C18H15Cl2FN4O2+ found 409.0625, calcd. 409.0634.
Degradation Studies of 2
To a 10 mL RBF was added 2 (97.8 mg, 0.231 mmol, 1.0 equiv), sodium acetate (19.0 mg, 0.231 mmol, 1.0 equiv), Ir(ppy)3 (1.50 mg, 2.31 μmol, 0.01 equiv), DMA (1.50 mL) and N-methylmorpholine (510 μL, 4.63 mmol, 20 equiv) to give a heterogeneous yellow solution. The flask was placed in a 25 mL jacketed beaker with iPrOH as the internal coolant connected to a recirculating heater chiller set to 5 °C. The flask was stirred for 2 min open to air before switching on 2 × 4W LED strips and stirring for 8 h. UPLC analysis (Figure S13) showed 27% a/a of starting material 1, 33% a/a of the reduced product 9 and also included the C6 carbon-chlorine bond reduction [(4-chloro-2-fluorophenyl)(2-methyl-8-(morpholinomethyl)imidazo[1,2-b]pyridazin-3-yl)methanone] 10% a/a and the C–N bond fragmentation product 6 9% a/a. While 9 could not be efficiently isolated via chromatography, analysis of the 1H NMR spectrum of the crude reaction mixture following work up also confirmed its presence as the major compound among a complex mixture.
To a 10 mL RBF was added 2 (97.8 mg, 0.231 mmol, 1.0 equiv), sodium acetate (19.0 mg, 0.231 mmol, 1.0 equiv), Ir(ppy)3 (1.50 mg, 2.31 μmol, 0.01 equiv), DMA (1.50 mL), N-methylmorpholine (510 μL, 4.63 mmol, 20 equiv) and formic acid (44.0 μL, 1.16 mmol, 5 equiv) to give a heterogeneous bright yellow/green solution. The flask was placed in a 25 mL jacketed beaker with iPrOH as the internal coolant connected to a recirculating heater chiller set to 5 °C. The flask was stirred for 2 min open to air before switching on 2 × 4W LED strips and stirring for 22 h. UPLC analysis (Figure S14) showed 34% a/a of starting material 2 and 42% a/a of the carbon–nitrogen reduced product 6. The reaction was worked up via the addition of EtOAC (50 mL) followed by washing with water (50 mL) and 5% LiCl (2 × 25 mL). The organic phase was then dried (Na2SO4) and concentrated in vacuo to give a crude product that was purified via automated column chromatography (12 g silica column, 70:10:10 Hexanes:EtOAc:CH2Cl2) to give 6 (16.0 mg, 20% yield) as a white solid with spectroscopic data in accordance with that reported.
Optimized Synthesis of 2
To four identical 10 mL RBFs was added 1 (75.0 mg, 0.231 mmol, 1.0 equiv), sodium acetate (19.0 mg, 0.231 mmol, 1.0 equiv), Ir(ppy)3 (0.8 mg, 1.16 μmol, 0.005 equiv), DMA (1.50 mL) and N-methylmorpholine (510 μL, 4.63 mmol, 20 equiv) to give a heterogeneous yellow solution. The flask was placed in a 25 mL jacketed beaker with iPrOH as the internal coolant connected to a recirculating heater chiller set to 0 °C. The flask was stirred for 2 min open to air before switching on a 1 W LED strips. The reaction was stirred for 32 h before (3:1 water:EtOH, 2.0 mL) was added and the reaction was stirred for a further 30 min. The solids were filtered, washed with water then dissolved in CH2Cl2 and dried with Na2SO4. The result material was dried overnight under a vacuum (<1 mbar) to give 2 (52–56 mg, 56% average yield). A portion of this solid (50 mg) was dissolved in EtAOc (30 mL) and extracted twice with 1 N HCl (25 mL). The aqueous HCl was basified to pH 12 with NaOH and extracted twice with CH2Cl2, dried (Na2SO4), filtered and concentrated to give 2 (29 mg, 58% recovery, >98% a/a), in accordance with the literature.321H NMR (500 MHz, CDCl3) δH 7.62 (1H, t, J 7.9), 7.38 (1H, td, J 0.7, 0.3), 7.30 (1H, dd, J 8.3), 7.12 (1H, dd, J 10.0, 1.9), 4.00 (2H, s), 3.79 (4H, br s), 2.68 (3 H, s) and 2.61 (3H, s).
General Procedure for the Synthesis of 10, 11, 12–21 and 24, 25 and 27
Because of the limited scalability of the reaction employing NMM, reactions employing other amines were conducted at 0.231 mmol scale in triplicate and combined prior to work up and isolation.
To a 10 mL RBF was added 1 (75.0 mg, 0.231 mmol, 1.0 equiv), sodium acetate (19.0 mg, 0.231 mmol, 1.0 equiv), Ir(ppy)3 (0.8 mg, 1.16 μmol, 0.005 equiv), DMA (1.50 mL) and amine (4.63 mmol, 20 equiv) to give a heterogeneous yellow solution. The flask was placed in a 25 mL jacketed beaker with iPrOH as the internal coolant connected to a recirculating heater chiller set to 5 °C. The flask was stirred for 2 min open to air before switching on a 1W LED strip. The reaction was monitored via UPLC and after a given time worked up with the addition of EtOAC (50 mL) followed by washing with water (2 × 50 mL) and 5% LiCl (2 × 25 mL). The organic phase was then dried (Na2SO4) and concentrated in vacuo to give a crude product that was purified via automated column chromatography under the stated conditions.
1-((6-Chloro-3-(4-chloro-2-fluorobenzoyl)-2-methylimidazo[1,2-b]pyridazin-8-yl)methyl)-3-methyltetrahydropyrimidin-2(1H)-one 10 and 4-(6-Chloro-3-(4-chloro-2-fluorobenzoyl)-2-methylimidazo[1,2-b]pyridazin-8-yl)-1,3-dimethyltetrahydropyrimidin-2(1H)-one 11
Following the general procedure with the absence of amine; three identical reactions using 2 (75.0 mg, 0.231 mmol, 1.0 equiv), sodium acetate (19.0 mg, 0.231 mmol, 1.0 equiv), Ir(ppy)3 (1.52 mg, 2.31 μmol, 0.01 equiv), DMPU (1.50 mL) for 41 h at 5 °C were combined and worked up together. The resulting crude material (1:5 exo:endo) was purified via automated column chromatography (12 g silica column, 50:50 EtOAc:Hexanes to EtOAc followed by a second column 10:90 Acetone:CH2Cl2 to 20:80 Acetone:CH2Cl2) to give 10 and 11 (164 mg, 53% yield, 1:5 exo:endo) as white solid.
