

Abstract
Accumulation of abnormal tau aggregates in neuron is an important pathological signature in multiple neurodegenerative disorders including Alzheimer's disease. Tau is a neuron specific microtubule-associated protein that regulates microtubule stability, which is critical for axonal outgrowth and synaptic plasticity. In a pathological condition, tau dissociates from microtubules and forms insoluble aggregates called neurofibrillary tangles (NFTs). The accumulation of NFTs in neuron directly correlates with microtubule dysfunction and neuronal degeneration. Due to the pathophysiological importance of tau, great efforts have been made to understand tau aggregation processes and find therapeutics to halt or reverse the processes. However, progress has been slow due to the lack of a suitable method for monitoring tau aggregation. In this mini-review, we will review the conventional methods for studying tau aggregation, and introduce recent cell-based sensor approaches that allow monitoring tau aggregation in living cells.
Keywords: Tauopathy, Tau phosphorylation, Tau oligomerization, In vitro assay, Cell-based sensor, BiFC
1. Introduction
Tau is a neuron specific microtubule-associated protein [1–3]. In a healthy neuron, tau binds to microtubules and regulates microtubule stability, which is critical for axonal outgrowth and neuronal plasticity [4–6]. When pathologically altered, tau molecules are not able to stabilize microtubules and become insoluble aggregates [3,7–9]. Since Alois Alzheimer discovered the abnormal tau inclusions in a patient's brain, the presence of tau aggregates is a critical biomarker for making the pathological diagnosis of AD [10]. In AD patients, three forms of tau aggregates occur; neurofibrillary tangles (NFTs) in neuronal somata, neuropil threads (NTs) in neuronal dendrites, and neuritic plaques (NPs). These tau aggregates induce neuronal degeneration. Especially, the density of NFTs correlates fairly well with regional and global aspects of cognitive decline during the progression of AD [10]. Hence, there has been great effort to understand how the deposition of NFT causes neurodegeneration (Fig. 1). NFT may damage neurons and glial cells in a number of ways [11]. First, aggregation of tau causes neuronal toxicity by reducing normal function of tau promoting microtubule stability. Also, the large filamentary tangles might be toxic to neurons by acting as physical barriers in the cytoplasm. Therefore, neurons containing tau tangles actively activate diverse cell metabolisms to get rid of the abnormal protein aggregates from cytoplasm [14,15]. This might be a great burden to a neuron that results in neuronal toxicity and neurodegeneration.
Fig. 1.

Tau aggregation and neuronal degeneration [18]. (a) In a healthy neuron, tau stabilizes microtubules promoting axonal outgrowth and synaptic vesicle transport. (b) When tau goes bad, tau becomes neurotoxic aggregates and microtubules become dissociates.
More recent studies suggest that, instead of the large insoluble filaments, soluble tau aggregates might play more critical roles in the onset and progression of disease prior to the development of NFT-induced neurotoxicity [6]. Especially hyperphosphorylated tau before NFT formation leads to microtubule disassembly, impairment of axonal transport, and organelle dysfunctions in neurons, leading to the neuronal cell apoptosis [3]. Also, the oligomeric species of tau may act as seeds for the aggregation of native tau, thereby promoting neurotoxic tau aggregation [8,16]. Accumulating evidence has suggested that tau aggregates are transmittable in neurons propagating as a prion-like manner [7,17].
Due to the pathological significance, tau becomes an important therapeutic target. Preventing tau aggregation becomes a potential strategy to cure neurodegenerative disorders associated with tau. So far, great effort has been made to identify molecular mechanism of tau aggregation and to reverse the processes. However, progress has been slow due to the lack of understanding the tau aggregation mechanism. Development of a reliable model for tau aggregation would be beneficial not only for identifying new therapeutic biomarkers but also for screening and evaluating drug candidates. Toward that, diverse tau aggregation methods have been developed: in vitro, cell-based, and in vivo models. Among the diverse models here we will review the in vitro and cell-based models for tau aggregation. In vitro tau aggregation methods have long been used for elucidating structural assembly of tau in the formation of PHFs. Cell-based models have recently developed to investigate the intracellular tau aggregation mechanism.
2. Multi-step Processes of Tau Aggregation
Contradictory to its pathological aggregation, tau is a naturally ‘unfolded’ protein, which is highly soluble in physiological condition [8,9]. To be a susceptible intermediate for aggregation tau molecules undergo a series of post-translational modifications and conformational changes in a neuron [19]. It is generally believed that tau aggregation is initiated by hyperphosphorylation (Fig. 2). Microtubule binding domains of tau contain a number of lysine residues, of which positive charges drive tau to bind negatively charged microtubules [20]. When tau is abnormally hyperphosphorylated, the balance is disrupted and tau dissociates from microtubules. Then, unbound tau undergoes conformational change to form a compact structure, called “Alz50 state” [21]. In this state, tau begins to aggregate and the further fibrillization is facilitated with proteolytic cleavages [1,13,22]. The truncated tau, named tau-66, assembles much faster than its native form [23]. NFTs are predominantly composed of paired helical filaments which appear to be made up of 10-nm filaments helically twisted with each other [24].
Fig. 2.
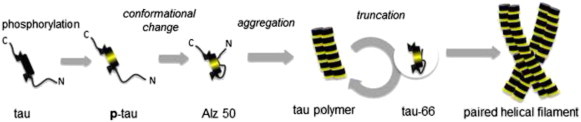
Diagrammatic representation of tau aggregation [25].
