

Abstract
The trafficking of AMPA receptors (AMPARs) to and from synapses is crucial for synaptic plasticity. Previous work has demonstrated that AMPARs undergo activity-dependent ubiquitination by the E3 ubiquitin ligase Nedd4-1, which promotes their internalization and degradation in lysosomes. Here, we define the molecular mechanisms involved in ubiquitination and deubiquitination of AMPARs. We report that Nedd4-1 is rapidly redistributed to dendritic spines in response to AMPAR activation and not in response to NMDA receptor (NMDAR) activation in cultured rat neurons. In contrast, NMDAR activation directly antagonizes Nedd4-1 function by promoting the deubiquitination of AMPARs. We show that NMDAR activation causes the rapid dephosphorylation and activation of the deubiquitinating enzyme (DUB) USP8. Surface AMPAR levels and synaptic strength are inversely regulated by Nedd4-1 and USP8. Strikingly, we show that homeostatic downscaling of synaptic strength is accompanied by an increase and decrease in Nedd4-1 and USP8 protein levels, respectively. Furthermore, we show that Nedd4-1 is required for homeostatic loss of surface AMPARs and downscaling of synaptic strength. This study provides the first mechanistic evidence for rapid and opposing activity-dependent control of a ubiquitin ligase and DUB at mammalian CNS synapses. We propose that the dynamic regulation of these opposing forces is critical in maintaining synapses and scaling them during homeostatic plasticity.
Keywords: AMPA receptors, deubiquitination, E3 ubiquitin ligase, endocytosis, lysosome, ubiquitin
Introduction
The insertion and removal of AMPA receptors (AMPARs) at the postsynaptic membrane is thought to underlie both rapid Hebbian-based synaptic plasticity and also to maintain stable excitability levels in a slow, homeostatic manner through positive and negative feedback mechanisms, respectively (Shepherd and Huganir, 2007; Huganir and Nicoll, 2013). AMPARs, comprised of GluA1-4 subunits, are subject to a number of modifications (e.g., phosphorylation) that tightly regulate their levels at the postsynaptic membrane and thus allow for activity-dependent control of synaptic strength (Lu and Roche, 2012).
Ubiquitination has emerged as a functionally relevant posttranslational modification of many synaptic proteins (Patrick, 2006; Mabb and Ehlers, 2010; Yamada et al., 2013). Ubiquitination mediates the targeting of protein substrates to the 26S proteasome and the lysosome, the major sites of protein degradation in eukaryotic cells. Ubiquitination in the form of single (mono) or short-chain ubiquitin modifications can promote the internalization and downregulation of integral membrane proteins (Hicke, 1997). In early endosomes, nonubiquitinated proteins are recycled back to the plasma membrane or directed to other intracellular compartments. In contrast, ubiquitinated proteins are sorted into multivesicular bodies, which eventually fuse with lysosomes for degradation (Hicke and Dunn, 2003; Piper and Luzio, 2007). Deubiquitinating enzymes (DUBs) are ubiquitin proteases that reverse the ubiquitination reaction. Although there are close to 100 DUBs expressed in mammals, very few have been characterized in neurons (Todi and Paulson, 2011).
We and others have shown that mammalian AMPARs are directly modified by ubiquitin to promote their internalization and degradation (Schwarz et al., 2010; Lin et al., 2011; Lussier et al., 2011). We found that AMPARs are ubiquitinated by the HECT E3 ligase Nedd4-1 in response to AMPAR but not NMDAR activation (Schwarz et al., 2010) even though the application of both AMPA and NMDA promotes their internalization (Shepherd and Huganir, 2007). This suggests that AMPAR ubiquitination is regulated with a high degree of specificity. Here, we sought to elucidate the mechanism of this specificity. We observed that Nedd4-1 is redistributed to dendritic spines in response to AMPAR but not NMDAR activation in a rapid and persistent manner involving its Ca2+ and lipid-binding C2 domain. Intriguingly, we show that NMDAR activity negatively regulates AMPAR ubiquitination. Furthermore, we identified a DUB, USP8/UBPY, which is rapidly dephosphorylated and activated selectively in response to NMDAR but not AMPAR activation. Overexpression and shRNA-mediated knockdown of USP8 protein increased and decreased synaptic strength, respectively, and also altered the ubiquitination of AMPARs. Therefore, Nedd4-1 and USP8 are regulated at synapses to control synaptic strength in an opposite fashion. Moreover, we found that bicuculline-induced downscaling of AMPARs and synaptic strength is accompanied by an increase and decrease in Nedd4-1 and USP8 protein levels, respectively. Functionally, we show that shRNA-mediated knockdown of Nedd4-1 prevents bicuculline-induced loss of surface AMPARs and downscaling of synaptic strength, as does overexpression of USP8. This study provides the first evidence for how activity-dependent and diametric control of a ubiquitin ligase and DUB at mammalian synapses is essential for dynamic control of synaptic strength.
Materials and Methods
Antibodies and reagents.
Antibodies were as follows: pAb (C-term) GluA1, pAb surface (N-terminal) GluA1, pAb GluA2/3, and pAb Nedd4-1 (Millipore); mAb PSD-95 and pAb synapsin I (Calbiochem); mAb tubulin (Sigma); mAb actin (Cytoskeleton); pAb USP8 (Sigma-Aldrich); mAb ubiquitin (P4D1; Santa Cruz Biotechnology); mAb GFP (NeuroMab, UC Davis); pAb GFP (Invitrogen); mAb hemagglutinin (HA; Covance); mAb synaptophysin (Synaptic Systems). Reagents were as follows: l-glutamic acid and glycine (Fisher); AMPA, NMDA, CNQX, DL-2-amino-5-phosphonopentanoic acid (APV), tetrodotoxin (TTX), bicuculline, ionomycin, and BAPTA-AM (Tocris Bioscience); sodium orthovanadate, N-ethylmaleimide (NEM), and leupeptin (Sigma); EGTA (Calbiochem); lambda protein phosphatase (Millipore); and HA-tagged ubiquitin probe for active DUB labeling (HAUb-VME; Enzo Life Sciences).
Neuronal cultures.
Rat dissociated hippocampal or cortical neurons from postnatal day 1 pups of either sex were plated at a density of 45,000 cells/cm2 onto poly-d-lysine-coated coverslips, glass-bottomed 35 mm dishes (hippocampal cultures) (Mattek) or poly-d-lysine-coated 6-well plastic dishes at ∼500,000 cells per well (cortical cultures) and were maintained in B27 supplemented neurobasal medium (Invitrogen) until ≥14 d in vitro (DIV), as described previously (Djakovic et al., 2009; Schwarz et al., 2010; Djakovic et al., 2012).
Recombinant DNA, Sindbis, and lentiviral constructs.
Mouse HA-tagged Nedd4-1 in pCDNA3.1(-) was purchased from the Addgene DNA repository. This original clone, isolated and reported by Kumar et al. in 1992, is considered to be a “near full-length” clone of Nedd4-1 that has the minimal C2 domain intact because its size on SDS PAGE is the same as endogenous Nedd4-1 protein (Kumar et al., 1992; Kumar et al., 1997). To create the HA-Nedd4-1 ΔC2 deletion mutant, we first cloned an NheI-HA-tag-XbaI sequence into the XbaI site of a double subgenomic Sindbis DNA vector (2Gene Sindbis). PCR deletion mutagenesis was then used to clone DNA sequence of mouse Nedd4-1 lacking the first 180 amino acids (ΔC2 deletion) into the XbaI site downstream and in frame with the HA-tag. The HA-Nedd4-1 ΔC2 deletion mutant was then subcloned out of the Sindbis construct into pCDNA3.1(-). Human GFP-USP8 WT and catalytically inactive C786S mutant were kind gifts from Sylvie Urbé (University of Liverpool). Human GFP-USP8 WT and C786S mutant were subcloned into pSinRep5 (Sindbis) vector. Rat USP8 was PCR amplified in two fragments, sequentially cloning them into pEGFP-N1 (SacII-BamHI and BamHI-AgeI fragments 1 and 2, respectively) placing GFP downstream and in frame with USP8. Rat USP8-GFP was additionally subcloned into the FG12 lentivirus vector with concomitant removal of the preexisting GFP sequence. Flag-tagged USP8 was created by inserting Flag epitope sequence to the N terminus of USP8 and subsequently cloned into pSinRep5. Production of recombinant Sindbis virus was performed as described previously (Djakovic et al., 2009). All DNA and viral constructs were verified by sequencing.
Transfections and infections.
HEK293T cells, maintained in DMEM plus 10% serum and penicillin/streptomycin, were transfected with Lipofectamine 2000 (Invitrogen) or polyethyleneimine (Polysciences) using recommended protocols. Hippocampal or cortical cultures were infected with Sindbis virion at DIV 16–22 and allowed to express for 14–22 h. For RNAi experiments, hippocampal cultures were infected with lentivirus expressing the RNAi constructs for 5–7 d. Viral titer and transduction efficiency were monitored for all viruses made to ensure equal expression of constructs.
