

Abstract
Susceptibility to type 1 diabetes is attributable to genes that link disease progression to distinct steps in immune activation, expansion, and regulation. Recent studies illustrate examples of disease-associated variants that function in multiple cell types and independent pathways, some that impact different steps of a single mechanistic pathway, and some that are functionally interactive for deterministic events in setting thresholds for immune response.
Introduction
Genes associated with common immune mediated diseases are normal genes. They vary as a natural consequence of the challenging and changing immunological environment of the human population. We begin this brief review with three key concepts that underlie this basic tenet:
Genetic variation drives functional diversity fundamental to creating a broad range of immune response within the human population. Hundreds of genes of immunological relevance participate in creating this spectrum of response, sometimes with multiple variants of each participating gene. The resulting profile of genetic diversity represents a dynamic mosaic that is manifest as a distribution of immune responses within any population dependent on the type of stimulus.
Biologically critical functions are often protected by redundancy. As a result, deviation in function of a single element in the immunological mosaic is often tolerated, even when there is a corresponding change in some important parameter, such as an activation threshold or in the level of an effector response.
Pathways that drive most immunological functions are composed of interdependent molecules such that a change in one molecule within a pathway can be functionally equivalent to a change in another molecule elsewhere within that pathway; conversely, a change in one molecule can be compensated for by another. As a result, there may be genetically distinct variants that have similar consequences, and functional outcomes depend on interactions among multiple genetic variants in a single pathway.
Some of these normal genetic variants achieve the distinction of being called disease-associated genes, such as a genome-wide association study (GWAS) “hit” by virtue of being common in the population and statistically associated with a clinical diagnosis. In this review, we highlight selected examples of genes strongly associated with type 1 diabetes (T1D that illustrate these points.
HLA
Extensive population genetic studies over the last 30 years have documented the primary role of two particular HLA-DQ heterodimers, DQA301/DQB302 and DQA501/DQB302, responsible for the strongest associations with T1D [1–3]. Numerous other HLA genes modify the strength of disease association [4–7], presumably due to their ability to bind and present specific peptides to antigen-restricted T cells, directly linking precise genetic polymorphisms to the functional attribute of antigen recognition.
In a number of experimental systems, the trimolecular structures for autoreactive T cell receptor (TCR) engaging cognate disease-associated major histocompatibility complex (MHC) and peptide molecules assumes a skewed or incomplete interface, potentially indicating that recognition occurs via a nontraditional interaction with low native avidity [8–10]. Consistent with this interpretation, visualization studies of the molecular architecture for disease-associated trimolecular interactions, using three T1D-related T cell clones, found initial MHC-peptide contacts failed to lead to sustained clustering within the T cell supramolecular activation complex (SMAC), and were instead correlated with incomplete activation and retention signals [11]. In another set of functional studies, MHC-defective mice were transgenically modified to express both human T1D-associated HLA genes and also human TCR from T1D-associated T cell clones [12;13]. Even in the face of thymic negative selection events, the peripheral repertoire of these animals was reconstituted with large numbers of surviving autoreactive T cells, suggesting an inefficient threshold for deletional tolerance consistent with a lower avidity TCR recognition profile.
There are a variety of potential antigenic targets in T1D, including T cell immunity to islet proteins, such as proinsulin, GAD, IA2, IGRP, ZnT8, and others [14;15]. It is not known whether this trimolecular model for HLA avidity skewing applies to all, or whether there are differences during the early stages of disease initiation compared to later during disease progression, following antigenic determinant spreading of the immune response.
INS
Polymorphisms in the proinsulin promoter region are associated with susceptibility to T1D and correlate with the presence of autoantibodies, even in individuals who do not have clinically evident disease [16;17]. Short length of a repeating sequence (VNTR I) within the proinsulin promoter a role for this genetic variation is associated with T1D, whereas longer length (VNTR III) is not. A higher level of thymic insulin expression, relative to pancreatic insulin gene expression, is found in individuals who carry the disease-protective VNTR III variant, consistent with the hypothesis that a lower level of specific antigen during thymic selection is permissive for the escape of autoreactive T cells [18;19]. In a direct test of this model, two types of insulin-HLA tetramers were developed and used to profile CD4 T cell recognition in subjects with different forms of the INS disease-associated polymorphism. These two tetramer reagents contained either native or a modified proINS peptide bound by disease-associated MHC molecules, and distinguished between recognition of high and low avidity T cells [20;21]. In these studies, individuals with disease-protective INS haplotypes (VNTR III) had significantly lower numbers of the high avidity T cells specific for insulin in the peripheral blood compared to HLA-matched individuals with VNTR I.