2.1. Tau Aggregation Assays In Vitro
To identify the ultra-molecular structure of PHFs, it is of obvious interest to reconstitute tau assembly process in vitro [12–14]. However, recombinant tau, which is purified from Escherichia coli, shows very little intrinsic tendency to aggregate in vitro due to the lack of a series of post-translational modifications required for aggregation. Over the last thirty years, the slow aggregation rate of purified tau has been improved by a combination of diverse advances (Table 1). First, recombinant truncated tau isoforms (e.g., the repeat domain alone aggregates) are more frequently used for in vitro tau aggregation, instead of full-length tau. Tau-repeat domains such as K18 or K19 alone aggregate much faster than the full-length tau (Fig. 3) [25,26]. Second, tau mutations such as P301L or ΔK280 occurring in FTDP-17 are known to enhance the β-sheet propensity to increase the aggregation reaction [27–31]. Third, addition of artificial cofactors also facilitate tau aggregation rate. Lysine-rich tau protein is extremely soluble in physiological conditions and it has very little intrinsic tendency to aggregate into filaments. So, the anionic cofactors screen the basic charges of tau and facilitate aggregation. There are two major classes of cofactors; polyanions (e.g., heparin, polyglutamate, or RNA) and fatty acids or fatty acid-like molecules (e.g., arachidonic acid, docosahexaenoic acid and alkyl sulfonate detergents) [26,28,32–34]. Poly-anionic cofactors such as heparin have a great efficiency in promoting the polymerization of tau fragments containing microtubule-binding domains (K18 or K19). Arachidonic acid also increases polymerization of full-length tau more rapidly than heparin.
Table 1.
Tau aggregation assay in vitro.
| Tau isoform | Mutants | Aggregation inducer | Incubation |
Detection | Ref. | |
|---|---|---|---|---|---|---|
| Temp | Time | |||||
| Tau-40 | – | Free fatty acid | 37 °C | 24–100 h | TEM | [38] |
| Arachidonic acid | 37 °C | 3–25 h | ThS, TEM | [28,32] | ||
| Heparin | 30 °C | 48–72 h | ThS, TEM | [33,34] | ||
| Polyglutamate | 30 °C | 48–72 h | ThS | [34,37] | ||
| Tau-23 | – | Arachidonic acid | 37 °C | 3–25 h | TEM | [26] |
| Pre-aggregated PHFs | 37 °C | 20 h | ThS | [40] | ||
| PHFs from AD patient | 37 °C | 20 h | ThS, TEM | [40] | ||
| K18 | – | Heparin | 37 °C | 3–25 h | ThS, TEM | [26,34] |
| Arachidonic acid | 37 °C | 3–25 h | ThS, TEM | [28,32] | ||
| RNA | 30 °C | − 7 weeks | TEM | [41] | ||
| Zinc | 37 °C | 1 h | ThS, TEM | [42] | ||
| K19 | – | Heparin RNA |
37 °C 30 °C |
72 h − 7 weeks |
ThS, TEM TEM |
[43] [41] |
| K18 | ΔK280 | – | 37 °C | 2–4 days | ThS, TEM | [29,30] |
| K18 | P301L | – | RT | 7 h | LLS | [31] |
Fig. 3.

Illustration represents human tau isoforms and truncated repeat domains. Human tau has six isoforms resulting from alternative splicing. The truncated tau repeat domains (K18 or K19) are known to facilitate tau aggregation.
These in vitro tau aggregation methods have been used in identifying structural assembly of tau in the formation of PHFs. Transmission electron microscopy technique (TEM) identifies that NFTs are predominantly composed of paired helical filaments [35]. Also, whether the fibers are paired helical filaments or not, PHFs isolated from Alzheimer's patients showed typical 80 nm crossover repeats. Whether it is artificial or not, these in vitro assays are commonly used to estimate the amount of tau aggregation in real time by using fluorescence probes such as thioflavin S (ThS); ThS is a widely used fluorescence probe to sensing the relative amount of β-sheet aggregates in solution [36,37].
3. Intracellular Modifications of Tau
Though the pathophysiological importance of NFTs in tauopathies, little progress has been made in understanding the packing of tau in the fibrils. The in vitro studies only provided the simple framework of tau aggregation hypothesis [38,44,45]. The cause and molecular mechanisms underlying tau aggregation remains still largely unknown. Progress has been slow because there is no reliable method for monitoring tau aggregation in physiological conditions, where tau is spontaneously altered and aggregated. Prior to or during NFT formation, tau undergoes numerous, and potentially harmful, modifications [39].
Among the diverse modifications of tau, phosphorylation induces the most critical change of tau leading to tau aggregation (Fig. 4) [46,47]. Full-length tau protein has 80 serine and threonine residues and 5 tyrosine residues (Fig. 4b). In an intact cell, tau is constantly phosphorylated and dephosphorylated for the regulation of microtubule assembly. Once the balance is disrupted, tau is highly phosphorylated. The additional phosphates disrupt the charge balance between tau and microtubules, and tau dissociates from microtubules. Therefore, hyper-phosphorylation is the critical event in the initiation of tau pathology [48]. Tau phosphorylation is tightly regulated by kinases or phosphatases [49,50]. Among diverse enzymes, GSK3β is the most effective tau kinase in the human brain (Fig. 4a). Increased GSK3β activity is directly linked to elevated levels of tau phosphorylation in AD patients [51–53].