RNAi.
Lentivirus expressing Nedd4-1 shRNA hairpin was described previously (Schwarz et al., 2010). To knock down expression of USP8 in rat hippocampal neurons, the oligo AGGTGAAGTGGCAGAAGAA was synthesized and inserted into the pSuper-eGFP vector and then subsequently mobilized the H1 promoter and hairpin out of pSuper and into FG-12 vector (which coexpresses GFP from a second promoter). Dissociated hippocampal cultures were infected with FG-12-Nedd4-1 or USP8 shRNA at DIV 9–12 and experiments were conducted 5–7 d later.
Immunoprecipitations.
Cultured rat cortical neurons were lysed in precipitation buffer containing the following (in mm): 100 NaCl, 10 Na2HPO4, 5 EDTA, and 5 EGTA with 1% Triton X-100, 0.1% SDS, 25 μm MG-132, 25 mm NEM, and protease inhibitors. Homogenates were cleared by centrifugation at 14,000 rpm at 4°C. For immunoprecipitations (IPs), cleared lysates were incubated with primary antibodies at 4°C for 1.5 h or overnight, after which protein A or protein G agarose beads were added for an additional 1 h (Pierce). Immunoprecipitates were then washed, boiled in sample buffer, resolved on SDS PAGE, and probed in Western blot analysis. For AMPAR ubiquitination assays, we used anti-GluA2/3 pAb antibodies because this antibody efficiently immunoprecipitates both GluA1 and GluA2 in our lysis buffer conditions, which do not disrupt the tetramer receptor complex. In biochemical assays in which neuronal cultures were evaluated for ubiquitination status, 100 μg/ml Leupeptin (Millipore) was administered 1 h before treatments unless otherwise specified.
Western blot analysis.
Total protein lysates were generated by scraping cells into RIPA buffer (50 mm Tric-HCl, 150 mm NaCl, 1% NP-40, 0.5% Na-deoxycholate, and 0.1% SDS) with protease inhibitors and incubating for 20 min at 4°C. Fractions from adult rat brain tissue were prepared as described previously (Carlin et al., 1980; Cho et al., 1992; Ehlers, 2003). Protein concentration was determined by BCA protein assay (Pierce) and equal protein amounts were loaded. Samples were boiled with sample buffer, resolved on 8% SDS page, and probed with primary antibodies. Blots were digitized and band intensities quantitated using ImageJ. For quantification of GluA1, Nedd4-1, and USP8 levels after chronic treatments of neuronal cultures with bicuculline, band intensities in each condition were normalized to tubulin band mean intensity from the same sample.
Active DUB activity labeling assay.
We measured activity of USP8 transfected in HEK293T cells after immunopreciptation of GFP-USP8 with anti-GFP antibody. The resulting precipitates were washed 2× in lysis buffer and then 2× with DUB labeling buffer (50 mm Tris, pH 7.4, 5 mm MgCl2, 250 mm sucrose, 1 mm DTT, and 1 mm ATP). Next, the precipitates were incubated with the active DUB labeling HAUb-VME substrate in labeling buffer for 1 h at 37°C. Proteins were then resolved by 8% SDS-PAGE and blots were subsequently probed with anti-HA monoclonal antibody. USP8 activity was calculated by dividing the HA signal by the immunoprecipitated USP8 signal.
Immunostaining.
After infections and drug treatments, neurons were washed with cold PBS-MC and fixed with a solution containing 4% paraformaldehyde and 4% sucrose for 10 min. Cells were then permeabilized with 0.2% Triton X-100 and 2% BSA in PBS-MC for 20 min, followed by a 1 h block in 5% BSA in PBS-MC. Primary and secondary antibodies were diluted into 2% BSA in PBS-MC and applied to neurons for 1 h at room temperature. Coverslips were mounted onto glass slides for confocal imaging. For surface immunostaining, neurons were fixed, blocked in 5% BSA for 12–20 h at 4°C, and incubated with surface GluA1 antibody for 12–20 h at 4°C. Cells were then permeabilized, blocked, incubated with anti-GFP antibody, and incubated with secondary antibodies.
Confocal microscopy.
All images were acquired with a Leica DMI6000 inverted microscope equipped with a Yokogawa Nipkon spinning disk confocal head, an Orca ER high-resolution black and white cooled CCD camera (6.45 μm/pixel at 1×), Plan Apochromat 63×/1.4 numerical aperture objective, and an argon/krypton 100 mW air-cooled laser for 488/568/647 nm excitations. Maximum projected confocal Z-stacks were analyzed with ImageJ. For redistribution experiments, dendrites were straightened and a custom macro was used to run the Find Maxima function through a range of noise tolerance levels ranging from 10 to 255. For further analysis of differences between conditions, a tolerance level between 70 and 100 was chosen for all images and statistical significance was determined through unpaired t tests or ANOVA with specified post hoc multiple-comparisons test using Prism software (GraphPad).
Electrophysiology.
For whole-cell recordings of miniature EPSCs (mEPSCs), dissociated rat hippocampal neurons were bathed at room temperature in a HEPES-buffered saline recording solution containing the following (in mm): 119 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 30 glucose, and 10 HEPES, pH 7.2, along with 1 μm TTX, and 10 μm bicuculline. The electrode recording solution contained the following (in mm): 10 CsCl, 105 CsMeSO3, 0.5 ATP, 0.3 GTP, 10 HEPES, 5 glucose, 2 MgCl2, and 1 EGTA, pH 7.2. Electrode resistances ranged from 2.5 to 4.5 MΩ and access resistances ranged from 10 to 25 MΩ. Signals were amplified, filtered to 2 or 5 kHz, and digitized at 10 kHz sampling frequency. Holding potential for all traces was −70 mV. mEPSCs were analyzed using ClampFit 10.3 (Molecular Devices). Infected cells were identified through GFP fluorescence. Statistical differences between conditions were analyzed through either unpaired t tests (two groups) or by ANOVA and indicated post hoc multiple-comparison test (>2 groups) conducted in Prism software (GraphPad).
Results
Nedd4-1 is rapidly redistributed to synapses in response to AMPAR activation
It has been shown previously that the application of AMPA or NMDA to hippocampal neurons promotes the internalization of AMPARs (Ehlers, 2000; Lin et al., 2000; Lee et al., 2004). However, we and others recently showed that these stimuli differ in their ability to induce the ubiquitination of AMPARs (Schwarz et al., 2010; Lussier et al., 2011). We predicted that Nedd4-1, a ubiquitin ligase that targets AMPARs, might be itself differentially regulated by specific synaptic cues. Therefore, we hypothesized that rapid regulation of Nedd4-1 could be achieved by control of its localization in neurons. To visualize clearly the cellular distribution of Nedd4-1 under various conditions, we infected dissociated hippocampal neurons with a Sindbis virus expressing cell-filling GFP and a HA-tagged Nedd4-1. As described previously, this tagged Nedd4-1 displays a similar cellular distribution to endogenous Nedd4-1 (Schwarz et al., 2010). We then investigated whether synaptic activity induces any change to the distribution of Nedd4-1 by bath-applying glutamate and glycine to neurons expressing HA-Nedd4-1 (Fig. 1A). We observed a robust change in distribution of Nedd4-1 in the treated condition compared with untreated neurons, quantified through a custom macro (see Materials and Methods). Specifically, the distribution of HA-Nedd4-1 changed from a diffuse pattern throughout dendrites and soma into a punctate pattern with tightly accumulated signal in dendritic spines (p < 0.001; Fig. 1A,C). Moreover, we found extensive colocalization of HA-Nedd4-1 with the postsynaptic density protein PSD-95 and juxtaposition to the presynaptic protein synapsin I (Fig. 1H,I), indicating a recruitment to synapses. Because glutamate activates both AMPA and NMDA receptors at synapses, we next tested the ability of AMPA and NMDA to induce these changes. AMPA induced a robust redistribution of Nedd4-1, whereas NMDA produced no effect (p < 0.001, p = 0.44, respectively; Fig. 1B,C), indicating that activation of surface AMPARs is necessary for the recruitment of Nedd4-1.
Figure 1.

