

Abstract
Compelling evidence shows that chemokine CXCL12 drives metastasis in multiple malignancies. Similar to other key cytokines in cancer, CXCL12 exists as several isoforms with distinct biophysical properties that may alter signaling and functional outputs. However, effects of CXCL12 isoforms in cancer remain unknown. CXCL12-α, β, and γ showed cell-type specific differences in activating signaling through G protein-dependent pathways in cell-based assays, while CXCL12-γ had greatest effects on recruitment of the adapter protein β-arrestin 2. CXCL12-β and γ also stimulated endothelial tube formation to a greater extent than CXCL12-α. To investigate effects of CXCL12 isoforms on tumor growth and metastasis, we used a mouse xenograft model of metastatic human breast cancer combining CXCR4+ breast cancer cells and mammary fibroblasts secreting an isoform of CXCL12. While all CXCL12 isoforms produced comparable growth of mammary tumors, CXCL12-γ significantly increased metastasis to bone marrow and other sites. Breast cancer cells originating from tumors with CXCL12-γ fibroblasts upregulated RANKL, contributing to bone marrow tropism of metastatic cancer cells. CXCL12-γ was expressed in metastatic tissues in mice, and we also detected CXCL12-γ in malignant pleural effusions from patients with breast cancer. In our mouse model, mammary fibroblasts disseminated to sites of breast cancer metastases, providing another mechanism to increase levels of CXCL12 in metastatic environments. These studies identify CXCL12-γ as a potent pro-metastatic molecule with important implications for cancer biology and effective therapeutic targeting of CXCL12 pathways.
Keywords: chemokine, bioluminescence, imaging, bone marrow
Introduction
Paget's “seed and soil” hypothesis of metastasis proposes that interactions between malignant cells and stromal environments control trafficking, survival, and proliferation of disseminated tumor cells. Differences among organs, including cytokines, stromal cells, and extracellular matrix, contribute to specific patterns of metastasis for various types of cancers (1, 2). Studies also show that cells in a primary tumor actively regulate metastatic potential of malignant cells and secondary organs (3-5). Cytokines in a primary tumor may select for cancer cells with tropism for specific sites and/or enhance angiogenesis as a route for vascular intravasation (6) (7). Stromal cells from a primary tumor may disseminate with cancer cells, directly remodeling secondary organs to promote metastasis (8). Through processes including secreted signaling molecules, extracellular vesicles, and/or recruitment of additional cell types, primary tumors also indirectly remodel organs to generate pre-metastatic niches (9, 10). Understanding mechanisms of metastasis is critical to effective use of approved drugs and development of new treatments for metastatic disease, which causes death of ≈ 90% of patients with cancer.
Chemokine CXCL12 and its receptor CXCR4 govern primary and metastatic tumor microenvironments and organ-specific metastases in breast cancer and over 20 different malignancies (11) (12). Elevated levels of CXCL12 in common sites of metastatic breast cancer, such as bone marrow, lung, and liver, are postulated to attract CXCR4+ cancer cells and/or facilitate cell survival and proliferation (13). Inhibiting CXCL12-CXCR4 signaling reduces metastases in mouse models, reinforcing functions of this signaling pathway in the metastatic process (14-16). Beyond chemotaxis and proliferation of CXCR4+ cancer cells, CXCL12 secreted by carcinoma associated fibroblasts stimulates angiogenesis, which is essential to metastasis (6, 17). In addition, CXCL12 in a primary tumor may select for cancer cells capable of metastasizing, highlighting effects of CXCL12 to drive tumor evolution (7). These observations underscore a critical role of CXCL12-CXCR4 signaling in metastatic cancer.
Studies of CXCL12 typically focus solely on CXCL12-α. However, like vascular endothelial growth factor (VEGF), TGF-β, and many other important cytokines in cancer, there are multiple isoforms of CXCL12 (18) (19). Mice, rats, and humans have three CXCL12 isoforms (α, β, and γ), and humans have three additional minor isoforms (δ, ε, and φ). Amino acids in human and mouse CXCL12-α, β, and γ are 99% identical, allowing inter-species signaling with no known barriers (20). All CXCL12 isoforms share the same N-terminal 68 amino acids, which comprise the entirety of CXCL12-α; β and γ isoforms add an additional four and 30 carboxy-terminal amino acids, respectively (21). While all isoforms bind heparan sulfate through a BBXB motif (B, basic amino acid; X, any amino acid) in the core of the protein, the carboxy-terminal extensions of CXCL12-β and CXCL12-γ add one or three more BBXB motifs, respectively. Additional BBXB motifs in CXCL12-γ confer one of the highest known binding affinities for the extracellular matrix molecule heparan sulfate (0.9 nM) (22). Compared with CXCL12-α, CXCL12-γ has substantially lower affinity for CXCR4 (15 versus 350 nM) (22). CXCL12 isoforms show distinct tissue- and time-dependent expression under physiologic and pathologic conditions, suggesting unique outputs of each isoform (23, 24). Functions of CXCL12 isoforms in primary and metastatic cancer remain unknown.
To investigate CXCL12-α, β, and γ in tumor growth and metastasis, we expressed individual isoforms of each chemokine in human mammary fibroblasts that were co-implanted with CXCR4+ human breast cancer cells in an orthotopic mouse model. This model reproduces primary human breast cancer in which carcinoma-associated fibroblasts secrete CXCL12 (6). Fibroblasts secreting CXCL12-γ increased metastasis of CXCR4+ breast cancer cells to multiple sites including bone marrow without altering overall growth of the orthotopic tumor. Bone marrow metastatic breast cancer cells from tumors with CXCL12-γ fibroblasts upregulated RANKL, a molecule associated with tropism of cancer cells for bone (25). We show that orthotopically-implanted mammary fibroblasts co-localize to sites of breast cancer metastases, providing another mechanism by which tumors regulate metastatic microenvironments. Furthermore, we demonstrate expression of CXCL12 isoforms in human breast cancer metastases, indicating that differences among isoforms of this chemokine extend to human disease.
Results
Isoform-specific activation of CXCR4 signaling
We analyzed pathways downstream of G protein signaling (ERK1/2, AKT, and inhibition of cAMP) and recruitment of the adapter protein β-arrestin 2. We used MDA-MB-231 breast cancer cells, a well-established model of triple negative breast cancer, transduced stably with CXCR4 (231-CXCR4) for all signaling assays except activation of ERK1/2. Mutant KRas in MDA-MB-231 cells constitutively activates ERK1/2, so we used 293T-CXCR4 cells for this assay. We used CXCL12 isoforms secreted from either stably transduced 293T cells or immortalized human mammary fibroblasts to link our cell-based studies to mouse models. We fused each isoform to Gaussia luciferase (GL) so we readily could quantify isoforms and use equal amounts for assays. The GL fusion also enables sensitive detection of cells secreting different isoforms of CXCL12 ex vivo. We previously demonstrated that CXCL12-α, β, and γ maintain signaling activity when fused to GL (26, 27).