Fig. 4.
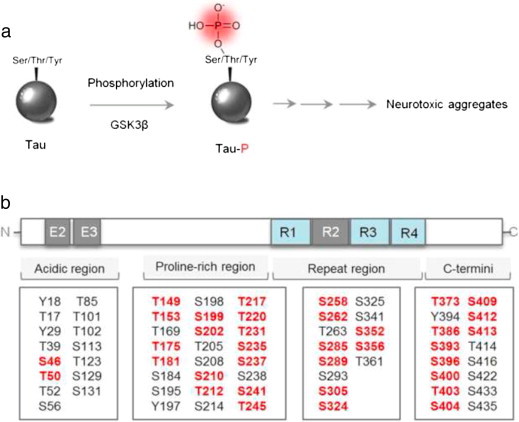
Tau phosphorylation. (a) Initiation of tau phosphorylation by GSK3β. (b) Putative tau phosphorylation residues. The red colored residues indicate GSK3β mediated tau phosphorylation sites.
Tau contains a number of lysine residues, of which positive charges are critical for binding to negatively charged microtubules. Similar to tau phosphorylation, acetylation of those lysine residues disrupts the charge balance between tau and microtubules resulting in tau aggregation. Lys280 is known as the major acetylation site and the increased acetylation promotes pathological tau aggregation [54]. Biochemical analysis of tau isolated from patient's brain also indicated the increased level of tau acetylation in AD [55]. Due to the diverse intracellular modifications of tau, tau aggregation has to be investigated in the complex cellular system. In the next section, we will introduce diverse cell-based approaches to induce and visualize tau aggregation in cells (Table 2).
Table 2.
Induction of tau aggregation in cells.
| Tau isoform | Mutants | Host cell | Expression | Aggregation inducer | Detection | Ref. |
|---|---|---|---|---|---|---|
| K18 | ΔK280 ΔK280/PP (I277P/I308P) |
N2a | Stable doxicyclin inducible | – | ThS antibody | [57] |
| Tau-40 | – | HEK293 | Stable tetracycline inducible | Congo red | Antibody | [59] |
| Tau-40 | N279K ΔK280 P301L S305N V337M R406W |
CHO | Stable expression | – | Antibody | [63] |
| Tau-40 | QBI–293 | Transient expression | Exogenous tau | Antibody | [60] | |
| Tau-46 Tau-37 |
SH-SY5Y | Transient expression | Exogenous tau | Antibody GFP |
[62] | |
| Tau-40 Tau-39 |
P301L V337M R406W |
NIH 3T3 | Stable expression | – | GFP CFP YFP |
[61] |
3.1. Tau Aggregation Assays in Cells
As the simplest model of tau expression, Pouplana and co-workers expressed tau in E. coli and confirmed its aggregation in inclusion body by ThS stain [56]. Although it is an efficient way to enrich tau protein, bacterial system does not allow post-translational modifications required for spontaneous tau aggregation in cells. For the mammalian expression, Mandelkow's group generated tau-inducible cell lines [57,58]. In their study, tau overexpression was toxic to the N2a neuroblastoma cells. Thus they expressed tau by using doxycycline-inducible system. The N2a cells expressing tau showed robust tau aggregation, which was detected by a fluorescence dye, ThS. Similarly, Jeff Kuret's group generated a tetracycline-insoluble tau cell line for tau aggregates. In their study, tau aggregation was promoted by the treatment of congo red, which is a known small-molecule agonist of tau aggregation, within cells expressing full length tau isoform [59]. In addition, Lee's group demonstrated intracellular tau aggregation that can be facilitated by the treatment of exogenous tau fibrils [60]. In their study, full-length of tau 40 was expressed in QBI-293 cells and tau aggregation was induced by the treatment of preformed tau fibrils, which act as a seed for intracellular tau aggregation. These studies successfully demonstrated that overexpressed tau could be aggregated in cells and also the aggregated tau induces cellular toxicity. However, these approaches require secondary methods to confirm intracellular tau aggregation such as or ThS or immune-stains against phosphorylated tau. To monitoring intracellular tau aggregation without any secondary detection methods, diverse fluorescence proteins (GFP, CFP, and YFP) are introduced to label tau and showed tau aggregation in living cells [61,62]. These cellular systems provide evidence of tau aggregation in cells and also enable the investigation of pathological mechanism of tau aggregation. However, these cell-based systems are not validated enough to quantify tau aggregation in living cells.
4. Cell-based Sensor for Tau Aggregation
A cell-based model that could monitor and quantify tau assembly in living cells would be a useful tool to investigate tau pathology and to discover methods to prevent and reverse the process. In this regards, diverse fluorescence protein technologies such as fluorescence resonance energy transfer (FRET) or bimolecular fluorescence complementation (BiFC) have been introduced to investigate tau–tau interaction in cells (Table. 3).
Table 3.
