

Abstract
Study Objectives:
To determine prevalence and heritability of insomnia during middle/late childhood and adolescence; examine longitudinal associations in insomnia over time; and assess the extent to which genetic and environmental factors on insomnia remain stable, or whether new factors come into play, across this developmental period.
Design:
Longitudinal twin study.
Setting:
Academic medical center.
Patients or Participants:
There were 739 complete monozygotic twin pairs (52%) and 672 complete dizygotic twin pairs (48%) initially enrolled and were followed up at three additional time points (waves). Mode ages at each wave were 8, 10, 14, and 15 y (ages ranged from 8–18 y).
Interventions:
None.
Measurements and Results:
Clinical ratings of insomnia symptoms were assessed using the Child and Adolescent Psychiatric Assessment (CAPA) by trained clinicians, and rated according to Diagnostic and Statistical Manual of Mental Disorders, 3rd Edition—Revised criteria for presence of “clinically significant insomnia,” over four sequential waves. Insomnia symptoms were prevalent but significantly decreased across the four waves (ranging from 16.6% to 31.2%). “Clinically significant insomnia” was moderately heritable at all waves (h2 range = 14% to 38%), and the remaining source of variance was the nonshared environment. Multivariate models indicated that genetic influences at wave 1 contributed to insomnia at all subsequent waves, and that new genetic influences came into play at wave 2, which further contributed to stability of symptoms. Nonshared environmental influences were time-specific.
Conclusion:
Insomnia is prevalent in childhood and adolescence, and is moderately heritable. The progression of insomnia across this developmental time period is influenced by stable as well as new genetic factors that come into play at wave 2 (modal age 10 y). Molecular genetic studies should now identify genes related to insomnia progression during childhood and adolescence.
Citation:
Barclay NL, Gehrman PR, Gregory AM, Eaves LJ, Silberg JL. The heritability of insomnia progression during childhood/adolescence: results from a longitudinal twin study. SLEEP 2015;38(1):109–118.
Keywords: genetics, insomnia, sleep, twins
INTRODUCTION
The transition from childhood to adolescence is accompanied by numerous physiological and social changes. During this time, it is perhaps not surprising that sleep disturbances are common.1 Indeed, prevalence of insomnia symptoms has been estimated to range from 4% to 41% in early childhood and adolescence, depending on sample, age, and mode of assessment.2–8 Insomnia manifests as difficulties initiating or maintaining sleep, early morning awakening, or feeling that the sleep period was non-restorative or unrefreshing, with the sleep problem causing significant distress or impairment.9 Despite our knowledge that insomnia exists in early childhood and adolescence, we know relatively little about its developmental course. In adults it has been demonstrated that when insomnia reaches clinical significance, it is likely to persist over time.10 Longitudinal studies in young children and adolescents have provided mixed results regarding the persistence of insomnia over time. A study of individuals aged 12 to 18 y demonstrated that more than 50% of adolescents who reported insomnia symptoms at baseline continued to exhibit insomnia at 4-y follow-up.11 A similar pattern of symptom persistence was observed over the course of 2 y between the ages of 13 to 15 y.12 Studies of younger children, however, find little degree of persistence of insomnia symptoms,13,14 although one study demonstrated that approximately 60% of children between the ages of 9 and 11 y reported persistent difficulties initiating sleep over 1 y.5 Studies spanning childhood and adolescence are also mixed. Over the course of 5 y, Strauch and colleagues reported some degree of stability of insomnia symptoms from age 10 to 14 y,15 although only 2% exhibited symptoms at all time points. Likewise, Gregory and colleagues observed some stability of sleep disturbance in children aged 4 y who were followed up in midadolescence (r = 0.29), although sleep disturbances largely decreased over time.16 One source of the inconsistencies in persistence rates may be differences in mode of assessment (i.e. parent report versus child report). Regardless of these inconsistencies, it is unequivocal that sleep disturbances in early childhood and adolescence may have detrimental effects on brain development, and long-term physical and mental health, given the role of sleep in synaptic homeostasis,17 brain plasticity,18 brain maturation,19 and immune function.20 For these reasons, it is important to understand factors contributing to insomnia and its potential persistence over time in early childhood and adolescence.
