

Abstract
Depletion of DNA replication initiation factors such as CDC7 kinase triggers the origin activation checkpoint in healthy cells and leads to a protective cell cycle arrest at the G1 phase of the mitotic cell division cycle. This protective mechanism is thought to be defective in cancer cells. To investigate how this checkpoint is activated and maintained in healthy cells, we conducted a quantitative SILAC analysis of the nuclear- and cytoplasmic-enriched compartments of CDC7-depleted fibroblasts and compared them to a total cell lysate preparation. Substantial changes in total abundance and/or subcellular location were detected for 124 proteins, including many essential proteins associated with DNA replication/cell cycle. Similar changes in protein abundance and subcellular distribution were observed for various metabolic processes, including oxidative stress, iron metabolism, protein translation and the tricarboxylic acid cycle. This is accompanied by reduced abundance of two karyopherin proteins, suggestive of reduced nuclear import. We propose that altered nucleo-cytoplasmic trafficking plays a key role in the regulation of cell cycle arrest. The results increase understanding of the mechanisms underlying maintenance of the DNA replication origin activation checkpoint and are consistent with our proposal that cell cycle arrest is an actively maintained process that appears to be distributed over various subcellular locations.

Keywords: quantitative proteomics, mass spectrometry, SILAC, CDC7, DNA replication, cell cycle, checkpoint, oxidative stress, metabolism, nucleus-cytoplasm trafficking
1. INTRODUCTION
DNA replication initiation in the mitotic cell division cycle occurs at thousands of replication origins spread along the length of eukaryotic chromosomes.1,2 During DNA replication licensing in early G1 phase, several factors colocalize at origin sites, including the origin recognition complex (ORC), CDC6 and CDT1 which together recruit the MCM helicase leading to the formation of the prereplicative complex (pre-RC). In late G1, the pre-RC is “fired” by CDKs and CDC7 kinase which phosphorylate MCM subunits and stimulate helicase activity. This allows loading of auxiliary factors, CDC45 and the replisome which contains RPA, PCNA, and DNA polymerase, leading to entry into S phase for DNA synthesis.2 Somatic cells have evolved “checkpoint” mechanisms both within and at the boundary of each cell cycle phase to ensure that genomic duplication occurs only once per cell cycle and that the next phase is only entered following successful completion of the previous one. Aberrant regulation of these protective cell cycle mechanisms can result in uncontrolled proliferation, incorporation of genomic defects and the development of cancer.
A novel, protective cell cycle checkpoint has recently been identified at the G1/S transition in mammalian diploid cells, and functions to monitor the competency of replication origins.1,3-5 If not enough origins are competent for DNA replication initiation, this “origin activation checkpoint” (OAC) will prevent DNA synthesis (S phase) and will instead promote cellular viability by prolonging the gap phase (G1). In this way, a compromised cell will not undergo a potentially lethal round of incomplete DNA replication and will instead remain arrested in G1. Cancer cells, however, appear to have lost this protective checkpoint control and continue with an incomplete round of DNA replication, leading to fork stalling/collapse, strand breaks and ultimately apoptosis.1,3,6
It has been shown that the origin activation checkpoint can be triggered by perturbation of the DNA replication initiation pathway and our previous work has revealed that a specific reduction in the CDC7 kinase alone is sufficient to activate this checkpoint mechanism in normal cells.5 Furthermore, Montagnoli et al.3 have also shown that RNAi against CDC7 blocked the activation of the MCM helicase and led to G1 arrest through induction of p53, p21 and accumulation of hypophosphorylated Rb. The CDC7 kinase is a highly conserved, pleiotropic molecule with many diverse roles in cellular regulation.7 In addition to promoting S phase entry by phosphorylating the MCM2–7 helicase,8 CDC7 can activate DNA damage repair responses and checkpoints9-12 and is involved in cohesin loading onto chromatin for chromosome segregation in mitosis,13 in the regulation of meiotic transcription14 as well as double strand break formation during meiotic recombination.15
Because various tumor types overexpress CDC7,16-18 this kinase has now become a key cell cycle target for cancer therapy.19,20 Inhibition of CDC7 appears to have the dual beneficial effect of triggering a protective nongenotoxic cell cycle arrest in healthy cells, while sending cancer cells into an abortive S-phase, resulting in cancer cell-specific death.3,4 Therefore, pharmacological CDC7 small molecule inhibitors are currently under investigation as potent and selective anticancer therapeutics.19-21 It is predicted that tumors exhibiting high CDC7 levels, with accelerated cell cycle progression and an underlying defect in the origin activation checkpoint machinery will respond optimally to a clinical CDC7-targeted approach.6 Importantly, it has been demonstrated in cell line models that the G1 arrest triggered by CDC7 knockdown is fully reversible on recovery of CDC7 protein levels,4 which suggests that normal tissue can resume proliferative activity following elimination of cancer cells. This implies that self-renewing tissues characterized by high turnover (such as gut mucosa) may respond well to CDC7 small molecule inhibitors and will be less prone to the toxicity associated with classical chemotherapeutic agents.
If CDC7 is to become a successful cancer target, it is imperative that we understand the molecular mechanisms underlying the origin activation checkpoint: how it triggers cell cycle arrest in normal cells and how this checkpoint mechanism is circumvented by cancer cells under CDC7 rate-limiting conditions. To elucidate the mechanisms involved in regulation of this checkpoint, we have previously used a combination of cell biology, transcriptomics and a global quantitative proteomics approach.5,22 Specifically, we triggered the activation of this novel checkpoint by RNAi depletion of CDC7 in IMR90 normal human diploid fibroblasts. We have shown that this checkpoint-activated cell cycle arrest is coordinated by the transcription factor FOXO3a, which undergoes subcellular redistribution to the nucleus for activation of the p53-p21 axis and for upregulation of p15-INK4B and p21. p53 can then induce the expression of a WNT pathway antagonist DKK3, which in turn downregulates Myc and cyclin D1, leading to inactivation of the Rb-E2F pathway and overriding the entire G1/S transcriptional program.5
To further investigate how the arrested phenotype is maintained and how it promotes cellular survival, we employed a global quantitative proteomics strategy. RNAi suppression of CDC7 in combination with SILAC-based high resolution mass spectrometry was coupled with downstream bioinformatics.22 This approach revealed that many dynamic processes distributed across the mitochondria, lysosomes and the cell surface are also involved in maintaining a quiescent, (G0)-like arrested state, including altered metabolic flux, moderate stress response and reduced proliferative capacity.
In the present study, the coverage of our quantitative proteomics strategy is improved by focusing on two subcellular locations critical for cell cycle control: the cytoplasm and the nucleus. This strategy gave much greater detail on changes in abundance of nuclear proteins involved in DNA replication and cell cycle control and revealed previously unidentified participants in the checkpoint. Our previous work established that nucleo-cytoplasmic redistribution of FOXO3a is important for co-ordinating the checkpoint response.5 The present results indicate that dynamic redistribution of proteins between the nucleus and the cytoplasm, with or without changes in overall protein abundance, is an integral aspect of the checkpoint that may underlie the spatially distributed cellular response in mitochondria, lysosomes and at the cell surface that we detected previously.22
2. MATERIAL AND METHODS
2.1. Cell Culture and SILAC Labeling
IMR90 (ATCC CCL-186), a human primary diploid fibroblast adherent cell line, was obtained from LGC Standards (Middlesex, U.K.) at population doubling (PD) 12. All experiments with IMR90 cells were performed with a PD of less than 22. IMR90 cells were cultured at 37 °C with 5% CO2 in SILAC-specific DMEM media (Invitrogen, Paisley, U.K.), supplemented with 10% dialyzed FBS (Invitrogen), 100 U/mL penicillin and 100 μg/mL streptomycin (Sigma Aldrich, Poole, U.K.) according to manufacturer’s instructions (Invitrogen, Paisley, U.K.). For quantitative SILAC analysis of two labeling states, the cells were divided into two populations, labeled with either “light” l-Lysine and l-Arginine (Lys0, Arg0) or “heavy” 13C6-Lysine and 13C6,15N4–Arginine (Lys6, Arg10) (Invitrogen, Paisley, U.K.) and cultured for six passages to achieve full incorporation of the SILAC amino acids.
2.2. RNA Interference
Small interfering RNA (siRNA) depletion of CDC7 was performed on the “light” population, while the “heavy” cells were transfected with a control oligo for three biological replicates. CDC7 expression was inhibited in SILAC-labeled IMR90 cells with custom double-stranded RNA oligonucleotides (Ambion, Warrington, U.K.) at 10 nM concentration for 72 h (5′→3′ sense oligo: GCUCAGCAGGAAAGGUGUUUU; antisense oligo: AACACCUUUCCUGCUGAGCUU). The CDC7 siRNA used in this study has previously been extensively characterized for transfection efficiency, off-target effects, BrdU incorporation assays and flow cytometry studies.5 Nontargeting siRNA was used as the negative control.
2.3. Sample Preparation
Total cell lysates (T) as well as nuclear and cytoplasmic-enriched subcellular fractions (N, C) were prepared as described in Tudzarova et al.5 Briefly, SILAC-labeled IMR90 fibroblasts were lysed for 45 min on ice in modified RIPA lysis buffer (50 mM Tris–HCl, 300 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA with protease inhibitors) and sonicated for 10 s to obtain the total lysate samples (T). For subcellular fractionation, cells were lysed in a hypotonic buffer containing 10 mM HEPES pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 0.1% Triton X-100, 1.0 mM DTT with protease inhibitors and carefully homogenized by several inversions of the tube. Nuclei were precipitated by centrifugation at 1000× g for 5 min at 4 °C and the cytoplasmic-enriched fraction (C) was removed. For preparation of nuclear-enriched (N) fractions, the crude nuclear pellets were washed twice with the hypotonic buffer, lysed in the modified RIPA buffer for 30 min, sonicated and centrifuged at 13000× g.
2.4. Protein Separation, Western Blotting and In-gel Digestion
Protein concentration was evaluated by the Bio-Rad protein assay kit (Bio-Rad, Hemel Hempstead, U.K.). For Western blot analysis, 60 μg of total protein was loaded in each lane and separated by 4% to 20% SDS-PAGE. Proteins were transferred onto PVDF membranes (Bio-Rad) by semidry electroblotting. Blocking, antibody incubations and washing steps were performed as described.5 Antibodies used were: CDC7 (DCS-342) from Caltag-MedSystems Ltd., U.K.; MCM2-pS53 (A300–756A) from Universal Biologicals Ltd., Cambridge, U.K.; MCM2-pS108 (A300–094A) Universal Biologicals Ltd., Cambridge, U.K.; FoxO3A (07–702) from Millipore (U.K.) Ltd.; Nucleolin/C23 (sc-13057) from Insight Biotechnology Ltd., U.K.; Ku80 (05–393) Millipore Ltd., U.K.; Ku70 (Ab-1358MI) Fisher Scientific UK Ltd.; GLS-1 (ab93434) VWR International Ltd., U.K.; PC4/SUB1 (ab72132) VWR International Ltd., UK; SET (ab92872) VWR International Ltd., U.K.; ORC4 (sc-19726) Insight Biotechnology Ltd., UK; MEK2 (610236) Insight Biotechnology Ltd., U.K.; FTH1 (3998) New England Biolabs Ltd., UK; HO1 (5141) New England Biolabs Ltd., U.K.; KPNA2 (A300–484A) Cambridge Bioscience Ltd., UK; HAT1 (PA1–25446) Thermo Scientific, UK; p53 (645702) Insight Biotechnology Ltd., U.K.
For comparative SILAC analysis, “heavy” and “light” nuclear or cytoplasmic extracts were combined in a 1:1 ratio based on protein concentration (30 μg each) and separated by SDS-PAGE under reducing conditions. Proteins were visualized by silver-staining (ProteoSilver Plus, Sigma Aldrich, Poole, U.K.), equally sized bands were excised from the gel lane and processed with the Progest Investigator (Digilab, Huntingdon, U.K.) using established protocols for reduction and alkylation.23 Finally, gel plugs were rehydrated in 20 μg/mL sequencing grade modified trypsin (Promega, Southampton, UK) and incubated overnight at 37 °C. Tryptic peptides were eluted, vacuum-dried, resuspended in 0.1% formic acid and analyzed by LC–MS/MS.
2.5. Mass Spectrometry
LC–MS/MS was performed with an LTQ-Orbitrap Classic mass spectrometer (Thermo Fisher Scientific, UK). Peptide samples were loaded using a Surveyor MS Pump and Micro AS autosampler (Thermo Fisher Scientific, UK) onto a MiChrom C18 CapTrap for desalting and then introduced into the MS via a nanoelectrospray ion source consisting of a fused silica capillary column (I.D. 100 μm; O.D. 360 μm; length 20 cm; 5 μm C18 Reprosil, Nikkyo Technos Co., Japan). Separation was achieved by a dual gradient, formed of 5–23% Buffer B for 65 min, followed by 23–40% Buffer B for 30 min, and a step gradient to 60% Buffer B for 5 min (Buffer A = 0.1% formic acid; Buffer B = 100% acetonitrile (ACN), 0.1% formic acid). The pump rate was reduced via a splitter to 0.9 μL/min. Measurements were performed in the positive ion mode. The full scan precursor MS spectra (450–1600 m/z) were acquired in the Orbitrap analyzer with a resolution of r = 60000. This was followed by data dependent MS/MS fragmentation of the most intense ion from the survey scan using collision induced dissociation (CID) in the linear ion trap (normalized collision energy 35%, activation Q 0.25; electrospray voltage 1.4 kV; capillary temperature 200 °C; isolation width 2.00). This MS/MS scan event was repeated for the top 6 peaks in the MS survey scan. Target ions already selected for MS/MS were dynamically excluded for 40 s. Singly charged ions were excluded from MS/MS analysis. Total lysate, nuclear and cytoplasmic samples were all acquired using the same mass spectrometric parameters and instrument methods. XCalibur software version 2.0.7 (Thermo Fisher Scientific, U.K.) was used for data acquisition.