We treated 231-CXCR4 or 293T-CXCR4 cells with equal amounts of each CXCL12 isoform (≈ 15 ng/ml) and measured activation of AKT and ERK1/2 by Western blotting. For 231-CXCR4 cells, CXCL12-α and -β produced comparable activation of AKT, while CXCL12-γ produced only minimal phosphorylation (Fig 1A, B). We observed a similar pattern for 293T-CXCR4 cells with ERK1/2 (Fig 1C, D). By comparison, all CXCL12 isoforms stimulated only modest activation of AKT in 293T-CXCR4 cells with slightly higher AKT phosphorylation from CXCL12-γ (Fig S1). These results demonstrate pathway-specific responses to CXCL12 isoforms among different cell types and within a single cell type. To measure inhibition of cAMP, we treated 231-CXCR4 cells with forskolin without or with an isoform of CXCL12 (≈ 15 ng/ml). CXCL12-α blocked the forskolin-dependent increase in cAMP by ≈ 85%, while CXCL12-β and -γ were less effective (Fig 1E). We also measured recruitment of the adapter protein β-arrestin 2 to CXCR4, which causes receptor internalization to endosomes and initiates arrestin-biased signaling. Using a luciferase complementation assay, we showed that CXCL12-α and -β increased association of CXCR4 and β-arrestin 2 by only two-fold, while CXCL12-γ enhanced the signal by almost four-fold (Fig 1F (28)). These data demonstrate an isoform-specific dichotomy with CXCL12-α and β generally activating G protein pathways to a greater extent than CXCL12-γ with reversal of the pattern for β-arrestin 2.
Figure 1. Preferential activation of CXCR4 G protein versus β-arrestin 2 pathways by CXCL12 isoforms.
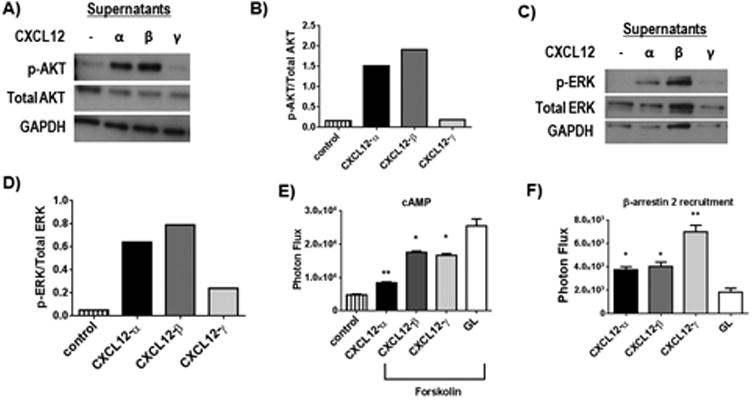
A) Western blot shows phosphorylation of AKT (p-AKT) in 231-CXCR4 cells treated with equal amounts of cell supernatants with CXCL12-α, β, γ or control for 10 minutes. Total AKT and GAPDH are shown as loading controls. B) Quantified data from panel A for ratios of p-AKT to total AKT. C) Western blot shows phosphorylation of ERK1/2 (p-ERK) in 239T-CXCR4 cells treated for 5 minutes with various CXCL12 isoforms as shown. Total ERK and GAPDH were used as loading controls. D) Graph depicts quantified data from C for ratios of phosphorylated to total ERK. E) 231-CXCR4 cells with a luciferase biosensor for cAMP were treated with forskolin to increase cAMP in the presence of listed CXCL12 isoforms or Gaussia luciferase (GL) as a control. Graph shows mean values + SEM (n = 4 per condition). Greater activation of CXCR4 corresponds with lower levels of cAMP present in control cells not treated with forskolin. F) 231 cells expressing a luciferase complementation reporter for recruitment of β-arrestin 2 to CXCR4 were treated with equal amounts of a CXCL12 isoform or GL control. Graph shows mean values +SEM for each condition with higher photon flux values corresponding with greater interaction between CXCR4 and β-arrestin 2. *, p < 0.05; **, p < 0.01.
CXCL12-β and CXCL12-γ enhance angiogenesis in vitro
CXCL12-CXCR4 signaling promotes angiogenesis under physiologic and multiple pathologic conditions (29, 30). To test effects on angiogenesis, we quantified endothelial tube formation in co-cultures of human umbilical vein endothelial cells (HUVEC) and 293T cells secreting an isoform of CXCL12 fused to GL. Control 293T cells secreted unfused GL. CXCL12-β and -γ stimulated significantly more endothelial tube formation than control 293T cells (p < 0.05) (Fig 2A-C), while differences between CXCL12-α and control were not significant. We observed similar results when we treated HUVECs with 100 ng/ml recombinant CXCL12-α, β, or γ and omitted 293T cells, although recombinant CXCL12-γ drove greater tube formation than other isoforms (Fig S2). These data show enhanced effects of CXCL12-β and CXCL12-γ to drive angiogenesis in vitro.
Figure 2. CXCL12-β and CXCL12-γ promote endothelial tube formation in vitro.
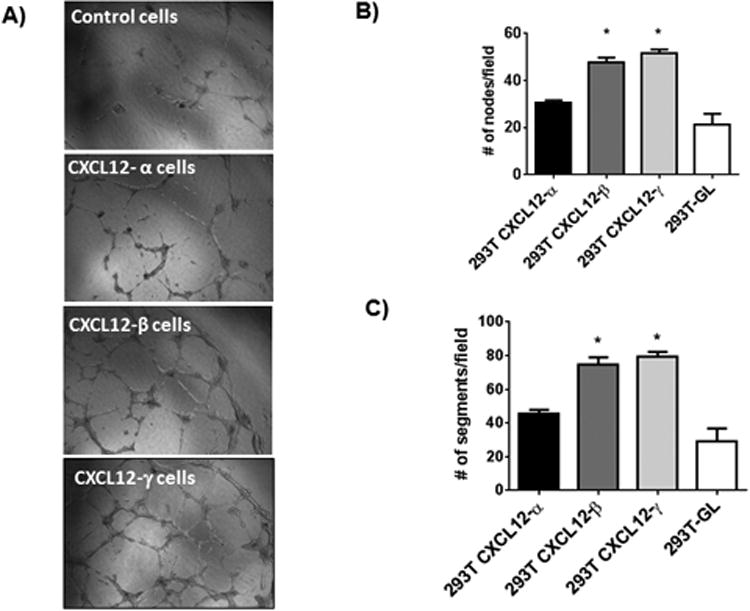
A) Representative photographs of tube formation by HUVECs co-cultured with 293T cells secreting CXCL12-α, β, or γ or GL control. B, C) Graphs show mean values + SEM for numbers of nodes (B) and segments (C) formed by endothelial cells under each experimental condition. *, p < 0.05.
Tumor-associated fibroblasts secreting CXCL12-γ promote metastasis of CXCR4+ breast cancer cells
We recently demonstrated that CXCL12-α, β, and γ are detectable in orthotopic xenograft and syngeneic mouse models of breast cancer (27). These isoforms also were present in human primary breast cancers with relative frequencies of CXCL12-α = CXCL12-β> CXCL12-γ with the latter only expressed in advanced stage cancers. To investigate tumor growth and metastasis, we co-implanted 231-CXCR4 breast cancer cells with human mammary fibroblasts secreting CXCL12-α, CXCL12-β, CXCL12-γ, or unfused GL control cells as orthotopic tumor xenografts in NSG mice (31). Based on in vitro bioluminescence from GL fusions with each isoform, human mammary fibroblasts transduced with CXCL12-α, β, or γ secreted approximately 4.5, 5, and 1 ng/ml of chemokine, respectively. 231-CXCR4 cells also expressed firefly luciferase for bioluminescence imaging.