These findings support the notion that a consequence of the INS gene polymorphism is to influence the strength of negative selection during T cell development, and directly links a tissue-specific susceptibility gene locus to a putative immunological mechanism for disease association. This also implicates a susceptibility modification role for thymic gene expression in autoimmunity, a key immunological checkpoint more generally controlled by tissue regulator factors, such as AIRE [22;23].
PTPN22
A variant of PTPN22 characterized by a single nucleotide polymorphism (SNP) at position 1858, resulting in a change from an Arg at position 620 of the protein to Trp (Lyp620W), is associated with T1D as well as with rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), Graves disease, and myasthenia gravis [24–26]. Further analysis of the PTPN22 gene has confirmed that the 1858 C/T variant is the major risk variant in the PTPN22 locus [27]; however, rare missense variants which may contribute to risk have been identified [28;29]. PTPN22 encodes a protein tyrosine phosphatase, lymphocyte tyrosine phosphatase (Lyp), that has been shown to be a negative regulator of T cell activation [30], confirmed by studies of PEP−/− mice (the murine orthologue of Lyp) [31]. When expressed in Jurkat cells, Lyp620W has been shown to confer a dominant gain of function, resulting in blunted TCR activation [32], but studies by others indicated that Lyp620W results in increased responsiveness to TCR stimulation [33]. A recently described murine model which expresses the orthologue of Lyp620W demonstrates enhanced TCR signaling, which was linked to the rapid degradation of the phosphatase [34].
To reconcile these findings, it is instructive to look beyond the T cell, since Lyp is expressed in T cells, B cells, and myeloid lineages, and it is likely that the disease-associated function of this variant is due to its involvement in more than one disease-related pathway. The presence of autoantibodies is a prominent feature of the autoimmune diseases associated with this variant, and defects in B cell tolerance are present, leading to an increase in the escape of autoreactive B cells into the periphery [35;36]. Lyp620W is also associated with blunted B cell receptor signaling and enhanced survival of transitional B cells, which may be due in part to a novel function of the variant protein in these individuals. As shown in Figure 1, both T and B cell functional measures are directly correlated with PTPN22 genotype, and this impact on multiple pathways in immunity, possibly also including innate responses, may account for the broad relationship between Lyp620W and autoimmune diseases.
Figure 1. A susceptibility gene can affect multiple pathways.
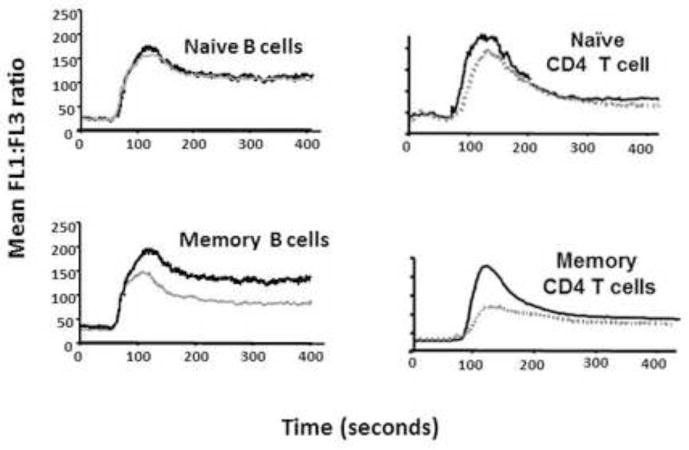
PTPN22 variants that are associated with T1D and other autoimmune diseases account for blunted signaling, as shown in plots of calcium flux for human B cells (left) and T cells (right), adapted from Rieck et al. [45]. The grey lines indicate individuals carrying the 1858T variant, demonstrating reduced BCR- and TCR-mediated calcium mobilization after stimulation with anti-IgM or anti-CD3, respectively.