Data for 11-endo
1H NMR (700 MHz, CDCl3) δH 7.62 (1H, t, J 7.9), 7.30 (1H, dd, J 8.2, 1.7), 7.11 (1H, dd, J 10.1, 1.6), 6.97 (1H, s), 5.19 (1H, d, J 3.9), 3.14–3.07 (2H, m), 2.99 (3H, s), 2.98 (3H, s), 2.66 (3H, s), 2.57–2.53 (1H, m) and 2.38 (1H, dd, J 13.8, 2.9); 13C NMR (176 MHz, CDCl3) δc 180.1 160.5 (d, J 255), 152.1, 147.0, 140.0, 139.2, (d, J 10.6), 138.8, 131.4 (d, J 3.6), 126.7 (d, J 14.1), 125.5, 125.3 (d, J 3.2), 120.0, 116.5 (d, J 25.6), 65.4, 64.6, 55.4, 54.8, 27.3, 23.7, and 16.3. 19F NMR (377 MHz, CDCl3) δF −112.4 (t, J 8.8); m/z (ESI+) HRMS [M + H] C20H19Cl2FN5O2+ found 450.0894, calcd. 450.0894.
(4-Chloro-2-fluorophenyl)(6-chloro-2-methyl-8-(piperidin-1-ylmethyl)imidazo[1,2-b]pyridazin-3-yl)methanone 12
Following the general procedure three identical reactions using 1 (75.0 mg, 0.231 mmol, 1.0 equiv), sodium acetate (19.0 mg, 0.231 mmol, 1.0 equiv), Ir(ppy)3 (0.8 mg, 1.16 μmol, 0.005 equiv), DMA (1.50 mL) and N-methylpiperidine (562 μL, 4.63 mmol, 20 equiv) for 40 h at 5 °C were combined and worked up together. Prior to work up 12 was present at ∼45% a/a (UPLC PDA) with >10:1 exo:endo (UPLC MS). The resulting crude material was purified via automated column chromatography (24 g silica column, 80:20 hexanes:EtOAc 1% Et3N) to give 12 (87 mg, 30% yield) as a white solid.
νmax (ATR)/cm–1 2938, 2360, 2336, 1636, 1603, 1417, 1277, 1074, 918, 856 and 622; 1H NMR (500 MHz, CDCl3) δH 7.61 (1H, t, J 7.9), 7.39 (1H, s), 7.30 (1H, dd, J 8.3, 1.3), 7.12 (1H, dd, J 10.0, 1.7), 3.94 (2H, s), 2.68 (4H, s), 2.53 (4H, br), 1.65 (4H, app quintet, J 5.6) and 1.52–1.50 (2H, m); 13C NMR (176 MHz, CDCl3) δc 180.5, 160.3 (d, J 255), 152.0, 147.3, 140.0, 138.9 (d, J 10.6), 138.5, 131.2 (d, J 3.5), 126.6 (d, J 14.1), 125.5, 125.1 (d, J 3.2), 119.1, 116.3 (d, J 25.7), 56.4, 55.0, 26.0, 23.9, and 16.3. 19F NMR (471 MHz, CDCl3) δF −112.4 (t, J 8.5); m/z (ESI+) HRMS [M + H] C20H20Cl2FN4O+ found 421.0992, calcd. 421.0998.
Methyl-N-((6-chloro-3-(4-chloro-2-fluorobenzoyl)-2-methylimidazo[1,2-b]pyridazin-8-yl)methyl)-N-methylglycinate 13
Following the general procedure three identical reactions using 1 (75.0 mg, 0.231 mmol, 1.0 equiv), sodium acetate (19.0 mg, 0.231 mmol, 1.0 equiv), Ir(ppy)3 (0.8 mg, 1.16 μmol, 0.005 equiv), DMA (1.50 mL) and N,N-dimethylglycine methylester (557 μL, 4.63 mmol, 20 equiv) for 28 h at 5 °C were combined and worked up together. Prior to work up 13 was present at ∼60% a/a (UPLC PDA) with 10:1 exo:endo (UPLC MS). The resulting crude material was purified via automated column chromatography (24 g silica column, 70:30 hexanes:EtOAc 1% Et3N) to give 13 (172 mg, 56% yield) as a cream solid.
νmax (ATR)/cm–1 2948, 2359, 2335, 1741, 1634, 1604, 1505, 1479, 1415, 1275, 1074, 917, 872, 817, 736, and 622; 1H NMR (500 MHz, CDCl3) δH 7.62 (1H, t, J 7.9), 7.45 (1H, t, J 1.1), 7.30 (1H, dd, J 8.3, 1.9), 7.12 (1H, dd, J 10.0, 1.9), 4.19 (2H, d J 1.1), 3.74 (3H, s) 3.45 (2H, s) 2.67 (3H, s) and 2.51 (3H, s); 13C NMR (176 MHz, CDCl3) δc 180.0, 171.0, 160.3 (d, J 255), 152.1, 147.3, 139.0 (d, J 10.6), 138.9, 138.3, 131.2 (d, J 3.5), 126.6 (d, J 14.1), 125.5, 125.1(d, J 3.2), 119.3, 116.3 (d, J 25.7), 58.2, 54.5, 51.7, 42.7, and 16.3; 19F NMR (471 MHz, CDCl3) δF −112.4 (t, J 8.9); m/z (ESI+) HRMS [M + H] C19H18Cl2FN4O3+ found 439.0736, calcd. 439.0740.
(4-Chloro-2-fluorophenyl)(6-chloro-8-(((2-(dimethylamino)ethyl)(methyl)amino)methyl)-2-methylimidazo[1,2-b]pyridazin-3-yl)methanone 14
Following the general procedure two identical reactions using 1 (75.0 mg, 0.231 mmol, 1.0 equiv), sodium acetate (19.0 mg, 0.231 mmol, 1.0 equiv), Ir(ppy)3 (0.8 mg, 1.16 μmol, 0.005 equiv), DMA (1.50 mL) and tetramethylenediamine (349 μL, 2.31 mmol, 10 equiv) for 7 h at 5 °C were combined and worked up together. Prior to work up 14 was present at ∼70% a/a (UPLC PDA) with >10:1 exo:endo (UPLC MS). The resulting crude material was purified via automated column chromatography (24 g silica column, 10:90 hexanes:EtOAc 1% Et3N) to give 14 (88 mg, 44% yield) as a light yellow solid.