Cell-based sensor for tau aggregation.
| Tau isoform | Mutants | Tag | Host cell | Expression | Aggregation inducer | Detection | Ref. |
|---|---|---|---|---|---|---|---|
| Tau-40 Tau (1–421) |
– | HEK293 | Transient expression | GSK3β | FRET | [65] | |
| K18 | ΔK280 P301L V337M I277P I308P |
HEK293 | Transient expression | K18 | FRET | [66] | |
| Tau-40 K18 |
ΔK280 I277P I308P |
HEK293 | Transient expression | GSK3β | BiFC | [71] | |
| Tau-40 | – | HEK293 | Stable expression | Forskolin okadaic acid | BiFC | [72] |
4.1. FRET-based Tau Aggregation Sensor
First, Johnson's group introduced FRET technique to investigate tau–tau interaction in living cells. Fluorescence resonance energy transfer (FRET) is a process by which energy is transferred from a donor fluorophore (CFP) to an acceptor fluorophore (YFP) (Fig. 5a). The CFP and YFP are tagged to the proteins of interest [64]. Energy transfer between CFP and YFP occurs only when those proteins of interest are close enough (typically 2–6 nm). In their study, full-length tau and caspase-cleaved tau were labeled respectively with CFP and YFP, and then co-expressed in HEK293 cells [65]. When tau aggregation was induced by GSK3β, the two different tau isoforms bind to each other resulting in the energy transfer between CFP and YFP. By measuring the FRET intensity, the level of tau aggregation could be quantified in a living cell. More interestingly, Diamond's group used this FRET technique to identify trans-cellular propagation of tau aggregation [66]. In their study various tau mutants (K18) was labeled with YFP or CFP, and then expressed separately. Then, cell-medium was collected from donor cells expressing tau–CFP and treated to adopter cells expressing tau–YFP. FRET microscopy showed that tau fibrils secreted into extracellular space could be taken up by other cells and aggregate with intracellular tau. FRET technology has great advantage in differentiating the aggregated tau species from non-aggregated tau in living cells.
Fig. 5.
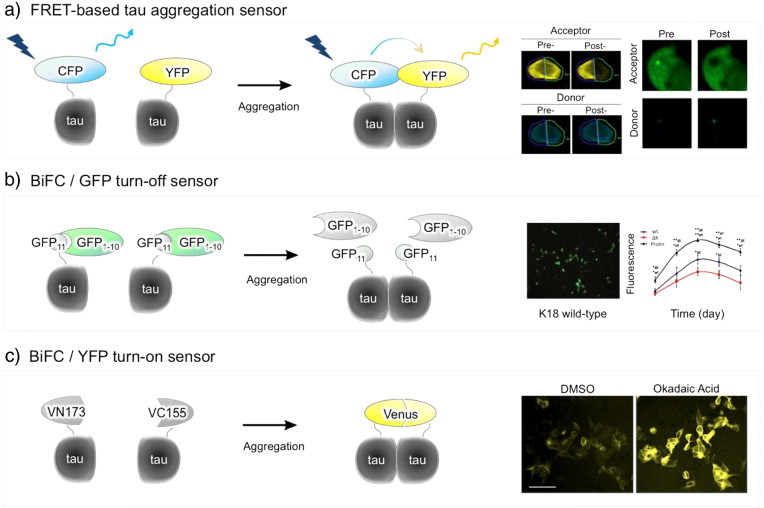
Cell-based sensors for tau aggregation. (a) FRET-based sensor. Tau protein is fused to CFP or YFP. In the system, the FRET sensor is activated only when tau assembles. (b) BiFC/GFP turn-off sensor. Tau is fused to a smaller fragment of GFP (GFP11) and co-expressed with a large GFP fragments (GFP1–10). When tau exists as a monomer, GFP1–10 freely binds to GFP11 giving the strong GFP fluorescence. When tau aggregates, GFP1–10 cannot access to GFP11, resulting in the decrease of GFP fluorescence intensity. (c) Tau is fused to non-fluorescent N- or C-terminal fragment of Venus fluorescence protein (VN173 or VC155). The Venus fluorescence turns on only when tau assembles together.
4.2. BiFC: Turn-off Sensor
It is clear that FRET is one of the outstanding methods that can quantify protein–protein interactions in living cells. However, one drawback of the approach is that the use of quite huge fluorescence protein tagging that might interfere with the interaction between the proteins of interest. Therefore, there have been efforts to minimize the size of fluorescence protein tagging. The best example is bimolecular fluorescence complementation (BiFC) technique. To reduce the size of tagging, a fluorescence protein is split into two non-fluorescent fragments in the BiFC approach. Then the non-fluorescent constituents are tagged to the proteins of interest. Fluorescent turns on only when those proteins of interest are associated together [67–69].
Johnson's group applied this BiFC technique to visualize tau aggregation in living cells. A split green fluorescent protein (GFP) complementation technique was used to quantify tau aggregation in situ [70,71] (Fig. 5b). In this assay, full-length tau protein was directly fused to a smaller GFP fragment (GFP11), and co-expressed in cells with a larger GFP fragment (GFP1–10). When tau exists as a monomer or low degree aggregate, the complementary large GFP fragment is able to access the small GFP fragment fused to tau, leading to the association of the fluorescently active GFP. As a fluorescence “turn-off” approach, when tau aggregates, the reconstitution of active GFP is prohibited and then, intensity of GFP fluorescence decreases in cells.