PTPN2/CD25
The IL-2/IL-2R signaling pathway is implicated in the development of autoimmunity due to the association of multiple disease-associated genetic variants that appear in different components of that pathway. In the case of T1D, the IL-2/IL-21 gene locus is modestly associated with disease [37], but more significant associations are found in components of the IL-2R signaling pathway, including the receptor itself and a phosphatase (PTPN2) that participates in molecular interactions governing phosphorylation of the transcription factor STAT5 [38]. Multiple non-coding SNPs in the high affinity IL-2 receptor, CD25 (IL-2RA), have been associated with T1D [39], some of which correlate with levels of CD25 expression on CD4 T cells [40] or the level of soluble CD25 produced by T cells [39]. Several non-coding SNPs in the PTPN2 gene are associated with T1D, Crohn’s disease, and RA [39;41], and it has been suggested that these SNPs affect expression or splicing of the PTPN2 isoforms [42].
The general picture emerging from these studies is the notion that disease-associated variants in the IL-2 pathway confer a decreased ability to respond to IL-2 or limit the availability of IL-2 at sites of inflammation. In a recent study of STAT5 phosphorylation in human T cells, one of the PTPN2 disease-associated SNPs correlated with decreased phosphorylated (p)STAT5, decreased IL-2R signaling in CD4(+) T cells, and reduced FOXP3 expression in activated cells [42]. A similar relationship has been reported for risk variants in CD25 [43], consistent with the hypothesis that IL-2-dependent T cells, particularly regulatory FOXP3-positive cells, may be impaired. Notably in T1D, diminished IL-2R signaling is a common feature of disease, even in those subjects who do not have the known PTPN2 or CD25 variants in this pathway, suggesting that additional, rare variants may contribute to the phenotype of impaired IL-2 signaling in T1D [44].
Concluding Remarks
Different disease-associated genetic variants impact different stages of the immunological life cycle, including development and selection of the adaptive response, engaging activation thresholds, guiding cell fate and commitment, and assembling a regulated set of interactions (Figure 2). Genotypic variation encompasses different categories of immunological function, listed in Figure 3, emphasizing the challenge of linking specific genes to specific disease pathways. Disease-associated variants persist in the population in the form of subclinical traits that bias towards disease initiation and progression, offering the hope that identifying the pathways impacted by genetic variation will also identify a large number of rational therapeutic targets, although not necessarily the same molecule impacted by the susceptibility gene itself. For example, IL-2-directed therapies may compensate for and “correct” the IL-2-signaling defect common to T1D subjects, and antigen delivery in the context of regulatory signals may compensate for the biased selection of specific autoreactive T cells. This linkage between therapeutic intervention and genotype highlights the opportunity to use genetic stratification, such as during clinical trials, as a window into pathway analysis and disease mechanisms.
Figure 2. Building blocks of T1D susceptibility.
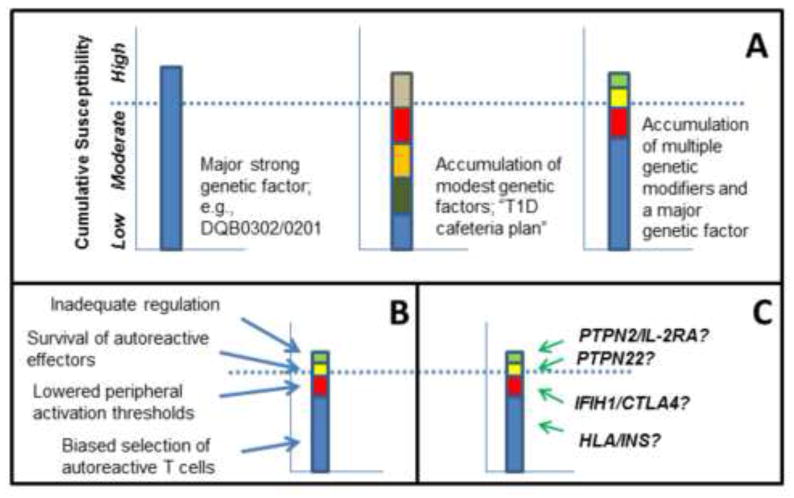
T1D, like most other autoimmune diseases, occurs predominantly on a background of genetic susceptibility that is a result of several genetic elements. Presence of a high risk genotype, such as HLA-DQB1*03:02/*02:01 is sufficient to pass a susceptibility threshold (A, left panel). An alternative scenario involves multiple genes, each with moderate contributions to functional pathways, which in combination surpass the threshold barrier (A, middle panel). The most common combination of genetic risk in T1D is represented by the third model, in which HLA contributes most of the risk, but other cumulative contributions from moderate risk genes also participate (A, right panel). Functional mechanisms that facilitate the generation, survival, and activation of autoreactive cells are represented in (B), illustrating the concept that multiple steps in the progression of autoimmune disease are required and are cumulative or synergistic in reaching a clinical threshold. Specific genetic variation is tied to particular steps in this functional progression, as indicated in the speculative, but plausible, relationships shown in (C).