Accumulating evidence from large-scale twin datasets points to the possibility that, in adults, insomnia is to some extent heritable, with genetic factors accounting for approximately 30–60% of variability.21 Twin studies in early childhood and adolescence, however, have largely focused on broadly defined sleep disturbances rather than specifically investigating the heritability of insomnia per se. For example, Van den Oord and colleagues estimated that genetic influences contributed approximately 60% of variance in sleep disturbances assessed by the Child Behavior Checklist (CBCL) in 3-y-old twins.22 Gregory and colleagues have repeatedly demonstrated the heritability of broadly defined sleep disturbances in early childhood, ranging from 18–20% in 3- to 4-y-old twins,23 to ∼60–70% at 8–10 y of age.24–26 Studies of adolescents report heritability estimates more akin to adult estimates.27–30
Although these studies identify the presence of genetic factors on sleep disturbances during discrete time points, they tell us little about its developmental course. Longitudinal genetically informative designs allow us to examine the extent to which genetic and environmental factors contribute to the associations in a phenotype over time, as well as examining the extent of stability (overlap) and change in the contribution of such influences. Using such methodology, Gregory and colleagues reported that the association between sleep disturbances at 8 and 10 y of age share some genetic overlap (46% shared genetic effects).26 Although this suggests some degree of stability in the genetic influences on sleep disturbances in this age group, it also suggests that new genetic influences come into play at 10 y.
Longitudinal twin studies mapping the developmental course of insomnia from early childhood through adolescence are lacking. Because of this, the question of whether genetic and environmental factors contributing to insomnia in early childhood and adolescence remain stable over time, or whether new etiological factors come into play during this period, remains unknown. It is possible that specific genes contribute to the initial onset of insomnia. However, it is also possible that different genes are partially responsible for its maintenance, given the many physiological and social changes that occur from the transition from early childhood to adolescence through puberty, including changes in the organization of the circadian system, as well as sleep timing, quality, and architecture.1 Moreover, pubertal development has been associated with increases in sleep disturbances,31 making it likely that genetic factors controlling puberty contribute to sleep disturbances occurring during this developmental age. Examining the extent of stability and change in the genetic and environmental influences on insomnia over time will allow us to further progress toward identifying specific genetic mechanisms underlying insomnia. In addition, such examination will enable us to identify specific environmental factors contributing to insomnia, given that they may be time specific.
With these considerations in mind, the objectives of the current study were to determine the prevalence and heritability of insomnia symptoms, including difficulties initiating sleep, maintaining sleep and early morning awakenings, across four time points spanning the period of middle/late childhood to adolescence in a longitudinal sequential sample of twins aged 8–18 y. Further, this study examined the longitudinal associations in insomnia over time, and assessed the extent to which genetic and environmental factors on insomnia remained stable, or whether new factors came into play, across this developmental time period.
METHODS
Participants
The data for this study are derived from the Virginia Twin Study of Adolescent Behavioral Development (VTSABD), a longitudinal sequential cohort of 8- to 17-y-old Caucasian twins born between 1974–1983 focused on developmental trajectories of adolescent psychopathology and associated risk factors,32,33 as well as the Young Adult Follow-Up (YAFU) study,34 of the same twins when they were 18 y of age. Twin pairs were identified through the state school system and participating private schools in the state of Virginia in 1989–1990 and were then contacted by mail. Interested families were scheduled for detailed assessments of behavioral development and psychopathology, and were invited to participate in up to two comprehensive interview-based follow-up assessments. At wave 1, 1,412 twin families participated (2,822 individual twins aged 8–18 y). At wave 2, which took place on average 1.52 y following wave 1, 1,047 families participated whose children continued to meet age and residence requirements for the study (80% of those targeted). At wave 3, which took place on average 3.3 years following wave 2, 628 families participated (81% of those targeted). All twins who participated at wave 1 were recontacted as young adults (when aged 18 y or older) to participate in a telephone interview as part of the YAFU study (termed wave 4 in the current analyses). Wave 4 took place over a variable number of years from the study's conception as participants were contacted when they reached 18 y of age. At wave 4, 1,185 twin families participated (84% of those targeted). Twenty-four percent of those who participated in the YAFU participated in only the first wave of the VTSABD, 32% participated in two waves, 31% in three waves, and 13% in all four waves.35 Participating families were representative of the Virginia population in terms of socioeconomic status.36 More details of sample ascertainment, participation rates, ages of assessment, and socioeconomic bias for the four waves of the study have been reported elsewhere.32,35–37 Because this is a sequential longitudinal cohort that contains individuals spanning the ages of 8 to 18 y at all waves, results are interpreted in terms of the progression across time, rather than differences between discrete age groups. Every family provided signed consent forms, which were completed by parents when twins were younger than 14 y, and by the twins themselves when aged 14 y or older. Ethical approval was granted by the Institutional Review Board at Virginia Commonwealth University, consistent with US federal guidelines.