νmax (ATR)/cm–1 2767, 2361, 1622, 1601, 1558, 1507, 1414, 1289, 1086, 919, 879, 816, 761, and 596; 1H NMR (700 MHz, CDCl3) δH 7.62 (1H, t), 7.48 (1H, s), 7.30 (1H, dd, J 8.2, 1.8), 7.12 (1H, dd, J 9.9), 4.03 (2H, s), 2.68 (3H, s), 2.63 (2H, t, J 6.8,), 2.47 (2H, t, J 6.8), 2.38 (3H, s) and 2.25 (6H, s); 13C NMR (176 MHz, CDCl3) δc 180.1, 160.3 (d, J 255), 152.1, 147.4, 140.1, 139.0 (d, J 10.4), 138.5, 131.2 (d, J 3.5), 126.6 (d, J 14.2), 125.6, 125.1 (d, J 3.2), 119.4, 116.4 (d, J 25.6), 57.6, 56.0, 55.7, 46.0, 43.2, and 16.3; 19F NMR (471 MHz, CDCl3) δF −112.4 (t, J 8.7); m/z (ESI+) HRMS [M + H] C20H23Cl2FN5O+ found 438.1259, calcd. 438.1264.
(4-Chloro-2-fluorophenyl)(6-chloro-8-(((6-(dimethylamino)hexyl)(methyl)amino)methyl)-2-methylimidazo[1,2-b]pyridazin-3-yl)methanone 15
Following the general procedure three identical reactions using 1 (75.0 mg, 0.231 mmol, 1.0 equiv), sodium acetate (19.0 mg, 0.231 mmol, 1.0 equiv), Ir(ppy)3 (0.8 mg, 1.16 μmol, 0.005 equiv), DMA (1.50 mL) and tetramethyl-1,6-hexanediamine (991 μL, 4.63 mmol, 20 equiv) for 20 h at 5 °C were combined and worked up together. Prior to work up 15 was present at ∼55% a/a (UPLC PDA) with >10:1 exo:endo (UPLC MS). Excess tetramethyl-1,6-hexanediamine was removed via Kugelrohr distillation at 50–60 °C and <1 mbar. The resulting crude material was purified via automated column chromatography (24 g silica column, EtOAc 1% Et3N) to give 15 (82 mg, 24% yield) as a light brown solid.
νmax (ATR)/cm–1 2923, 2775, 1636, 1610, 1506, 1414, 1214, 1074, 920 and 818; 1H NMR (700 MHz, CDCl3) δH 7.60 (1H, t, J 7.9), 7.38 (1H, s), 7.28 (1H, dd, J 8.3, 1.87.11 (1H, dd, J 9.9), 3.95 (2H, s), 2.49 (2H, t, J 7.4) 2.29 (3H, s), 2.22 (2H, t, J 7.4), 2.21 (6H, s), 1.53 (2H, dt, J 14.7, 7.3), 1.45 (2H, dt, J 14.7, 7.3) and 1.36–1.29 (4H, m); 13C NMR (176 MHz, CDCl3) δc 180.2, 160.4 (d, J 255), 152.1, 147.4, 140.5, 138.9 (d, J 10.3), 138.5, 131.2 (d, J 3.5), 126.6 (d, J 14.0), 125.5, 125.1 (d, J 3.3), 119.2, 116.3 (d, J 25.4), 59.8, 58.1, 55.3, 45.5, 42.8, 27.7, 27.4, 27.4, 27.3 and 16.3; 19F NMR (471 MHz, CDCl3) δF −112.4 (t, J 8.9); m/z (ESI+) HRMS [M + H] C24H31Cl2FN5O+ found 494.1890, calcd. 494.1884.
(4-Chloro-2-fluorophenyl)(6-chloro-8-((ethyl(methyl)amino)methyl)-2-methylimidazo[1,2-b]pyridazin-3-yl)methanone 16
Following the general procedure three identical reactions using 1 (75.0 mg, 0.231 mmol, 1.0 equiv), sodium acetate (19.0 mg, 0.231 mmol, 1.0 equiv), Ir(ppy)3 (1.52 mg, 2.31 μmol, 0.01 equiv), DMA (1.50 mL) and Dimethylethylamine (502 μL, 4.63 mmol, 20 equiv) for 8 h at 5 °C were combined and worked up together. Prior to work up 16 was present at ∼63% a/a (UPLC PDA) with ∼6–7:1 exo:endo (UPLC MS). The resulting crude material was purified via automated column chromatography (24 g silica column, 80:20 Hexanes:EtOAc 1% Et3N) to give 16 (145 mg, 53% yield) as a cream solid.
νmax (ATR)/cm–1 2804, 1634, 1603, 1507, 1414, 1278, 1213, 1077, 1039, 919, 859, and 734; 1H NMR (500 MHz, CDCl3) δH 7.62 (1H, t, J 7.9), 7.40 (1H, t, J 1.3), 7.30 (1H, dd, J 8.3, 1.9), 7.12 (1H, dd, J 9.9, 1.9), 3.98 (2H, d, J 1.3), 2.68 (3H, s), 2.59 (2H, q, J 7.1), 2.33 (3H, s) and 1.14 (3H, t, J 7.1); 13C NMR (176 MHz, CDCl3) δc 180.2, 160.3 (d, J 255), 152.1, 147.4, 140.5, 139.0 (d, J 10.4), 138.5, 131.2 (d, J 3.5), 126.6 (d, J 14.0), 125.5, 125.1 (d, J 3.3), 119.3, 116.4 (d, J 25.6), 54.9, 52.0, 42.3, 16.3, and 12.6; 19F NMR (471 MHz, CDCl3) δF −112.4 (t, J 8.5); m/z (ESI+) HRMS [M + H] C18H18Cl2FN4O+ found 395.0840, calcd. 395.0842.
(4-Chloro-2-fluorophenyl)(6-chloro-2-methyl-8-((4-methylpiperazin-1-yl)methyl)imidazo[1,2-b]pyridazin-3-yl)methanone 17
Following the general procedure three identical reactions using 1 (75.0 mg, 0.231 mmol, 1.0 equiv), sodium acetate (19.0 mg, 0.231 mmol, 1.0 equiv), Ir(ppy)3 (0.8 mg, 1.16 μmol, 0.005 equiv), DMA (1.50 mL) and 1,4-Dimethylpiperizine (621 μL, 4.63 mmol, 20 equiv) for 18 h at 5 °C were combined and worked up together. Prior to work up 17 was present at ∼55% a/a (UPLC PDA) with ∼5–6:1 exo:endo (UPLC MS). The resulting crude material was purified via automated column chromatography (12 g silica column, EtOAc 1% Et3N) to give 17 (109 mg, 36% yield) as a light brown solid.