4.3. BiFC: Turn-on Sensor
As a method of quantifying tau aggregation in living cells, the split-GFP complementation assay has been highlighted. However, as a fluorescence “turn-off” sensor, the split-GFP technique has an intrinsic limitation to monitoring the initial tau aggregation processes such as soluble tau intermediates. This limitation was overcome by using a Venus-based BiFC system. In this approach, Venus fluorescence protein is split into two non-fluorescent N- and C-terminal fragments (VN173 and VC155) and used to label tau [72] (Fig. 5c). As a fluorescence “turn-on” approach, Venus fluorescence turns on only when tau assembles together. There is little fluorescence background in basal condition, suggesting that most tau molecules exist as a monomer. Tau–BiFC fluorescence turns on dramatically when tau aggregation was stimulated by the treatment of small molecules inducing tau phosphorylation. As fluorescence ‘turn-on’ technology, tau–BiFC approach enables to achieve spatial and temporal resolution of tau aggregation in living cells.
5. Concluding Remark
Due to the implications of tau pathology in diverse neurodegenerative disorders, tau becomes an important therapeutic target and great effort has been made to develop tau-targeted therapy. The primary therapeutic tactics considered include; (i) reduction of tau hyperphosphorylation using kinase inhibitors and phosphatase activators, (ii) activation of proteosomal degradation pathways of tau, (iii) tau clearance by immunotherapy, (iv) inhibition of tau aggregation using small molecules, and stabilizing microtubules. A reliable method to detect and monitor tau aggregation would accelerate understating tau aggregation mechanism and also expedite the development of tau-targeted therapeutics. Although innovative, the FRET-based tau aggregation sensor needs fine control to measure tau aggregation, thus it is not applicable for massive drug screening that needs robust screening platform. In contrast, the recently developed tau–BiFC “Turn-On” system allows simple and quantitative measurement of tau aggregation by measuring the increased YFP intensity in cells as an indication of tau aggregation. Also, as an established cell line, it allows 384-well based high-throughput drug screening providing reliable results. While the fluorescence protein tagging approaches enable monitoring intracellular tau aggregation, the use of a large tagging protein itself is a limitation of the approaches by adding an artificial manipulation of the system. We believe that this limitation can be overcome by developing a tau-selective fluorescence probe that can detect intracellular tau aggregation without the need for genetic manipulation. In this regards, the recently developed tau imaging probes give us a hope to facilitate the entire tau research [73,74].
Acknowledgments
This work was supported by an intramural funding from Korea Institute of Science and Technology (2E25023 and 2V03400).
References
- 1.Binder L.I., Guillozet-Bongaarts A.L., Garcia-Sierra F., Berry R.W. Tau, tangles, and Alzheimer's disease. Biochim Biophys Acta. 2005;1739:216–223. doi: 10.1016/j.bbadis.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 2.Gendron T.F., Petrucelli L. The role of tau in neurodegeneration. Mol Neurodegener. 2009;4:13. doi: 10.1186/1750-1326-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reddy P.H. Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer's disease. Brain Res. 2011;1415:136–148. doi: 10.1016/j.brainres.2011.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson G.V., Hartigan J.A. Tau protein in normal and Alzheimer's disease brain: an update. J Alzheimers Dis. 1999;1:329–351. doi: 10.3233/jad-1999-14-512. [DOI] [PubMed] [Google Scholar]
- 5.Brandt R., Hundelt M., Shahani N. Tau alteration and neuronal degeneration in tauopathies: mechanisms and models. Biochim Biophys Acta. 2005;1739:331–354. doi: 10.1016/j.bbadis.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 6.Alonso A.C., Zaidi T., Grundke-Iqbal I., Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:5562–5566. doi: 10.1073/pnas.91.12.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunden K.R., Trojanowski J.Q., Lee V.M. Evidence that non-fibrillar tau causes pathology linked to neurodegeneration and behavioral impairments. J Alzheimers Dis. 2008;14:393–399. doi: 10.3233/jad-2008-14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lasagna-Reeves C.A., Castillo-Carranza D.L., Sengupta U., Clos A.L., Jackson G.R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener. 2011;6:1–14. doi: 10.1186/1750-1326-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iqbal K., Liu F., Gong C.-X., Alonso AdC, Grundke-Iqbal I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009;118:53–69. doi: 10.1007/s00401-009-0486-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perl D.P. Neuropathology of Alzheimer's disease. Mt Sinai J Med: J Transl Pers Med. 2010;77:32–42. doi: 10.1002/msj.20157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gendron T.F., Petrucelli L. The role of tau in neurodegeneration. Mol Neurodegener. 