Measures
Child and Adolescent Psychiatric Assessment
Insomnia symptoms were assessed by the Child and Adolescent Psychiatric Assessment (CAPA), a semistructured interview designed to assess a number of behavioral and psychological symptoms based on the Diagnostic and Statistical Manual of Mental Disorders, 3rd Edition—Revised (DSM-III-R),38 as this was the current diagnostic manual in use at the time of data collection. The modules for sleep problems by child/adolescent report were used for the current analyses. The DSM-III-R criteria for insomnia vary to some extent to the current criteria for insomnia disorder set forth in DSM-5,9 and consist of (1) difficulty initiating or maintaining sleep, or nonrestorative sleep; (2) sleep difficulty that occurs three or more times per week for at least 1 mo, and (3) clinically significant distress or impairment. The sleep module of the CAPA interview taps into these criteria, although it is more aligned to DSM-5 in terms of duration (i.e., it focuses on a period of 3 mo), and includes a series of questions about the child's/adolescent's current sleep patterns, including whether the child has difficulty falling asleep or waking up too early in the morning, and then makes a clinical judgment of whether or not “clinically significant insomnia” symptoms are present. In each area, the presence of symptoms over the past 3 mo was ascertained, along with the frequency of occurrence, duration, and earliest age of onset (if symptoms were present). For all questions, a rating of 0 was used if it was determined that a disorder was not present. A rating of 2 indicated that the disorder was present at least at the minimum level of severity (if the insomnia covers a period between 1 and 2 h), and a rating of 3 that the disorder was present at a higher level of severity (if the insomnia duration was greater than or equal to 2 h per night). A rating of 1 was discouraged because it indicated that the rater was not able to determine whether criteria were met, in which case the rater was supposed to continue to query the respondent until a determination could be made. For these analyses, ratings of 2 and 3 were combined to create a dichotomous (yes/no) insomnia rating, henceforth referred to as “clinically significant insomnia.” For descriptive purposes, additional ratings of the timing of insomnia during the night (difficulty initiating sleep [initial insomnia], difficulty maintaining sleep [middle insomnia], or early morning awakening [late insomnia]), and the presence of any insomnia symptom were examined in the current analyses. These ratings were repeated within the same sample at four time points (waves) from age 8–18 y (only participants aged 18 y or younger were retained in the study at each wave).
Zygosity
Zygosity was inferred using an algorithm that incorporates data from parental responses to a questionnaire and ratings of photographs, and validated in a subset of 231 twin pairs for whom zygosity was confirmed by blood group typing or DNA polymorphisms. Additional details of zygosity determination in this sample have been published previously.30,32
Data Analyses
Descriptive statistics and tetrachoric correlations between waves were computed. Significant differences in the proportion of dichotomous insomnia symptom variables across waves were tested using χ2 tests. Similarly, significant differences between males and females on dichotomous insomnia symptom variables were tested using χ2 tests. Differences in age between cases of insomnia symptoms versus no symptoms at each wave were computed using t-tests. Twin model fitting was performed in Mx (computer software designed to analyse genetically informative designs) using structural equation modeling and the method of maximum likelihood estimation,39 on “clinically significant insomnia.” Twin studies allow us to estimate the relative contribution of genetic and environmental influences upon traits by comparing the similarity between monozygotic (MZ) twins, who share almost 100% of their genetic material, and dizygotic (DZ) twins, who share on average 50% of their segregating genes. Using this information it is possible to parse the variance in a phenotype into additive genetic influences (A), dominant (nonadditive/interactive) genetic influences (D), shared/common environmental influences (C) (which act so as to make family members more similar), and nonshared environmental influences (E) (unique environmental influences that contribute to dissimilarity between family members).27 It is not possible to examine D and C simultaneously because they predict different MZ:DZ correlation ratios, which are confounded if examined together.40 Accordingly, it is typical to examine separate ACE and ADE models if data suggest that nonadditive genetic effects may be likely. Nonadditive genetic effects are implied if MZ twin correlations are greater than double DZ twin correlations.