νmax (ATR)/cm–1 2797, 1629, 1603, 1498, 1414, 1271, 1224, 1212, 1075, 1013, 917, 826 and 764; 1H NMR (700 MHz, CDCl3) δH 7.63 (1H, t, J 7.9), 7.36 (1H, s), 7.29 (1H, dd, J 8.3, 1.7), 7.11 (1H, dd, J 9.9, 1.7), 4.00 (2H, d, J 1.0), 2.67 (3H, s) overlapping 2.64 (4H, br s), 2.51 (4H, br s) and 2.32 (3H, s); 13C NMR (176 MHz, CDCl3) δc 180.2, 160.4 (d, J 255), 152.1, 147.3, 139.3, 139.0 (d, J 10.3), 138.4, 131.3 (d, J 3.5), 126.6 (d, J 14.1), 125.6, 125.1 (d, J 3.3), 119.1, 116.4 (d, J 25.6), 55.5, 55.0, 53.4, 46.0 and 16.3; 19F NMR (471 MHz, CDCl3) δF −112.4 (t, J 8.8); m/z (ESI+) HRMS [M + H] C20H21Cl2FN5O+ found 436.1102, calcd. 436.1107.
tert-Butyl 4-((6-chloro-3-(4-chloro-2-fluorobenzoyl)-2-methylimidazo[1,2-b]pyridazin-8-yl)methyl)piperazine-1-carboxylate 18
Following the general procedure three identical reactions using 1 (75.0 mg, 0.231 mmol, 1.0 equiv), sodium acetate (19.0 mg, 0.231 mmol, 1.0 equiv), Ir(ppy)3 (0.8 mg, 1.16 μmol, 0.005 equiv), DMA (1.50 mL) and tBu-4-methylpiperazine-1-carboxylate (695 mg, 3.47 mmol, 15 equiv) for 20 h at 5 °C were combined and worked up together. Prior to work up 18 was present at ∼55% a/a (UPLC PDA) with ∼3–4:1 exo:endo (UPLC PDA). The resulting crude material was purified via automated column chromatography (12 g silica column, 90:10 DCM:EtOAc to 50:50 EtOAc:DCM) to give 18-exo (99 mg, 27% yield) as a light cream solid and 18-endo (56 mg, 15% yield) as a cream solid.
Data for exo-18
νmax (ATR)/cm–1 1694, 1634, 1604, 1418, 1238, 1164, 1128, 1073, 1008, 920, 850, and 831; 1H NMR (400 MHz, CDCl3) δH 7.62 (1H, t, J 7.9), 7.37 (1H, s), 7.30 (1H, dd, J 8.2, 1.9), 7.12 (1H, dd, J 10.0, 1.8), 4.01 (2H, d, J 0.9), 3.51 (4H, t, J 4.8) 2.68 (3H, s), 2.55 (4H, t, J 4.8) and 1.47 (9H, s); 13C NMR (176 MHz, CDCl3) δc 180.2, 160.5 (d, J 255), 154.8, 152.3, 147.4, 139.3 (d, J 10.4), 139.0, 138.5, 131.4 (d, J 3.5), 126.7 (d, J 14.0), 125.8, 125.3 (d, J 3.3), 119.2, 116.5 (d, J 25.5), 80.1, 55.8, 53.4, 20.6, and 16.4; 19F NMR (471 MHz, CDCl3) δF −112.4 (t, J 8.6); m/z (ESI+) HRMS [M + H] C24H26Cl2FN5O3+ found 522.1481, calcd. 522.1469.
Data for endo-18
νmax (ATR)/cm–1 1696, 1636, 1604, 1415, 1270, 1147, 1077, 920, 860, and 728; 1H NMR (500 MHz, CDCl3) δH 7.62 (1H, t, J 7.9), 7.34 (1H, s), 7.31 (1H, dd, J 8.3, 1.9), 7.13 (1H, dd, J 9.9, 1.8), 4.16 (2H, s), 3.96 (1H, dd, J 10.2, 2.8), 3.05 (1H, br s), 2.94 (1H, d, J 11.4), 2.77 (1H, br s), 2.68 (3H, s), 2.39 (1H, td, J 11.9, 3.3), 2.17 (3H, s), 1.63 (1H, br s) and 1.46 (9H, s); 13C NMR (176 MHz, CDCl3) δc 180.2, 160.5 (d, J 255), 152.7 (2 × C), 147.3, 140.6, 139.2 (d, J 10.4), 138.0, 131.4 (d, J 3.5), 126.7 (d, J 13.9), 126.0, 125.3 (d, J 3.2), 119.1, 116.6 (d, J 25.6), 61.2, 54.6, 43.8, 28.5, 28.5, and 16.5; 19F NMR (471 MHz, CDCl3) δF −112.4 (app m); m/z (ESI+) HRMS [M + H] C24H26Cl2FN5O3+ found 522.1479, calcd. 522.1469.
(4-Chloro-2-fluorophenyl)(6-chloro-2-methyl-8-((methyl(phenyl)amino)methyl)imidazo[1,2-b]pyridazin-3-yl)methanone 19
Following the general procedure three identical reactions using 1 (75.0 mg, 0.231 mmol, 1.0 equiv), sodium acetate (19.0 mg, 0.231 mmol, 1.0 equiv), Ir(ppy)3 (0.8 mg, 1.16 μmol, 0.005 equiv), DMA (1.50 mL) and dimethylanaline (441 μL, 3.47 mmol, 15 equiv) for 15 h at 5 °C were combined and worked up together. Prior to work up 19 was present at ∼60% a/a (UPLC PDA). The resulting crude material was purified via automated column chromatography (12 g silica column, 0% EtOAc to 30% EtOAc) to give 19 (123 mg, 40% yield) as a light yellow solid that was contaminated with ∼5% of N-methyl-N-phenylformamide.