Imaging data and tumor weights showed that the type of co-implanted human mammary fibroblasts did not alter growth of 231-CXCR4 cells in mammary tumors (Fig 3A, B). Excised tumors showed comparatively more CD31+ blood vessels in tumors with human mammary fibroblasts secreting CXCL12-γ, and these tumors also had reduced staining for cleaved caspase 3, a marker of apoptosis (Fig 4A-C). However, we did not observe differences in cell proliferation as assessed by immunohistochemistry for Ki67. These data establish that CXCL12-γ alters angiogenesis and cell survival in the tumor environment, even though overall tumor growth was unaffected.
Figure 3. CXCL12 isoforms do not alter growth of primary tumor xenografts.

A) Mice with orthotopic tumor xenografts of 231-CXCR4 human breast cancer cells and listed human mammary fibroblasts secreting a CXCL12 isoform or GL control were imaged over 40 days. Graph shows mean values + SEM for photon flux (n = 8 per group). B) Combined weights of two tumor xenografts per mouse from animals along with listed types of human mammary fibroblasts. Graphs shows mean values + SEM (n = 8 each).
Figure 4. CXCL12-γ promotes tumor angiogenesis and limits apoptosis in orthotopic breast cancer xenografts.
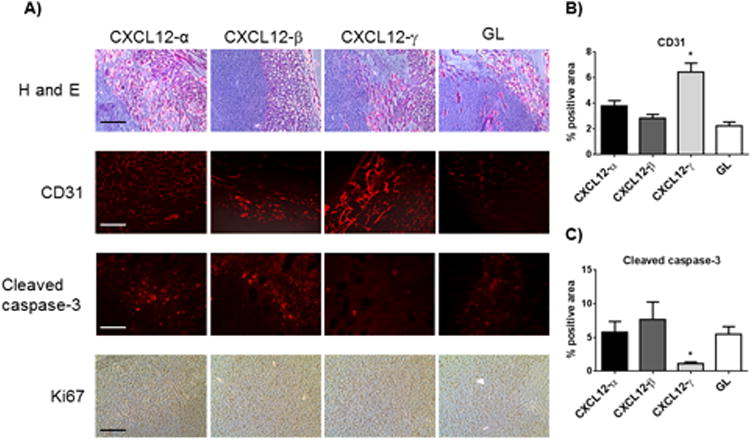
A) Images show representative histology and immunostaining from orthotopic tumors of 231-CXCR4 cells and listed human mammary fibroblasts. Rows display images from staining with hematoxylin and eosin (H and E), CD31 tumor endothelial cells, cleaved caspase-3 for apoptosis, and immunohistochemistry for Ki67 as a marker of proliferation. B, C) Graphs show quantified data for area occupied by staining for CD31 (B) or cleaved caspase-3 (C), respectively. Each bar represents data from 4 randomly selected fields per mouse and eight mice total. *, p < 0.05.
Since a primary tumor environment can control metastasis, we also quantified total and site-specific metastases 42 days after implanting tumors. Mice with implants of 231-CXCR4 cells and human mammary fibroblasts secreting CXCL12-γ had significantly more metastases measured by region-of-interest analysis of the entire animal and multiple anatomic sites (Fig 5A-C) (p < 0.01). We also quantified relative numbers of viable 231-CXCR4 cancer cells in bone marrow by ex vivo bioluminescence, revealing 231-CXCR4 cells in bone marrow of 81% of mice with CXCL12-γ fibroblasts and 13-27% of all other human mammary fibroblasts (Table 1). These data show that expression of CXCL12-γ by fibroblasts in an orthotopic tumor implant dramatically increases breast cancer metastasis.
Figure 5. CXCL12-γ promotes metastasis of CXCR4+ breast cancer cells.
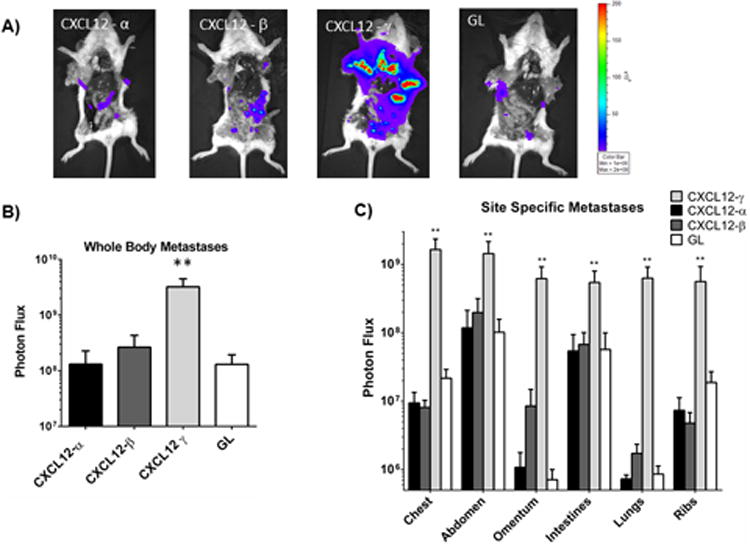
A) Representative bioluminescence images of euthanized mice show metastases in each mouse. Labels correspond with the type of human mammary fibroblast implanted orthotopically with 231-CXCR4 cells. Pseudocolor scale shows blue as lowest and red as highest values for photon flux. B) Graph shows mean values + SEM for total body metastases in each group (n = 8 each). **, p < 0.01. C) Graph depicts mean values + SEM for metastatic 231-CXCR4 cells in specific anatomic sites. All values for CXCL12-γ are significantly greater (**, p < 0.01) than all other conditions.
Table 1.
Bone marrow metastases (cumulative data from 4 independent experiments with CXCL12-α, CXCL12-γ, and GL fibroblasts; 2 experiments with CXCL12-β fibroblasts).
| Fibroblast | Number with metastases/Total | % with Metastases |
|---|---|---|
| CXCL12-α | 7/27 | 26 |
| CXCL12-β | 2/15 | 13 |
| CXCL12-γ | 21/26 | 81 |
| GL | 7/26 | 27 |
CXCL12-γ expression in human breast cancer metastases
To link these studies with human breast cancer, we analyzed CXCL12 isoforms in total cells recovered from malignant pleural effusions in patients with metastatic breast cancer. By RT-PCR we identified CXCL12-α, β, and/or γ in some patients with CXCL12-α and CXCL12-β present more commonly (Table 2, Fig S3). Since malignant pleural effusions contain a variety of cell types, these analyses did not define sources of CXCL12. Nevertheless, the results show that CXCL12-γ may be expressed in human metastatic breast cancer, suggesting that this isoform contributes to functions of CXCL12-CXCR4 signaling in metastasis.
Table 2. RT-PCR detection of CXCL12 isoforms in metastatic pleural effusions from patients.
| Isoform | Number with detectable isoform/Total |
|---|---|
| CXCL12-α | 6/9 |
| CXCL12-β | 7/9 |
| CXCL12-γ | 3/9 |
CXCL12-γupregulates RANK ligand (RANKL) in bone marrow metastatic breast cancer cells
Bone is the most common site of metastatic breast cancer with disseminated tumor cells in bone marrow progressing to osteolytic or osteoblastic metastases through a multi-step process. Given associations of CXCL12-CXCR4 with bone metastases, we further investigated processes by which CXCL12-γ increases the frequency of 231-CXCR4 cells in bone marrow. We initially analyzed expression of CXCL12-α, β, and γ in total populations of bone marrow cells recovered from NSG mice without tumor xenografts. By qRT-PCR we detected these three isoforms in bone marrow with relative abundance of CXCL12-α > β > γ (Fig 6A). These data suggest that higher levels of CXCL12-γ in bone marrow do not account for greater metastasis of 231-CXCR4 cells in this site.