2009;4:13. doi: 10.1186/1750-1326-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Canu N., Dus L., Barbato C., Ciotti M.T., Brancolini C. Tau cleavage and dephosphorylation in cerebellar granule neurons undergoing apoptosis. J Neurosci. 1998;18:7061–7074. doi: 10.1523/JNEUROSCI.18-18-07061.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gamblin T.C., Chen F., Zambrano A., Abraha A., Lagalwar S. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci. 2003;100:10032–10037. doi: 10.1073/pnas.1630428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cripps D., Thomas S.N., Jeng Y., Yang F., Davies P. Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-Tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J Biol Chem. 2006;281:10825–10838. doi: 10.1074/jbc.M512786200. [DOI] [PubMed] [Google Scholar]
- 15.Morishima-Kawashima M., Hasegawa M., Takio K., Suzuki M., Titani K. Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron. 1993;10:1151–1160. doi: 10.1016/0896-6273(93)90063-w. [DOI] [PubMed] [Google Scholar]
- 16.Guzman-Martinez L., Farias G.A., Maccioni R.B. Tau oligomers as potential targets for Alzheimer's diagnosis and novel drugs. Front Neurol. 2013;4:167. doi: 10.3389/fneur.2013.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lasagna-Reeves C.A., Castillo-Carranza D.L., Sengupta U., Clos A.L., Jackson G.R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener. 2011;6:39. doi: 10.1186/1750-1326-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brunden K.R., Trojanowski J.Q., Lee V.M. Advances in tau-focused drug discovery for Alzheimer's disease and related tauopathies. Nat Rev Drug Discov. 2009;8:783–793. doi: 10.1038/nrd2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia-Sierra F., Ghoshal N., Quinn B., Berry R.W., Binder L.I. Conformational changes and truncation of tau protein during tangle evolution in Alzheimer's disease. J Alzheimers Dis. 2003;5:65–77. doi: 10.3233/jad-2003-5201. [DOI] [PubMed] [Google Scholar]
- 20.Kolarova M., García-Sierra F., Bartos A., Ricny J., Ripova D. Structure and pathology of tau protein in Alzheimer disease. Int J Alzheimers Dis. 2012;2012:731526. doi: 10.1155/2012/731526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guillozet-Bongaarts A.L., Garcia-Sierra F., Reynolds M.R., Horowitz P.M., Fu Y. Tau truncation during neurofibrillary tangle evolution in Alzheimer's disease. Neurobiol Aging. 2005;26:1015–1022. doi: 10.1016/j.neurobiolaging.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 22.Fasulo L., Ugolini G., Visintin M., Bradbury A., Brancolini C. The neuronal microtubule associated protein tau is a substrate for caspase 3 and an effector of apoptosis. J Neurochem. 2000;75:624–633. doi: 10.1046/j.1471-4159.2000.0750624.x. [DOI] [PubMed] [Google Scholar]
- 23.Wischik C.M. Cell biology of the Alzheimer tangle. Curr Opin Cell Biol. 1989;1:115–122. doi: 10.1016/s0955-0674(89)80047-x. [DOI] [PubMed] [Google Scholar]
- 24.Perry G., Rizzuto N., Autilio-Gambetti L., Gambetti P. Paired helical filaments from Alzheimer disease patients contain cytoskeletal components. Proc Natl Acad Sci U S A. 1985;82:3916–3920. doi: 10.1073/pnas.82.11.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim Y., Kim Y., Hwang O., Kim D.J. Pathology of neurodegenerative diseases. In: Gonzalez-Quevedo D.A., editor. Brain damage — bridging between basic research and clinics: InTech. 2012. pp. 99–138. [Google Scholar]
- 26.Barghorn S., Mandelkow E. Toward a unified scheme for the aggregation of tau into Alzheimer paired helical filaments. Biochemistry. 2002;41:14885–14896. doi: 10.1021/bi026469j. [DOI] [PubMed] [Google Scholar]
- 27.Schweers O., Mandelkow E.M., Biernat J., Mandelkow E. Oxidation of cysteine-322 in the repeat domain of microtubule-associated protein tau controls the in vitro assembly of paired helical filaments. Proc Natl Acad Sci U S A. 1995;92:8463–8467. doi: 10.1073/pnas.92.18.8463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.King M.E., Ahuja V., Binder L.I., Kuret J. Ligand-dependent tau filament formation. Implications Alzheimers Dis. 1999;50:14851–14859. doi: 10.1021/bi9911839. [DOI] [PubMed] [Google Scholar]
- 29.Okamura N., Furumoto S., Harada R., Tago T., Yoshikawa T. Novel 18 F-labeled arylquinoline derivatives for noninvasive imaging of tau pathology in Alzheimer disease. J Nucl Med: Off Publ Soc Nucl Med. 2013;54:1420–1427. doi: 10.2967/jnumed.112.117341. [DOI] [PubMed] [Google Scholar]
- 30.Fodero-Tavoletti M.T., Okamura N., Furumoto S., Mulligan R.S., Connor A.R. F-18-THK523: a novel in vivo tau imaging ligand for Alzheimer's disease. Brain. 2011;134:1089–1100. doi: 10.1093/brain/awr038. [DOI] [PubMed] [Google Scholar]
- 31.Gamblin T.C., King M.E., Dawson H., Vitek M.P., Kuret J. In vitro polymerization of tau protein monitored by laser light scattering: method and application to the study of FTDP-17 mutants. Biochemistry. 2000;39:6136–6144. doi: 10.1021/bi000201f. [DOI] [PubMed] [Google Scholar]
- 32.King M.E., Gamblin T.C., Kuret J., Binder L.I. Differential assembly of human tau isoforms in the presence of arachidonic acid. J Neurochem. 2000;74:1749–1757. doi: 10.1046/j.1471-4159.2000.0741749.x. [DOI] [PubMed] [Google Scholar]