νmax (ATR)/cm–1 2888, 2360, 2330, 1632, 1599, 1505, 1417, 1204, 1075, 917, 848, 817, and 748; 1H NMR (700 MHz, CDCl3) δH 7.64 (1H, t, J 7.9), 7.31 (1H, dd, J 8.3, 1.8), 7.27–7.24 (2H, m), 7.13 (1H, dd, J 9.9, 1.7), 7.00 (1H, s), 6.80 (1H, t, J 7.3), 6.71 (2H, d, J 8.2), 4.95 (2H, d, J 0.7), 3.15 (3H, s) and 2.72 (3H, s); 13C NMR (176 MHz, CDCl3) δc 180.3, 160.4 (d, J 255), 152.3, 148.7, 147.4, 139.5, 139.1 (d, J 10.7), 137.7, 131.3 (d, J 3.7), 126.5 (d, J 14.0), 125.9, 125.2 (d, J 3.3), 118.1, 117.9, 116.4 (d, J 25.6), 112.3, 52.4, 39.4, and 16.4 (one C was not be resolved); 19F NMR (377 MHz, CDCl3) δF −112.3 (t, J 8.8); m/z (ESI+) HRMS [M + H] C22H18Cl2FN4O+ found 443.0837, calcd. 443.0836.
(8-((Benzyl(methyl)amino)methyl)-6-chloro-2-methylimidazo[1,2-b]pyridazin-3-yl)(4-chloro-2-fluorophenyl)methanone 20
Following the general procedure three identical reactions using 1 (75.0 mg, 0.231 mmol, 1.0 equiv), sodium acetate (19.0 mg, 0.231 mmol, 1.0 equiv), Ir(ppy)3 (0.8 mg, 1.16 μmol, 0.005 equiv), DMA (1.50 mL) and dimethylbenzylamine (346 μL, 2.31 mmol, 10 equiv) for 15 h at 5 °C were combined and worked up together. Prior to work up 20 was present at ∼70% a/a (UPLC PDA) and in >10:1 exo:endo (UPLC MS). The resulting crude material was purified via automated column chromatography (12 g silica column, 50:50 CH2Cl2:Hexanes then a gradient of EtOAc:Hexanes) to give 20 (171 mg, 54% yield) as a light yellow solid.
νmax (ATR)/cm–1 2794, 2361, 1634, 1604, 1413, 1285, 1076, 919, 732, and 698; 1H NMR (500 MHz, CDCl3) δH 7.61 (1H, t, J 7.9), 7.46 (1H, dt, J, 1.9, 1.3), 7.39–7.37 (2H, m), 7.33 (2H, dd, J 7.0, 1.6), 7.31–7.24 (3H, m), 7.11 (1H, dd, J 9.9, 1.9), 4.00 (2H, s), 3.70 (2H, s), 2.67 (3H, s) and 2.35 (3H, s); 13C NMR (176 MHz, CDCl3) δc 180.1 160.3 (d, J 255), 152.1, 147.4, 140.4, 139.0 (d, J 10.6), 138.5, 131.2 (d, J 3.6), 126.6 (d, J 13.9), 127.5, 126.7, 126.3, 125.6, 125.1 (d, J 3.2), 119.2, 116.3 (d, J 25.6), 62.6, 54.5, 43.2, and 16.4; 19F NMR (377 MHz, CDCl3) δF −112.4 (t, J 8.8); m/z (ESI+) HRMS [M + H] C23H19Cl2FN4O+ found 457.0993, calcd. 457.0993.
(S)-(4-Chloro-2-fluorophenyl)(6-chloro-8-((2-(hydroxymethyl)pyrrolidin-1-yl)methyl)-2-methylimidazo[1,2-b]pyridazin-3-yl)methanone 21
Following the general procedure three identical reactions using 1 (75.0 mg, 0.231 mmol, 1.0 equiv), sodium acetate (19.0 mg, 0.231 mmol, 1.0 equiv), Ir(ppy)3 (0.8 mg, 1.16 μmol, 0.005 equiv), DMA (1.50 mL) and N-methyl-l-prolinol (555 μL, 4.63 mmol, 20 equiv) for 15 h at 5 °C were combined and worked up together. Prior to work up 21 was present at ∼25% a/a (UPLC PDA) and in >10:1 exo:endo (UPLC MS). The resulting crude material was purified via automated column chromatography (12 g silica column, 20:80 EtOAc:Hexanes to 40:60 EtOAc:Hexanes) to give 21 (60 mg, 20% yield) as a light brown oil. +19.6 (c 1.0, CHCl3); Chiral HPLC analysis Chiralpak OD-H (5% IPA:hexane, flow rate 1.0 mL min–1, 254 nm) tR major (S) 13.8 min, tR minor (R) 16.5 min 96:4 er. The racemic standard was prepared in an identical fashion from (rac)-N-methyl-prolinol to give a light brown solid.
νmax (ATR)/cm–1 2946, 2361, 1335, 1634, 1602, 1558, 1506, 1418, 1286, 213, 1077, 919, 821, and 735; 1H NMR (700 MHz, CDCl3) δH 7.60 (1H, t, J 7.8), 7.29 (1H, dd, J 8.2, 1.2), 7.11 (1H, dd, J 9.9, 1.3), 7.10 (1H, s), 5.32 (1H, br s), 4.63 (1H, d, J 14.1), 3.82 (1H, dd, J 11.7, 2.7), 3.58 (1H, d, J 14.1), 3.52 (1H, dd, J 11.7, 5.5), 2.99–2.96 (1H, m), 2.95–2.92 (1H, m), 2.68 (3H, s), 2.39–2.36 (1H, m), 1.94–1.89 (1H, m), 1.79–1.71 (2H, m), 1.70–1.64 (1H, m); 13C NMR (176 MHz, CDCl3) δc 180.1 160.5 (d, J 255), 152.1, 147.0, 140.0, 139.2, (d, J 10.6), 138.8, 131.4 (d, J 3.6), 126.7 (d, J 14.1), 125.5, 125.3 (d, J 3.2), 120.0, 116.5 (d, J 25.6), 65.4, 64.6, 55.4, 54.8, 27.3, 23.7, and 16.3; 19F NMR (377 MHz, CDCl3) δF −112.2 (t, J 8.7); m/z (ESI+) HRMS [M + H] C20H20Cl2FN4O2+ found 437.0943, calcd. 437.0942.