2.6. Protein Identification and Quantification
Raw MS files from all replicate SILAC experiments were uploaded onto the MaxQuant software platform (version 1.2.0.18) for peaklist generation, quantification of SILAC pairs, identification of individual peptides and assembly into protein groups.24 The Andromeda search engine was used25 and searched against a concatenated International Protein Index (IPI) human protein database (version 3.68; containing 87,061entries as well as commonly observed contaminants such as porcine trypsin and some human keratins). Selected MaxQuant analysis parameters included: trypsin enzyme specificity, SILAC doublet measurements of Lys6 and Arg10, 2 missed cleavages, minimum peptide length of 6 amino acids, minimum of 2 peptides (1 of which is unique), top 6 MS/MS peaks per 100 Da, peptide mass tolerance of 20 ppm for precursor ions and MS/MS tolerance of 0.8 Da. Oxidation of methionine and N-terminal protein acetylation were selected as variable modifications and cysteine carbamidomethylation was selected as a fixed modification. All proteins were filtered according to a false discovery rate (FDR) of 1% applied at both peptide and protein levels and a maximum peptide posterior error probability (PEP) of 0.5. Proteins were automatically quantified in the MaxQuant software: a minimum of 3 peptide ratio counts from razor and unique peptides were necessary for protein quantification and the “requantification” option was enabled. An Experimental Design template was used to specify individual replicate experiments (each data set contained three biological replicate experiments). In addition to the cytoplasmic and nuclear extract data sets, three of the previously analyzed total cell lysate data sets22 were reanalyzed under the same parameters in order to make a valid comparison between the three data sets (T, C and N). MaxQuant output files were subsequently uploaded into Perseus (version 1.2.0.17) for calculation of Significance B scores as well as the GO term association for each Protein Group.
2.7. Correlation of Proteins Across Different Samples
The Max Quant analysis gave a total of 2598, 2996, and 2001 protein sequence groups for the cytoplasm (C), nucleus (N) and total lysate (T) samples, respectively. The data for each sample type, including each replicate, is shown in Supplementary Tables 1-3, Supporting Information. These corresponded to 4940 different protein sequence groups across the three samples. For the same underlying gene, slightly different protein sequence groups were sometimes observed in the different samples depending on the exact set of peptides detected in each sample. Using the principle that sequence groups in different samples that correspond to the same underlying protein(s) must have protein sequences in common, the union of the unique peptides for protein sequence groups with shared protein sequences was formed and used to requery the full human IPI v3.68 sequence data set to identify “consensus” sequence groups across the three sample types. This correlated the initial 4940 protein sequence groups to give 3684 “consensus” protein sequence groups across the three samples. All of the consensus sequence groups contained at least one consensus protein sequence that contained all unique peptides in the group and all were independent, that is, there were no protein sequences shared between different consensus protein sequence groups. Of these consensus protein sequence groups, 10 corresponded to protein sequences without an assigned gene name. For 55 genes involving 113 protein sequence groups, more than one independent protein sequence group was required, that is, the peptides gave direct evidence for more than one protein isoform and the different isoforms could be resolved across the three samples. For 57 protein sequence groups involving 27 genes, mixtures of multiple isoforms could not be unravelled across the three different samples. Overall, 3627 independent, unambiguous consensus protein sequence groups were established across the three samples, of which 2554, 2736, and 1985 were observed in the cytoplasm (C), nucleus (N) and total lysate (T) samples respectively. The data for these 3627 consensus protein sequence groups is shown in Supplementary Table 4, Supporting Information.
2.8. Selection of Significantly Changed Proteins
Three types of selection limits were applied to the MaxQuant Significance B (SigB) measures of significance of the SILAC ratios (i.e., Sn, nucleus; Sc, cytoplasm; and St, total lysate) to delineate proteins with significant changes in abundance and/or in nuclear/cytoplasm subcellular distribution. (1) SigB (union) < limit-1, where SigB (union) denotes the aggregate SigB for all three biological replicates, for example, SigB (Sn, union) denotes SigB calculated over the SILAC ratio Sn measured in all three nucleus samples. (2) For each biological replicate, SigB < limit-2 and a minimum number of replicates, for example, SigB (Sn) < limit-2 in all three nucleus replicates. These two criteria were combined (see below) to ensure both overall significance and reproducibility between replicates. (3) |log2(Sn/Sc)| > limit-3, which detects redistribution of proteins even without major changes in total abundance (see Supplementary Text, Supporting Information). For any individual protein, only some of the SILAC ratios Sn, Sc and St may have been measured in all replicates/samples. Furthermore, because it is independent of subcellular fractionation, St is probably inherently more reproducible than Sn or Sc. We therefore selected a “stringent” set of 95 reproducibly significant proteins (see text) using the criteria: (a) SigB (St, union) < 0.001 AND SigB (St) < 0.003 for at least 2 replicates; (b) SigB (Sn or Sc, union) < 0.001 AND SigB (Sn or Sc) < 0.003 for all three replicates; (c) SigB (Sn) < 0.003 AND SigB (Sc) < 0.003 for at least 2 replicates (to handle cases where the reduced coverage of the T sample led to Sn and Sc, but not St being measured—conservation of mass requires that if Sn and Sc show major changes, St should also, see Supplementary Text, Supporting Information); (d) |log2(Sn/Sc)| > 1, that is, 2 < Sn/Sc < 0.5, for both the union and at least 2 replicates (conservation of mass requires Sn/Sc = 1 if there is no redistribution, see Supplementary Text).
We also selected a slightly larger “strict” set of 124 proteins showing significant changes using the selection criteria: (a) SigB (St, union) < 0.003 AND SigB (St) < 0.009 for at least 2 replicates; (b) SigB (Sn or Sc, union) < 0.003 AND SigB (Sn or Sc) < 0.009 for at least 2 replicates; (c) SigB (Sn) < 0.009 AND SigB (Sc) < 0.009 for at least 2 replicates; (d) |log2(Sn/Sc)| > 0.85, that is, 1.8 < Sn/Sc < 0.55, for both the union and at least 2 replicates. The use of both the union and individual replicates in the selection criteria ensured reproducibility and that only proteins with appreciable numbers of ratio counts (e.g., a minimum of 8 in a single sample, with a median of 41 for the strict set) were included among proteins designated as showing significant changes. Overall, both sets of selection criteria were quite stringent with only 2.6% (stringent set) or 3.4% (strict set) of the 3627 identified proteins designated as showing the most significant changes.
2.9. Bioinformatics Analysis of Functional Networks
For the identification of functional annotations, associations, interactions and networks within our data set, a combination of several data analysis tools were used. Initial tests with the stringent and strict sets of most significantly changed proteins indicated that very similar networks were identified and the set of 124 strict proteins is therefore used in this paper. The consensus Gene Name identifiers for the strict set of 124 most significantly changed proteins were uploaded into STRING version 9.0 (Search Tool for the Retrieval of Interacting Genes/Proteins), to create a protein interaction network based on known and predicted protein–protein interactions.26,27 A threshold confidence score of 0.7 was used to ensure that only highly confident protein interactions were considered for inclusion in the network. Seven types of protein interaction information were used for network generation, including neighborhood, gene fusion, co-occurrence, coexpression, experimental, database knowledge and text mining. The network was imported into Cytoscape (version 2.8.2) for further analysis and visualization.28,29
Alternatively, the list of consensus Gene Name identifiers for the 124-protein set was uploaded to the GeneMania plugin (v2.5) via the Cytoscape platform for functional biological prediction and network integration.30,31 The data set was searched against the Homo sapiens core network data (version 2012–05–25-core) allowing for all types of network interactions (coexpression, colocalization, genetic interactions, pathway, physical interactions, predicted and shared protein domains) and also identifying the top-20 “related” proteins using automatic weighting. The resulting network was visualized in Cytoscape (v2.8.2).28,29 The list of 124 proteins were also uploaded into the Reactome pathway analysis plugin for Cytoscape32 and analyzed for significantly overrepresented pathways, processes and locations.
Within the protein interaction networks, proteins (nodes) are linked by edges representing types of interaction which have been experimentally validated or for which there is strong evidence in the literature. The String and GeneMania networks were analyzed further using the Cytoscape plugins MCODE and BiNGO. MCODE (v1.32) was used to identify clusters of densely interconnected regions within the protein networks33 and BiNGO (v2.44) was used to determine Gene Ontology term enrichment associated with protein lists such as the MCODE cluster groups.34 These clusters could represent a protein complex, pathway or functional process. We subsequently expanded these clusters to look for other proteins detected in our data sets that had fewer ratio counts, were quantified in fewer replicates and/or showed smaller changes in abundance or spatial redistribution but were densely connected to the primary seed clusters in the STRING interaction network.
3. RESULTS
Depletion of CDC7 kinase by siRNA methods has been shown to trigger a novel DNA replication origin activation checkpoint in mammalian cells3 and we have developed and rigorously validated this CDC7-depleted system in IMR90 human primary diploid fibroblasts by using complementary molecular biology and proteomics tools.5,22 Efficiency, specificity and validation of the knockdown have all been previously addressed by our group and are detailed elsewhere.5 The quantitative proteomic approach used for the present study is shown in Figure 1a. IMR90 fibroblasts grown in either light or heavy SILAC medium were treated with CDC7 siRNA or with a control oligo. Cells were then harvested and subjected to crude fractionation to give nucleus-enriched and cytoplasm-enriched subcellular samples. Three biological replicate experiments were conducted for each sample type. To compare this subcellular fractionation approach to an unfractionated sample, three biological replicate data sets were taken from our previous SILAC analysis of CDC7-depleted total cell lysate samples (a subset of the samples published in ref 22) and reanalysed under identical parameters.
Figure 1.

Comparative analysis of CDC7-depleted nuclear-enriched and cytoplasmic-enriched fractions. (a) SILAC workflow: SILAC-labeled IMR90 fibroblasts were treated with CDC7 siRNA or control oligo and fractionated into cytoplasmic, nuclear or total cell lysates. MaxQuant software was used for protein inference and quantitation.24 STRING,26 GeneMania30 and Reactome32 software were used for protein networks, functional annotation enrichment and pathway analysis. Cytoscape28 was used for network visualization and for further analysis with BiNGO34 and MCODE33 plugins. (b) Cells treated with either control oligo or CDC7-siRNA were fractionated into cytoplasmic and nuclear-enriched fractions and subjected to Western blot analysis for CDC7, phospho-MCM2 (pS53 and pS108 sites), p53, FOXO3a, ORC4 and MEK2. (c) Venn diagram showing: (A) distribution over the three sample types for the 3627 proteins identified; (B) distribution over the three sample types for the strict set of 124 significantly changed proteins (see text).
Previous gene microarray experiments with the negative control siRNA5 and reverse SILAC labeling experiments22 indicated that there are no detected general (e.g., interferon-like/virus-like off-target effects), nonspecific differences in cellular response to the CDC7 siRNA and the nontargetting control siRNA.
Stringent purification of organelles was not attempted in this study as we believe it is not feasible to purify dynamic organelles to homogeneity and that unnecessary protein loss is incurred during such organelle isolation methods. We preferred to obtain “enriched” rather than “highly purified” subcellular regions as a way to reduce sample complexity compared to total lysate samples and to explore the possibility of protein redistribution following activation of the checkpoint. As shown in the following, this crude subcellular fractionation approach was found to be an effective strategy for the detection of changes in protein abundance in the nuclear and cytoplasmic regions.
Western blot analysis of the nuclear-enriched and cytoplasm-enriched preparations (Figure 1b) demonstrated that CDC7 was primarily localized to the nucleus in control fibroblasts and was undetectable in samples treated with CDC7 siRNA. The CDC7 substrate MCM2 showed a loss of phosphorylation at two CDC7 responsive sites (p-S53 and p-S108) confirming a reduction in CDC7 kinase activity in the siRNA treated cells. We estimated from antibody measurements that the abundance/activity of CDC7 was reduced to less than 10%. This was confirmed at the mRNA level by qRT-PCR. A shift of FOXO3a from the cytoplasm to the nucleus with CDC7 suppression indicated that the origin activation checkpoint has been engaged in the CDC7-depleted cells, as FOXO3a is one of the three major axes known to coordinate this checkpoint response.5 ORC4 and MEK2 were used as marker proteins for the nuclear and cytoplasmic regions respectively and confirmed the overall enrichment of the two fractions.35
3.1. Crude Subcellular Fractionation of CDC7-depleted Fibroblasts Enhances Proteomic Coverage of the DNA Replication Origin Activation Checkpoint
The MS data for the cytoplasm (C), nucleus (N) and total lysate (T) samples, including each replicate, is given in Supplementary Tables 1-3, Supporting Information. Correlation of the 4940 distinct protein sequence groups contained in the three data sets identified 3627 unambiguous, independent, consensus protein sequence groups across the three samples (see Material and Methods), of which 2494, 2886, and 1922 were observed in the C, N and T samples respectively. Data for the consensus protein sequence groups is shown in Supplementary Table 4, Supporting Information. The three biological replicate experiments for each sample type were combined in a single analysis using the MaxQuant quantitative analysis suite and each data set was evaluated for quality and reproducibility (Supplementary Figure S1, Supporting Information). With requirement of a minimum of three peptide ratio counts per protein, 2196 C, 2526 N and 1723 T proteins were quantified. Overall, 3203 distinct proteins were quantified in at least one of the three samples; 56, 53 and 58% of the proteins for the C, N and T samples respectively were quantified in all three biological replicates.