AMPAR activation leads to rapid redistribution and accumulation of Nedd4-1 at synapses. A, B, Representative immunofluorescent images of dissociated hippocampal neurons 19–22 DIV expressing HA-tagged Nedd4-1 (Sindbis) for 18–20 h and treated with glutamate and glycine (100 μm/10 μm), AMPA (10 μm), or NMDA and glycine (25 μm/10 μm) for 10 min or left untreated and then stained with anti-HA (red) and anti-GFP (green) antibodies. Representative maximum z-projected confocal images of whole cell and dendrite (A) or dendrite alone (B) are depicted. C–G, Quantification of HA-Nedd4-1 fluorescence local maxima: after various drug treatments described in A (C), time course of AMPA treatments (in minutes) (D), persistence of effect 5 or 10 min after washout of 10 min 10 μm AMPA treatment (indicated by arrowhead) (E), effect of calcium chelation with EGTA (2.5 mm, 15 min pretreatment) (F), and treatments with CNQX (40 μm, 10 min) and ionomycin (10 μm, 15 min) (G); n > 20 dendrites per condition over 3–4 independent experiments. H, Representative immunofluorescent images of dendrites from dissociated hippocampal neurons expressing HA-tagged Nedd4-1 (Sindbis) after AMPA stimulation (10 μm, 10 min) and immunostained with anti-HA (red) and anti-PSD-95 (blue) antibodies. Colocalization of HA and PSD-95 punctate clusters is highlighted by arrowheads. I, Representative immunofluorescent images of dendrites after AMPA stimulation (10 μm, 10 min), immunostained with anti-HA (red) and anti-synapsin I (blue) antibodies. Areas of close HA and synapsin proximity are highlighted by arrowheads. **p < 0.01, ***p < 0.001; ANOVA with Tukey's post hoc analysis. Graphs show mean ± SEM. Scale bar, 10 and 5 μm for whole-cell and dendrite images, respectively.
We next explored the properties of this activity-induced redistribution. Nedd4-1 was observed to relocalize rapidly, with significant changes apparent after 2 min of AMPA treatment and a more robust effect at 5 and 10 min (p < 0.01 and p < 0.001, respectively; Fig. 1D). In addition, we found that HA-Nedd4-1 remains localized to synapses after washout of AMPA, with significant redistribution evident for at least 20 min after washout (p < 0.001; Fig. 1E). Because previous studies indicated that the AMPA-induced ubiquitination of AMPARs is calcium dependent (Schwarz et al., 2010; Lussier et al., 2011), we next investigated whether the rapid redistribution of Nedd4-1 requires calcium as well. AMPA treatments after a pretreatment with the calcium buffer EGTA induced a significantly smaller redistribution (two-way ANOVA, interaction: F(1,209) = 13.59, p < 0.001), although the effect was not completely abolished (AMPA vs EGTA + AMPA: p < 0.01; Fig. 1F). Interestingly, raising intracellular calcium levels by ionomycin treatment produced no changes to Nedd4-1's localization, indicating that calcium alone is not sufficient to recruit Nedd4-1 to synapses. We hypothesized that redistribution requires both a rise in calcium and ligand binding at AMPA receptors. To test this, we applied the AMPAR antagonist CNQX to serve as a nonactivating ligand and ionomycin to raise intracellular calcium. As predicted, application of both CNQX and ionomycin was sufficient to promote significant redistribution (p < 0.001) of Nedd4-1, whereas the application of either alone had no effect (p = 0.93 and p = 0.99, respectively; Fig. 1G). This indicates that ligand binding plus a rise in intracellular calcium is necessary and sufficient to induce recruitment of Nedd4-1 to synapses. Together, these data indicate that the subcellular distribution of Nedd4-1 is dynamically regulated by specific synaptic cues.
Nedd4-1's C2 domain regulates its trafficking and function in neurons
In addition to the enzymatic HECT domain and four WW domains, Nedd4-1 contains a C2 domain. In a number of other proteins, this domain serves as a calcium sensor and a regulator of membrane interactions (Nalefski and Falke, 1996). In the yeast homolog Rsp5, the C2 domain is critical for proper membrane localization (Dunn et al., 2004). In MDCK cells, increased calcium causes a shift in localization of mammalian Nedd4-1 to membrane compartments, an effect that requires the C2 domain (Plant et al., 1997; Plant et al., 2000). Interestingly, this domain has also been found to be auto-inhibitory, because a Nedd4-1 mutant lacking this region displayed dramatically enhanced ubiquitination activity (Wang et al., 2010). It has been proposed that calcium reduces the inhibitory control by the C2 domain, possibly by promoting membrane association. To test the role of this domain in neurons, we generated an HA-tagged mutant Nedd4-1 with a heavily truncated C2 domain and expressed this mutant variant in hippocampal neurons via Sindbis virus. Under control conditions, Nedd4-1ΔC2 appears to have a very similar cellular distribution to the wild-type form (Fig. 2A). However, we observed no differences in AMPA-treated neurons compared with untreated neurons (p = 0.72), indicating that this domain is necessary for AMPA-induced redistribution of Nedd4-1 (Fig. 2A,B).
Figure 2.

Nedd4-1's C2 domain regulates its trafficking and function in neurons. A, Representative immunofluorescent images of dissociated hippocampal neurons 19–22 DIV expressing HA-tagged Nedd4-1ΔC2 (Sindbis) for 18–20 h and treated with AMPA (10 μm, 10 min) or left untreated and stained for HA (red) and GFP (green). Representative maximum z-projected confocal images of whole cell and dendrite are depicted. Scale bar, 10 and 5 μm for whole cell and dendrite images, respectively. B, Quantification of images from HA-Nedd4-1 wild-type and ΔC2-expressing cells with and without AMPA (10 μm, 10 min); ***p < 0.001, Student's t test; n > 18 dendrites per condition over 3 independent experiments. C, Representative traces of mEPSCs recorded from dissociated hippocampal neurons 18–24 DIV expressing control GFP, HA-Nedd4-1 wild-type, or HA-Nedd4-1ΔC2 (Sindbis) for 18–22 h. Scale bar, 500 ms and 20 pA. D, Cumulative probability distributions of amplitudes of all mEPSCs recorded from neurons infected with Sindbis GFP (control), HA-Nedd4-1 wild-type, or HA-Nedd4-1ΔC2. n = 2739, 1781, and 2426 events, respectively. Inset displays averaged waveform of all events in each condition. Scale bar represents 5 ms, 5 pA. E, Mean mEPSC amplitude. *p < 0.05, ANOVA with Tukey's post hoc analysis; n = 20–25 cells; data collected across 3 independent experiments. F, Representative immunofluorescent images of hippocampal neurons 18–24 DIV expressing GFP (control), HA-Nedd4-1 wild-type, or HA-Nedd4-1ΔC2 for 18–22 h and stained for surface GluA1-containing AMPARs (red) and GFP (green). Scale bar, 5 μm. G, Quantification of surface GluA1 signal in straightened dendrites of GFP-positive cells in all three conditions. *p < 0.05, ANOVA with Tukey's post hoc analysis; n = 57–59 cells; data collected across 3 independent experiments. H, Immunoblots showing reduced coimmunoprecipitation between GFP-tagged GluA1 and HA-tagged Nedd4-1ΔC2 compared with HA-tagged Nedd4-1 WT. HEK293 cells were cotransfected with GFP-GluA1 and either HA-Nedd4-1ΔC2 or HA-Nedd4-1 WT for 18–24 h. HA-Nedd4-1 was immunoprecipitated with anti-HA antibody and amount of coprecipitating GluA1 was determined through anti-GFP blotting. Graphs show mean ± SEM.
To delineate between its role in trafficking and an auto-inhibitory role of Nedd4-1's C2 domain, we compared the level of decreased synaptic strength between overexpressed wild-type Nedd4-1 and Nedd4-1ΔC2 in cultured hippocampal neurons by recording mEPSCs. As previously reported, overexpression of wild-type Nedd4-1 caused a significant decrease in the amplitude of mEPSCs (Schwarz et al., 2010) compared with control GFP-expressing cells (Fig. 2C–E). In contrast, expression of the Nedd4-1ΔC2 mutant did not cause any reduction of mEPSC amplitude (GFP: 15.8 ± 0.6 pA, n = 25 cells; WT: 13.2 ± 0.6 pA, n = 20 cells; ΔC2: 16.2 ± 0.7 pA, n = 23 cells; p < 0.05; Fig. 2C–E). There was no observable difference in the frequency of mEPSCs (GFP: 373.2 ± 70.0 ms; WT: 806.5 ± 203.1 ms; ΔC2: 650.1 ± 155.6 ms). To verify that these changes in synaptic strength reflect changes in surface AMPAR expression, we expressed these constructs in hippocampal neurons and immunostained for surface GluA1-containing AMPARs. We observed a reduction in surface GluA1 in Nedd4-1 wild-type expressing cells, but no reduction was observed in Nedd4-1ΔC2-expressing cells (GFP, normalized surface signal: 1.0 ± 0.05, n = 59 cells; WT: 0.82 ± 0.05, n = 57 cells; ΔC2: 0.95 ± 0.06, n = 59 cells; p < 0.05; Fig. 2F,G). Therefore, although the Nedd4-1ΔC2 mutant has been reported to have dramatically enhanced ligase activity (Wang et al., 2010), these data indicate that the C2 domain is required for downregulating synaptic strength upon overexpression. We further corroborated this by biochemically assessing whether the C2 domain is critical for interaction with GluA1 in cotransfected HEK293 cells. We detected a much stronger interaction of GFP-GluA1 with wild-type HA-Nedd4-1 compared with the Nedd4-1ΔC2 mutant, suggesting that the C2 domain is also required for normal binding of Nedd4-1 to AMPA receptors (Fig. 2H).