Ethyl 6-chloro-2-methyl-8-(morpholinomethyl)imidazo[1,2-b]pyridazine-3-carboxylate 23 and Ethyl 6-chloro-2-methyl-8-(4-methylmorpholin-3-yl)imidazo[1,2-b]pyridazine-3-carboxylate 24
Following the general procedure two identical reactions using 1 (55.4 mg, 0.231 mmol, 1.0 equiv), sodium acetate (19.0 mg, 0.231 mmol, 1.0 equiv), Ir(ppy)3 (0.8 mg, 1.16 μmol, 0.005 equiv), DMA (1.50 mL) and N-methylmorpholine (510 μL, 4.63 mmol, 20 equiv) for 12 h at 5 °C were combined and worked up together. The resulting crude material (∼1:1 exo:endo) was purified via automated column chromatography (12 g silica column, 50:20:30 Hexanes:EtOAc:CH2Cl2) to give exo-23 (57 mg, 36% yield) as a white solid and endo-24 that was further purified via acid base extraction to give (29 mg, 19%)
Data for exo-23 in Accordance with the Literature32
1H NMR (500 MHz, CDCl3) δH 7.40 (1H, s), 4.46 (2H, q, J 7.1), 3.98 (2H, s), 3.78 (4H, t, J 4.4), 2.72 (3H, s), 2.60 (4H, t, J 4.4) and 1.43 (3H, t, J 7.1).
Data for endo-24
νmax (ATR)/cm–1 2982, 2851, 1696, 1230, 1417, 1288, 1225, 1115, 989, 952, 876, 758, and 636; 1H NMR (500 MHz, CDCl3) δH 7.39 (1H, s), 4.46 (2H, q, J 7.1), 4.13 (1H, d, J 8.2), 3.99 (1H, dd, J 11.2, 2.9), 3.96 (1H, d, J 11.4), 3.74 (1H, t J 11.4), 3.29 (1H, t, J 9.8), 2.88 (1H, d, J 11.7), 2.73 (3H, s), 2.56 (1H, d, J 10.6), 2.17 (3H, s) and 1.43 (3H, t, J 7.1); 13C NMR (176 MHz, CDCl3) δc 159.5, 151.6, 147.9, 139.3, 139.1, 118.8, 117.9, 71.5, 67.4, 61.0, 54.6, 43.8, 16.8, and 14.5 (one C was not resolved); m/z (ESI+) HRMS [M + H] C15H20ClN4O3+ found 339.1226, calcd. 339.1218.
Ethyl 6-chloro-2,8-dimethylimidazo[1,2-b]pyridazine-3-carboxylate 25 and Ethyl 2-methylimidazo[1,2-b]pyridazine-3-carboxylate S3
Following the general procedure two identical reactions using 22 (55.4 mg, 0.231 mmol, 1.0 equiv), sodium acetate (19,0 mg, 0.231 mmol, 1.0 equiv), Ir(ppy)3 (0.8 mg, 1.16 μmol, 0.005 equiv), DMA (1365 μL), water (137 μL) and N-methylmorpholine (510 μL, 4.63 mmol, 20 equiv) for 16 h at 5 °C were combined and worked up together. The resulting crude material (1:1.3 exo:endo) was purified via automated column chromatography (12 g silica column, 50:20:30 Hexanes:EtOAc:CH2Cl2) to give 25 (7 mg, 6% yield), 23-exo that was further purified via acid base extraction to remove ethyl 2-methylimidazo[1,2-b]pyridazine-3-carboxylate (S3) to give (26 mg, 17%) and 24-endo (37 mg, 24% yield)
Data for 25
νmax (ATR)/cm–1 1679, 1528, 1417, 1347, 1295, 1252, 1143, 1130, 11063, 930, 884 765 and 715 1H NMR (500 MHz, CDCl3) δH 7.04 (1H, d, J 1.1), 4.46 (2H, q, J 7.1), 2.73 (3H, s), 2.66 (3H, d, J 1.1) and 1.44 (3H, t, J 7.1); 13C NMR (176 MHz, CDCl3) δc 159.6, 151.1, 147.3, 140.2, 138.2, 120.4, 117.6, 60.9, 16.7, 16.7, and 14.5; m/z (ESI+) HRMS [M + H] C11H13ClN3O2+ found 254.0693, calcd. 254.0691.
Data for S3
νmax (ATR)/cm–1 3113, 2934, 1703, 1425, 1385, 1323, 1266, 1188, 1083, 1040, 805, and 768 1H NMR (500 MHz, CDCl3) δH 8.56 (1H, dd, J 4.5, 1.7), 7.97 (1 H, dd, J 9.1, 1.7), 7.22 (1H, dd, J 9.1, 4.5), 4.49 (2H, q, J 7.1), 2.77 (3H, s) and 1.45 (3H, t, J 7.1); 13C NMR (176 MHz, CDCl3) δc 159.8, 151.8, 143.6, 140.7, 124.9, 119.1, 117.0, 60.8, 16.9, and 14.5 m/z (ESI+) HRMS [M + H] C10H12N3O2+ found 206.0926, calcd. 206.0924.
4-((6-Chloro-3-(4-chloro-2-fluorobenzyl)-2-methylimidazo[1,2-b]pyridazin-8-yl)methyl)morpholine 27
Following the general procedure five identical reactions using 27 (71.6 mg, 0.231 mmol, 1.0 equiv), sodium acetate (19,0 mg, 0.231 mmol, 1.0 equiv), Ir(ppy)3 (0.8 mg, 1.16 μmol, 0.01 equiv), DMA (1365 μL), water (137 μL) and N-methylmorpholine (510 μL, 4.63 mmol, 20 equiv) for 20 h at 5 °C were combined and worked up together. The resulting crude material (1.2:1 exo:endo) was purified via automated column chromatography (12 g silica column, 50:40:10 Hexanes:EtOAc:CH2Cl2) to give 27 (64 mg, 14% yield in accordance with the literature.321H NMR (500 MHz, CDCl3) δH 7.19 (1H, t, J 1.3), 7.16 (1H, d, J 8.2), 7.06 (1H, dd, J 9.7 and 2.1), 7.01 (1H, ddd, J 8.2, 2.1 and 0.7), 4.27 (2 H, s), 3.96 (2H, d, J 1.3,), 3.77 (4H, t, J 4.6), 2.59 (4H, t, J 4.6), 2.46 (3H, d, J 0.5).
Use of Iminium 31
Following the general procedure 1 (75.0 mg, 0.231 mmol, 1.0 equiv), Ir(ppy)3 (2.27 mg, 3.47 μmol, 0.015 equiv), 31 (627 mg, 4.63 mmol, 20 equiv), made via the literature report of Shustov and Khlebnikov42 anhydrous DMA (5 mL), gave a slurry that was sparged with nitrogen for 15 min and then sealed. A 1 W LED strip was turned on for 24 h at 22 °C before the reaction was worked up and purified to give 2 (13 mg, 13% yield). Prior to purification 1H NMR spectroscopic analysis and UPLC PDA analysis of the crude reaction mixture showed a exo:endo ratio of >20:1.