The distribution of the different protein sequence groups across the three samples is shown in Figure 1c. Only 47 (1.3%) of the 3627 proteins were observed solely in the T sample, mostly with small numbers of counts, indicating that there were no large, systematic losses of proteins during the fractionation of the C and N samples. Of the 3580 proteins identified in the C and N samples, 694 and 1086 were observed exclusively in C or N respectively. This included many proteins with large numbers of peptides/counts, thereby further indicating that nuclear enrichment/depletion of the N and C fractions was efficient and that there are many proteins that are characteristic of one or the other subcellular location. On the other hand, 1800 proteins (50.3%) were detected in both the C and N samples, which is consistent with increasing evidence that many proteins have multiple subcellular locations.36-39 Overall, coverage of the proteome was increased from 1922 to 3580 proteins by the subcellular fractionation. For the 1875 T proteins observed in N, C or both, this was generally associated with increases both in sequence coverage and the number of SILAC ratio counts, i.e. with better identification and quantitation of the protein (see Supplementary Figure S2, Supporting Information).
MaxQuant normalizes protein ratios for each data set and employs an algorithm within the Perseus program to calculate an outlier probability score (Significance B score, hereafter SigB) in order to find significantly altered proteins.24 For each sample, four sets of SigB values were calculated using the MaxQuant quantitative analysis suite: one for each of the three replicates and one for the union over all three replicates. These SigB values and the parameter log2(Sn/Sc), which detects redistribution between C and N even in the absence of total abundance changes (see Supplementary Text, Supporting Information), were used to select sets of 95 proteins (stringent set, 2.6% of all identified proteins) or of 124 proteins (strict set, 3.4%) that showed the strongest, reproducible changes in abundance and/or redistribution between the cytoplasm and nucleus upon CDC7 suppression (see Material and Methods). The inclusion of reproducibility over replicates meant that the selection criteria were quite stringent for both protein sets and tended to reject proteins with few ratio counts, for example, a minimum of 8 ratio counts (in a single sample) and a median of 41 ratio counts per protein were used in classifying the proteins in the strict set (see Material and Methods). For the samples in which they were quantified, the distribution over the C, N and T samples of the 124 proteins in the strict set is shown in Figure 1c. A summary of the stringent and strict protein sets is presented in Supplementary Table 5, Supporting Information. Complete data for these proteins (sequence groups), including alternate sequences/names where appropriate, is contained in Supplementary Table 4, Supporting Information.
3.2. Identification of Functional Networks Associated with DNA-Replication and Cell Cycle
Initial analysis revealed that both the GeneMania functional prediction tool30,31 and the STRING database for protein–protein interactions26,27 identified similar networks using both the 95-protein stringent and 124-protein strict sets of most significantly changed proteins – results using the strict set are presented in the following. As described below, both programmes identified “DNA Replication” as the top-scoring process associated with the set of 124 most significantly changed proteins.
The GeneMania network contained 142 nodes linked by 3796 edges representing various types of functional association evidence. The software identified DNA replication as the top-scoring functional GO term (q-value = 3.2 × 10−22), with 26 of the 142 network nodes annotated to this term. The GeneMania network, including these 26 DNA replication proteins is presented in Supplementary Figure S3, Supporting Information. DNA replication was also identified by MCODE analysis, which found a primary cluster composed of 43 nodes and 1429 edges within the GeneMania network (MCODE score 33.23) that was highly enriched for the GOBP term DNA replication (corr p-value: 1.1 × 10−39). This top-scoring cluster is shown in Figure 2a.
Figure 2.

DNA replication is the most significantly affected process following depletion of CDC7. DNA replication was identified as differentially regulated by the GeneMania network tool. (a) Top-scoring interaction cluster within the GeneMania network contained 43 nodes and 1429 edges (MCODE score 33.23) and was highly enriched for the GOBP term DNA replication. (b) CDC7 “interactome”. Fifty-eight nodes from our GeneMania network were linked to CDC7 via direct edges. The gray nodes are proteins from our 124 strict data set, the yellow nodes are from the list of software “suggested” proteins which were potentially affected by CDC7 depletion and observed in our data set (see text) and the white nodes are suggested proteins that were not identified in our data set. The sizes of white and yellow nodes correspond to the strength of association with the data set. Edges are colored by interaction evidence (see key) and the edge thickness represents the significance of the association.
Although CDC7 itself was not quantified in our SILAC data set (most likely due to its absence in the siRNA treated sample), it appeared in the list of “related” proteins suggested by GeneMania. In fact, 58 nodes within our GeneMania network were linked to CDC7 via direct edges, including 40 proteins from the 124-protein search set, 5 “related” nodes suggested by the software which were quantified in our data set, but were not among the proteins showing the strongest changes (RFC3, RFC4, DHFR, H2AFZ, SSRP1), and 13 nodes which were suggested by the software but which were not identified in this study (Figure 2b). CDC7 was linked to these 58 nodes via 92 distinct edges (8 physical interactions; 14 genetic interactions; 43 coexpression; 11 colocalization; 10 pathway; 5 predicted; 1 shared protein domains). The group of 58 nodes is referred to as the “CDC7 interactome” in the following.
A similar analysis was carried out using MCODE to identify densely interconnected clusters in the STRING interaction network. The top-scoring MCODE cluster containing 16 nodes and 327 edges (MCODE score of 20.43) is shown encircled within the whole STRING network in Figure 3 (see Supplementary Figure S4 for primary clusters without MCODE expansion, Supporting Information). This cluster was enriched for the GOBP annotation term “DNA replication” (corrected p-value = 4.56 × 10−20), and all 16 nodes were included in the top-scoring GeneMania cluster presented in Figure 2a. To further compare the GeneMania and STRING results and to identify other proteins related to DNA-replication and cell cycle that were quantified in our data sets, the entire list of protein annotations associated with the GOBP terms “DNA replication” (GO:0006260) and “cell cycle” (GO:0007049) were downloaded from the QuickGO reference set at EBI (http://www.ebi.ac.uk/QuickGO). ID mapping was used to select IPI identifiers filtered for human taxonomy, and network connectivity was identified with MCODE/STRING.
Figure 3.

STRING origin activation checkpoint protein interaction network. STRING software was used to identify highly confident protein interactions between our significant set. The resulting protein interaction network containing 91 nodes and 752 edges was visualized in Cytoscape. Nodes representing proteins are colored by the type of selection criteria used for inclusion in the significant set (Supplementary Table S5, Supporting Information). These criteria detect changes in abundance and/or subcellular distribution and are detailed in the Materials and Methods section. Briefly, blue nodes correspond to changes in St, green nodes to changes in Sc and/or Sn, red nodes to the redistribution criterion |log2(Sn/Sc)| and yellow nodes are proteins which qualified by matching multiple criteria. The edges linking the nodes represent the types of supporting interaction data used by STRING. The edges are colored by interaction type, which are detailed in the figure key. The top-scoring MCODE cluster for DNA replication is outlined in a black circle.
The MS data set contained a total of 322 and 70 proteins annotated to cell cycle and DNA replication respectively. Of these, 12 cell cycle and 16 DNA replication-associated proteins were included in the 124-protein strict search set of most strongly changed proteins. Although they had smaller numbers of ratio counts and/or were quantified in fewer replicates, an additional 26 proteins showed appreciable changes in abundance (0.6 > normalized CDC7KD/CO ratio > 1.5) with a minimum of 6 ratio counts in the union over the three biological replicates for the sample type (C, N or T). This “relaxed” set particularly highlighted two protein complexes, namely the Replication Factor C (subunits RFC 1, 2, 3, 4, 5) and the Replication Protein A (subunits RPA 1, 2, 3), all of which showed reduced nuclear abundance. From the three network analyses (GeneMania, String and Reactome), we extracted a total set of 50 “consensus” proteins to characterize changes in abundance and subcellular distribution connected with DNA replication and cell cycle. The data for these proteins is summarized in Table 1 and Figure 4 shows Western blots used to confirm the SILAC results for ten of the strict proteins. Figure 5 shows a visual summary of changes in abundance or subcellular distribution for the strict proteins found to be differentially expressed between cytoplasm and nucleus in response to CDC7 suppression. The functional significance of the altered abundance and/or distribution of these proteins are considered further in the Discussion.
Table 1. Proteins Associated with DNA Replication that Respond Strongly to CDC7 Knockdowna.
| Consensus | DNA Replication Network | Cytoplasm | Nucleus | Total lysate | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene Name | Unique Peptides | Gene Mania | String | Reactome | SILAC Ratio | SigB | Ratio Count | SILAC Ratio | SigB | Ratio Count | SILAC Ratio | SigB | Ratio Count |
|
Strict proteins found by programmatic network analysis
| |||||||||||||
| CDK1 (CDC2) | 11 | • | • | • | 0.39 | 5.19E-11 | 33 | 0.40 | 6.66E-04 | 22 | ND | ND | ND |
| CDKN1A | 4 | • | • | • | 3.47 | 1.16E-06 | 18 | 1.16 | 3.91E-01 | 16 | ND | ND | ND |
| DUT | 8 | • | • | • | 0.34 | 1.66E-05 | 44 | 0.28 | 5.16E-06 | 16 | NaN | NaN | 2 |
| FEN1 | 7 | • | • | • | 0.92 | 7.18E-01 | 12 | 0.43 | 1.62E-03 | 17 | 0.43 | 2.83E-03 | 3 |
| HAT1 | 9 | • | 1.21 | 4.51E-01 | 13 | 0.34 | 7.88E-06 | 12 | ND | ND | ND | ||
| KPNA2 | 13 | • | • | 0.37 | 1.36E-06 | 40 | 0.36 | 1.04E-04 | 28 | 0.38 | 4.95E-04 | 13 | |
| MAD2L1 | 6 | • | • | • | 0.38 | 7.87E-06 | 11 | 0.52 | 3.87E-02 | 10 | ND | ND | ND |
| MCM2 | 21 | • | • | • | 1.78 | 2.75E-02 | 11 | 0.36 | 8.09E-07 | 45 | NaN | NaN | 2 |
| MCM3 | 24 | • | • | • | 0.47 | 5.12E-04 | 4 | 0.39 | 4.22E-04 | 46 | ND | ND | ND |
| MCM4 | 19 | • | • | • | 0.74 | 1.43E-01 | 3 | 0.42 | 1.26E-03 | 33 | ND | ND | ND |
| MCM5 | 15 | • | • | • | 0.98 | 9.14E-01 | 7 | 0.39 | 3.84E-04 | 21 | ND | ND | ND |
| MCM6 | 24 | • | • | • | 0.53 | 3.13E-03 | 6 | 0.35 | 8.85E-05 | 39 | ND | ND | ND |
| MCM7 | 21 | • | • | • | 0.60 | 4.05E-02 | 7 | 0.35 | 5.93E-05 | 37 | 0.26 | 1.89E-06 | 3 |
| NASP | 16 | • | • | 0.67 | 5.99E-02 | 28 | 0.30 | 4.60E-06 | 27 | 0.51 | 1.64E-02 | 5 | |