The deubiquitinating enzyme USP8 is activated by NMDA receptor activation and regulates synaptic strength in an opposite fashion to Nedd4-1
Nedd4-1 is rapidly redistributed to synapses in response to AMPA (Fig. 1), which is consistent with the fact that AMPA induces robust AMPAR ubiquitination (Schwarz et al., 2010; Lussier et al., 2011). Curiously, however, although the application of glutamate and glycine caused a robust recruitment of Nedd4-1 to synapses similar to AMPA, we found that it did not induce AMPAR ubiquitination (Fig. 3A). Because glutamate also activates NMDA receptors, we hypothesized that NMDAR activation could negatively regulate AMPAR ubiquitination to counteract the action of Nedd4-1. Indeed, NMDAR activation does not induce the ubiquitination of AMPARs (Schwarz et al., 2010; Lussier et al., 2011). Surprisingly, however, under conditions in which NMDAR activity is blocked, we found that glutamate and glycine induced robust AMPAR ubiquitination (Fig. 3A). Furthermore, we found that AMPAR ubiquitination was dramatically reduced in cells exposed to both AMPA and NMDA concomitantly compared with AMPA alone (Fig. 3B). One possible explanation for this observation is that NMDAR activation counteracts Nedd4-1 function by rapidly enhancing the deubiquitination of AMPARs (Fig. 3C).
Figure 3.

Rapid NMDAR-dependent dephosphorylation and activation USP8. A, B, Representative Western blots depicting the antagonistic effects of NMDAR activation on the ubiquitination of AMPARs. A, Dissociated cortical neurons (18–21 DIV) were treated for 10 min with AMPA (100 μm), glutamate and glycine (100 μm/10 μm), glutamate and glycine (100 μm/10 μm) with APV (50 μm, 60 min pretreatment), or left untreated before IP with anti-GluA2/3 antibodies in nondenaturing IP conditions. IPs were resolved by SDS-PAGE and Western blot was probed with anti-ubiquitin, anti-GluA1, and anti-GluA2/3 antibodies. B, Coapplication of NMDA/glycine (50 μm/10 μm, 10 min) and AMPA (100 μm, 10 min) indicates that NMDAR activation directly antagonizes AMPA-dependent AMPAR ubiquitination. Shown is a representative blot from 2 or 3 independent experiments. C, Model depicting reversibility of ubiquitination by action of DUBs. D, E, Rapid dephosphorylation of USP8 is induced by NMDA/glycine but not by AMPA (D); there was no detectable modification of AMSH in neurons treated with NMDA/glycine or AMPA (E). Dissociated cortical cultures (DIV > 18) were treated for indicated times (minutes) with either NMDA and glycine (50 μm/10 μm) or AMPA (100 μm). Cell lysates were resolved by SDS-PAGE and probed with anti-USP8, anti-AMSH, or anti-actin (loading control) antibodies. Shown is a representative blot from > 3 independent experiments per condition. F, Lysates resulting from control or NMDA/glycine (50 μm/10 μm, 10 min.) treated dissociated cortical neurons were treated with lambda phosphatase (1 h, 30°C) and exhibit similarly faster migrating band to NMDA/glycine treatments by SDS-PAGE. Shown is a representative blot from 2 independent experiments. G, Dephosphorylation of USP8 induced by NMDA/glycine requires external Ca2+ influx through NMDA receptors. Dissociated cortical neuronal cultures (DIV > 18) were treated for 10 min with either control (untreated) or NMDA/glycine (50 μm/10 μm) plus or minus APV (50 μm, 60 min pretreatment) in HBS or Ca2+-free HBS. Cell lysates were resolved by SDS-PAGE and probed with anti-USP8 antibodies. Shown is a representative blot from 3 independent experiments. H, The general tyrosine phosphatase inhibitor blocks NMDA-dependent dephosphorylation of USP8. Dissociated cortical neuronal cultures (DIV > 18) were treated for 10 min with either control (untreated) or NMDA/glycine (50 μm/10 μm) plus increasing concentrations of sodium orthovanadate (Na3VO4). Cell lysates were resolved by SDS-PAGE and probed with anti-USP8 antibodies. Shown is a representative blot from 3 independent experiments. I, Ionomycin promotes dephosphorylation of USP8 in a similar fashion to NMDAR activation. Dissociated cortical neuronal cultures (DIV > 18) were treated for 10 min with either control (untreated), ionomycin (10 μm), NMDA/glycine (50 μm/10 μm), or AMPA (100 μm). Cell lysates were resolved by SDS-PAGE and probed with anti-USP8 antibodies. Shown is a representative blot from 3 independent experiments. J, Activity assay of USP8. Lysates from HEK293 cells transfected for 18–24 h (PEI) with either WT GFP-tagged USP8 or C746S catalytically inactive mutant were immunoprecipitated with anti-GFP antibodies. Immunoprecipitates were labeled with active DUB labeling reagent HAUb-VME, resolved by SDS-PAGE, and probed with anti-HA and anti-USP8 antibodies. Shown is a representative blot. K, L, Representative blot depicting USP8 activity is enhanced by the Ca2+ ionophore ionomycin. K, HEK293 cells transfected for 18–24 h (PEI) with WT GFP-tagged USP8 were treated for 10 min with ionomycin (10 μm) or left untreated. Lysates were immunoprecipitated with anti-GFP antibodies. Immunoprecipitates were labeled with HAUb-VME, resolved by SDS-PAGE, and probed with anti-HA and anti-USP8 antibodies. L, Graphs showing quantification of USP8 activity (HA signal) normalized to USP8 levels (in IP) relative to control treated cells. *p < 0.05, ANOVA with Dunnett's post hoc analysis; n = 4 independent experiments. Graph shows mean ± SEM.
We therefore sought to identify a deubiquitinating enzyme that would be preferentially regulated by NMDA and not AMPA. USP8/UBPY and AMSH are two DUBs that are known to participate in the endosomal sorting complex required for transport (ESCRT) pathway. These DUBs have been shown to act at various stages in the ESCRT pathway to facilitate recycling to the plasma membrane as well as degradation in lysosomes (Wright et al., 2011). We first evaluated any obvious differences in the mobility of USP8 or AMSH by SDS-PAGE in lysates from neuronal cultures treated with either AMPA or NMDA and glycine (Fig. 3D). In response to NMDA and glycine, USP8 migrated slightly faster by SDS-PAGE, whereas no observable difference was seen in response to AMPA. This phenomenon was specific to USP8 because no detectable changes were observed in AMSH mobility by SDS-PAGE (Fig. 3E). We predicted that this NMDA-induced modification of USP8 could be due to actions of a phosphatase. Indeed, when we treated lysates from control or NMDA/glycine-treated dissociated neurons with lambda phosphatase, we observed a similarly faster migrating band by SDS-PAGE, suggesting that NMDA receptor activation leads to the rapid dephosphorylation of USP8 (Fig. 3F). This modification required external calcium entry, likely through NMDARs because the NMDA receptor antagonist APV completely prevented the modification of USP8 (Fig. 3G). Furthermore, we found that pretreating neuronal cultures with sodium orthovanadate (Na3VO4), a general inhibitor of tyrosine phosphatase activity, completely blocked NMDA-induced dephosphorylation of USP8 (Fig. 3H). In contrast, inhibition of PP1, PP2A, or PP2B with calyculin A, okadaic acid, or FK506, respectively, had no effect on the NMDA-induced dephosphorylation of USP8 (data not shown). The involvement of calcium in USP8 dephosphorylation is further underscored by the fact that the calcium ionophore ionomycin also promotes the dephosphorylation of USP8 in a similar manner as NMDAR activation (Fig. 3I). These data suggest that the calcium- and NMDAR-dependent dephosphorylation of USP8 involves an as yet unidentified tyrosine phosphatase.
USP8 has previously been reported to be phosphorylated on both serine and tyrosine residues (Mizuno et al., 2007; Meijer et al., 2013). Interestingly, these studies determined that increased USP8 activity was associated with the dephosphorylation of USP8 (Mizuno et al., 2007). To determine whether the observed dephosphorylation was related to a change in activity, we evaluated the effects of ionomycin on ectopically expressed GFP-tagged USP8 in HEK293 cells. We first demonstrated that we could efficiently label immunoprecipitated GFP-USP8 with a DUB activity probe, HAUb-VME. We found that active GFP-USP8 was readily labeled with HAUb-VME (Fig. 3J). In contrast, the C746S catalytically inactive mutant of USP8 was devoid of any labeling despite being efficiently immunoprecipitated (Fig. 3J). We then investigated whether ionomycin, which promotes the dephosphorylation of USP8, alters USP8 activity. Relative to control treated cells, ionomycin increased GFP-USP8 activity by more than 2-fold (p < 0.05; Fig. 3K,L). Furthermore, pretreating GFP-USP8-expressing cells with Na3VO4, which blocks the dephosphorylation of endogenous USP8 in response to ionomycin (Fig. 3H), completely prevented the ionomycin-induced increase in activity (Fig. 3K,L). Together, these data suggest that the calcium-dependent dephosphorylation of USP8 increases its activity.