- 33.Taniguchi S., Suzuki N., Masuda M., Hisanaga S., Iwatsubo T. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J Biol Chem. 2005;280:7614–7623. doi: 10.1074/jbc.M408714200. [DOI] [PubMed] [Google Scholar]
- 34.Goedert M., Jakes R., Spillantini M.G., Hasegawa M., Smith M.J. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature. 1996;383:550–553. doi: 10.1038/383550a0. [DOI] [PubMed] [Google Scholar]
- 35.Ksiezak‐Reding H., Wall J.S. Characterization of paired helical filaments by scanning transmission electron microscopy. Microsc Res Tech. 2005;67:126–140. doi: 10.1002/jemt.20188. [DOI] [PubMed] [Google Scholar]
- 36.Bulic B., Pickhardt M., Khlistunova I., Biernat J., Mandelkow E.M. Rhodanine-based tau aggregation inhibitors in cell models of tauopathy. Angew Chem. 2007;119:9375–9379. doi: 10.1002/anie.200704051. [DOI] [PubMed] [Google Scholar]
- 37.Friedhoff P., Schneider A., Mandelkow E.-M., Mandelkow E. Rapid assembly of Alzheimer-like paired helical filaments from microtubule-associated protein tau monitored by fluorescence in solution. Biochemistry. 1998;37:10223–10230. doi: 10.1021/bi980537d. [DOI] [PubMed] [Google Scholar]
- 38.Wilson D.M., Bindert L. In vitro evidence for a common effector of pathogenesis in Alzheimer's disease. Vol. 150. 1997. Free fatty acids stimulate the polymerization of tau and amyloid beta peptides; pp. 2181–2195. [PMC free article] [PubMed] [Google Scholar]
- 39.Martin L., Latypova X., Terro F. Post-translational moifications of tau protein: implications for Alzheimer's disease. Neurochem Int. 2001;58(4):458-457. doi: 10.1016/j.neuint.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 40.Friedhoff P., von Bergen M., Mandelkow E.M., Davies P., Mandelkow E. A nucleated assembly mechanism of Alzheimer paired helical filaments. Proc Natl Acad Sci U S A. 1998;95:15712–15717. doi: 10.1073/pnas.95.26.15712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kampers T., Friedhoff P., Biernat J., Mandelkow E.M. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996;399:344–349. doi: 10.1016/s0014-5793(96)01386-5. [DOI] [PubMed] [Google Scholar]
- 42.Mo Z.Y., Zhu Y.Z., Zhu H.L., Fan J.B., Chen J. Low micromolar zinc accelerates the fibrillization of human tau via bridging of Cys-291 and Cys-322. J Biol Chem. 2009;284:34648–34657. doi: 10.1074/jbc.M109.058883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pickhardt M., Gazova Z., von Bergen M., Khlistunova I., Wang Y. Anthraquinones inhibit tau aggregation and dissolve Alzheimer's paired helical filaments in vitro and in cells. J Biol Chem. 2005;280:3628–3635. doi: 10.1074/jbc.M410984200. [DOI] [PubMed] [Google Scholar]
- 44.Tatebayashi Y., Miyasaka T., Chui D.-H., Akagi T., Mishima K.-I. Tau filament formation and associative memory deficit in aged mice expressing mutant (R406W) human tau. Proc Natl Acad Sci. 2002;99:13896–13901. doi: 10.1073/pnas.202205599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wille H., Drewes G., Biernat J., Mandelkow E.M., Mandelkow E. Alzheimer-like paired helical filaments and antiparallel dimers formed from microtubule-associated protein tau in vitro. J Cell Biol. 1992;118:573–584. doi: 10.1083/jcb.118.3.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drewes G., Trinczek B., Illenberger S., Biernat J., Schmitt-Ulms G. Microtubule-associated protein/microtubule affinity-regulating kinase (p110mark). A novel protein kinase that regulates tau–microtubule interactions and dynamic instability by phosphorylation at the Alzheimer-specific site serine 262. J Biol Chem. 1995;270:7679–7688. doi: 10.1074/jbc.270.13.7679. [DOI] [PubMed] [Google Scholar]
- 47.Biernat J., Gustke N., Drewes G., Mandelkow E.M., Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron. 1993;11:153–163. doi: 10.1016/0896-6273(93)90279-z. [DOI] [PubMed] [Google Scholar]
- 48.Mazanetz M.P., Fischer P.M. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat Rev Drug Discov. 2007;6:464–479. doi: 10.1038/nrd2111. [DOI] [PubMed] [Google Scholar]
- 49.Trojanowski J.Q., Lee V.M. Phosphorylation of paired helical filament tau in Alzheimer's disease neurofibrillary lesions: focusing on phosphatases. FASEB J. 1995;9:1570–1576. doi: 10.1096/fasebj.9.15.8529836. [DOI] [PubMed] [Google Scholar]
- 50.Buée L., Bussiere T., Buée-Scherrer V., Delacourte A., Hof P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Rev. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- 51.Hurtado D.E., Molina-Porcel L., Carroll J.C., MacDonald C., Aboagye A.K. Selectively silencing GSK-3 isoforms reduces plaques and tangles in mouse models of Alzheimer's disease. J Neurosci. 2012;32:7392–7402. doi: 10.1523/JNEUROSCI.0889-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hernández F., EGD Barreda, Fuster-Matanzo A., Goni-Oliver P., Lucas J.J. The role of GSK3 in Alzheimer disease. Brain Res Bull. 2009;80:248–250. doi: 10.1016/j.brainresbull.2009.05.017. [DOI] [PubMed] [Google Scholar]