Acknowledgments
The authors thank Dr. Scott May, Dr. James Devery, Dr. Elizabeth Swift, Mr. Joel Beatty, Dr. John Nguyen and Mr. Bryan Matsuura for helpful discussion. Research was supported by a Lilly Innovation Fellowship Award to J.J.D. from Eli Lilly and Company. Financial support from the NIH (GM096129 to CRJS), the Alfred P. Sloan Foundation, the Camille and Henry Dreyfus Foundation, and the University of Michigan is gratefully acknowledged.
Supporting Information Available
Further optimization and reaction data; UPLC chromatograms; quenching studies, copies of NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Wilks A. F. Proc. Natl. Acad. Sci. U. S. A. 1989, 86, 1603–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Silvennoinen O.; Witthuhn B. A.; Quelle F. W.; Cleveland J. L.; Yi T.; Ihle J. N. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 8429–8433. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kralovics R.; Passamonti F.; Buser A. S.; Teo S.-S.; Tiedt R.; Passweg J. R.; Tichelli A.; Cazzola M.; Skoda R. C. N. Engl. J. Med. 2005, 352, 1779–1790. [DOI] [PubMed] [Google Scholar]; d Burkholder T. P.; Clayton J. R. U.S. Pat. Appl. Publ. US 20100152181, A1 20100617; CAN 153:97762, AN 2010: 753991.2010.
- Duncton M. A. J. MedChemComm 2011, 2, 1135–1161. [Google Scholar]
- Mitchell D.; Cole K. P.; Pollock P. M.; Coppert D. M.; Burkholder T. P.; Clayton J. R. Org. Process Res. Dev. 2011, 16, 70–81. [Google Scholar]
- Accelerating rate calorimetry (ARC) onset for NMO was measured at 140 °C with a very high decomposition energy of 1370 J/g as measured by differential scanning calorimetry (DSC), and thus, NMO was considered a potential explosive risk on large scale.
- a Tucker J. W.; Stephenson C. R. J. J. Org. Chem. 2012, 77, 1617–1622. [DOI] [PubMed] [Google Scholar]; b Prier C. K.; Rankic D. A.; MacMillan D. W. C. Chem. Rev. 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Douglas J. J.; Nguyen J. D.; Cole K. P.; Stephenson C. R. J. Aldrichimica Acta 2014, 47, 15–25. [Google Scholar]
- (6); a Shi L.; Xia W. Chem. Soc. Rev. 2012, 41, 7687–7697. [DOI] [PubMed] [Google Scholar]; b Hu J.; Wang J.; Nguyen T. H.; Zheng N. Beilstein J. Org. Chem. 2013, 9, 1977–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renaud P.; Giraud L. Synthesis 1996, 8, 913–926. [Google Scholar]
- a Kohls P.; Jadhav D.; Pandey G.; Reiser O. Org. Lett. 2012, 14, 672–675. [DOI] [PubMed] [Google Scholar]; b Ruiz Espelt L.; Wiensch E. M.; Yoon T. P. J. Org. Chem. 2013, 78, 4107–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Singh A.; Arora A.; Weaver J. D. Org. Lett. 2013, 15, 5390–5393. [DOI] [PubMed] [Google Scholar]; d Zhou H.; Lu P.; Gu X.; Li P. Org. Lett. 2013, 15, 5646–5649. [DOI] [PubMed] [Google Scholar]; e Zhu S.; Das A.; Bui L.; Zhou H.; Curran D. P.; Rueping M. J. Am. Chem. Soc. 2013, 135, 1823–1829. [DOI] [PubMed] [Google Scholar]; f Miyake Y.; Nakajima K.; Nishibayashi Y. J. Am. Chem. Soc. 2012, 134, 3338–3341. [DOI] [PubMed] [Google Scholar]; g Miyake Y.; Nakajima K.; Nishibayashi Y. Chem.—Eur. J. 2012, 18, 16473–16477. [DOI] [PubMed] [Google Scholar]; h McNally A.; Prier C. K.; MacMillan D. W. C. Science 2011, 334, 1114–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Prier C. K.; MacMillan D. W. C. Chem. Sci. 2014, 5, 4173–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cassani C.; Bergonzini G.; Wallentin C.-J. Org. Lett. 2014, 16, 4228–4231. [DOI] [PubMed] [Google Scholar]; b Chu L.; Ohta C.; Zuo Z.; MacMillan D. W. C. J. Am. Chem. Soc. 2014, 136, 10886–10889. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Nobel A.; MacMillan D. W. C. J. Am. Chem. Soc. 2014, 136, 11602–11605. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Zuo Z.; MacMillan D. W. C. J. Am. Chem. Soc. 2014, 136, 5257–5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Miyake Y.; Ashida Y.; Nakajima K.; Nishibayashi Y. Chem. Commun. 2012, 48, 6966–6968. [DOI] [PubMed] [Google Scholar]; b Miyake Y.; Ashida Y.; Nakajima K.; Nishibayashi Y. Chem.—Eur. J. 2014, 20, 6120–6125. [DOI] [PubMed] [Google Scholar]
- Nagib D. A.; MacMillan D. W. C. Nature 2011, 480, 224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiRocco D. A.; Dykstra K.; Krska S.; Vachal P.; Conway D. V.; Tudge M. Angew. Chem., Int. Ed 2014, 53, 4802–4806. [DOI] [PubMed] [Google Scholar]
- a Xue D.; Jia Z. H.; Zhao C. J.; Zhang Y. Y.; Wang C.; Xiao J. Chem.—Eur. J. 2014, 20, 2960–2965. [DOI] [PubMed] [Google Scholar]; b Allen L. J.; Cabrera P. J.; Lee M.; Sanford M. S. J. Am. Chem. Soc. 2014, 136, 5607–5610. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Qvortrup K.; Rankic D. A.; Macmillan D. W. C. J. Am. Chem. Soc. 2014, 136, 626–629. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hari D. P.; Schroll P.; König B. J. Am. Chem. Soc. 2012, 134, 2958–2961. [DOI] [PubMed] [Google Scholar]; e Furst L.; Matsuura B. S.; Narayanam J. M. R.; Tucker J. W.; Stephenson C. R. J. Org. Lett. 2010, 12, 3104–3107. [DOI] [PubMed] [Google Scholar]
- Shih H.-W.; Vander Wal M. N.; Grange R. L.; MacMillan D. W. C. J. Am. Chem. Soc. 2010, 132, 13600–13603Weaver has shown the synthesis of a Nizatidine analogue; see ref (8c). [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEDs were purchased from creative lighting solutions (http://www.creativelightings.com).