To begin to ascertain the function of USP8 in neurons, we first evaluated the distribution of endogenous USP8 in various subcellular compartments using biochemical fractionation of rat brain tissue. USP8 is present in all fractions and is notably abundant in postsynaptic density fractions (Fig. 4A). Using immunofluorescence, we observed that endogenous USP8 is present in both somatic and dendritic compartments in cultured hippocampal neurons (Fig. 4B). We also found that, in a subpopulation of neurons, USP8 was located in punctate clusters and colocalized with the postsynaptic density protein PSD-95 (Fig. 4B), suggesting that it may function at synapses. We assessed whether manipulating USP8 affects the ubiquitination of AMPARs and found that the overexpression of USP8 caused a significant reduction in AMPA-induced ubiquitination of AMPARs (Fig. 4C). In addition, we observed that shRNA-mediated knockdown of USP8 (Fig. 4E) increases basal AMPAR ubiquitination (Fig. 4D). To determine whether manipulations of USP8 affect synaptic strength, we recorded mEPSCs in neurons overexpressing USP8 and observed that overexpression significantly increases mEPSC amplitude. This effect was dependent on USP8 activity, because overexpression of the C746S catalytic inactive mutant had no effect on synaptic strength (GFP, amplitude: 13.9 ± 0.7 pA, n = 12 cells; WT: 17.8 ± 1.5 pA, n = 12 cells; CS: 12.98 ± 0.5 pA, n = 13 cells; p < 0.05; Fig. 4F–H). Similarly, expression of wild-type but not catalytically inactive USP8 led to an increase in surface GluA1 levels, as assessed by immunofluorescence (GFP, normalized signal: 1.00 ± 0.09, n = 36 cells; WT: 1.34 ± 0.08, n = 36 cells; CS: 1.02 ± 0.11, n = 36 cells; p < 0.05; Fig. 4I,J). In contrast, we found that expression of a USP8-targeted shRNA hairpin (Fig. 4E) significantly decreased mEPSC amplitude (control: 16.5 ± 0.8 pA, n = 33 cells; shRNA: 14.2 ± 0.7 pA, n = 31 cells; p < 0.05; Fig. 4K,L). Interevent interval was not significantly affected by either USP8 overexpression (GFP: 1561 ± 351.9 ms, n = 12 cells; WT: 761 ± 185.6 ms, n = 12 cells; CS: 1639 ± 292.4 ms, n = 13 cells) or knockdown (control: 334.6 ± 37.8 ms, n = 33 cells; shRNA: 300.3 ± 37.16 ms, n = 31 cells). These data indicate that USP8 alters synaptic strength in an opposite fashion to Nedd4-1 (Schwarz et al., 2010).
Figure 4.
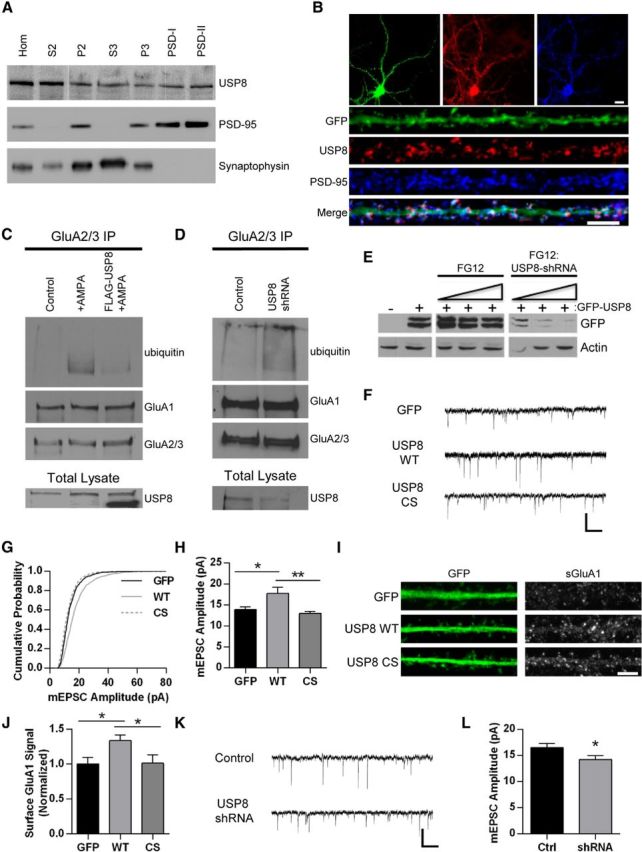
USP8 regulates synaptic strength. A, Western blot depicting distribution of USP8 across subcellular fractions in rat brain tissue. Brains were homogenized and subjected to biochemical fractionation. Samples of each fraction (homogenate, S2, P2, S3, P3, and postsynaptic density I and II) were resolved by SDS-PAGE and probed for USP8, PSD-95, and synaptophysin. B, Representative immunofluorescent images of dissociated hippocampal neurons 21 DIV expressing GFP (Sindbis, 16–18 h) stained with anti-USP8 (red) and anti-PSD-95 (blue) antibodies. GFP signal in green. Representative maximum z-projected confocal images of whole cell and dendrites are depicted. C, Representative Western blot displaying effect of USP8 overexpression on AMPA-induced AMPAR ubiquitination. Cortical neurons DIV 18–22 were infected with Sindbis USP8-FLAG for 16–18 h or left uninfected and were then treated with AMPA (50 μm, 10 min) or left untreated. Lysates were immunoprecipitated with GluA2/3 antibodies in nondenaturing conditions. IPs were resolved by SDS-PAGE and probed with anti-ubiquitin, anti-GluA1, and anti-GluA2/3 antibodies; total lysates were probed for USP8 (endogenous USP8 running slightly larger by SDS-PAGE). Shown is a representative blot from two independent experiments. D, Representative Western blot depicting increased basal AMPAR ubiquitination with USP8 knockdown. Cortical neurons DIV 14-16 were infected with control FG12 or USP8 shRNA (lentivirus) for 5–6 d before immunoprecipitation with anti-GluA2/3 antibodies. IPs were resolved by SDS-PAGE and probed for ubiquitin, GluA1, and GluA2/3; total lysates were probed for USP8. Shown is a representative blot from two independent experiments. E, Representative Western blot of lysates from HEK293 cells cotransfected with rat GFP-USP8 and FG12 (control vector coexpressing GFP) or shRNA USP8 hairpin (coexpressing GFP). Increasing ratio of FG12 control or shRNA USP8 hairpin (from right to left) is indicated. Actin and GFP blot shown as loading control. F–H, Representative traces of mEPSCs recorded from control GFP-, GFP-USP8 WT-, and GFP-USP8 C746S-expressing hippocampal neurons (18–22 DIV, 18–22 h Sindbis infection) (F); cumulative probability distributions of amplitudes of all mEPSCs recorded from neurons, n = 1206, 1210, 1305 events, respectively (G); mean mEPSC amplitude *p < 0.05, **p < 0.01, ANOVA with Tukey's post hoc analysis; n = 12–13 cells per condition (H). I, Representative images of straightened dendrites from hippocampal neurons 18–22 DIV expressing GFP (control), USP8 WT, or USP8 CS (Sindbis, 18–22 h) and stained for GluA1-containing surface AMPARs (red) and GFP (green). J, Quantification of surface GluA1 signal in all dendrites expressing GFP, USP8 WT, and USP8 CS. *p < 0.05, ANOVA with Tukey's post hoc analysis; n = 36 cells across two independent experiments. K–L, Representative traces of mEPSCs recorded from control FG12- and shRNA USP8 hairpin (coexpressing GFP)-expressing hippocampal neurons (18–22 DIV, lentiviral-based transduction) (K); mean mEPSC amplitude. *p < 0.05, Student's t test; n = 31 cells across four independent experiments (L). Graphs show mean ± SEM. Scale bar, 10 and 5 μm for whole cell and dendrite images, respectively; 500 ms and 20 pA for traces.