Figure 3. Classification of T1D susceptibility genes.
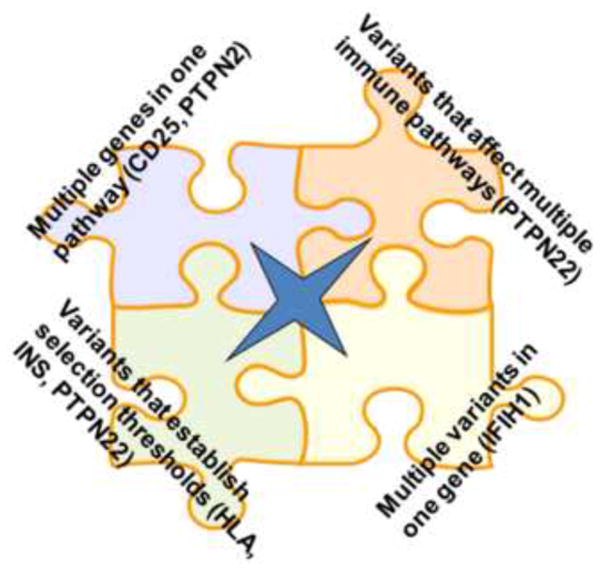
The examples discussed in the text illustrate different categories of genotype-phenotype relationships: (i) those genes that function in the same mechanistic pathway, such as CD25 and PTPN2; (ii) those genes that alter function in multiple different pathways or cells, such as PTPN22; (iii) those genetic variants that independently arise in the same gene but similarly affect function; and (iv) those that combine to establish key immunological thresholds, such as HLA and INS in thymic selection of proINS specific T cells.
Highlights.
Genetic susceptibility to T1D is a composite of risk factors contributed by the major histocompatibility complex with a large number of additional genetic modifiers;
The functional properties of each of these susceptibility genes vary due to subtle genetic variation that occurs normally in the population;
Identification of functional pathways impacted by specific GWAS “hits” link disease progression to distinct steps in immune activation, expansion, and regulation.
Stratification of risk based on genetic variation provides a classification scheme that implicates distinct functional pathways as therapeutic targets.