Because of the categorical nature of the variables, liability threshold models were used, which assume an underlying normal distribution to the categories, with thresholds that discriminate the classes (0, 1), estimated from the relative cell proportions of the data. Initially, univariate models were run to investigate the relative contribution of genetic and environmental influences on “clinically significant insomnia” at each wave. The fit statistic provided by Mx for raw data modeling is −2LL (minus twice the log likelihood of the observations). Saturated models, which provide a perfect fit to the data, were first approximated to the data, and the resulting -2LL was then subtracted from the -2LL of the genetic models. The difference between the -2LL for the saturated and genetic models is χ2 distributed with equal df and so provides a relative fit index. A nonsignificant difference in fit between the genetic and saturated models indicates that the genetic model does not fit the data significantly worse than the saturated model, thus providing a good description of the data. Akaike's Information Criterion (AIC) also provides information regarding fit (calculated as Δχ2 – 2 × Δdf), which accounts for the number of parameters being estimated and the goodness-of-fit. A good fit is indicated by lower, negative values of AIC.41 Both ACE and ADE models were tested as the pattern of twin correlations suggested possible nonadditive genetic effects, followed by more restricted models where one of the parameters was removed (i.e. the AE, DE, and CE models were run), and compared to the fuller models to determine their significance. For the best-fitting models (those that were the most parsimonious, that did not significantly deviate from the fit of the saturated model), likelihood-based 95% confidence intervals (CIs) on the parameter estimates were obtained in order to determine their precision.
Following the univariate analyses, multivariate Cholesky genetic models40 were used to model “clinically significant insomnia” at all four waves simultaneously. This model allows us to test the etiological specificity across the four waves. This model decomposes the variances and covariances between the phenotypes into latent common (shared between the phenotypes) and unique (specific to each phenotype) genetic and environmental components (see Figure 1 for an example of a DE model – A was dropped for simplicity of presentation). This model provides us with four pieces of information. First, it indicates the genetic and environmental influences common to “clinically significant insomnia” at all four waves (D1, E1). Second, it indicates whether a second set of genetic and environmental influences come into play that are common to the second, third, and fourth waves (D2, E2). Third, it indicates whether a third set of influences are common to the third and fourth waves (D3, E3). Finally it indicates whether a unique set of influences contribute to the fourth wave (D4, E4). In all cases, the model allows the estimation of unique genetic factors, indicated by the significance of the diagonal elements (e.g. d11, d22, d33, d44). If there are common genetic factors influencing more than one wave, the off-diagonal parameter estimates would be significantly distinguishable from zero (e.g., d21, d31, d41, d32, etc.). The same logic applies to the environmental factors. Each of the parameter estimates can be squared to estimate the proportion of the variance at each wave accounted for by the genetic and environmental factors. As with the univariate analyses, the fit of the full model was compared to more restricted models that sequentially dropped individual parameter estimates. Both ACE and ADE models were tested. The most parsimonious model that did not fit significantly worse than the saturated model was selected for interpretation. Sex differences in the etiological influences in both univariate and multivariate models were not computed given the small cell sizes by sex/zygosity group for later waves (see Table 2). The analyses are performed on raw data.
Figure 1.

Multivariate Cholesky decomposition with parameter estimates (95% confidence interval) from best-fitting DE model. Figure is shown for one twin only. D = nonadditive genetic influence; E = nonshared environmental influence. Figure displays unsquared parameter estimates for significant paths. The parameter estimates can be squared to indicate relative proportions of variance (%). The extent to which genetic influences account for the phenotypic correlations between variables can be calculated as follows: (d11*d21)/r(wave 1 and wave 2); (d11*d31)/r(wave 1 and wave 3); (d11*d41)/r(wave 1 and wave 4); (d21*d31) + (d22*d32)/r(wave 2 and wave 3); (d21*d41) + (d22*d42)/r(wave 2 and wave 4); (d31*d41) + (d32*d42) + (d33*d43)/r(wave 3 and wave 4). The same principles apply for calculating the relative proportions of variance accounted for by non-shared environmental influences.