Nedd4-1 and USP8 are involved in homeostatic downscaling of synaptic strength
Homeostatic plasticity involves the ability of synapses to dynamically scale their strength up or down in response to prolonged changes in activity levels (Pozo and Goda, 2010; Davis, 2013). Raising activity levels through long-term treatment with the GABAA receptor antagonist bicuculline leads to a downscaling of synaptic strength due to the removal of synaptic AMPARs (Turrigiano et al., 1998). Several proteins have been shown to be directly or indirectly involved in homeostatic downscaling of AMPARs. In many cases, these molecules have themselves been shown to be regulated by synaptic activity (Ehlers, 2003; Seeburg et al., 2008; Fu et al., 2011). Due to Nedd4-1's role in the internalization of AMPARs, we sought to explore whether this E3 ligase plays a role in homeostatic downscaling. We first examined whether a chronic increase in activity would lead to enhanced AMPAR ubiquitination. Indeed, we observed that neuronal cultures treated with bicuculline for 24 h had significantly increased AMPAR ubiquitination (Fig. 5A). We next investigated whether chronic activity elevation would alter Nedd4-1 protein levels. As expected, total GluA1 levels were significantly reduced in neurons after a 3 d bicuculline treatment (normalized bicuculline level: 0.68 ± 0.07; p < 0.01; Fig. 5B,C). In contrast, bicuculline caused a significant increase in total Nedd4-1 protein levels (1.26 ± 0.09; p < 0.01; Fig. 5B,C). Interestingly, we additionally observed a large reduction in total USP8 protein (0.67 ± 0.09; p < 0.05; Fig. 5B,C). Because we found that the distribution of Nedd4-1 can be modulated by AMPAR activation, we tested whether chronic application of bicuculline induces a change in subcellular distribution of Nedd4-1. We treated HA-Nedd4-1-expressing hippocampal neurons with bicuculline (24 h) and observed a significant redistribution of Nedd4-1 to dendritic spines (p < 0.001; Fig. 5D,E) comparable to that observed with AMPAR activation (Fig. 1B). Therefore, prolonged bicuculline-induced activity elevation increases AMPAR ubiquitination and upregulates total and synaptic Nedd4-1 protein levels while concomitantly downregulating USP8 total protein levels.
Figure 5.

Synaptic activity levels regulate Nedd4-1 and USP8 in opposing fashion. A, Ubiquitination of AMPARs after chronic bicuculline treatment. Cultured cortical neurons 18–22 DIV were treated with bicuculline (20 μm) for 24 h before IP with GluA2/3 antibodies in nondenaturing IP conditions. IPs were resolved by SDS-PAGE and probed with anti-ubiquitin, anti-GluA1, and anti-GluA2/3 antibodies. B, Chronic bicuculline-induced changes in Nedd4-1 and USP8 protein levels. Representative Western blots from total lysate of cortical neurons treated with bicuculline (20 μm) for 72 h compared with untreated neurons. Cell lysates were resolved by SDS-PAGE and probed anti-GluA1, anti-Nedd4-1, anti-USP8, and anti-tubulin antibodies. C, Quantification of immunoblots for total GluA1, Nedd4-1, and USP8 levels normalized to tubulin and control condition, n ≥ 8 samples per condition. D, E, Chronic application of bicuculline promotes the redistribution of Nedd4-1. Dissociated hippocampal neurons 18–22 DIV expressing GFP HA-Nedd4-1 (Sindbis, 24 h) were treated with bicuculline (20 μm, 24 h) or left untreated and stained with anti-HA (red) and anti-GFP (green) antibodies. Redistribution was quantified using local maxima macro; n ≥ 20 dendrites per condition over 2 independent experiments (D); representative maximum z-projected confocal images of whole cell and dendrite are depicted (E). Scale bar, 10 and 5 μm for whole cell and dendrite images, respectively. *p < 0.05, **p < 0.01, ***p < 0.001; Student's t test. Graphs show mean ± SEM.
We therefore investigated whether Nedd4-1 is necessary for homeostatic loss of surface AMPARs and downscaling of synaptic strength. We used a Nedd4-1 shRNA hairpin that we previously showed to efficiently knock down endogenous Nedd4-1 protein levels (Schwarz et al., 2010). Hippocampal neurons transduced with control or Nedd4-1 shRNA hairpin lentivirus (coexpressing GFP) for 5 d were subsequently treated with bicuculline or vehicle (DMSO) for 3 d. GFP-positive cells were identified by epifluorescence and whole-cell mEPSCs were recorded. As previously reported, chronic treatment with bicuculline caused a significant reduction in both mEPSC amplitude and frequency in control cells (Ctrl-Veh, amplitude: 16.1 ± 0.5 pA, IEI: 403.1 ± 38.9 ms, n = 46 cells; Ctrl-Bic, amplitude: 12.7 ± 0.5 pA, IEI: 876.9 ± 77.7 ms, n = 46 cells; p < 0.001; Fig. 6A–C). Strikingly, we observed that knockdown of Nedd4-1 prevented the bicuculline-induced decrease in mEPSC amplitude (RNAi-Veh, amplitude: 15.9 ± 0.5 pA, n = 42 cells; RNAi-Bic, amplitude: 15.3 ± 0.8 pA, n = 41 cells; p = 0.50; Fig. 6A–C). Nedd4-1 knockdown only partially reduced the increase in interevent interval (RNAi-Veh, IEI: 500.8 ± 68.7 ms; RNAi-Bic: 729.8 ± 86.2 ms; p < 0.05). We additionally evaluated whether loss of Nedd4-1 could prevent bicuculline-induced loss of surface AMPARs. Although bicuculline caused a significant decrease in surface GluA1 levels in control cells (Ctrl-Veh, normalized surface GluA1 expression: 1.00 ± 0.05, n = 57 cells; Ctrl-Bic: 0.63 ± 0.03, n = 60 cells; p < 0.001), the knockdown of Nedd4-1 completely prevented this decrease (RNAi-Veh: 0.88 ± 0.05, n = 62 cells; RNAi-Bic: 0.79 ± 0.05, n = 59 cells; p = 0.18; Fig. 6E,F). These data indicate Nedd4-1 is required for homeostatic downscaling of synaptic strength.
Figure 6.

Nedd4-1 and USP8 are regulators of homeostatic downscaling. A–C, Hippocampal neurons DIV 14-15 were infected with FG12 control vector (expressing GFP) or shRNA Nedd4-1 hairpin (coexpressing GFP) for 5 d and then treated with bicuculline (20 μm) or vehicle (control, DMSO) for 72 h. A, Representative traces of mEPSCs recorded from each condition as indicated. Scale bar, 500 ms, 20 pA. B, Mean mEPSC amplitude for all conditions; n = 41–46 cells across 4 independent experiments. C, Cumulative probability distributions of amplitudes of all events recorded from neurons in each condition. n = 3532, 3180, 3205, and 3140 events. Inset displays averaged waveform of all events in each condition. Scale bar, 5 ms, 5 pA. D, Representative immunofluorescence images of straightened dendrites from hippocampal neurons from each condition described in A, stained for surface GluA1 (red) and GFP (green). E, Quantification of images from dendrites in all conditions, normalized to Ctrl-Veh; n = 57–60 cells across 3 independent experiments. F–H, Hippocampal neurons DIV 21–23 were infected with control GFP or Sindbis USP8-FLAG (coexpressing GFP) for 24 h and concurrently treated with 20 μm bicuculline or DMSO for 22 h. F, Representative traces of mEPSCs recorded from each condition. Scale bar, 500 ms, 20 pA. G, Cumulative probability distributions of amplitudes of all mEPSCs recorded in each condition. n = 3535, 3237, 3743, and 2798 events. Inset depicts averaged waveform of all mEPSCs. Scale bar, 5 ms, 5 pA. H, Mean mEPSC amplitude for neurons in each condition; n = 28–34 neurons across 3 independent experiments. *p < 0.05, ***p < 0.001; Student's t test. Graphs show mean ± SEM.
Because our findings suggest that USP8 works to oppose Nedd4-1's actions at the synapse and can control synaptic strength in an opposite fashion, we hypothesized that overexpression of USP8 would also prevent bicuculline-induced downscaling of synaptic strength. Using a shorter (22 h) bicuculline treatment, we again observed that increased activity causes a decrease in mEPSC amplitude (Ctrl-Veh: 16.77 ± 0.49 pA, n = 34 cells; Ctrl-Bic: 13.99 ± 0.60 pA, n = 32 cells; p < 0.001; Fig. 6F–H), but we did not observe an effect on interevent interval at this earlier time point (Ctrl-Veh: 354.2 ± 39.3 ms; Ctrl-Bic: 457.1 ± 87.8 ms; p = 0.29). In neurons overexpressing USP8, we found that bicuculline no longer produced downscaling of mEPSC amplitude (USP8-Veh: 18.55 ± 0.44 pA, n = 35 cells; USP8-Bic: 18.24 ± 0.85 pA, n = 28 cells; p = 0.74; Fig. 6F–H), but we did observe an increase in interevent interval in these cells (USP8-Veh: 249.1 ± 20.8 ms; USP8-Bic: 737.8 ± 132. 1 ms; p < 0.001).