Acknowledgments
We thank our many colleagues and research subjects who have contributed to our studies. The authors acknowledge the support of grants from the National Institutes of Health, Juvenile Diabetes Research Foundation, and the Washington State Life Sciences Discovery Fund for work cited in this article.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Robinson DM, Holbeck S, Palmer J, Nepom GT. HLA DQ beta 3.2 identifies subtypes of DR4+ haplotypes permissive for IDDM. Genet Epidemiol. 1989;6:149–154. doi: 10.1002/gepi.1370060128. [DOI] [PubMed] [Google Scholar]
- 2.Nepom GT, Kwok WW. Molecular basis for HLA-DQ associations with IDDM. Diabetes. 1998;47:1177–1184. doi: 10.2337/diab.47.8.1177. [DOI] [PubMed] [Google Scholar]
- 3.Erlich H, Valdes AM, Noble J, Carlson JA, Varney M, Concannon P, Mychaleckyj JC, Todd JA, Bonella P, Fear AL, Lavant E, Louey A, Moonsamy P. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes. 2008;57:1084–1092. doi: 10.2337/db07-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noble JA, Valdes AM, Thomson G, Erlich HA. The HLA class II locus DPB1 can influence susceptibility to type 1 diabetes. Diabetes. 2000;49:121–125. doi: 10.2337/diabetes.49.1.121. [DOI] [PubMed] [Google Scholar]
- 5.Cucca F, Dudbridge F, Loddo M, Mulargia AP, Lampis R, Angius E, De VS, Koeleman BP, Bain SC, Barnett AH, Gilchrist F, Cordell H, Welsh K, Todd JA. The HLA-DPB1--associated component of the IDDM1 and its relationship to the major loci HLA-DQB1, -DQA1, and -DRB1. Diabetes. 2001;50:1200–1205. doi: 10.2337/diabetes.50.5.1200. [DOI] [PubMed] [Google Scholar]
- 6.Nejentsev S, Howson JM, Walker NM, Szeszko J, Field SF, Stevens HE, Reynolds P, Hardy M, King E, Masters J, Hulme J, Maier LM, Smyth D, Bailey R, Cooper JD, Ribas G, Campbell RD, Clayton DG, Todd JA. Localization of type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature. 2007;450:887–892. doi: 10.1038/nature06406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Noble JA, Valdes AM, Varney MD, Carlson JA, Moonsamy P, Fear AL, Lane JA, Lavant E, Rappner R, Louey A, Concannon P, Mychaleckyj JC, Erlich HA. HLA class I and genetic susceptibility to type 1 diabetes: results from the Type 1 Diabetes Genetics Consortium. Diabetes. 2010;59:2972–2979. doi: 10.2337/db10-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Y, Huang Y, Lue J, Quandt JA, Martin R, Mariuzza RA. Structure of a human autoimmune TCR bound to a myelin basic protein self-peptide and a multiple sclerosis-associated MHC class II molecule. EMBO J. 2005;24:2968–2979. doi: 10.1038/sj.emboj.7600771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hahn M, Nicholson MJ, Pyrdol J, Wucherpfennig KW. Unconventional topology of self peptide-major histocompatibility complex binding by a human autoimmune T cell receptor. Nat Immunol. 2005;6:490–496. doi: 10.1038/ni1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sethi DK, Schubert DA, Anders AK, Heroux A, Bonsor DA, Thomas CP, Sundberg EJ, Pyrdol J, Wucherpfennig KW. A highly tilted binding mode by a self-reactive T cell receptor results in altered engagement of peptide and MHC. J Exp Med. 2011;208:91–102. doi: 10.1084/jem.20100725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11**.Schubert DA, Gordo S, Sabatino JJ, Jr, Vardhana S, Gagnon E, Sethi DK, Seth NP, Choudhuri K, Reijonen H, Nepom GT, Evavold BD, Dustin ML, Wucherpfennig KW. Self-reactive human CD4 T cell clones form unusual immunological synapses. J Exp Med. 2012;209:335–352. doi: 10.1084/jem.20111485. Clustering of TCR molecules at the interface with peptide-MHC ligands on lipid bilayers is found to be defective or delayed in T cells with autoantigen specificity. As a consequence, these T cells display high mobility properties, failing to arrest after recognition of cognate target molecules. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gebe JA, Unrath KA, Yue BB, Miyake T, Falk BA, Nepom GT. Autoreactive human T-cell receptor initiates insulitis and impaired glucose tolerance in HLA DR4 transgenic mice. J Autoimmun. 2008;30:197–206. doi: 10.1016/j.jaut.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gebe JA, Yue BB, Unrath KA, Falk BA, Nepom GT. Restricted autoantigen recognition associated with deletional and adaptive regulatory mechanisms. J Immunol. 2009;183:59–65. doi: 10.4049/jimmunol.0804046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Lorenzo TP, Peakman M, Roep BO. Translational mini-review series on type 1 diabetes: Systematic analysis of T cell epitopes in autoimmune diabetes. Clin Exp Immunol. 2007;148:1–16. doi: 10.1111/j.1365-2249.2006.03244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hondowicz BD, Schwedhelm KV, Kas A, Tasch MA, Rawlings C, Ramchurren N, McIntosh M, D’Amico LA, Sanda S, Standifer NE, Shendure J, Stone B. Discovery of T cell antigens by high-throughput screening of synthetic minigene libraries. PLoS One. 2012;7:e29949. doi: 10.1371/journal.pone.0029949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bennett ST, Lucassen AM, Gough SC, Powell EE, Undlien DE, Pritchard LE, Merriman ME, Kawaguchi Y, Dronsfield MJ, Pociot F. Susceptibility to human type 1 diabetes at IDDM2 is determined by tandem repeat variation at the insulin gene minisatellite locus. Nat Genet. 1995;9:284–292. doi: 10.1038/ng0395-284. [DOI] [PubMed] [Google Scholar]
- 17.Walter M, Albert E, Conrad M, Keller E, Hummel M, Ferber K, Barratt BJ, Todd JA, Ziegler AG, Bonifacio E. IDDM2/insulin VNTR modifies risk conferred by IDDM1/HLA for development of Type 1 diabetes and associated autoimmunity. Diabetologia. 2003;46:712–720. doi: 10.1007/s00125-003-1082-z. [DOI] [PubMed] [Google Scholar]
- 18.Pugliese A, Zeller M, Fernandez A, Jr, Zalcberg LJ, Bartlett RJ, Ricordi C, Pietropaolo M, Eisenbarth GS, Bennett ST, Patel DD. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat Genet. 1997;15:293–297. doi: 10.1038/ng0397-293. [DOI] [PubMed] [Google Scholar]
- 19.Vafiadis P, Bennett ST, Todd JA, Nadeau J, Grabs R, Goodyer CG, Wickramasinghe S, Colle E, Polychronakos C. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat Genet. 1997;15:289–292. doi: 10.1038/ng0397-289. [DOI] [PubMed] [Google Scholar]
- 20.Yang J, Danke N, Roti M, Huston L, Greenbaum C, Pihoker C, James E, Kwok WW. CD4+ T cells from type 1 diabetic and healthy subjects exhibit different thresholds of activation to a naturally processed proinsulin epitope. J Autoimmun. 2008;31:30–41. doi: 10.1016/j.jaut.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 21*.Durinovic-Bello I, Wu RP, Gersuk VH, Sanda S, Shilling HG, Nepom GT. Insulin gene VNTR genotype associates with frequency and phenotype of the autoimmune response to proinsulin. Genes Immun. 2010;11:188–193. doi: 10.1038/gene.2009.108. Polymorphisms in the proinsulin promoter are associated with T1D in the population, and are shown in this study to correlate with the frequency of proinsulin-specific CD4 T cells detected using modified HLA class II tetramers, consistent with a role in T cell selection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, von BH, Bronson R, Dierich A, Benoist C, Mathis D. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002;298:1395–1401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 23.Anderson MS, Venanzi ES, Chen Z, Berzins SP, Benoist C, Mathis D. The cellular mechanism of Aire control of T cell tolerance. Immunity. 2005;23:227–239. doi: 10.1016/j.immuni.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 24.Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, Ardlie KG, Huang Q, Smith AM, Spoerke JM, Conn MT, Chang M, Chang SY, Saiki RK, Catanese JJ, Leong DU, Garcia VE, McAllister LB, Jeffery DA, Lee AT, Batliwalla F, Remmers E, Criswell LA, Seldin MF, Kastner DL, Amos CI, Sninsky JJ, Gregersen PK. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet. 2004;75:330–337. doi: 10.1086/422827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, MacMurray J, Meloni GF, Lucarelli P, Pellecchia M, Eisenbarth GS, Comings D, Mustelin T. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet. 2004;36:337–338. doi: 10.1038/ng1323. [DOI] [PubMed] [Google Scholar]