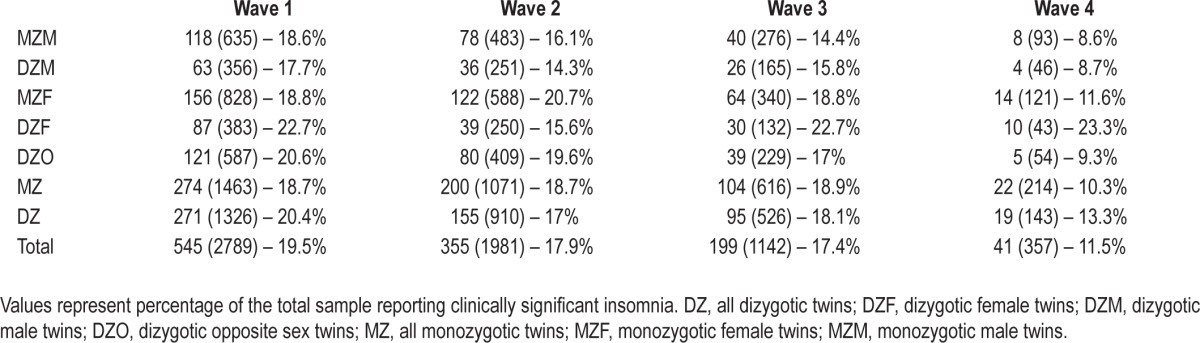
Table 2.
Frequencies of self-reported “clinically significant insomnia” by zygosity (total n of those with and without insomnia in parentheses).
RESULTS
Descriptives
A total of 1,412 complete twin pairs participated in the study at wave 1. Overall, there were 46% males and 54% females at wave 1. Zygosity was available from 1,411 twin pairs. The sex/ zygosity groups for the twin pairs were as follows: 322 MZ male (MZM), 417 MZ females (MZF), 180 DZ males (DZM), 194 DZ females (DZF), and 298 DZ opposite sex pairs (DZO). The modal ages at the different waves were as follows: 8.3 y (range, 8–18 y) at wave 1; 10.7 y (range, 9–18 y) at wave 2; 14.2 y (range, 12–18 y) at wave 3; and 15.3 y (range, 14–18 years) at wave 4. Age spread at each wave was largely homogenously distributed (e.g., although age 8.3 y was most common at wave 1, there was a relatively similar distribution of other ages within this wave). Complete data on all sleep variables were available from 2,789 individuals at wave 1 (98.8% of those targeted); 1,981 (94.6% of those targeted) at wave 2; 1,142 (90.9% of those targeted) at wave 3; and 357 (15.1% of those targeted) at wave 4. In total, 325 individuals provided complete sleep data at all four waves. There did not appear to be significant differences in insomnia ratings at wave 1 among those who did and did not participate at all four waves (χ2[1] = 0.15, P = 0.70), suggesting no evidence of selective attrition. Retrospective reports from parents indicated that MZ twins were significantly more likely than DZ twins to share a bedroom with their co-twin in young childhood (99% of MZ twins shared a bedroom always, usually or sometimes; compared to 94% of DZs: χ2[3] = 143.66, P = 0.00) as well as at the time of initial assessment (64% of MZ twins shared a bedroom always, usually, or sometimes; compared to 35% of DZs: χ2[3] = 117.61, P = 0.00). Whether or not twins shared a bedroom did not reliably contribute to twin similarity on our measure of clinically significant insomnia (analyses available upon request from the first author).
Frequency of insomnia symptoms for the total sample and categorized by sex are shown in Table 1. The proportion of individuals meeting criteria for a rating of “clinically significant insomnia” based on child/adolescent ratings significantly decreased across all waves, from 19.5% at wave 1 to 11.5% at wave 4 (overall: χ2[3] = 14.58, P = 0.00; all χ2 individually comparing waves 1–3 versus wave 4: P < 0.05). Significance of the decrease in the proportion of individual insomnia symptoms across waves is shown in Table 1.
Table 1.
Prevalence of child/adolescent reported insomnia symptoms (n cases in parentheses).