| PCNA | 12 | • | • | • | 0.93 | 7.42E-01 | 60 | 0.37 | 1.77E-06 | 49 | 0.49 | 2.37E-04 | 40 |
| PSME3 | 9 | • | • | • | 2.35 | 1.10E-04 | 29 | 0.61 | 8.03E-02 | 45 | 0.92 | 7.94E-01 | 17 |
| RBM14 | 13 | • | • | ND | ND | ND | 0.65 | 1.28E-01 | 31 | 0.30 | 1.88E-05 | 8 | |
| RRM1 | 17 | • | • | • | 0.52 | 1.46E-03 | 29 | 0.45 | 4.12E-03 | 5 | 0.59 | 5.16E-02 | 5 |
| RRM2 | 6 | • | • | • | 0.25 | 5.39E-08 | 19 | ND | ND | ND | ND | ND | ND |
| SET | 6 | • | • | 3.14 | 1.08E-10 | 41 | 0.60 | 1.99E-02 | 43 | 0.97 | 8.67E-01 | 39 | |
| STMN1 | 8 | • | • | 0.52 | 4.84E-06 | 70 | 0.41 | 8.59E-04 | 34 | 0.68 | 7.22E-02 | 31 | |
| TK1 | 8 | • | • | • | 0.27 | 2.79E-07 | 15 | NaN | NaN | 2 | ND | ND | ND |
| TYMS | 9 | • | • | • | 0.31 | 4.38E-06 | 23 | 0.27 | 1.78E-05 | 3 | ND | ND | ND |
| YWHAZ | 13 | • | 0.61 | 8.66E-04 | 178 | 0.56 | 7.96E-03 | 95 | 0.90 | 8.02E-01 | 131 | ||
|
| |||||||||||||
|
Strict proteins based on literature evidence
| |||||||||||||
| KPNB1 | 32 | 0.59 | 3.04E-04 | 153 | 0.65 | 6.06E-02 | 119 | 0.84 | 5.37E-01 | 90 | |||
| PC4 (SUB1) | 6 | 0.46 | 1.72E-04 | 32 | 1.53 | 7.94E-03 | 35 | 1.16 | 4.78E-01 | 22 | |||
| XRCC5 | 31 | 2.28 | 2.80E-06 | 53 | 0.68 | 9.62E-02 | 108 | 0.89 | 6.85E-01 | 37 | |||
| XRCC6 | 29 | 2.43 | 4.78E-07 | 72 | 0.66 | 6.32E-02 | 133 | 0.85 | 5.68E-01 | 83 | |||
|
| |||||||||||||
|
Additional proteins under “relaxed” selection criteria
| |||||||||||||
| CDK2 | 7 | • | • | 0.61 | 4.93E-02 | 15 | 0.44 | 4.07E-03 | 11 | ND | ND | ND | |
| CDK4 | 7 | • | • | 1.67 | 4.99E-02 | 9 | ND | ND | ND | 2.14 | 1.73E-02 | 5 | |
| CDK6 | 6 | • | • | 0.43 | 2.02E-03 | 6 | NaN | NaN | 2 | ND | ND | ND | |
| CDKN2A | 3 | • | • | 1.32 | 4.49E-01 | 5 | ND | ND | ND | 6.23 | 2.81E-09 | 6 | |
| DHFR | 3 | • | • | • | 0.31 | 2.79E-05 | 10 | NaN | NaN | 2 | ND | ND | ND |
| DTYMK | 15 | • | 0.64 | 3.62E-02 | 40 | 0.60 | 7.71E-02 | 16 | 0.68 | 1.81E-01 | 18 | ||
| H2AFV,H2AFZ | 2 | • | 0.65 | 8.73E-02 | 10 | 0.81 | 4.28E-01 | 67 | 0.59 | 5.86E-02 | 18 | ||
| MSH2 | 7 | • | ND | ND | ND | 0.65 | 1.50E-01 | 12 | ND | ND | ND | ||
| MSH6 | 9 | • | ND | ND | ND | 0.57 | 2.98E-02 | 10 | ND | ND | ND | ||
| NCAPD2 | 9 | • | 0.56 | 5.86E-03 | 9 | ND | ND | ND | ND | ND | ND | ||
| NCAPG | 10 | • | 0.53 | 2.46E-03 | 12 | 0.65 | 1.74E-01 | 3 | ND | ND | ND | ||
| NES | 1 | • | ND | ND | ND | 1.01 | 7.38E-01 | 7 | 0.57 | 2.62E-02 | 14 | ||
| PBK | 6 | • | 0.35 | 9.58E-07 | 7 | NaN | NaN | 1 | ND | ND | ND | ||
| PDS5A | 10 | • | ND | ND | ND | 0.63 | 9.74E-02 | 15 | ND | ND | ND | ||
| RANGAP1 | 24 | • | 0.94 | 7.87E-01 | 47 | 0.79 | 3.51E-01 | 52 | 0.60 | 1.45E-02 | 74 | ||
| RECQL | 20 | • | 1.85 | 1.02E-02 | 7 | 0.87 | 6.81E-01 | 55 | ND | ND | ND | ||
| SMC2 | 19 | • | • | 0.58 | 2.97E-02 | 17 | 0.43 | 4.70E-04 | 11 | ND | ND | ND | |
| T0P2A | 11 | • | • | ND | ND | ND | 0.34 | 5.42E-05 | 14 | ND | ND | ND | |
|
| |||||||||||||
|
Protein complexes
| |||||||||||||
| RFC1 | 2 | • | • | ND | ND | ND | 0.56 | 6.43E-02 | 4 | ND | ND | ND | |
| RFC2 | 8 | • | • | 0.96 | 7.61E-01 | 8 | 0.62 | 7.40E-02 | 14 | ND | ND | ND | |
| RFC3 | 7 | • | • | • | ND | ND | ND | 0.55 | 2.06E-02 | 13 | ND | ND | ND |
| RFC4 | 10 | • | • | • | NaN | NaN | 2 | 0.61 | 6.06E-02 | 18 | NaN | NaN | 0 |
| RFC5 | 12 | • | • | NaN | NaN | 2 | 0.59 | 6.03E-02 | 25 | ND | ND | ND | |
|
| |||||||||||||
| RPA1 | 11 | • | • | 0.82 | 3.17E-01 | 8 | 0.62 | 1.36E-01 | 5 | ND | ND | ND | |
| RPA2 | 6 | • | • | 0.78 | 2.26E-01 | 7 | 0.66 | 1.63E-01 | 7 | ND | ND | ND | |
| RPA3 | 4 | • | • | • | 0.81 | 3.91E-01 | 9 | 0.55 | 6.02E-02 | 6 | ND | ND | ND |
Details of protein names, sequences, etc. are given in Supplementary Table 4, Supporting Information. Shading indicates the criterion used to include the protein (see Material and Methods): (blue) St, (green) Sn and/or Sc, (red) log2(Sn/Sc), (orange) relaxed (see text). ND denotes proteins not detected. NaN denotes proteins not quantified.
Figure 4.

Western blot validation of nuclear/cytoplasm distribution for 10 proteins following CDC7 depletion. (a) Western blots for ten representative proteins from the 124 protein strict group. (b) Corresponding normalized SILAC protein ratios (CDC7KD/CO) in the cytoplasmic and nuclear enriched samples.
Figure 5.

Protein redistribution between subcellular compartments in response to CDC7 inhibition. Summary of proteins from the strict set that showed altered localization between the nucleus and cytoplasm in response to CDC7 depletion. Proteins that showed the strongest changes in abundance in the nucleus or cytoplasm according to the strict set selection criteria are indicated by red (increased abundance) and green (decreased abundance). Blue arrows indicate redistribution of proteins according to the strict set selection criterion. Proteins are grouped by function.
3.3. Altered Metabolic Processes Are Involved in Maintenance of the Origin Activation Checkpoint
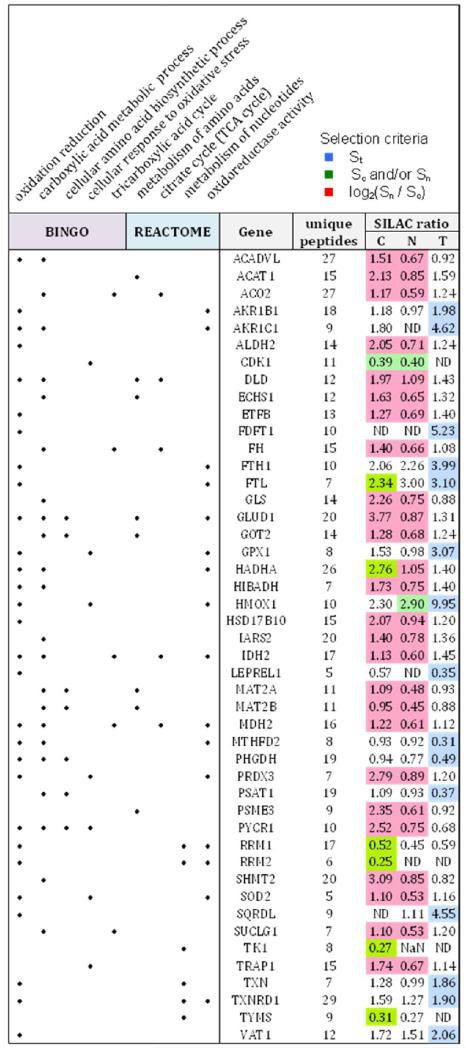
The STRING/MCODE analysis of subnetworks, pathway analysis with Reactome and BINGO/MCODE enrichment of GO terms all identified other groups of proteins which show changes in abundance and/or redistribution, but are involved in metabolic functions rather than DNA-replication/cell cycle (Table 2). These are smaller sets of proteins for which the dominant function associated with the group is not primarily associated with the nucleus and which may be involved in integration of function over different subcellular locations (see Discussion).
Table 2. Metabolic Proteins that Respond Strongly to CDC7 Knockdowna.
|
Details of protein names, sequences, etc. are given in Supplementary Table 4, Supporting Information. Shading of the selection criteria for the proteins is the same as in Table 1.
GO term enrichment analysis identified 78 of our 124 strict proteins as belonging to the high-level GO term “metabolic processes”. More specifically, such metabolic processes include “oxidation reduction” (29 proteins identified by BiNGO), “oxidoreductase activity” (17 proteins identified by Reactome) and “oxidative stress response” (7 proteins identified by BiNGO). This corroborates our previous findings that a mild NRF2-driven oxidative stress response contributes to the maintenance of cell survival at the DNA replication origin activation checkpoint22 and there is ample evidence in the literature to support a role for oxidative processes in cell cycle control.40,41 In contrast to the DNA-replication network, many of these metabolic proteins showed increases in abundance, with some showing redistribution from the nucleus to the cytoplasm (Table 2, Figure 5).
Several proteins involved in the citrate (TCA) cycle were found to be altered by CDC7 depletion, with redistribution to the cytoplasm (ACO2, DLD, FH, IDH2, MDH2 and SUCLG1). Both oxidation reduction and the TCA cycle are mitochondrial processes and this subcellular organelle was identified as a major region associated with our search set. In total, 34 mitochondrial proteins were identified by Reactome, BiNGO or GeneMania, from our set of 124. Interestingly, 91% (31/34) of these “mitochondrial” proteins (i.e., excluding MTHFD2, YWHAZ and DUT) showed statistically significant redistribution between the N and C regions and are key candidates for subcellular relocalization and/or trafficking. 70% (24/34) of these “mitochondrial” proteins are known to have alternative localizations (www.hprd.org), 21 of which can be attributed directly to the cytoplasm or nucleus. Overall 90 of the 124 strict proteins (73%) have established multiple subcellular locations (www.hprd.org), which is consistent with the concept that eukaryotic organelle composition is dynamic and context-dependent.
4. DISCUSSION
The subcellular fractionation approach used here achieved a major increase in proteome coverage compared to analysis of total cell lysate alone, and was a successful strategy for detecting changes in protein abundance in the nuclear and cytoplasmic regions. In the following we discuss three aspects of the results: (a) core changes in DNA-replication and related cell cycle functions in the nucleus, (b) indications for involvement of other functional networks, especially those connected to oxidative stress and iron metabolism, and (c) evidence for altered nucleo-cytoplasmic distribution as an integral component of the cell cycle.
4.1. Activation of the DNA Replication Origin Activation Checkpoint Leads to Strong Reduction in Key DNA Replication and Cell Cycle Regulators—the “CDC7 Interactome”
As shown in Table 1, several aspects of the DNA replication machinery are repressed during CDC7-depleted cell cycle arrest. The down-regulated proteins include all six subunits of the MCM2–7 helicase complex, which is an essential requirement for pre-RC loading at DNA replication origins and is a direct substrate of CDC7 kinase activity.2,8 The DNA polymerase cofactor PCNA (proliferating cell nuclear antigen) is known to directly bind and stimulate the endonuclease activity of FEN1, a key enzyme involved in the synthesis of the DNA lagging strand and in DNA repair mechanisms.42 Both of these essential DNA replication molecules were strongly reduced in the nuclear sample while remaining unchanged in the cytoplasmic portion. Furthermore, replication factor C has been found to also stimulate the activity of FEN1,43 and therefore our results suggest a critical reduction in FEN1, PCNA and RFC, which is indicative of reduced DNA synthesis at the origin activation checkpoint.
Various DNA replication molecules involved in many aspects of replication and repair were also found to be altered in response to CDC7 inhibition. The small and large subunits of ribonucleotide reductase (RRM1 and RRM2) play a role in biosynthesis of deoxyribonucleotides for DNA synthesis and were downregulated by CDC7 depletion. Both histone acetyltransferase 1 (HAT1) which is involved in chromatin assembly, and NASP which is a H1 linker histone chaperone protein required for chromatin assembly, cell cycle progression and proliferation, were sharply reduced in response to CDC7 inhibition. A conserved nuclear histone acetyltransferase has previously been found to associate with the origin recognition complex,44 which suggests a novel link between histone modification and DNA replication, and indicates that HAT1 and NASP may also play a direct role in replication control.
Cyclin-dependent kinases (CDKs) are extremely important in the regulation of the eukaryotic cell cycle. Fluctuating levels of cyclins, CDKs and CDK-inhibitors are all involved in progression through the cell cycle. We have shown that loss of CDC7 initiates G1 arrest accompanied by a strict reduction in levels of CDK1 as well as “relaxed” indications for reduction in CDK2 and CDK6 (Table 1). Additionally, we detected strict upregulation of the CDK-inhibitor CDKN1A and “relaxed” upregulation of CDKN2A, in keeping with inhibition of CDK activity during cell cycle arrest. We have previously performed gene expression microarray (GEM) analysis on the CDC7-depleted IMR90 model system of the origin activation checkpoint, which demonstrated down-regulation of MCM2–7, CDK1 and CDK6 at the transcriptomic level and corroborates our proteomics findings.5
Our results also demonstrate a significant alteration in many proteins involved in folate metabolism and in the supply of one-carbon units for the biosynthesis of nucleotides and amino acids. These molecules include DUT, TK1 and TYMS, as well as DHFR and DTYMK (Tables 1 and 2). MTHFD2 and SHMT2 are also significantly changed in abundance and involved in folate metabolism, but were not attributed by the software programs to the DNA replication network.