Figure 6. CXCL12-γ upregulates RANKL in bone marrow metastatic 231-CXCR4 cells.
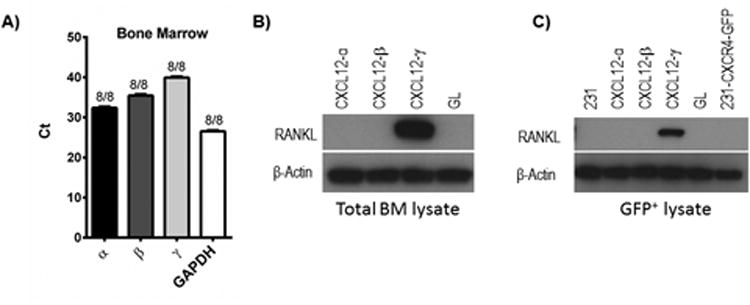
A) QRT-PCR data for expression of CXCL12 isoforms and GAPDH in total bone marrow cells collected from non-tumor bearing NSG mice. Numbers denote numbers of mice with detectable expression of each target mRNA and total numbers of mice. Ct value on Y-axis designates PCR cycle number at which amplification exceeds threshold. B) Western blot for RANKL in lysates of total bone marrow cells recovered from mice with orthotopic tumor implants of 231-CXCR4 cells and listed human mammary fibroblasts. β-actin is shown as a loading control. C) Bone marrow cells were sorted by flow cytometry for GFP+ breast cancer cells and then lysed for analysis by Western blot as in panel B. 231 cells and 231-CXCR4-GFP cells represent lysates from cells maintained only in cell culture.
We next investigated expression of RANKL, a molecule with multiple functions in bone metastasis. Cancer cells that metastasize to bone have higher levels of RANKL than cells that metastasize to other sites, suggesting that RANKL and its receptor RANK promote trafficking and/or survival of cancer cells in bone marrow (32). RANKL also activates osteoclasts, resulting in bone resorption and lytic metastases (33). We recovered total bone marrow cells from tumor bearing mice and expanded cells in culture for one week. After this time, we prepared cell lysates from total bone marrow cells or sorted cells by flow cytometry to separate GFP+ 231-CXCR4 cells from remaining bone marrow cells. Western blotting of lysates from total bone marrow cells revealed marked upregulation of RANKL only in mice with CXCL12-γ human mammary fibroblasts in tumor xenografts (Fig 6B). Lysates from sorted cell populations showed RANKL in 231-CXCR4 cells recovered from bone marrow of mice with CXCL12-γ human mammary fibroblasts in xenografts (Fig 6C). We did not detect RANKL in normal bone marrow cells from mice with any other type of human mammary fibroblast in tumor xenografts, metastases from other sites, or 231 cells maintained solely in culture (Fig 6C and data not shown). RANKL also was undetectable in 231-CXCR4 cells cultured in vitro with any CXCL12 isoform (data not shown). These data suggest that CXCL12-γ in combination with other molecules in bone marrow upregulates persistent expression of RANKL in 231-CXCR4 cells, providing a potential mechanism for greater frequency of bone marrow metastases.
Tumor-associated fibroblasts at sites of metastasis
In addition to local effects in an orthotopic tumor, stromal cells may increase metastasis by trafficking with malignant cells to secondary organs (8). We capitalized on the GL fusion to isoforms of CXCL12 or unfused GL itself to test the hypothesis that human mammary fibroblasts from a primary tumor implant entered the circulation and arrived at sites with 231-CXCR4 metastases. From mice with xenografts of 231-CXCR4 cells and CXCL12-α, CXCL12-γ, or GL human mammary fibroblasts, we collected blood, bone marrow, and lung metastases with surrounding normal tissue. We cultured cells for one week and then measured bioluminescence from GL in culture supernatants. These assay conditions require cells to be present in a given tissue sample; adhere and survive in culture; and secrete GL above background bioluminescence from cultures of these same tissues and organs obtained from normal NSG mice.
We detected GL bioluminescence in cells cultured from blood, lung, and bone marrow samples from mice with each type of implanted human mammary fibroblast (Fig 7A-C). Signals from CXCL12-α and CXCL12-γ were higher than GL in bone marrow, while all were comparable in blood and lung metastases. To verify these data, we also performed RT-PCR on cDNA prepared from total cells from each culture. We identified human CXCL12 or unfused GL in samples that showed GL bioluminescence (Fig S4). These data demonstrate that human mammary fibroblasts implanted in a tumor xenograft disseminate to organs with metastatic breast cancer cells.
Figure 7. Human mammary fibroblasts disseminate to sites of 231-CXCR4 metastases.
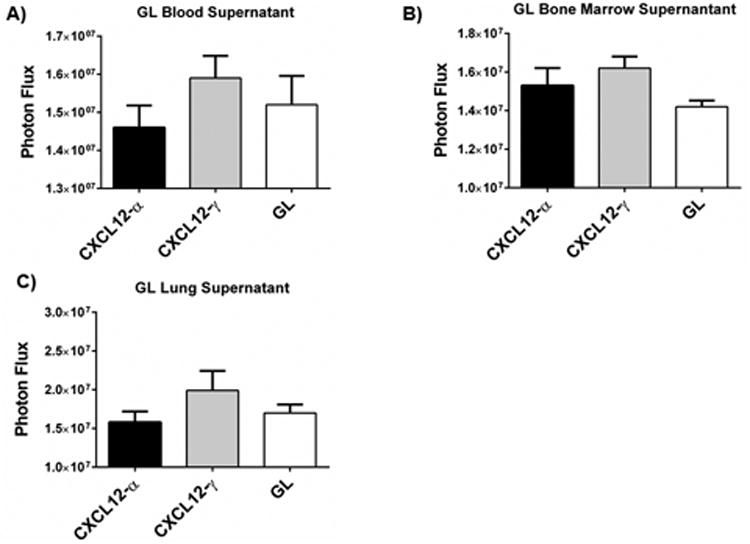
A-C) We recovered blood (A), bone marrow (B), and lung (C) metastases from mice with CXCL12-α, CXCL12-γ, or GL fibroblasts implanted in orthotopic tumors. We quantified Gaussia luciferase activity in supernatants from these cultures and graphed mean values + SEM (n = 4-6 each). Photon flux values from the non-tumor bearing mice were subtracted from presented values.
Discussion
Studies of CXCL12 in normal physiology, cancer, and other diseases typically focus on the α isoform or do not specify an isoform. However, isoforms of CXCL12 have differing potencies and functions in processes such as angiogenesis and chemotaxis. CXCL12-β stimulates proliferation and limits death of cultured endothelial cells to a greater extent than CXCL12-α (30). Mice engineered to express only CXCL12-α have significantly reduced angiogenesis in response to leg ischemia, and these defects only can be rescued by wild-type CXCL12-γ (34). While CXCL12-γ drives substantially less cell migration than other isoforms in standard cell-based assays, this isoform much more potently stimulates migration of leukocytes in mice (35)(36). These results highlight functional differences among CXCL12 isoforms in processes relevant to cancer biology and metastasis.