- 53.Reddy P.H. Amyloid beta-induced glycogen synthase kinase 3β phosphorylated VDAC1 in Alzheimer's disease: implications for synaptic dysfunction and neuronal damage. Biochim Biophys Acta (BBA) — Mol Basis Dis. 2013;1832:1913–1921. doi: 10.1016/j.bbadis.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cohen T.J., Guo J.L., Hurtado D.E., Kwong L.K., Mills I.P. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. 2011;2:252. doi: 10.1038/ncomms1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Min S.W., Cho S.H., Zhou Y., Schroeder S., Haroutunian V. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010;67:953–966. doi: 10.1016/j.neuron.2010.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pouplana S., Espargaro A., Galdeano C., Viayna E., Sola I. 2014. Thioflavin-S staining of bacterial inclusion bodies for the fast, simple and inexpensive screening of amyloid aggregation inhibitors. [DOI] [PubMed] [Google Scholar]
- 57.Khlistunova I., Biernat J., Wang Y., Pickhardt M., von Bergen M. Inducible expression of Tau repeat domain in cell models of tauopathy: aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem. 2006;281:1205–1214. doi: 10.1074/jbc.M507753200. [DOI] [PubMed] [Google Scholar]
- 58.Gossen M., Bujard H. Studying gene function in eukaryotes by conditional gene inactivation. Annu Rev Genet. 2002;36:153–173. doi: 10.1146/annurev.genet.36.041002.120114. [DOI] [PubMed] [Google Scholar]
- 59.Bandyopadhyay B., Li G., Yin H., Kuret J. Tau aggregation and toxicity in a cell culture model of tauopathy. J Biol Chem. 2007;282:16454–16464. doi: 10.1074/jbc.M700192200. [DOI] [PubMed] [Google Scholar]
- 60.Guo J.L., Lee V.M.Y. Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. 2011;286:15317–15331. doi: 10.1074/jbc.M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lu M., Kosik K.S. Vol. 12. 2001. Competition for microtubule-binding with dual expression of tau missense and splice isoforms; pp. 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nonaka T., Watanabe S.T., Iwatsubo T., Hasegawa M. Seeded aggregation and toxicity of {alpha}-synuclein and tau: cellular models of neurodegenerative diseases. J Biol Chem. 2010;285:34885–34898. doi: 10.1074/jbc.M110.148460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vogelsberg-Ragaglia V., Bruce J., Richter-Landsberg C., Zhang B., Hong M. Distinct FTDP-17 missense mutations in tau produce tau aggregates and other pathological phenotypes in transfected CHO cells. Mol Biol Cell. 2000;11:4093–4104. doi: 10.1091/mbc.11.12.4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ma Rizzo, Springer G.H., Granada B., Piston D.W. An improved cyan fluorescent protein variant useful for FRET. Nat Biotechnol. 2004;22:445–449. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- 65.Chun W., Johnson G.V.W. Activation of glycogen synthase kinase 3beta promotes the intermolecular association of tau. The use of fluorescence resonance energy transfer microscopy. J Biol Chem. 2007;282:23410–23417. doi: 10.1074/jbc.M703706200. [DOI] [PubMed] [Google Scholar]
- 66.Kfoury N., Holmes B.B., Jiang H., Holtzman D.M., Diamond M.I. Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem. 2012;287:19440–19451. doi: 10.1074/jbc.M112.346072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Outeiro T.F., Putcha P., Tetzlaff J.E., Spoelgen R., Koker M. Formation of toxic oligomeric alpha-synuclein species in living cells. PLoS ONE. 2008;3:e1867. doi: 10.1371/journal.pone.0001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kerppola T.K. Visualization of molecular interactions by fluorescence complementation. Nat Rev Mol Cell Biol. 2006;7:449–456. doi: 10.1038/nrm1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen C.-D., Oh S.-Y., Hinman J.D., Abraham C.R. Visualization of APP dimerization and APP-Notch2 heterodimerization in living cells using bimolecular fluorescence complementation. J Neurochem. 2006;97:30–43. doi: 10.1111/j.1471-4159.2006.03705.x. [DOI] [PubMed] [Google Scholar]
- 70.Chun W., Waldo G.S., Johnson G.V.W. Split GFP complementation assay for quantitative measurement of tau aggregation. In Situ. 2011:109–123. doi: 10.1007/978-1-60761-744-0_9. [DOI] [PubMed] [Google Scholar]
- 71.Chun W., Waldo G.S., Johnson G.V.W. Split GFP complementation assay: a novel approach to quantitatively measure aggregation of tau in situ: effects of GSK3beta activation and caspase 3 cleavage. J Neurochem. 2007;103:2529–2539. doi: 10.1111/j.1471-4159.2007.04941.x. [DOI] [PubMed] [Google Scholar]
- 72.Tak H., Haque M.M., Kim M.J., Lee J.H., Baik J.-H. Bimolecular fluorescence complementation; lighting-up tau–tau interaction in living cells. PloS ONE. 2013;8:e81682. doi: 10.1371/journal.pone.0081682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maruyama M., Shimada H., Suhara T., Shinotoh H., Ji B. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron. 2013;79:1094–1108. doi: 10.1016/j.neuron.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harada R., Okamura N., Furumoto S., Yoshikawa T., Arai H. Use of a benzimidazole derivative BF-188 in fluorescence multispectral imaging for selective visualization of tau protein fibrils in the Alzheimer's disease brain. Mol Imaging Biol. 2014;16:19–27. doi: 10.1007/s11307-013-0667-2. [DOI] [PubMed] [Google Scholar]