4.2. Maintenance of Cell Cycle Arrest Involves Diverse Signaling Networks Including Oxidative Stress Response Pathways and Altered Iron Homeostasis
Ferritin is a ubiquitously expressed, iron-sequestering molecule composed of multiple heavy and light subunits that can store several thousand atoms of iron per molecule.45 By buffering intracellular labile iron levels, ferritin actively reduces the generation of reactive oxygen species (ROS) and therefore is an important contributor to oxidative stress response pathways.46 In fact, in HeLa cells, overexpression of ferritin strongly reduced cell growth and increased resistance to H2O2 toxicity due to its increased ferroxidase activity.47 Inducible heme-oxygenase (HMOX1) also has important anti-inflammatory and antiproliferative effects, and it is an essential player in the protective NRF2-mediated oxidative stress response pathway. Recently, Nam et al.48 have shown that p53 promotes cellular survival by directly inducing the expression of HMOX1, providing an additional link between p53 and protective cell fate decisions. We have shown a very significant upregulation of both ferritin heavy and light chains (FTH, FTL) as well as heme oxygenase (HMOX1). Furthermore, we found evidence for upregulation of several antioxidant response molecules including EPHX1, GPX1, PRDX3, SOD2, SQSTM1, TXN and TXNRD1. These observations reconfirm our previous results, which found that NRF2-mediated oxidative stress response was one of the most significantly overrepresented pathways following CDC7 inhibition and activation of the CDC7-sensitive checkpoint.22
In addition to FTH, FTL and HMOX1, several other proteins involved in iron homeostasis were affected by cell cycle arrest at the DNA replication checkpoint, including the transferrin receptor (TFRC) and ribonucleotide reductase subunit 2 (RRM2). Iron metabolism is an essential aspect of cell proliferation and is critical for many cellular enzymatic processes, whereas iron depletion blocks progression through the cell cycle at the G1/S border, resulting in reduced proliferation.49 Iron is a cofactor for ribonucleotide reductase and many of the G1 phase CDKs, and is an essential component of DNA synthesis.50,51 The classic proto-oncogene c-myc can act by repressing ferritin and increasing the intracellular iron pool necessary for cell proliferation and transformation.52 Iron chelators are therefore used as a therapeutic strategy for cancer treatment, where they can be used to sequester iron and induce cell cycle arrest and apoptosis of highly proliferative cancer cells.53,54
We detected upregulation of both FTH and FTL in all CDC7-depleted sample types (N, C, T), as well as a reduction in nuclear and cytoplasmic TFRC and RRM2. These findings are in close agreement with the findings of Zhang et al.,55 who also demonstrated up-regulation of ferritin light and heavy subunits with a corresponding decline in transferrin receptor protein levels following induction of a p53-dependent cell cycle arrest in H1299 lung cancer cells. They propose that p53 initiates cell cycle arrest, not only by induction of CDK-inhibitors such as p21, but also by reducing the availability of intracellular iron in an antiproliferative mechanism that enhances iron storage and reduces iron uptake.
4.3. Indications that Cdc7 Depletion Results in Other Altered Cellular Metabolic Responses
There are indications in the present data for other metabolic changes that have been connected with the cell cycle and which have functions that are primarily associated with non-nuclear subcellular locations. For example, we detected potential nucleo-cytoplasmic trafficking for four proteins involved in protein translation (RPL11, RPL12, RPLP0, RPLP2, see Supplementary Text, Supplementary Table 5 and Supplementary Figure 4b, Supporting Information) and for six enzymes of the TCA-cycle (ACO2, DLD, FH, IDH2, MDH2, SUCLG1), as well as the mitochondrial TCA-cycle-shunt between Asp/Glu (GOT2) (Table 2 and Supplementary Figure 4c, Supporting Information). Such trafficking was not detected for the cytoplasmic analogues (ACO1, IDH1, MDH1, GOT1, Supplementary Table 4, Supporting Information). A number of the TCA-cycle enzymes have previously been detected in the nucleus of cancer and other cell types,39,56,57 but their nuclear function(s) remain unknown. Similar redistribution was detected for two enzymes at the fringe of the TCA cycle, GLUD1 (glutamate dehydrogenase) and GLS (glutamine synthase). The homeostatic balance between glycolysis and glutaminolysis varies over the cell cycle and both glucose and glutamine are required for progression through the G1 restriction point, whereas glutamine alone is essential for progression through the S phase.58
Further work will be needed to clarify the possible connection of these metabolic proteins to the cell cycle. It may be that they are involved in integrative feedback loops of the type that are increasingly apparent for other non-nuclear cellular functions. For example, apart from the role of the glycolysis-related enzyme PFKFB3 (fructose-2,6-biphosphatase 3) in increasing proliferation via CDKs59 and in altering the glutaminolysis/glycolysis balance through regulation of GLS as a function of the cell cycle,58 more than half of other glycolytic enzymes are now known to have nuclear functions, including in transcription feedback loops related to hypoxia and cancer.39
4.4. Differential Distribution of Many Proteins Suggests that Altered Nucleo-cytoplasmic Trafficking Plays a Major Role in the Coordination of Cell Cycle Arrest
A notable feature of the 124-protein search set was that redistribution between the nucleus and cytoplasm was detected for almost half of the proteins. This included proteins known to be involved in cell cycle control (Ku70/XRCC6, Ku80/XRCC5, NCL), proteins involved in regulation of p53-responsive DNA damage repair mechanisms (PC4/SUB1, PSME3, PRMT1) as well as proteins with oncogenic potential (SET, C1QBP). Other proteins involved in nucleo-cytoplasmic transport (importins KPNA2 and KPNB1) showed substantial reductions in abundance. As described in the following, this suggests nucleocytoplasmic trafficking is an important contributor to cell cycle control.
PC4 (SUB1)
The human transcriptional positive coactivator PC4 (mammalian orthologue of yeast SUB1) was found to be increased in abundance in the nuclear fraction with a corresponding significant reduction in its cytoplasmic portion following CDC7 depletion. This protein was found to be unchanged in abundance in the unfractionated total lysate sample which would imply that altered localization rather than altered total expression levels may underlie functional changes associated with this protein. PC4 is a highly abundant nuclear protein with an established role in multiple cellular functions involving stabilization of DNA–protein interactions, chromatin organization, transcriptional activation and repression, DNA elongation, reinitiation and DNA replication and repair.60-62 PC4 is a nonhistone chromatin-associated protein and stimulates double strand break repair at sites of DNA damage.63 Mortusewicz et al.64 showed that in response to DNA damage induction by oxidative stress (H2O2) or replication arrest (hydroxyurea), PC4 underwent relocalization from a diffuse nuclear distribution in untreated cells to discrete subnuclear foci in stressed cells. Furthermore, PC4 activates p53 function and induces the transcription of p53-responsive genes.65,66 PC4 itself is p53-inducible and enhances the recruitment of p53 to its own promoter region, demonstrating a positive autoregulatory feedback loop for p53 control.67 Our previous work has demonstrated that p53 lies on one of the key axes involved in regulation and activation of the DNA replication origin activation checkpoint.5 Therefore, as PC4 is implicated in the regulation of DNA replication, is a potent repressor of transcription during DNA repair and as it responds to genotoxic insult in a p53 dependent manner, it is plausible that the observed cytoplasmic to nuclear relocalization of PC4 is an important response to CDC7 depletion and contributes to activation or maintenance of the DNA replication checkpoint response.
PSME3
PSME3 (also known as PA28-gamma, REG gamma or Ki antigen) is a subunit of a proteasome regulator involved in the ATP- and ubiquitin-independent activation of the 20S proteasome.68 Following CDC7 depletion, we have shown that this protein potentially relocates from its typical nuclear position to the cytoplasm. No alteration in total cellular levels of PSME3 was observed. PSME3 has an established role in cell cycle control and facilitates the proteasomal degradation of many key cell cycle regulators, including the CDK-inhibitors p21CIP1, p16INK4a and p19ARF.69,70 It also promotes the interaction between MDM2 and its target p53, which leads to enhanced ubiquitination and degradation of p53.71 PSME3-mediated monoubiquitination of p53 may also induce antiapoptotic effects by promoting nuclear export of p53 away from its target genes.72 PSME3 also plays a direct role in DNA damage response and cell cycle control, as it rapidly accumulates at sites of DNA damage when phosphorylated73 and also interacts with the DNA damage checkpoint regulator Chk2.74
Although PSME3 is primarily located in the nucleus, it has been shown to relocalize to the cytoplasm in response to various conditions. For example, during mitosis or in cells mitotically arrested with nocodazole, breakdown of the nuclear envelope allowed PSME3 to translocate to the cytoplasm where it is subject to phosphorylation by the exclusively cytoplasmic MEKK3 kinase, linking stress-activated protein kinase signaling with proteasomal activity.75 Furthermore, PSME3 can undergo post-translational SUMOylation, which mediates cytoplasmic translocation and increased stability of PSME3.76 We propose that redistribution of PSME3 to the cytoplasm following CDC7 depletion may be a protective cellular response which sustains cell cycle arrest by sequestering this proteasomal activator away from its nuclear target proteins such as p53.
SET
We show that a portion of the SET nuclear oncogene protein is relocalized to the cytoplasm following inhibition of CDC7. SET has previously been shown to relocalize from the nucleus to the cytoplasm in response to genotoxic stress.77 This is a predominantly nuclear, multifunctional protein that plays a role in cell growth and proliferation, and is implicated in transformation and tumorigenesis.78 It has an established role in many processes including the inhibition of the tumor suppressor PP2A,79 cell cycle regulation,80,81 inhibition of histone acetyltransferases82 and apoptosis.83 SET binds the CDK inhibitor p21 and together they cooperate in the inhibition of cyclin B-CDK1 activity at the G2/M transition.80 SET may also regulate the G1/S transition by modulating the activity of cyclin E-CDK2 via its interaction with p21.81 Fukukawa et al.84 suggested that SET can inhibit cell proliferation at the G1/S transition by negatively regulating the MEK/ERK pathway.
C1QBP
C1QBP (p32) is primarily a mitochondrial protein, but has also been found in alternative locations including nucleus and cytoplasm.85 It plays a role in tumorigenesis and has been found to be upregulated in human breast, lung and colon cancer cell lines,86 as well as in prostate tumor samples.85 It has been suggested that C1QBP regulates the balance between glycolysis and oxidative phosphorylation in tumor cells.87 Overexpression of C1QBP protects against oxidative stress88 and also protects against apoptosis, increases proliferation and cell migration, suggesting that this protein may be a potential target for anticancer therapeutics.86 Amamoto et al.,85 demonstrated that RNAi knockdown of C1QBP led to growth inhibition, decreased cyclin D1, increased p21 expression and G1/S cell cycle arrest specifically in prostate cancer cell lines and not in a noncancerous cell line. It is possible that the nuclear to cytoplasmic redistribution of C1QBP we detected following CDC7 inhibition in normal fibroblasts may reflect the cyto-protective function of C1QBP and that a differential response may occur in cancer cells.
PRMT1
Protein arginine N-methyltransferase 1 was found to relocalize from the nucleus to the cytoplasm following CDC7 depletion. PRMT1 is known to be essential for cell proliferation, for maintaining genomic integrity and for DNA damage response.89 Protein arginine methylation by PRMT1 is required for DNA damage control at the intra-S phase checkpoint.90 Protein methylation has recently been identified as a key post-translational mechanism for regulation of p53, allowing for transcriptional activation or repression depending on the methylation site.91 PRMT1 interacts directly with p53 and acts as a coactivator of p53-mediated transcription by enhancing histone methylation.92 PRMT1 has also been shown to promote arginine methylation of FOXO3a.93
Ku70 and Ku80
Ku is a heterodimeric protein composed of two subunits, Ku70 (XRCC6) and Ku80 (XRCC5). The Ku dimer has ATP-dependent helicase activity and acts as the DNA-binding subunit of DNA protein kinase (DNA-PK).94 It is involved in many nuclear processes including DNA replication and repair, chromosomal maintenance and cell proliferation.95-97 Ku participates in pre-RC formation by associating with DNA replication origins.96,98 An acute reduction of Ku80 expression by RNAi directly impaired the loading of replication factors and reduced origin firing, leading to a reduction in CDK2 levels, increased p21 expression and subsequent activation of a DNA replication checkpoint and cell cycle arrest at the G1/S boundary.99 Our results demonstrate a shift in localization of both Ku subunits from the nucleus to the cytoplasm following CDC7 depletion. Although Ku appears to be primarily of nuclear localization, it has also been found throughout the cytoplasm and at the cell surface,100,101 and undergoes differential subcellular localization during mitosis.102 Therefore, our observed results could represent a mechanism employed by the cell to sustain or prolong DNA replication arrest, by removing Ku from the vicinity of its target replication origins.
NCL
Our data suggests that a portion of the nucleolar protein nucleolin may undergo redistribution from the nuclear fraction to the cytoplasm, which is demonstrated by a significant increase in the cytoplasmic abundance of this protein without an apparent change in nuclear levels. Nucleolin (NCL) is an abundant nucleolar chaperone protein which shuttles continuously between the nucleus and cytosol.103 Nucleolin is known to interact directly with another nucleolar chaperone, nucleophosmin.104 Nucleophosmin was also potentially redistributed by CDC7 RNAi in a similar manner to nucleolin, but its altered abundance did not reach the required threshold of significance, and was therefore not included in our changed set. Nucleolin and nucleophosmin are termed “hub” nucleolar proteins as they interact with multiple binding partners.105 Nucleolin is involved in many diverse processes including DNA replication and cell cycle progression.106,107 Nucleolin forms a complex with RPA in response to genotoxic stress, leading to inhibition of both S-phase entry and DNA replication.108 Heat shock and genotoxic stress cause NCL to relocalize from the nucleolus to the nucleoplasm and subsequently inhibit DNA replication initiation.108,109 Both nucleolin and nucleophosmin can enhance the stability and transcriptional activity of p53.110-112 Together, these results suggest a complex regulatory role for nucleolin and its partner nucleophosmin in proliferation and regulation of DNA integrity.