Our study advances knowledge of CXCL12 isoforms, particularly CXCL12-γ, metastasis. Using orthotopic tumor xenografts combining CXCR4+ breast cancer cells and human mammary fibroblasts, we show for the first time that mice develop significantly more metastases from tumors with fibroblasts secreting CXCL12-γ than other isoforms of CXCL12 or a control protein. Our results showing emphasize effects of a primary tumor to enrich for cancer cells with enhanced metastatic potential. A recent study determined that CXCL12 produced by carcinoma-associated fibroblasts in triple negative breast cancers selected for malignant cells with tropism for bone marrow (7). This study did not distinguish among isoforms of CXCL12, and metastases increased only in bone marrow. Despite differences in overall distribution of metastases between studies, our data suggest that CXCL12-γ in a mammary tumor may enrich for breast cancer cells with the ability to metastasize to sites with increased levels of CXCL12. Combined with our recent discovery that CXCL12-γ is expressed in syngeneic and human xenograft breast cancers in mice and subsets of primary human breast tumors in patients, these data suggest that CXCL12-γcould be a marker for aggressive breast cancer (27).
Given central functions of CXCL12 in breast cancer metastasis to bone and the frequency of metastatic disease at this site, we investigated how CXCL12-γ enhances bone marrow metastases relative to other isoforms. CXCL12-γ, but not other isoforms of this chemokine, caused bone-metastatic, CXCR4+ breast cancer cells to express RANKL. Previous studies establish that CXCL12 signaling drives expression of RANKL on normal cells exposed to chronic inflammation and malignant cells, and interactions with stromal cells also may upregulate RANKL on cancer cells (37, 38). RANKL on breast cancer cells has been associated with bone metastases (25), providing a mechanism for CXCL12-γ-dependent increases in bone marrow metastasis of CXCR4+ cancer cells.
CXCR4 on breast cancer cells often increases tumor growth at the primary tumor site as well as promoting metastases. While we observed relatively increased angiogenesis and reduced apoptosis in tumors with CXCL12-γ fibroblasts, we did not measure any significant effect of CXCL12 isoforms on overall size of orthotopic tumors. Potentially, CXCL12 did not increase tumor growth because of a lack of CXCR7 cells in tumors. Human breast cancers commonly contain malignant cells expressing either CXCR4 or CXCR7, and we have shown that CXCR7+ cells promote growth of CXCR4+ breast cancer cells in tumors (39). CXCR7 scavenges CXCL12 from the extracellular space, preventing desensitization of CXCR4 signaling (40, 41). At least under some conditions, CXCR7 also may contribute to tumor growth by directly activating signaling (42)(43). The fact that CXCL12-γ did not alter growth of orthotopic tumors in our mouse model establishes that larger tumors do not account for more metastases in these mice.
While concentrations of CXCL12 produced by human mammary fibroblasts are lower than typically used in cell culture assays, these amounts are comparable to total CXCL12 in serum (0.5 – 2 ng/ml) and greater than present in murine bone marrow (0.3-0.4 ng/ml) (44-49). Particularly for CXCL12-γ, local concentrations of CXCL12 in sites such as mammary fat pads may be higher because of binding to extracellular matrix molecules, although exact levels are not known. We note that effects of individual CXCL12 isoforms produced by implanted human mammary fibroblasts are superimposed upon endogenous, modest-to-low levels of CXCL12-α, β, and γ in mammary tumors in this xenograft system (27). Mechanisms that regulate changes in expression of endogenous isoforms in tumor environments will require additional investigation.
In addition to functional differences among CXCL12 isoforms, our data demonstrate that mammary fibroblasts travel to secondary sites. We detected fibroblasts several weeks after initial implantation, implying that these cells persisted in metastatic sites for extended periods of time. While our study did not determine if fibroblasts traveled to metastatic sites along with cancer cells or independently, prior research by Duda et al showed that cancer cells and fibroblasts metastasized together as aggregates (8). Co-metastasizing stromal cells also increased growth of disseminated cancer cells in secondary sites. We did not preferentially identify fibroblasts secreting CXCL12-γ in blood or metastases as compared with cells secreting CXCL12-α or GL, so pro-metastatic effects of CXCL12-γ are not due to greater numbers of these stromal cells in secondary sites. Therefore, CXCL12-γ from disseminated mammary fibroblasts may boost levels of this isoform already present in metastatic environments to reduce apoptosis and promote angiogenesis as observed in mammary tumors.
This study identifies CXCL12-γ as a potent driver of metastasis. Since CXCL12-γ preferentially activates recruitment of β-arrestin 2 relative to G protein pathways as compared with CXCL12-α and -β, these results suggest that arrestin-dependent signaling regulates pro-metastatic effects of CXCL12-γ. We currently are further investigating how differences in CXCL12-γ-CXCR4 signaling relative to other isoforms affect metastasis. Defining functions of specific isoforms of CXCL12 in pre-clinical and clinical studies, rather than considering this chemokine as a single entity, will advance understanding of CXCL12 in cancer and other diseases. Specific isoforms of CXCL12 also may provide new prognostic and predictive biomarkers for breast cancer and other malignancies (50)(51). It also will be important to test efficacy of existing and new CXCL12-CXCR4 inhibitors against different isoforms of this chemokine. This strategy will maximize success of ongoing efforts to translate agents targeting CXCL12 and/or CXCR4 to cancer therapy.
Materials and Methods
Cells
We cultured human umbilical vein endothelial cells (HUVEC) in endothelial basal medium with SingleQuot supplements (Lonza) (52) and other cells in DMEM with 10% fetal bovine serum and 1% glutamine/penicillin/streptomycin (Life Technologies). We previously described 293T (Open Biosystems) and MDA-MB-231 cells (ATCC) stably transduced with CXCR4 (293T-CXCR4 and 231-CXCR4, respectively) (28, 52). For animal studies, we used 231-CXCR4 cells that co-express GFP. We used immortalized human mammary fibroblasts (HMF) stably transduced with CXCL12-α fused to Gaussia luciferase (CXCL12-α-GL) or unfused Gaussia luciferase (GL) (26). We cultured all cells in a 37°C incubator with 5% CO2.
Plasmids and lentiviruses
To measure cAMP levels in intact cells, we used a firefly luciferase-based reporter (pGloSensor-20F, Promega). We excised the reporter with NheI and BamHI and transferred this by blunt end ligation to lentiviral vector FUW (53). We fused Gaussia luciferase (GL) to the C-termini of CXCL12-β and CXCL12-γ at XhoI and NotI sites. We transferred the fusion chemokines to vector FUFP650W by digesting with PacI (54). We generated recombinant lentiviruses through transient transfection of 293T (14). We used flow sorting for eqFP650 to isolate HMF cells with each CXCL12 fusion protein or GL.
Concentrations of CXCL12-GL
We quantified amounts of CXCL12-GL fusions by correlating bioluminescence from each isoform with a standard curve of photon flux versus protein concentration as described (26).