- 26.Kyogoku C, Langefeld CD, Ortmann WA, Lee A, Selby S, Carlton VE, Chang M, Ramos P, Baechler EC, Batliwalla FM, Novitzke J, Williams AH, Gillett C, Rodine P, Graham RR, Ardlie KG, Gaffney PM, Moser KL, Petri M, Begovich AB, Gregersen PK, Behrens TW. Genetic association of the R620W polymorphism of protein tyrosine phosphatase PTPN22 with human SLE. Am J Hum Genet. 2004;75:504–507. doi: 10.1086/423790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin JE, Alizadeh BZ, Gonzalez-Gay MA, Balsa A, Pascual-Salcedo D, Gonzalez-Escribano MF, Rodriguez-Rodriguez L, Fernandez-Gutierrez B, Raya E, Coenen MJ, van RP, Radstake TR, Kvien TK, Viken MK, Lie BA, Koeleman BP, Martin J. Evidence for PTPN22 R620W polymorphism as the sole common risk variant for rheumatoid arthritis in the 1p13.2 region. J Rheumatol. 2011;38:2290–2296. doi: 10.3899/jrheum.110361. [DOI] [PubMed] [Google Scholar]
- 28.Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, Zhang CK, Boucher G, Ripke S, Ellinghaus D, Burtt N, Fennell T, Kirby A, Latiano A, Goyette P, Green T, Halfvarson J, Haritunians T, Korn JM, Kuruvilla F, Lagace C, Neale B, Lo KS, Schumm P, Torkvist L, Dubinsky MC, Brant SR, Silverberg MS, Duerr RH, Altshuler D, Gabriel S, Lettre G, Franke A, D’Amato M, McGovern DP, Cho JH, Rioux JD, Xavier RJ, Daly MJ. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 2011;43:1066–1073. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Onengut-Gumuscu S, Buckner JH, Concannon P. A haplotype-based analysis of the PTPN22 locus in type 1 diabetes. Diabetes. 2006;55:2883–2889. doi: 10.2337/db06-0225. [DOI] [PubMed] [Google Scholar]
- 30.Cloutier JF, Veillette A. Cooperative inhibition of T-cell antigen receptor signaling by a complex between a kinase and a phosphatase. J Exp Med. 1999;189:111–121. doi: 10.1084/jem.189.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hasegawa K, Martin F, Huang G, Tumas D, Diehl L, Chan AC. PEST domain-enriched tyrosine phosphatase (PEP) regulation of effector/memory T cells. Science. 2004;303:685–689. doi: 10.1126/science.1092138. [DOI] [PubMed] [Google Scholar]
- 32.Vang T, Congia M, Macis MD, Musumeci L, Orru V, Zavattari P, Nika K, Tautz L, Tasken K, Cucca F, Mustelin T, Bottini N. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet. 2005;37:1317–1319. doi: 10.1038/ng1673. [DOI] [PubMed] [Google Scholar]
- 33.Zikherman J, Hermiston M, Steiner D, Hasegawa K, Chan A, Weiss A. PTPN22 deficiency cooperates with the CD45 E613R allele to break tolerance on a non-autoimmune background. J Immunol. 2009;182:4093–4106. doi: 10.4049/jimmunol.0803317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang J, Zahir N, Jiang Q, Miliotis H, Heyraud S, Meng X, Dong B, Xie G, Qiu F, Hao Z, McCulloch CA, Keystone EC, Peterson AC, Siminovitch KA. The autoimmune disease-associated PTPN22 variant promotes calpain-mediated Lyp/Pep degradation associated with lymphocyte and dendritic cell hyperresponsiveness. Nat Genet. 2011;43:902–907. doi: 10.1038/ng.904. [DOI] [PubMed] [Google Scholar]