Discussion
AMPAR activation-specific regulation of Nedd4-1 trafficking
The ubiquitination of several postsynaptic proteins has been reported to regulate their stability and function at synapses (Mabb and Ehlers, 2010). Although many proteins likely undergo constitutive ubiquitination, recent evidence suggests that ubiquitination and turnover of synaptic proteins can be regulated in part by synaptic activity (Ehlers, 2003). Ubiquitination can be rapidly reversed by the actions of DUBs. Furthermore, ubiquitination and deubiquitination can be regulated by other substrate-specific posttranslational modifications including phosphorylation (Pickart, 2004). Recently, ubiquitination has been shown to be important for the internalization, endocytic sorting, and degradation of AMPARs (Schwarz et al., 2010; Fu et al., 2011; Lin et al., 2011; Lussier et al., 2011). GluA1 and GluA2 subunits are ubiquitinated in response to the agonist AMPA, whereas the NMDAR agonist NMDA does not induce the ubiquitination of these subunits (Schwarz et al., 2010; Lussier et al., 2011). Therefore, although both AMPA and NMDA induce the internalization of AMPARs, ubiquitination appears to be a distinct feature of direct AMPAR activation.
We and others previously reported that Nedd4-1 is a HECT E3 ubiquitin ligase that ubiquitinates GluA1-containing AMPARs (Schwarz et al., 2010; Lin et al., 2011). The precise steps leading up to Nedd4-1-dependent ubiquitination of AMPARs have thus far been unknown. In this regard, we first set out to define the molecular mechanisms that regulate Nedd4-1 function at synapses. In this study, we provide evidence of rapid redistribution of Nedd4-1 to dendritic spines and synapses in response to AMPAR activation but not in response to NMDAR activation (Fig. 1). Nedd4-1 is rapidly relocalized, on the order of minutes, to dendritic spines in neurons treated with AMPA or glutamate/glycine. This redistribution requires both external calcium entry and ligand binding at the receptor (Fig. 1F,G). Hou et al. (2011) previously reported that Nedd4-1 and polyubiquitin conjugates accumulate at synapses after light-induced activation of AMPARs; however, the mechanism and functional relevance of this phenomenon has until now remained unknown. In addition, we have uncovered a critical role for the C2 domain in Nedd4-1 that has not previously been investigated in neurons. We show that the C2 domain is required for Nedd4-1's ability to traffic to dendritic spines and to reduce surface AMPAR expression (Fig. 2). Furthermore, application of CNQX and ionomycin was capable of mimicking AMPA-induced redistribution of Nedd4-1 to dendritic spines, whereas either alone was insufficient (Fig. 1G). Together, these data suggest that calcium-dependent and ligand-induced conformational changes in Nedd4-1 and AMPARs, respectively, are involved in the rapid redistribution of Nedd4-1 to dendritic spines in response to AMPAR activation.
Identification of the deubiquitinating enzyme USP8 as a regulator of synaptic strength via NMDAR-specific signaling
Although the application of glutamate and glycine caused the rapid recruitment of Nedd4-1 to dendritic spines (Fig. 1A,C), we found that it did not induce AMPAR ubiquitination (Fig. 3A). However, when we blocked NMDAR activity with the antagonist APV, we observed robust AMPAR ubiquitination in response to glutamate and glycine (Fig. 3A). In fact, we found that NMDAR activation directly antagonizes AMPA-dependent AMPAR ubiquitination (Fig. 3B). We predicted that NMDAR activation counteracts Nedd4-1 by rapidly enhancing the activity of a DUB at synapses (Fig. 3C). USP8 is a DUB that functions within the ESCRT pathway and has been shown to facilitate both recycling and degradation of previously ubiquitinated membrane cargo (Wright et al., 2011). We determined that USP8 undergoes rapid tyrosine dephosphorylation in response to NMDAR activation and not AMPAR activation and that this dephosphorylation leads to a sharp increase in its deubiquitinatase activity (Fig. 3). Mapping the phosphorylated tyrosine residue(s) and identifying the phosphatase responsible for its dephosphorylation in response to NMDAR activation will be important for a better functional understanding of USP8's role in endocytic sorting of AMPARs and other ubiquitinated membrane cargo in dendritic spines. To determine the functional relevance of USP8, we explored the possibility that USP8 could regulate synaptic strength in neurons. To date, a mammalian DUB that targets glutamate receptors has not been identified, although recent work in C. elegans has demonstrated a role for USP46 in the deubiquitination of AMPA-type glutamate receptors (GLR-1; Kowalski and Juo, 2012). USP8 is localized throughout somatic and dendritic compartments in neurons and is enriched at synapses, putting it in an ideal position to respond dynamically to neuronal activity and to regulate synaptic strength (Fig. 4A,B). Indeed, we observed that knockdown and overexpression of USP8 altered the ubiquitination of AMPARs and manipulated synaptic strength in an opposite fashion to Nedd4-1 misexpression (Schwarz et al., 2010; Figs. 2, 6). Specifically, knockdown of USP8 increased the basal ubiquitination of AMPARs and decreased synaptic strength, whereas overexpression decreased AMPA-induced ubiquitination and increased synaptic strength (Fig. 4), demonstrating a relationship between USP8 and ubiquitin-dependent AMPAR trafficking at synapses.
Nedd4-1 and USP8 regulate homeostatic downscaling of synaptic strength
Neurons have the capability to sense long-term changes in synaptic activity and scale their synaptic inputs to maintain an excitability set point (Turrigiano et al., 1998). Because Nedd4-1 and USP8 are both capable of altering synaptic strength in an opposing fashion, we hypothesized that chronic changes in activity levels could manipulate the balance of these opposing forces. Synaptic scaling has been the subject of numerous recent studies seeking to elucidate the molecular processes involved, although upscaling during activity blockade has been investigated far more thoroughly than downscaling. Although the ubiquitination and degradation of several synaptic proteins have been shown to be involved in homeostatic scaling (Seeburg et al., 2008; Shin et al., 2012; Mabb et al., 2014), to date, few E3 ubiquitin ligases have been implicated as regulators of downscaling (Fu et al., 2011; Hou et al., 2011). We found that long-term treatment with bicuculline led to a significant increase in total levels of Nedd4-1 and a concomitant decrease of USP8, suggesting a shift in the balance of ubiquitination and deubiquitination that overall favors the internalization of surface AMPARs (Figs. 5B,C, 7). Consistent with this hypothesis, we observed significantly increased AMPAR ubiquitination in neurons exposed to long-term treatment with bicuculline (Fig. 5A). Furthermore, similar treatments promoted the redistribution of Nedd4-1 to synapses (Fig. 5D,E). Together, these findings led us to hypothesize that Nedd4-1 could be in part responsible for the decrease in synaptic strength observed in bicuculline-induced downscaling. Indeed, knockdown of Nedd4-1 prevented bicuculline-induced scaling of both surface AMPARs and synaptic strength (Fig. 6), underscoring the importance of this E3 ligase in the dynamics of synaptic scaling (Fig. 7B). In addition, we further demonstrated the antagonistic relationship between Nedd4-1 and USP8 function by showing that USP8 overexpression similarly prevented activity-induced downscaling (Fig. 6). Interestingly, our manipulations of Nedd4-1 and USP8 did not eliminate reductions in frequency, which may suggest that there are alternate mechanisms being engaged in these neurons that control synapse number or presynaptic changes.
Figure 7.

Model depicting involvement of ubiquitin-dependent AMPAR trafficking in synaptic downscaling. A, AMPAR activation recruits Nedd4-1 to dendritic spines through a mechanism dependent on calcium and the C2 domain, leading to AMPAR internalization. NMDAR activation causes activation of USP8 through dephosphorylation by a protein tyrosine phosphatase (PTP), promoting the recycling of internalized AMPARs. B, Increased synaptic activity through prolonged bicuculline treatment leads to a decrease in USP8 levels and an increase in and recruitment of Nedd4-1 at synapses, causing a shift in the balance of AMPAR trafficking that overall favors their internalization, resulting in downscaling of synaptic strength.
Our results describe for the first time the activity-dependent and diametric control of an E3 ubiquitin ligase and DUB in the regulation of surface AMPARs and synaptic strength. These results do not preclude the involvement of other enzymes or even other E3 ligases, because it is known that other ligases can ubiquitinate AMPARs directly. Instead, it is likely that a shift in activity levels engages a number of molecular mechanisms at different time scales, all of which contribute to the observed changes in synaptic strength. Dynamic control of synaptic strength involves a number of complementary and competing mechanisms, and AMPARs in particular are subject to a wide variety of rapid posttranslational modifications that manipulate surface expression. Our results underscore the critical role of a fine-tunable balance between ubiquitination and deubiquitination that allows synapses to adjust to changing activity levels (Fig. 7). However, future studies describing the functional and likely dynamic relationship of these modifications (e.g., phosphorylation and ubiquitination) in the regulation of AMPAR trafficking and synaptic plasticity will be of great interest.
Footnotes
This work was supported by the National Science Foundation (Graduate Research Fellowships to S.L.S. and M.S.G.), the National Institutes of Health (Grant NS060847 to G.N.P., Training Grant T32 AG00216 to S.L.S.), and the Center for Systems Biology (Grant P50-GMO85764). We thank R. Malinow, B. Bloodgood, and members of the Patrick laboratory for helpful discussion and Lara Dozier for technical assistance.
The authors declare no competing financial interests.