Western blotting
We cultured 231-CXCR4 and 293T-CXCR4 cells overnight in serum free DMEM with 0.2% probumin bovine serum albumin (Millipore). We stimulated cells with equal amounts of bioluminescence for each chemokine isoform (CXCL12-α, β, or γ fused to GL)(26, 55). As a negative control, we used unfused GL. Based on prior studies with CXCL12 fusions to GL, we added ≈15ng/ml CXCL12 to 231-CXCR4 and 293T-CXCR4 cells. We lysed cells after 5 minutes (293T-CXCR4 cells for phosphorylated ERK1/2) or 10 minutes (231-CXCR4 cells for phosphorylated AKT) (26). We detected phosphorylated ERK1/2 or AKT (serine 473) in total cell lysates by Western blotting (26). Blots were stripped and re-probed with total ERK or total AKT and GAPDH (Cell Signaling). We recovered total bone marrow cells from lower extremities of tumor-bearing mice and sorted cells for GFP-positive cancer cells. We probed total cell lysates for RANKL (Biovision) followed by β-actin (Cell Signaling) as a loading control. We quantified relative intensities of bands with ImageJ.
Cell-based bioluminescence assays
To measure CXCL12-dependent effects on intracellular cAMP, we plated 2 × 104 231-CXCR4 cells stably expressing the firefly luciferase cAMP reporter (231-CXCR4-cAMP) in black wall 96 well plates one day before assays. We incubated cells for 10 minutes with equal amounts of CXCL12-α, β, γ; unfused GL; or DMEM with 0.2% probumin (Millipore). We then added 5 μM forskolin (Sigma) to increase intracellular cAMP or vehicle control in combination with 150 μg/ml luciferin (Promega) for imaging on an IVIS 100 (Perkin-Elmer, Waltham, MA, USA). Images (large binning, four minute acquisition) were obtained 12 minutes after adding forskolin.
We quantified recruitment of β-arrestin 2 to CXCR4 in response to different isoforms of CXCL12 fused to GL or GL control as described (28).
Endothelial tube formation
We used HUVECs at passage six or earlier. We coated 96 well plates with growth-factor reduced Matrigel (BD Biosciences) for 30 minutes at 37°C before adding 1.5 × 104 HUVECS in 100 μl EBM basal medium per well. We then added 1 × 104 293T cells expressing an isoform of CXCL12 fused to GL (α, β, or γ) or unfused GL as a control. We also performed experiments in two other formats: 1) adding 100ng/ml of a recombinant CXCL12 isoform (R&D Systems) or vehicle control; or 2) adding bioluminescent CXCL12 isoforms or GL to wells without adding the producing cells. All experimental conditions showed comparable results. We took images of all wells after eight hours of incubation (n = 4 per condition). A person blinded to experimental conditions manually quantified numbers of nodes and segments.
RNA isolation and RT-PCR
We isolated total RNA using Trizol (Life Technologies) followed by purification with RNeasy columns (Qiagen). To eliminate genomic DNA, we performed on column DNaseI digesition prior to eluting RNA from columns. We synthesized cDNA using a Reverse Transcription System (Promega). Primers for gene specific PCR are listed in Table 3.
Table 3. Primers for RT-PCR.
| CXCL12-α (mouse, human) | Forward (5′-3′) | tgcccttcagattgttgcacg |
| Reverse (5′-3′) | ggctgttgtgcttacttgtttaaagc | |
| CXCL12-β (mouse) | Forward (5′-3′) | tgcccttcagattgttgcacg |
| Reverse (5′-3′) | ctgactcacacctctcacatcttg | |
| CXCL12-β (human) | Forward (5′-3′) | tgcccttcagattgttgcacg |
| Reverse (5′-3′) | ggcgtctgaccctctcacatcttg | |
| CXCL12-γ (mouse, human) | Forward (5′-3′) | tgcccttcagattgttgcacg |
| Reverse (5′-3′) | gaactagtttttccttttctgggcagcc | |
| CXCL12 common | Forward (5′-3′) | tgcccttcagattgttgcacg |
| Reverse (5′-3′) | ctccaggtactcttggatccac | |
| Gaussia luciferase | Forward (5′-3′) | none |
| Reverse (5′-3′) | gttctcggtgggcttggc | |
| GAPDH (mouse) | Forward (5′-3′) | tatgtcgtggagtctactggt |
| Reverse (5′-3′) | gagttgtcatatttctcgtgg | |
| GAPDH (human) | Forward (5′-3′) | gaaggtgaaggtcggagt |
| Reverse (5′-3′) | gaagatggtgatgggatttc |
Animal studies
The University of Michigan Committee for the Use and Care of Animals approved all animal procedures. We established orthotopic tumor xenografts in the 4th inguinal mammary fat pads of 6-7 week old female NSG mice (Jackson Laboratory) by co-implanting 5 × 105 231-CXCR4-GFP-FL cells with an equal number of HMF expressing a CXCL12 isoform fused to GL or unfused GL (39). We quantified tumor growth and metastasis by bioluminescence imaging on an IVIS Spectrum (Perkin-Elmer) at approximately weekly intervals (56). We quantified bioluminescence data as photon flux with Living Image software (Perkin Elmer).
When mice were euthanized for tumor burden, we injected animals with luciferin prior to euthanization and then imaged metastases in dissected animals (39). In selected experiments, we collected 0.5 ml blood by cardiac puncture and cultured samples in DMEM growth medium for seven days prior to measuring numbers of viable tumor cells by bioluminescence. Based on bioluminescence imaging, we recovered metastatic 231-CXCR4-GFP-FL cancer cells in lung and liver to extract RNA. As a control, we used comparable weight tissues collected from lung and liver of NSG mice without tumor implants. We harvested bone marrow from both lower extremities and cultured total recovered cells in DMEM growth medium for seven days prior to quantifying numbers of viable cancer cells based on bioluminescence for firefly luciferase. We also quantified bioluminescence for Gaussia luciferase in bone marrow cells and culture supernatants using 1 μg/ml coelenterazine (Promega) (57). We subtracted background bioluminescence from comparable tissue sites obtained from NSG mice without tumors.
Human pleural effusion samples
We obtained malignant pleural effusion samples from women with metastatic breast cancer under a protocol approved by the University of Michigan Institutional Review Board. We centrifuged effusions at 500g × 10 minutes to pellet cells followed by lysis of red blood cells with ammonium chloride. We isolated RNA from total cells in pleural effusions and analyzed expression of CXCL12 isoforms or GADPH by RT-PCR.
Immunostaining
We fixed tumors in 4% buffered formalin overnight and then embedded tissues in paraffin blocks for sectioning. We stained sections with hematoxylin and eosin to define tumor histology. We detected tumor vasculature and apoptosis by staining for CD31 (eBioscience) or cleaved caspase-3 (Cell Signaling) and a fluorescent secondary antibody conjugated to Cy3 (Jackson ImmunoResearch). We used a 40× objective for epifluorescence microscopy of immunofluorescence staining and bright-field images of tissue sections stained by immunohistochemistry. We used ImageJ to quantify the percent area positive for fluorescent staining above background with each antibody on four different fields from one section each from two tumors per group. We identified proliferating cells by immunohistochemistry for Ki67 (Cell Signaling), which we assessed qualitatively.
Statistical analysis
We performed cell culture experiments 3-4 times each, while animal studies were done 2-4 times. We analyzed data using an unpaired T test (GraphPad Prism) with p ≤ 0.05 defining statistically significant differences.
Supplementary Material
Acknowledgments
This work was supported by United States National Institutes of Health grants R01CA136553, R01CA136829, R01CA142750, and P50CA093990. SPC was supported by a NSF predoctoral fellowship. Research also was supported by Fashion Footwear Association of New York (FFANY)/QVC presents Shoes on Sale.