KPNA2 and KPNB1
The cytoplasmic accumulation of several proteins following CDC7 depletion could potentially be due to either overactive translocation from the nucleus to the cytoplasm, or it could result from inefficient translocation into the nucleus by classic nuclear import mechanisms. Our discovery that cytoplasmic accumulation of several proteins occurs with a simultaneous reduction in the abundance of two key nuclear importins, could suggest that deregulated nuclear import is a mechanism underlying the retention of certain molecules in the cytoplasm.
The nuclear localization of proteins via the nuclear pore complex (NPC) is an important and highly selective cellular mechanism which is important in regulating aspects of the eukaryotic cell cycle.113 The karyopherin family of nuclear receptors (known as importins and exportins) use a classical import pathway for active transport of cargo proteins through the NPC.114 Importin-alpha recognizes nuclear localization sequence (NLS) domains within cargo proteins and then forms a trimeric complex with importin-beta to escort the cargo through the nuclear pore.115 Once inside the nucleus, importin-beta is bound by Ran GTP which causes dissociation of the import complex, release of the cargo protein and recycling of the importin dimer to the cytoplasm.
We show here that two specific subunits of the karyopherin family KPNA2 and KPNB1 are significantly reduced in abundance following CDC7 inhibition. KPNA2 and KPNB1 are importins, which can work in concert, and are increased together in expression and activity in cervical tumors and transformed cells, in a mechanism controlled by deregulated E2F/Rb activity.116 In fact, overexpression of the E2F inhibitor Rb directly decreased the activity of KPNA2 and KPNB1,116 implying cell cycle-dependent regulation of importin proteins. Nuclear import efficiency via the importin alpha/beta pathway has already been shown to be regulated in a cell cycle dependent manner117 and Pulliam et al.118 have demonstrated that importin-alpha is necessary for DNA replication and is essential for mediating the G1/S transition in S. cerevisiae. Furthermore, a reduction in the expression of distinct importin-alpha isoforms led to the strong inhibition of HeLa cell proliferation.119
We have previously shown that inactivation of the Rb-E2F transcriptional network is a major contributor to the G1 cell cycle arrest induced by depletion of CDC75 and therefore we propose that inhibition of the E2F transcriptional program could provide a potential mechanism for the reduction we see in KPNA2 and KPNB1 following CDC7 inhibition. In fact, the nuclear import of CDC7 is achieved by a direct association with importin-beta.120
We speculate that the reduction in these two importin molecules could lead to reduced nuclear import and cytoplasmic retention of several proteins known to be transported by this classical import mechanism. For example, Ku70 and Ku80 both contain NLS which allow independent translocation into the nucleus via the classical importin-alpha/beta mediated pathway.121,122 Song et al.123 showed that oxidative stress reduced the association between Ku and importins, and proposed that this disrupts the import mechanism leading to a nuclear loss of Ku, a slight alteration in cytoplasmic Ku and a subsequent apoptotic response. Furthermore, nucleolin and nucleophosmin utilize their NLS domains to assist the nucleo-cytoplasmic translocation of many cargo proteins, often via a direct association with the importin-alpha/beta nuclear import mechanism.124,125 Therefore, alterations in the major nuclear transport route involving importins may provide a powerful regulatory mechanism for cytoplasmic sequestration of key DNA replication molecules during the CDC7-inhibition checkpoint response. However, only a minority of the 322 and 70 proteins annotated to cell cycle and DNA replication that were monitored in our experiments showed appreciable redistribution. Furthermore, only small changes were observed in the abundances of other karyopherins (KPNA1, 3, 4, 6), suggesting that there are mechanisms for selective modulation of the nuclear import process in response to CDC7-suppression.
5. CONCLUSION
The high resolution, quantitative mass spectrometry approach presented here reveals that the DNA replication origin activation checkpoint may be activated and/or maintained by subcellular redistribution of many key proteins between the cytoplasm and nucleus. These proteins regulate diverse biological processes including DNA replication, cell cycle control, oxidative stress response, iron metabolism and the tricarboxylic acid cycle. Reduced nuclear import mechanisms may contribute to cytoplasmic sequestration of several proteins. Using strict acceptance criteria, 124 proteins demonstrated altered abundance levels and/or altered distribution following depletion of CDC7 and subsequent cell cycle arrest. Furthermore, 40 proteins in our changed protein set could be directly associated to CDC7 kinase, which we called the “CDC7 interactome”. We propose that the origin activation checkpoint mechanism provoked by depletion of the DNA replication initiation factor CDC7 is highly dynamic and regulated across many subcellular niches. These results will have important implications for the development of CDC7-targeted small molecule inhibitors as anticancer therapeutic strategies.
More generally, a notable feature of the present results is that a number of functionally important proteins showed subcellular redistribution without major changes in total abundance (e.g., PC4 and PSME3). This is in line with other work showing that large numbers of proteins have multiple subcellular locations,36,37 that many proteins are common to the nucleus and to mitochondria,39 and that large numbers of proteins show subcellular redistribution under conditions such as hypoxia.38 There is increasing evidence that the proteomes of eukaryotic subcellular organelles are dynamic and context dependent–the present work begins to provide methods to characterize the dynamic distribution of cellular proteomes with high-throughput proteomics methods.
Supplementary Material
ACKNOWLEDGMENTS
This study has been supported by Cancer Research UK scientific program grant C428/A6263 (K.S. and G.H.W.) and Wellcome Trust grant 081879/z/06/z (J.G.Z.)
Footnotes
ASSOCIATED CONTENT
Figure S1. Reproducibility and quality assessment of each data set. Figure S2. Subcellular fractionation substantially increases proteome coverage. Figure S3. GeneMania functional interaction network. Figure S4. String and MCODE analysis identify subclusters within the data set. Supplementary Text 1. Information on selection of significantly changed proteins. Table S1. Protein information for Cytoplasmic data set. Table S2: Protein information on nuclear data. Table S3: Protein information on total lysate. Table S4: Protein Consensus Characterization. Table S5. Summary of protein sets used for functional network searches. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).Blow JJ, Gillespie PJ. Replication licensing and cancer–a fatal entanglement? Nat. Rev. Cancer. 2008;8(10):799–806. doi: 10.1038/nrc2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Sclafani RA, Holzen TM. Cell cycle regulation of DNA replication. Annu. Rev. Genet. 2007;41:237–80. doi: 10.1146/annurev.genet.41.110306.130308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Montagnoli A, Tenca P, Sola F, Carpani D, Brotherton D, Albanese C, Santocanale C. Cdc7 inhibition reveals a p53-dependent replication checkpoint that is defective in cancer cells. Cancer Res. 2004;64(19):7110–6. doi: 10.1158/0008-5472.CAN-04-1547. [DOI] [PubMed] [Google Scholar]
- (4).Rodriguez-Acebes S, Proctor I, Loddo M, Wollenschlaeger A, Rashid M, Falzon M, Prevost AT, Sainsbury R, Stoeber K, Williams GH. Targeting DNA replication before it starts: Cdc7 as a therapeutic target in p53-mutant breast cancers. Am. J. Pathol. 2010;177(4):2034–45.5. doi: 10.2353/ajpath.2010.100421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Tudzarova S, Trotter MW, Wollenschlaeger A, Mulvey C, Godovac-Zimmermann J, Williams GH, Stoeber K. Molecular architecture of the DNA replication origin activation checkpoint. EMBO J. 2010;29(19):3381–94. doi: 10.1038/emboj.2010.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Williams GH, Stoeber K. The cell cycle and cancer. J. Pathol. 2012;226(2):352–64. doi: 10.1002/path.3022. [DOI] [PubMed] [Google Scholar]
- (7).Kim JM, Yamada M, Masai H. Functions of mammalian Cdc7 kinase in initiation/monitoring of DNA replication and development. Mutat. Res. 2003;532(1-2):29–40. doi: 10.1016/j.mrfmmm.2003.08.008. [DOI] [PubMed] [Google Scholar]
- (8).Sheu YJ, Stillman B. Cdc7-Dbf4 phosphorylates MCM proteins via a docking site-mediated mechanism to promote S phase progression. Mol. Cell. 2006;24(1):101–13. doi: 10.1016/j.molcel.2006.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kim JM, Kakusho N, Yamada M, Kanoh Y, Takemoto N, Masai H. Cdc7 kinase mediates Claspin phosphorylation in DNA replication checkpoint. Oncogene. 2008;27(24):3475–82. doi: 10.1038/sj.onc.1210994. [DOI] [PubMed] [Google Scholar]
- (10).Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, Gautier J. An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol. Cell. 2003;11(1):203–13. doi: 10.1016/s1097-2765(02)00799-2. [DOI] [PubMed] [Google Scholar]
- (11).Furuya K, Miyabe I, Tsutsui Y, Paderi F, Kakusho N, Masai H, Niki H, Carr AM. DDK phosphorylates checkpoint clamp component Rad9 and promotes its release from damaged chromatin. Mol. Cell. 2010;40(4):606–18. doi: 10.1016/j.molcel.2010.10.026. [DOI] [PubMed] [Google Scholar]
- (12).Matsumoto S, Shimmoto M, Kakusho N, Yokoyama M, Kanoh Y, Hayano M, Russell P, Masai H. Hsk1 kinase and Cdc45 regulate replication stress-induced checkpoint responses in fission yeast. Cell Cycle. 2010;9(23):4627–37. doi: 10.4161/cc.9.23.13937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Takahashi TS, Basu A, Bermudez V, Hurwitz J, Walter JC. Cdc7-Drf1 kinase links chromosome cohesion to the initiation of DNA replication in Xenopus egg extracts. Genes Dev. 2008;22(14):1894–905. doi: 10.1101/gad.1683308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lo HC, Kunz RC, Chen X, Marullo A, Gygi SP, Hollingsworth NM. Cdc7-Dbf4 is a gene-specific regulator of meiotic transcription in yeast. Mol. Cell. Biol. 2012;32(2):541–57. doi: 10.1128/MCB.06032-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Matos J, Lipp JJ, Bogdanova A, Guillot S, Okaz E, Junqueira M, Shevchenko A, Zachariae W. Dbf4-dependent CDC7 kinase links DNA replication to the segregation of homologous chromosomes in meiosis I. Cell. 2008;135(4):662–78. doi: 10.1016/j.cell.2008.10.026. [DOI] [PubMed] [Google Scholar]
- (16).Choschzick M, Lebeau A, Marx AH, Tharun L, Terracciano L, Heilenkotter U, Jaenicke F, Bokemeyer C, Simon R, Sauter G, Schwarz J. Overexpression of cell division cycle 7 homolog is associated with gene amplification frequency in breast cancer. Hum. Pathol. 2010;41(3):358–65. doi: 10.1016/j.humpath.2009.08.008. [DOI] [PubMed] [Google Scholar]
- (17).Bonte D, Lindvall C, Liu H, Dykema K, Furge K, Weinreich M. Cdc7-Dbf4 kinase overexpression in multiple cancers and tumor cell lines is correlated with p53 inactivation. Neoplasia. 2008;10(9):920–31. doi: 10.1593/neo.08216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kulkarni AA, Kingsbury SR, Tudzarova S, Hong HK, Loddo M, Rashid M, Rodriguez-Acebes S, Prevost AT, Ledermann JA, Stoeber K, Williams GH. Cdc7 kinase is a predictor of survival and a novel therapeutic target in epithelial ovarian carcinoma. Clin. Cancer Res. 2009;15(7):2417–25. doi: 10.1158/1078-0432.CCR-08-1276. [DOI] [PubMed] [Google Scholar]
- (19).Montagnoli A, Valsasina B, Croci V, Menichincheri M, Rainoldi S, Marchesi V, Tibolla M, Tenca P, Brotherton D, Albanese C, Patton V, Alzani R, Ciavolella A, Sola F, Molinari A, Volpi D, Avanzi N, Fiorentini F, Cattoni M, Healy S, Ballinari D, Pesenti E, Isacchi A, Moll J, Bensimon A, Vanotti E, Santocanale C. A Cdc7 kinase inhibitor restricts initiation of DNA replication and has antitumor activity. Nat. Chem. Biol. 2008;4(6):357–65. doi: 10.1038/nchembio.90. [DOI] [PubMed] [Google Scholar]
- (20).Swords R, Mahalingam D, O’Dwyer M, Santocanale C, Kelly K, Carew J, Giles F. Cdc7 kinase - a new target for drug development. Eur. J. Cancer. 2010;46(1):33–40. doi: 10.1016/j.ejca.2009.09.020. [DOI] [PubMed] [Google Scholar]