References
- Carlin RK, Grab DJ, Cohen RS, Siekevitz P. Isolation and characterization of postsynaptic densities from various brain regions: enrichment of different types of postsynaptic densities. J Cell Biol. 1980;86:831–845. doi: 10.1083/jcb.86.3.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KO, Hunt CA, Kennedy MB. The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron. 1992;9:929–942. doi: 10.1016/0896-6273(92)90245-9. [DOI] [PubMed] [Google Scholar]
- Davis GW. Homeostatic signaling and the stabilization of neural function. Neuron. 2013;80:718–728. doi: 10.1016/j.neuron.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djakovic SN, Schwarz LA, Barylko B, DeMartino GN, Patrick GN. Regulation of the proteasome by neuronal activity and calcium/calmodulin-dependent protein kinase II. J Biol Chem. 2009;284:26655–26665. doi: 10.1074/jbc.M109.021956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djakovic SN, Marquez-Lona EM, Jakawich SK, Wright R, Chu C, Sutton MA, Patrick GN. Phosphorylation of Rpt6 regulates synaptic strength in hippocampal neurons. J Neurosci. 2012;32:5126–5131. doi: 10.1523/JNEUROSCI.4427-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn R, Klos DA, Adler AS, Hicke L. The C2 domain of the Rsp5 ubiquitin ligase binds membrane phosphoinositides and directs ubiquitination of endosomal cargo. J Cell Biol. 2004;165:135–144. doi: 10.1083/jcb.200309026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/S0896-6273(00)00129-X. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Fu AK, Hung KW, Fu WY, Shen C, Chen Y, Xia J, Lai KO, Ip NY. APC(Cdh1) mediates EphA4-dependent downregulation of AMPA receptors in homeostatic plasticity. Nat Neurosci. 2011;14:181–189. doi: 10.1038/nn.2715. [DOI] [PubMed] [Google Scholar]
- Hicke L. Ubiquitin-dependent internalization and down-regulation of plasma membrane proteins. FASEB J. 1997;11:1215–1226. doi: 10.1096/fasebj.11.14.9409540. [DOI] [PubMed] [Google Scholar]
- Hicke L, Dunn R. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu Rev Cell Dev Biol. 2003;19:141–172. doi: 10.1146/annurev.cellbio.19.110701.154617. [DOI] [PubMed] [Google Scholar]
- Hou Q, Gilbert J, Man HY. Homeostatic regulation of AMPA receptor trafficking and degradation by light-controlled single-synaptic activation. Neuron. 2011;72:806–818. doi: 10.1016/j.neuron.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huganir RL, Nicoll RA. AMPARs and synaptic plasticity: the last 25 years. Neuron. 2013;80:704–717. doi: 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski JR, Juo P. The role of deubiquitinating enzymes in synaptic function and nervous system diseases. Neural Plast. 2012;2012:892749. doi: 10.1155/2012/892749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Tomooka Y, Noda M. Identification of a set of genes with developmentally down-regulated expression in the mouse brain. Biochem Biophys Res Commun. 1992;185:1155–1161. doi: 10.1016/0006-291X(92)91747-E. [DOI] [PubMed] [Google Scholar]
- Kumar S, Harvey KF, Kinoshita M, Copeland NG, Noda M, Jenkins NA. cDNA cloning, expression analysis, and mapping of the mouse Nedd4 gene. Genomics. 1997;40:435–443. doi: 10.1006/geno.1996.4582. [DOI] [PubMed] [Google Scholar]
- Lee SH, Simonetta A, Sheng M. Subunit rules governing the sorting of internalized AMPA receptors in hippocampal neurons. Neuron. 2004;43:221–236. doi: 10.1016/j.neuron.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Lin A, Hou Q, Jarzylo L, Amato S, Gilbert J, Shang F, Man HY. Nedd4-mediated AMPA receptor ubiquitination regulates receptor turnover and trafficking. J Neurochem. 2011;119:27–39. doi: 10.1111/j.1471-4159.2011.07221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JW, Ju W, Foster K, Lee SH, Ahmadian G, Wyszynski M, Wang YT, Sheng M. Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat Neurosci. 2000;3:1282–1290. doi: 10.1038/81814. [DOI] [PubMed] [Google Scholar]
- Lu W, Roche KW. Posttranslational regulation of AMPA receptor trafficking and function. Curr Opin Neurobiol. 2012;22:470–479. doi: 10.1016/j.conb.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lussier MP, Nasu-Nishimura Y, Roche KW. Activity-dependent ubiquitination of the AMPA receptor subunit GluA2. J Neurosci. 2011;31:3077–3081. doi: 10.1523/JNEUROSCI.5944-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabb AM, Ehlers MD. Ubiquitination in postsynaptic function and plasticity. Annu Rev Cell Dev Biol. 2010;26:179–210. doi: 10.1146/annurev-cellbio-100109-104129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabb AM, Je HS, Wall MJ, Robinson CG, Larsen RS, Qiang Y, Corrêa SA, Ehlers MD. Triad3A regulates synaptic strength by ubiquitination of Arc. Neuron. 2014;82:1299–1316. doi: 10.1016/j.neuron.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer IM, Kerperien J, Sotoca AM, van Zoelen EJ, van Leeuwen JE. The Usp8 deubiquitination enzyme is post-translationally modified by tyrosine and serine phosphorylation. Cell Signal. 2013;25:919–930. doi: 10.1016/j.cellsig.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Mizuno E, Kitamura N, Komada M. 14–3-3-dependent inhibition of the deubiquitinating activity of UBPY and its cancellation in the M phase. Exp Cell Res. 2007;313:3624–3634. doi: 10.1016/j.yexcr.2007.07.028. [DOI] [PubMed] [Google Scholar]
- Nalefski EA, Falke JJ. The C2 domain calcium-binding motif: structural and functional diversity. Protein Sci. 1996;5:2375–2390. doi: 10.1002/pro.5560051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick GN. Synapse formation and plasticity: recent insights from the perspective of the ubiquitin proteasome system. Curr Opin Neurobiol. 2006;16:90–94. doi: 10.1016/j.conb.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Pickart CM. Back to the future with ubiquitin. Cell. 2004;116:181–190. doi: 10.1016/S0092-8674(03)01074-2. [DOI] [PubMed] [Google Scholar]
- Piper RC, Luzio JP. Ubiquitin-dependent sorting of integral membrane proteins for degradation in lysosomes. Curr Opin Cell Biol. 2007;19:459–465. doi: 10.1016/j.ceb.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant PJ, Yeger H, Staub O, Howard P, Rotin D. The C2 domain of the ubiquitin protein ligase Nedd4 mediates Ca2+-dependent plasma membrane localization. J Biol Chem. 1997;272:32329–32336. doi: 10.1074/jbc.272.51.32329. [DOI] [PubMed] [Google Scholar]
- Plant PJ, Lafont F, Lecat S, Verkade P, Simons K, Rotin D. Apical membrane targeting of Nedd4 is mediated by an association of its C2 domain with annexin XIIIb. J Cell Biol. 2000;149:1473–1484. doi: 10.1083/jcb.149.7.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozo K, Goda Y. Unraveling mechanisms of homeostatic synaptic plasticity. Neuron. 2010;66:337–351. doi: 10.1016/j.neuron.2010.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz LA, Hall BJ, Patrick GN. Activity-dependent ubiquitination of GluA1 mediates a distinct AMPA receptor endocytosis and sorting pathway. J Neurosci. 2010;30:16718–16729. doi: 10.1523/JNEUROSCI.3686-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg DP, Feliu-Mojer M, Gaiottino J, Pak DT, Sheng M. Critical role of CDK5 and Polo-like kinase 2 in homeostatic synaptic plasticity during elevated activity. Neuron. 2008;58:571–583. doi: 10.1016/j.neuron.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Shin SM, Zhang N, Hansen J, Gerges NZ, Pak DT, Sheng M, Lee SH. GKAP orchestrates activity-dependent postsynaptic protein remodeling and homeostatic scaling. Nat Neurosci. 2012;15:1655–1666. doi: 10.1038/nn.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todi SV, Paulson HL. Balancing act: deubiquitinating enzymes in the nervous system. Trends Neurosci. 2011;34:370–382. doi: 10.1016/j.tins.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Wang J, Peng Q, Lin Q, Childress C, Carey D, Yang W. Calcium activates Nedd4 E3 ubiquitin ligases by releasing the C2 domain-mediated auto-inhibition. J Biol Chem. 2010;285:12279–12288. doi: 10.1074/jbc.M109.086405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright MH, Berlin I, Nash PD. Regulation of endocytic sorting by ESCRT-DUB-mediated deubiquitination. Cell Biochem Biophys. 2011;60:39–46. doi: 10.1007/s12013-011-9181-9. [DOI] [PubMed] [Google Scholar]
- Yamada T, Yang Y, Bonni A. Spatial organization of ubiquitin ligase pathways orchestrates neuronal connectivity. Trends Neurosci. 2013;36:218–226. doi: 10.1016/j.tins.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]