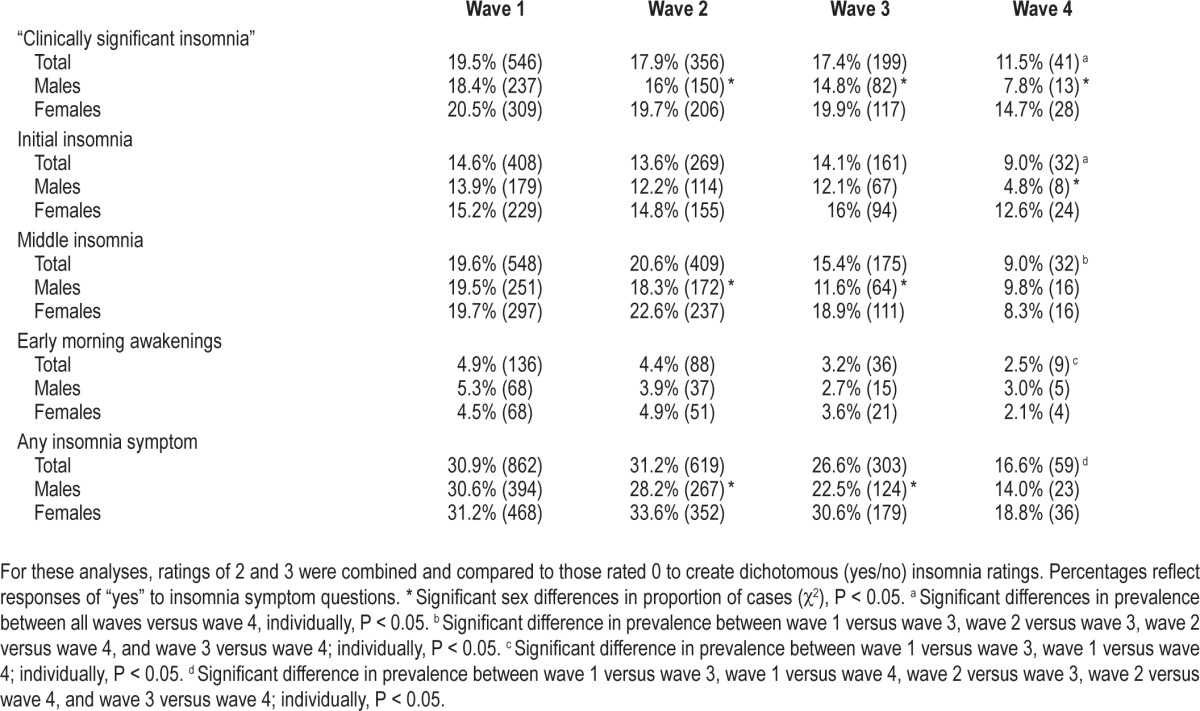
At wave 2, there were significant sex differences in ratings of “clinically significant insomnia” (χ2[1] = 4.43, P = 0.04), middle insomnia (χ2[1] = 5.54, P = 0.02), and the presence of any insomnia symptom (χ2[1] = 6.06, P = 0.02). At wave 3, there were significant sex differences in ratings of “clinically significant insomnia” (χ2[1] = 5.15, P = 0.02), middle insomnia (χ2[1] = 11.71, P = 0.001), and the presence of any insomnia symptom (χ2[1] = 9.61, P = 0.00). At wave 4, there were significant sex differences in ratings of “clinically significant insomnia” (χ2[1] = 4.07, P = 0.05), and initial insomnia (χ2[1] = 6.44, P = 0.02). In all cases, insomnia symptoms were more prevalent in females than males. There were no other significant sex differences in insomnia variables at all waves.
At wave 1 there were significant age differences in the presence of middle insomnia (t[2792] = 3.82, P = 0.00). At wave 2 there were significant age differences in the presence of middle insomnia (t[1984] = 2.53, P = 0.01) and any insomnia symptom (t[1984] = 2.38, P = 0.02). At wave 3 there were significant age differences in the presence of middle insomnia (t[1135] = 2.46, P = 0.01). In all cases, younger children were more likely than older children to experience insomnia symptoms at the same wave.
Frequency of cases (individuals categorized as “yes” for ratings of “clinically significant insomnia” present) split by sex and zygosity is shown in Table 2. Because of the small number of cases in each sex/zygosity group at each wave, it was not possible to perform genetic model fitting analyses by sex. As such, genetic model fitting analyses are performed for the total sample only.
Twin Correlations
Twin correlations for ratings of “clinically significant insomnia” at different waves are shown in Table 3A. MZ twin correlations were greater than corresponding DZ twin correlations for “clinically significant insomnia” at all waves, suggesting the influence of genetics on this phenotype. Because the MZ twin correlations were greater than double the DZ twin correlations, nonadditive genetic effects were implied and so ADE model fitting analyses were performed in addition to ACE models.
Table 3A.
Cross twin correlations (and 95% confidence intervals) for ratings of “clinically significant insomnia.”

Phenotypic Correlations
Tetrachoric correlations for ratings of “clinically significant insomnia” across waves are shown in Table 3B. There were significant associations between insomnia symptoms at adjacent waves (wave 1 with 2; wave 2 with 3; and wave 3 with 4) but not at nonadjacent waves (e.g., wave 1 with wave 3 or 4).
Table 3B.
Phenotypic correlations (and 95% confidence intervals) for ratings of “clinically significant insomnia.”