- (21).Montagnoli A, Moll J, Colotta F. Targeting cell division cycle 7 kinase: a new approach for cancer therapy. Clin. Cancer Res. 2010;16(18):4503–8.22. doi: 10.1158/1078-0432.CCR-10-0185. [DOI] [PubMed] [Google Scholar]
- (22).Mulvey C, Tudzarova S, Crawford M, Williams GH, Stoeber K, Godovac-Zimmermann J. Quantitative proteomics reveals a “poised quiescence” cellular state after triggering the DNA replication origin activation checkpoint. J. Proteome Res. 2010;9(10):5445–60. doi: 10.1021/pr100678k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006;1(6):2856–60. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- (24).Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26(12):1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- (25).Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011;10(4):1794–805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- (26).Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, Bork P, von Mering C. STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37(Database issue):D412–6. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, Jensen LJ, von Mering C. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39(Database issue):D561–8. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27(3):431–2. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Montojo J, Zuberi K, Rodriguez H, Kazi F, Wright G, Donaldson SL, Morris Q, Bader GD. GeneMANIA Cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics. 2010;26(22):2927–8. doi: 10.1093/bioinformatics/btq562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, Maitland A, Mostafavi S, Montojo J, Shao Q, Wright G, Bader GD, Morris Q. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38(Web Server issue):W214–20. doi: 10.1093/nar/gkq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Croft D, O’Kelly G, Wu G, Haw R, Gillespie M, Matthews L, Caudy M, Garapati P, Gopinath G, Jassal B, Jupe S, Kalatskaya I, Mahajan S, May B, Ndegwa N, Schmidt E, Shamovsky V, Yung C, Birney E, Hermjakob H, D’Eustachio P, Stein L. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 2011;39(Database issue):D691–7. doi: 10.1093/nar/gkq1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003;4:2. doi: 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21(16):3448–9. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- (35).Barkley LR, Hong HK, Kingsbury SR, James M, Stoeber K, Williams GH. Cdc6 is a rate-limiting factor for proliferative capacity during HL60 cell differentiation. Exp. Cell Res. 2007;313(17):3789–99. doi: 10.1016/j.yexcr.2007.07.004. [DOI] [PubMed] [Google Scholar]
- (36).Qattan AT, Mulvey C, Crawford M, Natale DA, Godovac-Zimmermann J. Quantitative organelle proteomics of MCF-7 breast cancer cells reveals multiple subcellular locations for proteins in cellular functional processes. J. Proteome Res. 2010;9(1):495–508. doi: 10.1021/pr9008332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ben-Menachem R, Tal M, Shadur T, Pines O. A third of the yeast mitochondrial proteome is dual localized: a question of evolution. Proteomics. 2011;11(23):4468–76. doi: 10.1002/pmic.201100199. [DOI] [PubMed] [Google Scholar]
- (38).Henke RM, Dastidar RG, Shah A, Cadinu D, Yao X, Hooda J, Zhang L. Hypoxia elicits broad and systematic changes in protein subcellular localization. Am. J. Physiol.: Cell Physiol. 2011;301(4):C913–28. doi: 10.1152/ajpcell.00481.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Qattan AT, Radulovic M, Crawford M, Godovac-Zimmermann J. Spatial distribution of cellular function: the partitioning of proteins between mitochondria and the nucleus in MCF7 breast cancer cells. J. Proteome Res. 2012;11(12):6080–101. doi: 10.1021/pr300736v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Burhans WC, Heintz NH. The cell cycle is a redox cycle: linking phase-specific targets to cell fate. Free Radic. Biol. Med. 2009;47(9):1282–93. doi: 10.1016/j.freeradbiomed.2009.05.026. [DOI] [PubMed] [Google Scholar]
- (41).Chiu J, Dawes IW. Redox control of cell proliferation. Trends Cell Biol. 2012;22(11):592–601. doi: 10.1016/j.tcb.2012.08.002. [DOI] [PubMed] [Google Scholar]
- (42).Sakurai S, Kitano K, Yamaguchi H, Hamada K, Okada K, Fukuda K, Uchida M, Ohtsuka E, Morioka H, Hakoshima T. Structural basis for recruitment of human flap endonuclease 1 to PCNA. EMBO J. 2005;24(4):683–93. doi: 10.1038/sj.emboj.7600519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Cho IT, Kim DH, Kang YH, Lee CH, Amangyelid T, Nguyen TA, Hurwitz J, Seo YS. Human replication factor C stimulates flap endonuclease 1. J. Biol. Chem. 2009;284(16):10387–99. doi: 10.1074/jbc.M808893200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Suter B, Pogoutse O, Guo X, Krogan N, Lewis P, Greenblatt JF, Rine J, Emili A. Association with the origin recognition complex suggests a novel role for histone acetyltransferase Hat1p/Hat2p. BMC Biol. 2007;5(1):38. doi: 10.1186/1741-7007-5-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Torti FM, Torti SV. Regulation of ferritin genes and protein. Blood. 2002;99(10):3505–16. doi: 10.1182/blood.v99.10.3505. [DOI] [PubMed] [Google Scholar]
- (46).Orino K, Lehman L, Tsuji Y, Ayaki H, Torti SV, Torti FM. Ferritin and the response to oxidative stress. Biochem. J. 2001;357(Pt 1):241–7. doi: 10.1042/0264-6021:3570241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Cozzi A, Corsi B, Levi S, Santambrogio P, Albertini A, Arosio P. Overexpression of wild type and mutated human ferritin H-chain in HeLa cells: in vivo role of ferritin ferroxidase activity. J. Biol. Chem. 2000;275(33):25122–9. doi: 10.1074/jbc.M003797200. [DOI] [PubMed] [Google Scholar]
- (48).Nam SY, Sabapathy K. p53 promotes cellular survival in a context-dependent manner by directly inducing the expression of haeme-oxygenase-1. Oncogene. 2011;30(44):4476–86. doi: 10.1038/onc.2011.150. [DOI] [PubMed] [Google Scholar]
- (49).Gao J, Richardson DR. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents, IV: The mechanisms involved in inhibiting cell-cycle progression. Blood. 2001;98(3):842–50. doi: 10.1182/blood.v98.3.842. [DOI] [PubMed] [Google Scholar]
- (50).Kulp KS, Green SL, Vulliet PR. Iron deprivation inhibits cyclin-dependent kinase activity and decreases cyclin D/CDK4 protein levels in asynchronous MDA-MB-453 human breast cancer cells. Exp. Cell Res. 1996;229(1):60–8. doi: 10.1006/excr.1996.0343. [DOI] [PubMed] [Google Scholar]
- (51).Yu Y, Kovacevic Z, Richardson DR. Tuning cell cycle regulation with an iron key. Cell Cycle. 2007;6(16):1982–94. doi: 10.4161/cc.6.16.4603. [DOI] [PubMed] [Google Scholar]
- (52).Wu KJ, Polack A, Dalla-Favera R. Coordinated regulation of iron-controlling genes, H-ferritin and IRP2, by c-MYC. Science. 1999;283(5402):676–9. doi: 10.1126/science.283.5402.676. [DOI] [PubMed] [Google Scholar]
- (53).Le NT, Richardson DR. The role of iron in cell cycle progression and the proliferation of neoplastic cells. Biochim. Biophys. Acta. 2002;1603(1):31–46. doi: 10.1016/s0304-419x(02)00068-9. [DOI] [PubMed] [Google Scholar]
- (54).Buss JL, Greene BT, Turner J, Torti FM, Torti SV. Iron chelators in cancer chemotherapy. Curr. Top. Med. Chem. 2004;4(15):1623–35. doi: 10.2174/1568026043387269. [DOI] [PubMed] [Google Scholar]
- (55).Zhang F, Wang W, Tsuji Y, Torti SV, Torti FM. Post-transcriptional modulation of iron homeostasis during p53-dependent growth arrest. J. Biol. Chem. 2008;283(49):33911–8. doi: 10.1074/jbc.M806432200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Yogev O, Naamati A, Pines O. Fumarase: a paradigm of dual targeting and dual localized functions. FEBS J. 2011;278(22):4230–42. doi: 10.1111/j.1742-4658.2011.08359.x. [DOI] [PubMed] [Google Scholar]
- (57).Yogev O, Pines O. Dual targeting of mitochondrial proteins: mechanism, regulation and function. Biochim. Biophys. Acta. 2011;1808(3):1012–20. doi: 10.1016/j.bbamem.2010.07.004. [DOI] [PubMed] [Google Scholar]
- (58).Colombo SL, Palacios-Callender M, Frakich N, Carcamo S, Kovacs I, Tudzarova S, Moncada S. Molecular basis for the differential use of glucose and glutamine in cell proliferation as revealed by synchronized HeLa cells. Proc. Natl. Acad. Sci. U.S.A. 2011;108(52):21069–74. doi: 10.1073/pnas.1117500108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Yalcin A, Clem BF, Simmons A, Lane A, Nelson K, Clem AL, Brock E, Siow D, Wattenberg B, Telang S, Chesney J. Nuclear targeting of 6-phosphofructo-2-kinase (PFKFB3) increases proliferation via cyclin-dependent kinases. J. Biol. Chem. 2009;284(36):24223–32. doi: 10.1074/jbc.M109.016816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Malik S, Guermah M, Roeder RG. A dynamic model for PC4 coactivator function in RNA polymerase II transcription. Proc. Natl. Acad. Sci. U.S.A. 1998;95(5):2192–7. doi: 10.1073/pnas.95.5.2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Conesa C, Acker J. Sub1/PC4 a chromatin associated protein with multiple functions in transcription. RNA Biol. 2010;7(3):287–90. doi: 10.4161/rna.7.3.11491. [DOI] [PubMed] [Google Scholar]
- (62).Das C, Hizume K, Batta K, Kumar BR, Gadad SS, Ganguly S, Lorain S, Verreault A, Sadhale PP, Takeyasu K, Kundu TK. Transcriptional coactivator PC4, a chromatin-associated protein, induces chromatin condensation. Mol. Cell. Biol. 2006;26(22):8303–15. doi: 10.1128/MCB.00887-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Batta K, Yokokawa M, Takeyasu K, Kundu TK. Human transcriptional coactivator PC4 stimulates DNA end joining and activates DSB repair activity. J. Mol. Biol. 2009;385(3):788–99. doi: 10.1016/j.jmb.2008.11.008. [DOI] [PubMed] [Google Scholar]
- (64).Mortusewicz O, Roth W, Li N, Cardoso MC, Meisterernst M, Leonhardt H. Recruitment of RNA polymerase II cofactor PC4 to DNA damage sites. J. Cell Biol. 2008;183(5):769–76. doi: 10.1083/jcb.200808097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Banerjee S, Kumar BR, Kundu TK. General transcriptional coactivator PC4 activates p53 function. Mol. Cell. Biol. 2004;24(5):2052–62. doi: 10.1128/MCB.24.5.2052-2062.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Batta K, Kundu TK. Activation of p53 function by human transcriptional coactivator PC4: role of protein-protein interaction, DNA bending, and posttranslational modifications. Mol. Cell. Biol. 2007;27(21):7603–14. doi: 10.1128/MCB.01064-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Kishore AH, Batta K, Das C, Agarwal S, Kundu TK. p53 regulates its own activator: transcriptional co-activator PC4, a new p53-responsive gene. Biochem. J. 2007;406(3):437–44. doi: 10.1042/BJ20070390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Mao I, Liu J, Li X, Luo H. REGgamma, a proteasome activator and beyond? Cell. Mol. Life Sci. 2008;65(24):3971–80. doi: 10.1007/s00018-008-8291-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Chen X, Barton LF, Chi Y, Clurman BE, Roberts JM. Ubiquitin-independent degradation of cell-cycle inhibitors by the REGgamma proteasome. Mol. Cell. 2007;26(6):843–52. doi: 10.1016/j.molcel.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Li X, Amazit L, Long W, Lonard DM, Monaco JJ, O’Malley BW. Ubiquitin- and ATP-independent proteolytic turnover of p21 by the REGgamma-proteasome pathway. Mol. Cell. 2007;26(6):831–42. doi: 10.1016/j.molcel.2007.05.028. [DOI] [PubMed] [Google Scholar]
- (71).Zhang Z, Zhang R. Proteasome activator PA28 gamma regulates p53 by enhancing its MDM2-mediated degradation. EMBO J. 2008;27(6):852–64. doi: 10.1038/emboj.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Liu J, Yu G, Zhao Y, Zhao D, Wang Y, Wang L, Li L, Zeng Y, Dang Y, Wang C, Gao G, Long W, Lonard DM, Qiao S, Tsai MJ, Zhang B, Luo H, Li X. REGgamma modulates p53 activity by regulating its cellular localization. J. Cell Sci. 2010;123(Pt 23):4076–84. doi: 10.1242/jcs.067405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Levy-Barda A, Lerenthal Y, Davis AJ, Chung YM, Essers J, Shao Z, van Vliet N, Chen DJ, Hu MC, Kanaar R, Ziv Y, Shiloh Y. Involvement of the nuclear proteasome activator PA28gamma in the cellular response to DNA double-strand breaks. Cell Cycle. 2011;10(24):4300–10. doi: 10.4161/cc.10.24.18642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Zannini L, Buscemi G, Fontanella E, Lisanti S, Delia D. REGgamma/PA28gamma proteasome activator interacts with PML and Chk2 and affects PML nuclear bodies number. Cell Cycle. 2009;8(15):2399–407. doi: 10.4161/cc.8.15.9084. [DOI] [PubMed] [Google Scholar]