Footnotes
Conflict of Interest: The authors declare no conflicts of interest relevant to this manuscript.
References
- 1.Lu P, Weaver V, Werb Z. The extracellular matrix: A dynamic niche in cancer progression. J Cell Biol. 2012;196(4):395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nguyen D, Bos P, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274–85. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 3.Deng J, Liu Y, Lee H, Herrmann A, Zhang W, C Z, et al. S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Cancer Cell. 2012;21(5):642–54. doi: 10.1016/j.ccr.2012.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peinado H, Aleckovic M, Lovotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18(883-891):883–91. doi: 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sceneay J, Smyth M, Moller A. The pre-metastatic niche: finding common ground. Cancer Metastasis Rev. 2013;32(3-4):449–64. doi: 10.1007/s10555-013-9420-1. [DOI] [PubMed] [Google Scholar]
- 6.Orimo A, Gupta P, Sgroi D, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–48. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 7.Zhang X, Jin X, Malladi S, Zou Y, Wen Y, Brogi E, et al. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell. 2013;154(5):1060–73. doi: 10.1016/j.cell.2013.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duda D, Duyverman A, Kohno M, Snuder M, Steller E, Fukumura D, et al. Malignant cells facilitate lung metastasis by bringing their own soil. Proc Natl Acad Sci USA. 2010;107(50):21677–82. doi: 10.1073/pnas.1016234107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Padua D, Zhang X, Wang Q, Nadal C, Gerald W, Gomis R, et al. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell. 2008;133(1):66–77. doi: 10.1016/j.cell.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erler J, Bennewith K, Cox T, Lang G, Bird D, Koong A, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the pre-metastatic niche. Cancer Cell. 2009;15(1):35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luker K, Luker G. Functions of CXCL12 and CXCR4 in breast cancer. Cancer Lett. 2006;238(1):30–41. doi: 10.1016/j.canlet.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 12.Duda D, Kozin S, Kirkpatrick N, Xu L, Fukumura D, Jain R. CXCL12 (SDF1{alpha}) - CXCR4/CXCR7 Pathway Inhibition: An Emerging Sensitizer for Anti-Cancer Therapies? Clin Cancer Res. 2011;17(8):2074–80. doi: 10.1158/1078-0432.CCR-10-2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanon M, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410(6824):50–6. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 14.Smith M, Luker K, Garbow J, Prior J, Jackson E, Piwnica-Worms D, et al. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004;64(23):8604–12. doi: 10.1158/0008-5472.CAN-04-1844. [DOI] [PubMed] [Google Scholar]
- 15.Zhang S, Han Z, Jing Y, Tao S, Li T, Wang H, et al. CD133+CXCR4+ colon cancer cells exhibit metastatic potential and predict poor prognosis of patients. BMC Medicine. 2012;10:85. doi: 10.1186/1741-7015-10-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jung Y, Kim J, Shiozawa Y, Wang J, Mishra A, Joseph J, et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat Commun. 2013;4:1795. doi: 10.1038/ncomms2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zetter B. Angiogenesis and tumor metastasis. Ann Rev Med. 1998;49:407–24. doi: 10.1146/annurev.med.49.1.407. [DOI] [PubMed] [Google Scholar]
- 18.Guo P, Xu L, Pan S, Brekken R, Yang S, Whitaker G, et al. Vascular endothelial growth factor isoforms display distinct activities in promoting tumor angiogenesis at different anatomic sites. Cancer Res. 2001;61(23):8569–77. [PubMed] [Google Scholar]
- 19.Kloen P, Gebhardt M, Perez-Atayde A, Rosenberg A, Springfield D, Gold L, et al. Expression of transforming growth factor-beta (TGF-beta) isoforms in osteosarcomas: TGF-beta3 is related to disease progression. Cancer. 1997;80(12):2230–9. [PubMed] [Google Scholar]
- 20.Shirozu M, Nakano T, Inazawa J, Tashiro K, Tada H, Shinohara K, et al. Structure and chromosomal localization of the human stromal cell-derived factor 1 (SDF1) gene. Genomics. 1995;28(3):495–500. doi: 10.1006/geno.1995.1180. [DOI] [PubMed] [Google Scholar]
- 21.Yu L, Cecil J, Peng S, Schrementi J, Kovacevic S, Paul D, et al. Identification and expression of novel isoforms of human stromal cell-derived factor 1. Gene. 2006;374:174–9. doi: 10.1016/j.gene.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 22.Laguri C, Sadir R, Rueda P, Baleux F, Gans P, Arenzana-Seisdedos F, et al. The novel CXCL12gamma isoform encodes an unstructured cationic domain which regulates bioactivity and interaction with both glycosaminoglycans and CXCR4. PLoS One. 2007;2(10):e1110. doi: 10.1371/journal.pone.0001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stumm R, Kolodziej A, Schulz S, Kohtz J, Hollt V. Patterns of SDF-1alpha and SDF-1gamma mRNAs, migration pathways, and phenotypes of CXCR4-expressing neurons in the developing rat telencephalon. J Comp Neurol. 2007;502(3):382–99. doi: 10.1002/cne.21336. [DOI] [PubMed] [Google Scholar]
- 24.Gahan J, Gasalbez M, T Y, Young E, Escudero D, Chi A, et al. Chemokine and chemokine receptor expression in kidney tumors: molecular profiling of histological subtypes and association with metastasis. J Urol. 2012;187(3):827–33. doi: 10.1016/j.juro.2011.10.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park H, Min S, Cho H, Kim D, Shin H, Park Y. Expression of osteoprotegerin and RANK ligand in breast cancer bone metastasis. J Korean Med Sci. 2003;18(4):541–6. doi: 10.3346/jkms.2003.18.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luker K, Gupta M, Luker G. Bioluminescent CXCL12 fusion protein for cellular studies of CXCR4 and CXCR7. Biotechniques. 2009;47(1):625–32. doi: 10.2144/000113126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cavnar S, Ray P, Moudgil P, Chang S, Luker K, Linderman J, et al. Microfluidic source-sink model reveals effects of biophysically distinct CXCL12-isoforms in breast cancer chemotaxis. Integr Biol. 2014 doi: 10.1039/c4ib00015c. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luker K, Gupta M, Luker G. Imaging CXCR4 signaling with firefly luciferase complementation. Anal Chem. 2008;80(14):5565–73. doi: 10.1021/ac8005457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salcedo R, Oppenheim J. Role of chemokines in angiogenesis: CXCL12/SDF-1 and CXCR4 interaction, a key regulator of endothelial cell responses. Microcirculation. 2003;10(3-4):359–70. doi: 10.1038/sj.mn.7800200. [DOI] [PubMed] [Google Scholar]
- 30.Ho T, Tsui J, Xu S, Abraham D, Baker D. Angiogenic effects of stromal cell-derived factor-1 (SDF-1/CXCL12) variants in vitro and the in vivo expressions of CXCL12 variants and CXCR4 in human critical leg ischemia. J Vasc Surg. 2010;51(3):689–99. doi: 10.1016/j.jvs.2009.10.044. [DOI] [PubMed] [Google Scholar]