- This photocataylst is commercially available CAS [94928–86–6] at approximately $1000/g.
- a Nguyen J. D.; D’Amato E. M.; Narayanam J. M. R.; Stephenson C. R. J. Nat. Chem. 2012, 4, 854–859. [DOI] [PubMed] [Google Scholar]; b Narayanam J. M. R.; Tucker J. W.; Stephenson C. R. J. J. Am. Chem. Soc. 2009, 131, 8756–8757. [DOI] [PubMed] [Google Scholar]
- Average of two independent reactions. All further optimization reactions were run in duplicate.
- The UPLC PDA method allowed for much greater sensitivity and highlighted a large number of small level impurities (typically <1% a/a) that collectively contribute to the moderate yield of the desired exo product 2.
- Battino R.; Rettich T. R.; Tominaga T. J. Phys. Chem. Ref. Data 1983, 12, 163–178. [Google Scholar]
- Evidence for this was obtained by conducting two reactions at different scales but with the same relative surface area (cm2/mmol) that provided identical reaction profiles. See the Supporting Information for more details.
- a Knowles J. P.; Elliott L. D.; Booker-Milburn K. I. Beilstein J. Org. Chem. 2012, 8, 2025–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Su Y.; Straathof N. J. W.; Hessel V.; Noël T. Chem.—Eur. J. 2014, 20, 10562–10589. [DOI] [PubMed] [Google Scholar]; c Andrews R. S.; Becker J. J.; Gagne M. R. Angew. Chem., Int. Ed 2012, 51, 4140–4143. [DOI] [PubMed] [Google Scholar]; d Tucker J. W.; Zhang Y.; Jamison T. F.; Stephenson C. R. J. Angew. Chem., Int. Ed 2012, 51, 4144–4147. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Bou-Hamdan F. R.; Seeberger P. H. Chem. Sci. 2012, 3, 1612–1616. [Google Scholar]; f Garlets Z. J.; Nguyen J. D.; Stephenson C. R. J. Isr. J. Chem. 2014, 54, 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See the Supporting Information for further information.
- Schönherr H.; Cernak T. Angew. Chem., Int. Ed. 2013, 52, 12256–12267. [DOI] [PubMed] [Google Scholar]
- The stability of 2 to the reaction conditions in Scheme 3 was potentially due to the decreased quantity of oxygen and or NMM in solution at 35 °C.
- a Wayner D. D. M.; Clark K. B.; Rauk A.; Yu D.; Armstrong D. A. J. Am. Chem. Soc. 1997, 119, 8925–8932. [Google Scholar]; b Dombrowski G. W.; Dinnocenzo J. P.; Farid S.; Goodman J. L.; Gould I. R. J. Org. Chem. 1998, 64, 427–431. [Google Scholar]
- Tamayo A. B.; Alleyne B. D.; Djurovich P. I.; Lamansky S.; Tsyba I.; Ho N. N.; Bau R.; Thompson M. E. J. Am. Chem. Soc. 2003, 125, 7377–7387. [DOI] [PubMed] [Google Scholar]
- Flamigni L.; Barbieri A.; Sabatini C.; Ventura B.; Barigelletti F. In Photochemistry and Photophysics of Coordination Compounds II; Balzani V., Campagna S., Eds.; Springer: Berlin, 2007; Vol. 281, pp 143–203. [Google Scholar]
- Other NMM related side products such as iminium species and morpholine-4-carbaldehyde were also tentatively assigned via 1H NMR.
- a Yoshimitsu T.; Arano Y.; Nagaoka H. J. Am. Chem. Soc. 2005, 127, 11610–11611. [DOI] [PubMed] [Google Scholar]; b Campos K. R. Chem. Soc. Rev. 2007, 36, 1069–1084. [DOI] [PubMed] [Google Scholar]; c Dai C.; Meschini F.; Narayanam J. M. R.; Stephenson C. R. J. J. Org. Chem. 2012, 77, 4425–4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formation of the radical at this position is also disfavoured kinetically due to steric interactions between the hydrogen at the 2 position of the phenyl and one of the N-methyl groups when adopting the required conformation for deprotonation. For further information, see ref (26b).
- Campbell A. N.; Cole K. P.; Martinelli J. R.; May S. A.; Mitchell D.; Pollock P. M.; Sullivan K. A. Org. Process Res. Dev. 2013, 17, 273–281. [Google Scholar]
- Grainger D. M.; Zanotti-Gerosa A.; Cole K. P.; Mitchell D.; May S. A.; Pollock P. M.; Calvin J. R. ChemCatChem 2013, 5, 1205–1210. [Google Scholar]
- Cowden C. J. Org. Lett. 2003, 5, 4497–4499. [DOI] [PubMed] [Google Scholar]
- The oxidation potential of NMM (E1/2red = +1.2 vs SCE) is significantly higher than that of Ir(ppy)3 (E1/2IV/III = +0.77 vs SCE).; Tajima T.; Fuchigami T. Angew. Chem., Int. Ed. 2005, 44, 4760–4763. [DOI] [PubMed] [Google Scholar]; For more discussion on process that operate outside of reactivity predicted by potentials, see ref (5c).
- Aromatization may be achieved via oxidation then deprotoantion or initial deprotonation followed by oxidation. For recent discussion of the latter, see:Studer A.; Curran D. P. Nat. Chem. 2014, 6, 765–773. [DOI] [PubMed] [Google Scholar]
- a Stern O.; Volmer M. Phys. Z. 1919, 20, 183–188. [Google Scholar]
- Imidazopyridazines 23 and 28, and NMM, were not efficient quenchers of Ir(ppy)3. See the Supporting Information for further details.
- For example, the excitation of 1 to its triplet state via energy transfer and subsequent reactivity analogous to that of benzophenone. For example, see:Hoffmann N.; Bertrand S.; Marinković S.; Pesch J. Pure Appl. Chem. 2006, 78, 2227. [Google Scholar]
- This type of experiment does not eliminate the possibility of radical propagation followed by a rapid termination event.
- Devery J. J.; Douglas J. J.; Nguyen J. D.; Cole K. P.; Flowers R. A; Stephenson C. R. J. Chem. Sci. 2014, 10.1039/c4sc03064h. [DOI] [Google Scholar]
- Shustov G.; Khlebnikov V. Can. J. Chem. 2011, 89, 1319–1324. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.