- (75).Hagemann C, Patel R, Blank JL. MEKK3 interacts with the PA28 gamma regulatory subunit of the proteasome. Biochem. J. 2003;373(Pt 1):71–9. doi: 10.1042/BJ20021758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Wu Y, Wang L, Zhou P, Wang G, Zeng Y, Wang Y, Liu J, Zhang B, Liu S, Luo H, Li X. Regulation of REGgamma cellular distribution and function by SUMO modification. Cell Res. 2011;21(5):807–16. doi: 10.1038/cr.2011.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Qu D, Zhang Y, Ma J, Guo K, Li R, Yin Y, Cao X, Park DS. The nuclear localization of SET mediated by impalpha3/impbeta attenuates its cytosolic toxicity in neurons. J. Neurochem. 2007;103(1):408–22. doi: 10.1111/j.1471-4159.2007.04747.x. [DOI] [PubMed] [Google Scholar]
- (78).Switzer CH, Cheng RY, Vitek TM, Christensen DJ, Wink DA, Vitek MP. Targeting SET/I(2)PP2A oncoprotein functions as a multi-pathway strategy for cancer therapy. Oncogene. 2011;30(22):2504–13. doi: 10.1038/onc.2010.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Li M, Makkinje A, Damuni Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J. Biol. Chem. 1996;271(19):11059–62. doi: 10.1074/jbc.271.19.11059. [DOI] [PubMed] [Google Scholar]
- (80).Canela N, Rodriguez-Vilarrupla A, Estanyol JM, Diaz C, Pujol MJ, Agell N, Bachs O. The SET protein regulates G2/M transition by modulating cyclin B-cyclin-dependent kinase 1 activity. J. Biol. Chem. 2003;278(2):1158–64. doi: 10.1074/jbc.M207497200. [DOI] [PubMed] [Google Scholar]
- (81).Estanyol JM, Jaumot M, Casanovas O, Rodriguez-Vilarrupla A, Agell N, Bachs O. The protein SET regulates the inhibitory effect of p21(Cip1) on cyclin E-cyclin-dependent kinase 2 activity. J. Biol. Chem. 1999;274(46):33161–5. doi: 10.1074/jbc.274.46.33161. [DOI] [PubMed] [Google Scholar]
- (82).Seo SB, McNamara P, Heo S, Turner A, Lane WS, Chakravarti D. Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the set oncoprotein. Cell. 2001;104(1):119–30. doi: 10.1016/s0092-8674(01)00196-9. [DOI] [PubMed] [Google Scholar]
- (83).Madeira A, Pommet JM, Prochiantz A, Allinquant B. SET protein (TAF1beta, I2PP2A) is involved in neuronal apoptosis induced by an amyloid precursor protein cytoplasmic subdomain. FASEB J. 2005;19(13):1905–7. doi: 10.1096/fj.05-3839fje. [DOI] [PubMed] [Google Scholar]
- (84).Fukukawa C, Shima H, Tanuma N, Okada T, Kato N, Adachi Y, Kikuchi K. The oncoprotein I-2PP2A/SET negatively regulates the MEK/ERK pathway and cell proliferation. Int. J. Oncol. 2005;26(3):751–6. [PubMed] [Google Scholar]
- (85).Amamoto R, Yagi M, Song Y, Oda Y, Tsuneyoshi M, Naito S, Yokomizo A, Kuroiwa K, Tokunaga S, Kato S, Hiura H, Samori T, Kang D, Uchiumi T. Mitochondrial p32/C1QBP is highly expressed in prostate cancer and is associated with shorter prostate-specific antigen relapse time after radical prostatectomy. Cancer Sci. 2011;102(3):639–47. doi: 10.1111/j.1349-7006.2010.01828.x. [DOI] [PubMed] [Google Scholar]
- (86).McGee AM, Douglas DL, Liang Y, Hyder SM, Baines CP. The mitochondrial protein C1qbp promotes cell proliferation, migration and resistance to cell death. Cell Cycle. 2011;10(23):4119–27. doi: 10.4161/cc.10.23.18287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Fogal V, Richardson AD, Karmali PP, Scheffler IE, Smith JW, Ruoslahti E. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol. Cell. Biol. 2010;30(6):1303–18. doi: 10.1128/MCB.01101-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).McGee AM, Baines CP. Complement 1q-binding protein inhibits the mitochondrial permeability transition pore and protects against oxidative stress-induced death. Biochem. J. 2011;433(1):119–25. doi: 10.1042/BJ20101431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Yu Z, Chen T, Hebert J, Li E, Richard S. A mouse PRMT1 null allele defines an essential role for arginine methylation in genome maintenance and cell proliferation. Mol. Cell. Biol. 2009;29(11):2982–96. doi: 10.1128/MCB.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Boisvert FM, Dery U, Masson JY, Richard S. Arginine methylation of MRE11 by PRMT1 is required for DNA damage checkpoint control. Genes Dev. 2005;19(6):671–6. doi: 10.1101/gad.1279805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Scoumanne A, Chen X. Protein methylation: a new mechanism of p53 tumor suppressor regulation. Histol. Histopathol. 2008;23(9):1143–9. doi: 10.14670/hh-23.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).An W, Kim J, Roeder RG. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell. 2004;117(6):735–48. doi: 10.1016/j.cell.2004.05.009. [DOI] [PubMed] [Google Scholar]
- (93).Yamagata K, Daitoku H, Takahashi Y, Namiki K, Hisatake K, Kako K, Mukai H, Kasuya Y, Fukamizu A. Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol. Cell. 2008;32(2):221–31. doi: 10.1016/j.molcel.2008.09.013. [DOI] [PubMed] [Google Scholar]
- (94).West RB, Yaneva M, Lieber MR. Productive and nonproductive complexes of Ku and DNA-dependent protein kinase at DNA termini. Mol. Cell. Biol. 1998;18(10):5908–20. doi: 10.1128/mcb.18.10.5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Park SJ, Ciccone SL, Freie B, Kurimasa A, Chen DJ, Li GC, Clapp DW, Lee SH. A positive role for the Ku complex in DNA replication following strand break damage in mammals. J. Biol. Chem. 2004;279(7):6046–55. doi: 10.1074/jbc.M311054200. [DOI] [PubMed] [Google Scholar]
- (96).Sibani S, Price GB, Zannis-Hadjopoulos M. Ku80 binds to human replication origins prior to the assembly of the ORC complex. Biochemistry. 2005;44(21):7885–96. doi: 10.1021/bi047327n. [DOI] [PubMed] [Google Scholar]
- (97).Jin S, Weaver DT. Double-strand break repair by Ku70 requires heterodimerization with Ku80 and DNA binding functions. EMBO J. 1997;16(22):6874–85. doi: 10.1093/emboj/16.22.6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Novac O, Matheos D, Araujo FD, Price GB, Zannis-Hadjopoulos M. In vivo association of Ku with mammalian origins of DNA replication. Mol. Biol. Cell. 2001;12(11):3386–401. doi: 10.1091/mbc.12.11.3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Rampakakis E, Di Paola D, Zannis-Hadjopoulos M. Ku is involved in cell growth, DNA replication and G1-S transition. J. Cell Sci. 2008;121(Pt 5):590–600. doi: 10.1242/jcs.021352. [DOI] [PubMed] [Google Scholar]
- (100).Fewell JW, Kuff EL. Intracellular redistribution of Ku immunoreactivity in response to cell-cell contact and growth modulating components in the medium. J. Cell Sci. 1996;109(Pt 7):1937–46. doi: 10.1242/jcs.109.7.1937. [DOI] [PubMed] [Google Scholar]
- (101).Koike M. Dimerization, translocation and localization of Ku70 and Ku80 proteins. J. Radiat. Res. 2002;43(3):223–36. doi: 10.1269/jrr.43.223. [DOI] [PubMed] [Google Scholar]
- (102).Koike M, Awaji T, Kataoka M, Tsujimoto G, Kartasova T, Koike A, Shiomi T. Differential subcellular localization of DNA-dependent protein kinase components Ku and DNA-PKcs during mitosis. J. Cell Sci. 1999;112(Pt 22):4031–9. doi: 10.1242/jcs.112.22.4031. [DOI] [PubMed] [Google Scholar]
- (103).Borer RA, Lehner CF, Eppenberger HM, Nigg EA. Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell. 1989;56(3):379–90. doi: 10.1016/0092-8674(89)90241-9. [DOI] [PubMed] [Google Scholar]
- (104).Li YP, Busch RK, Valdez BC, Busch H. C23 interacts with B23, a putative nucleolar-localization-signal-binding protein. Eur. J. Biochem. 1996;237(1):153–8. doi: 10.1111/j.1432-1033.1996.0153n.x. [DOI] [PubMed] [Google Scholar]
- (105).Emmott E, Hiscox JA. Nucleolar targeting: the hub of the matter. EMBO Rep. 2009;10(3):231–8. doi: 10.1038/embor.2009.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Srivastava M, Pollard HB. Molecular dissection of nucleolin’s role in growth and cell proliferation: new insights. FASEB J. 1999;13(14):1911–22. [PubMed] [Google Scholar]
- (107).Tuteja R, Tuteja N. Nucleolin: a multifunctional major nucleolar phosphoprotein. Crit. Rev. Biochem. Mol. Biol. 1998;33(6):407–36. doi: 10.1080/10409239891204260. [DOI] [PubMed] [Google Scholar]
- (108).Kim K, Dimitrova DD, Carta KM, Saxena A, Daras M, Borowiec JA. Novel checkpoint response to genotoxic stress mediated by nucleolin-replication protein a complex formation. Mol. Cell. Biol. 2005;25(6):2463–74. doi: 10.1128/MCB.25.6.2463-2474.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Daniely Y, Dimitrova DD, Borowiec JA. Stress-dependent nucleolin mobilization mediated by p53-nucleolin complex formation. Mol. Cell. Biol. 2002;22(16):6014–22. doi: 10.1128/MCB.22.16.6014-6022.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Saxena A, Rorie CJ, Dimitrova D, Daniely Y, Borowiec JA. Nucleolin inhibits Hdm2 by multiple pathways leading to p53 stabilization. Oncogene. 2006;25(55):7274–88. doi: 10.1038/sj.onc.1209714. [DOI] [PubMed] [Google Scholar]
- (111).Kurki S, Peltonen K, Latonen L, Kiviharju TM, Ojala PM, Meek D, Laiho M. Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell. 2004;5(5):465–75. doi: 10.1016/s1535-6108(04)00110-2. [DOI] [PubMed] [Google Scholar]
- (112).Bhatt P, d’Avout C, Kane NS, Borowiec JA, Saxena A. Specific domains of nucleolin interact with Hdm2 and antagonize Hdm2-mediated p53 ubiquitination. FEBS J. 2012;279(3):370–83. doi: 10.1111/j.1742-4658.2011.08430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Weis K. Regulating access to the genome: nucleocytoplasmic transport throughout the cell cycle. Cell. 2003;112(4):441–51. doi: 10.1016/s0092-8674(03)00082-5. [DOI] [PubMed] [Google Scholar]
- (114).Lange A, Mills RE, Lange CJ, Stewart M, Devine SE, Corbett AH. Classical nuclear localization signals: definition, function, and interaction with importin alpha. J. Biol. Chem. 2007;282(8):5101–5. doi: 10.1074/jbc.R600026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Gorlich D, Henklein P, Laskey RA, Hartmann E. A 41 amino acid motif in importin-alpha confers binding to importin-beta and hence transit into the nucleus. EMBO J. 1996;15(8):1810–7. [PMC free article] [PubMed] [Google Scholar]
- (116).Watt PJ, Ngarande E, Leaner VD. Overexpression of Kpnbeta1 and Kpnalpha2 importin proteins in cancer derives from deregulated E2F activity. PloS One. 2011;6(11):e27723. doi: 10.1371/journal.pone.0027723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Yasuhara N, Takeda E, Inoue H, Kotera I, Yoneda Y. Importin alpha/beta-mediated nuclear protein import is regulated in a cell cycle-dependent manner. Exp. Cell Res. 2004;297(1):285–93. doi: 10.1016/j.yexcr.2004.03.010. [DOI] [PubMed] [Google Scholar]
- (118).Pulliam KF, Fasken MB, McLane LM, Pulliam JV, Corbett AH. The classical nuclear localization signal receptor, importin-alpha, is required for efficient transition through the G1/S stage of the cell cycle in Saccharomyces cerevisiae. Genetics. 2009;181(1):105–18. doi: 10.1534/genetics.108.097303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Quensel C, Friedrich B, Sommer T, Hartmann E, Kohler M. In vivo analysis of importin alpha proteins reveals cellular proliferation inhibition and substrate specificity. Mol. Cell. Biol. 2004;24(23):10246–55. doi: 10.1128/MCB.24.23.10246-10255.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Kim BJ, Lee H. Importin-beta mediates Cdc7 nuclear import by binding to the kinase insert II domain, which can be antagonized by importin-alpha. J. Biol. Chem. 2006;281(17):12041–9. doi: 10.1074/jbc.M512630200. [DOI] [PubMed] [Google Scholar]
- (121).Koike M, Ikuta T, Miyasaka T, Shiomi T. Ku80 can translocate to the nucleus independent of the translocation of Ku70 using its own nuclear localization signal. Oncogene. 1999;18(52):7495–505. doi: 10.1038/sj.onc.1203247. [DOI] [PubMed] [Google Scholar]
- (122).Bertinato J, Schild-Poulter C, Hache RJ. Nuclear localization of Ku antigen is promoted independently by basic motifs in the Ku70 and Ku80 subunits. J. Cell Sci. 2001;114(Pt 1):89–99. doi: 10.1242/jcs.114.1.89. [DOI] [PubMed] [Google Scholar]
- (123).Song JY, Lim JW, Kim H, Morio T, Kim KH. Oxidative stress induces nuclear loss of DNA repair proteins Ku70 and Ku80 and apoptosis in pancreatic acinar AR42J cells. J. Biol. Chem. 2003;278(38):36676–87. doi: 10.1074/jbc.M303692200. [DOI] [PubMed] [Google Scholar]
- (124).Bier C, Knauer SK, Docter D, Schneider G, Kramer OH, Stauber RH. The importin-alpha/nucleophosmin switch controls taspase1 protease function. Traffic. 2011;12(6):703–14. doi: 10.1111/j.1600-0854.2011.01191.x. [DOI] [PubMed] [Google Scholar]
- (125).Bouvet P, Diaz JJ, Kindbeiter K, Madjar JJ, Amalric F. Nucleolin interacts with several ribosomal proteins through its RGG domain. J. Biol. Chem. 1998;273(30):19025–9. doi: 10.1074/jbc.273.30.19025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.