Cross-Twin Cross-Trait Correlations
Cross-twin cross-trait correlations (shown in Table 3C) were only significant for MZ twins on the association between ratings of “clinically significant insomnia” at waves 1 and 2. Despite the nonsignificance of the other associations, in all cases MZ twin correlations were greater than DZ correlations, suggesting possible genetic effects on the cross-wave associations.
Table 3C.
Cross-twin cross-trait correlations (and 95% confidence intervals) for ratings of “clinically significant insomnia” (MZ below diagonal, DZ above diagonal).

Univariate Genetic Model Fitting Analyses
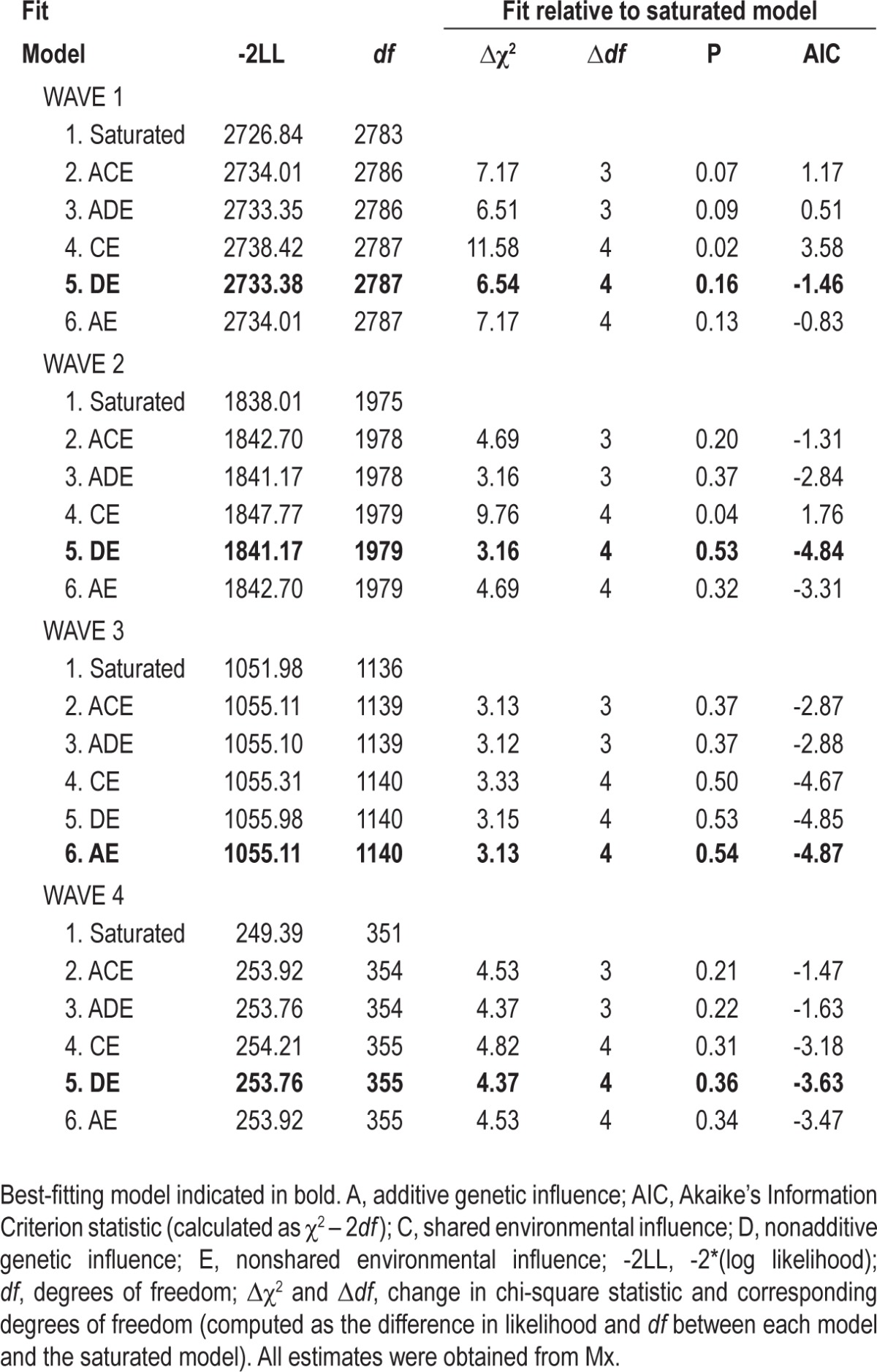
Model fitting analyses are shown in Table 4. At waves 1, 2, and 4, the best-fitting models were ones in which additive genetic influences were dropped, and shared environmental influences were replaced with nonadditive genetic effects (DE models). In these models, nonadditive genetic effects contributed 33%[95% CI 0.20–0.46], 38%[0.22–0.78] and 24%[0.00–0.66] of the total variance at waves 1, 2 and 4, respectively. At wave 3, the best-fitting model was one in which shared environmental influences were dropped (AE model). In this model, additive genetic influences contributed 14%[0.00–0.35] of the variance in ratings of “clinically significant insomnia.” At all waves, the remaining source of variance was the nonshared environment (accounting for 67%[0.54–0.80], 62%[0.48–0.78], 86%[0.65–1.00], and 76%[0.34–1.00] of variance explained for waves 1, 2, 3, and 4, respectively).