- 35*.Habib T, Funk A, Rieck M, Brahmandam A, Dai X, Panigrahi AK, Luning Prak ET, Meyer-Bahlburg A, Sanda S, Greenbaum C, Rawlings DJ, Buckner JH. Altered B cell homeostasis is associated with type I diabetes and carriers of the PTPN22 allelic variant. J Immunol. 2012;188:487–496. doi: 10.4049/jimmunol.1102176. Transitional B cells represent a poised autoreactive reservoir, and this study finds an accumulation of such cells in association with the PTPN22 susceptibility gene, correlating with defective BCR signaling that fails to promote the further maturation of these cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36**.Menard L, Saadoun D, Isnardi I, Ng YS, Meyers G, Massad C, Price C, Abraham C, Motaghedi R, Buckner JH, Gregersen PK, Meffre E. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J Clin Invest. 2011;121:3635–3644. doi: 10.1172/JCI45790. Using a molecular assay for autoreactive B cell repertoires, this study also identifies the transitional stage of B cell development as a defective checkpoint associated with the PTPN22 susceptibility gene. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Concannon P, Rich SS, Nepom GT. Genetics of type 1A diabetes. N Engl J Med. 2009;360:1646–1654. doi: 10.1056/NEJMra0808284. [DOI] [PubMed] [Google Scholar]
- 38.Simoncic PD, Bourdeau A, Lee-Loy A, Rohrschneider LR, Tremblay ML, Stanley ER, McGlade CJ. T-cell protein tyrosine phosphatase (Tcptp) is a negative regulator of colony-stimulating factor 1 signaling and macrophage differentiation. Mol Cell Biol. 2006;26:4149–4160. doi: 10.1128/MCB.01932-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F, Lowe CE, Szeszko JS, Hafler JP, Zeitels L, Yang JH, Vella A, Nutland S, Stevens HE, Schuilenburg H, Coleman G, Maisuria M, Meadows W, Smink LJ, Healy B, Burren OS, Lam AA, Ovington NR, Allen J, Adlem E, Leung HT, Wallace C, Howson JM, Guja C, Ionescu-Tirgoviste C, Simmonds MJ, Heward JM, Gough SC, Dunger DB, Wicker LS, Clayton DG. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dendrou CA, Plagnol V, Fung E, Yang JH, Downes K, Cooper JD, Nutland S, Coleman G, Himsworth M, Hardy M, Burren O, Healy B, Walker NM, Koch K, Ouwehand WH, Bradley JR, Wareham NJ, Todd JA, Wicker LS. Cell-specific protein phenotypes for the autoimmune locus IL2RA using a genotype-selectable human bioresource. Nat Genet. 2009;41:1011–1015. doi: 10.1038/ng.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Genome-wide association study of 14,000 cases of seven common diseases and 3, 000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42*.Long SA, Cerosaletti K, Wan JY, Ho JC, Tatum M, Wei S, Shilling HG, Buckner JH. An autoimmune-associated variant in PTPN2 reveals an impairment of IL-2R signaling in CD4(+) T cells. Genes Immun. 2011;12:116–125. doi: 10.1038/gene.2010.54. Measurements of STAT5 phosphorylation show variation in the population, with diminished signaling in subjects who carry the PTPN2 allele associated with T1D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43*.Garg G, Tyler JR, Yang JH, Cutler AJ, Downes K, Pekalski M, Bell GL, Nutland S, Peakman M, Todd JA, Wicker LS, Tree TI. Type 1 diabetes-associated IL2RA variation lowers IL-2 signaling and contributes to diminished CD4+CD25+ regulatory T cell function. J Immunol. 2012;188:4644–4653. doi: 10.4049/jimmunol.1100272. CD25-associated SNPs are found to diminish signals through the IL-2 receptor, correlating with a relative reduction in suppressive function of CD25+ T cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44*.Long SA, Cerosaletti K, Bollyky PL, Tatum M, Shilling H, Zhang S, Zhang ZY, Pihoker C, Sanda S, Greenbaum C, Buckner JH. Defects in IL-2R signaling contribute to diminished maintenance of FOXP3 expression in CD4(+)CD25(+) regulatory T-cells of type 1 diabetic subjects. Diabetes. 2010;59:407–415. doi: 10.2337/db09-0694. Regulatory T cell function is impaired in subjects with low phosphorylation of STAT5 following IL-2 receptor signaling, a consequence of the PTPN2 genetic variant associated with T1D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rieck M, Arechiga A, Onengut-Gumuscu S, Greenbaum C, Concannon P, Buckner JH. Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes. J Immunol. 2007;179:4704–4710. doi: 10.4049/jimmunol.179.7.4704. [DOI] [PubMed] [Google Scholar]