- 31.Luker K, Gupta M, Steele J, Foerster B, Luker G. Imaging Ligand-dependent Activation of CXCR7. Neoplasia. 2009;11(10):1022–35. doi: 10.1593/neo.09724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown J, Zhang J, Keller E. Opg, RANKL, and RANK in cancer metastasis: expression and regulation. Cancer Treat Res. 2004;118 doi: 10.1007/978-1-4419-9129-4_7. [DOI] [PubMed] [Google Scholar]
- 33.Udagawa N, Takahashi N, Jimi E, Matsuzaki K, Tsurukai T, Itoh K, et al. Osteoblasts/stromal cells stimulate osteoclast activation through expression of osteoclast differentiation factor/RANKL but not macrophage colony-stimulating factor: receptor activator of NF-kappa B ligand. Bone. 1999;25(5):517–23. doi: 10.1016/s8756-3282(99)00210-0. [DOI] [PubMed] [Google Scholar]
- 34.Rueda P, Richart A, Recalde A, Gasse P, Vilar J, Guerin C, et al. Homeostatic and tissue reparation defecs in mice carrying selective genetic invalidation of CXCL12/proteolycan interactions. Circulation. 2012;126(15):1882–95. doi: 10.1161/CIRCULATIONAHA.112.113290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Altenburg J, Broxmeyer H, Jin Q, Cooper S, Basu S, Alkhatib G. A naturally occurring splice variant of CXCL12/stromal cell-derived factor 1 is a potent human immunodeficiency virus type 1 inhibitor with weak chemotaxis and cell survival activities. J Virol. 2007;81(15):8140–8. doi: 10.1128/JVI.00268-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rueda P, Balabanian K, Lagane B, Staropoli I, Chow K, Levoye A, et al. The CXCL12gamma chemokine displays unprecedented structural and functional properties that make it a paradigm of chemoattractant proteins. PLoS One. 2008;3(7):e2543. doi: 10.1371/journal.pone.0002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zannettino A, Farrugia A, Kortesidis A, Manavis J, To L, Martin S, et al. Elevated serum levels of stromal-derived factor-1alpha are associated with increased osteoclast activity and osteolytic bone disease in multiple myeloma patients. Cancer Res. 2005;65(5):1700–9. doi: 10.1158/0008-5472.CAN-04-1687. [DOI] [PubMed] [Google Scholar]
- 38.Kim H, Kim K, Kim B, Jung H, Cho M, Lee S. Reciprocal activation of CD4+T cells and synovial fibroblasts by SDF-1 promotes RANKL expression and osteoclastogenesis in rheumatoid arthritis. Arthritis Rheum. 2013 Nov 18; doi: 10.1002/art.38286. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 39.Luker K, Lewin S, Mihalko L, Schmidt B, Winkler J, Coggins N, et al. Scavenging of CXCL12 by CXCR7 Regulates Tumor Growth and Metastasis of CXCR4-positive Breast Cancer Cells. Oncogene. 2012;31(45):4570–8. doi: 10.1038/onc.2011.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Venkiteswaran G, Lewellis S, Wang J, Reynolds E, Nicholson C, Knaut H. Generation and Dynamics of an Endogenous, Self-Generated Signaling Gradient across a Migrating Tissue. Cell. 2013;155(3):674–87. doi: 10.1016/j.cell.2013.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dona E, Barry J, Valentin G, Quirin C, Khmelinskii A, Kunze A, et al. Directional tissue migration through a self-generated chemokine gradient. Nature. 2013;503(7475):285–9. doi: 10.1038/nature12635. [DOI] [PubMed] [Google Scholar]
- 42.Odemis Y, Lipfert J, Kraft R, Hajek P, Abraham G, Hattermann K, et al. The presumed atypical chemokine receptor CXCR7 signals through G(i/o) proteins in primary rodent astrocytes and human glioma cells. Glia. 2012;60(3):372–81. doi: 10.1002/glia.22271. [DOI] [PubMed] [Google Scholar]
- 43.Rajagopal S, Kim J, Ahn S, Craig S, Lam C, Gerard N, et al. Beta-arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc Natl Acad Sci U S A. 2010;107(2):628–32. doi: 10.1073/pnas.0912852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levesque JP, Hendy J, Takamatsu Y, Simmons P, Bendall L. Disruption of the CXCR4/CXCL12 chemotactic interaction during hematopoietic stem cell mobilization induced by GCSF or cyclophosphamide. J Clin Invest. 2003;111(2):187–96. doi: 10.1172/JCI15994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Diamond P, Labrinidis A, Martin S, Farrugia A, Gronthos S, To L, et al. Targeted disruption of the CXCL12/CXCR4 axis inhibits osteolysis in a murine model of myeloma-associated bone loss. J Bone Miner Res. 2009;24(7):1150–61. doi: 10.1359/jbmr.090210. [DOI] [PubMed] [Google Scholar]
- 46.Shu H, Yoon Y, Hong S, Xu K, Gao H, Hao C, et al. Inhibition of the CXCL12/CXCR4-axis as preventive therapy for radiation-induced pulmonary fibrosis. PLoS One. 2013;8(11):e79768. doi: 10.1371/journal.pone.0079768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chu Q, Panu L, Holm N, Li B, Johnson L, Zhang S. High chemokine receptor CXCR4 level in triple negative breast cancer specimens predicts poor clinical outcome. J Surg Res. 2010;159(2):689–95. doi: 10.1016/j.jss.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 48.Chueh B, Huh D, Kyrtsos C, Houssin T, Futai N, Takayama S. Leakage-free bonding of porous membranes into layered microfluidic array systems. Anal Chem. 2007;79(9):3504–8. doi: 10.1021/ac062118p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ebos J, Lee C, Christensen J, Mutsaers A, Kerbel R. Multiple circulating proangiogenic factors induced by sunitinib malate are tumor-independent and correlate with antitumor efficacy. Proc Natl Acad Sci USA. 2007;104(43):17069–74. doi: 10.1073/pnas.0708148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holland J, Gyorffy B, Vogel R, Eckert K, Valenti G, Fang L, et al. Combined Wnt/β-catenin, met, and CXCL12/CXCR4 signals characterize basal breast cancer and predict disease outcome. Cell Rep. 2013;(13):S2211–1247. 00649–9. doi: 10.1016/j.celrep.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 51.Nixon A, Pang H, Starr M, Friedman P, Bertagnoli M, Kindler H, et al. Prognostic and Predictive Blood-Based Biomarkers in Patients with Advanced Pancreatic Cancer: Results from CALGB80303 (Alliance) Clin Cancer Res. 2013 Nov 12; doi: 10.1158/1078-0432.CCR-13-0926. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song J, Cavnar S, Walker A, Luker K, Gupta M, Tung Y, et al. Microfluidic endothelium for studying the intravascular adhesion of metastatic breast cancer cells. PLoS One. 2009;4(6):e5756. doi: 10.1371/journal.pone.0005756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lois C, Hong E, Pease S, Brown E, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295(5556):868–72. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- 54.Shcherbo D, Shemiakina I, Ryabova A, Luker K, Schmidt B, Souslova E, et al. Near infrared fluorescent proteins. Nat Methods. 2010;7(10):827–9. doi: 10.1038/nmeth.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ray P, Mihalko L, Coggins N, Moudgil P, Ehrlich A, Luker K, et al. Carboxy-terminus of CXCR7 regulates receptor localization and function. Int J Biochem Cell Biol. doi: 10.1016/j.biocel.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.