Table 4.
Fit statistics for univariate genetic model fitting analyses for ratings of “clinically significant insomnia.”
Multivariate Cholesky Model Fitting Analyses
For the fullest multivariate Cholesky models, an ADE model provided a better fit to the data than an ACE model (as indicated by lower AIC). In addition, a DE model including all four Cholesky factors provided a better fit to the data than the saturated ADE model (Δχ2(10) = 2.04, P = 0.99, ΔAIC = -17.96). Subsequent models were then run to test the significance of each of the Cholesky parameters. Dropping the unique genetic factor at wave 4 did not result in a significant loss of model fit (Δχ2(1) = 0.00, P = 1.00, ΔAIC = -2.00). Additionally dropping the genetic factors at wave 3 (both unique and shared with wave 4) did not result in a significant loss of model fit (Δχ2(3) = 0.56, P = 0.91, ΔAIC = -5.44). Further dropping the genetic factors at wave 2 (both unique and shared with subsequent waves) did not significantly reduce model fit (Δχ2(6) = 8.98, P = 0.17, ΔAIC = -3.02), but examination of the ΔAIC value indicated that the best-fitting model was the previous model, which allowed genetic factors from waves 1 and 2 to map onto subsequent waves. Removal of any of the non-shared environmental factors significantly reduced model fit in all cases (all P < 0.05). Standardized path coefficients for each of the significant paths from the best-fitting model are shown in Figure 1.
DISCUSSION
This set of analyses sought to determine the extent to which genetic and environmental influences on insomnia are stable across childhood and adolescence. Our analyses focus on data from a sequential sample of twins followed up across time, with time points representative of children and adolescents aged 8, 10, 14, and 15 y across the four waves. There are four noteworthy findings from this research. First, prevalence of “clinically significant insomnia” was relatively high compared to expected figures for adulthood in middle/late childhood, but significantly decreased to levels in line with adults by adolescence. Similarly, individual insomnia symptoms (initial insomnia, middle insomnia, and early morning awakening) significantly decreased across waves. This decrease in insomnia symptoms by adolescence is consistent with a study demonstrating a decrease in broadly defined sleeping difficulties in children age 4 y though adolescence.16 Another study documented a decrease in insomnia symptoms (specifically difficulties initiating sleep) from age 10 to 13 y in a longitudinal study of more than 1,000 children.42 The current study extends this previous work by demonstrating the continued decrease in insomnia symptoms throughout adolescence. One possibility for this greater proportion of insomnia symptoms in younger children in comparison with adolescents could be that insomnia symptoms in younger children may be largely behavioral in nature (i.e., behavioral insomnia of childhood), and stem from poor sleep hygiene and inappropriate associations of the bedroom environment with wakefulness (i.e., children often use their bedrooms for play), which may cease by adolescence. Alternatively, it is possible that as parents often set bedtimes in younger children, timing of sleep does not coincide with the child's feelings of tiredness or their optimal time for sleep onset as governed by that child's circadian rhythm. As a consequence, the child may lie awake, unable to sleep for periods of time. During adolescence, parents may be less stringent about bedtimes, allowing their children to go to bed at times more in line with their circadian rhythm, and as such adolescents may experience fewer sleep difficulties if they attempt sleep at times in line with circadian rhythmicity. The decrease in sleep disturbances may also reflect changes in maturation and sleep architecture, which occur during this time.43
Second, there were associations between “clinically significant insomnia” between adjacent waves, suggesting that within childhood and adolescence, insomnia persists, but that continuity of symptoms across time within childhood and adolescence is minimal. This is also reflected by the smaller phenotypic correlation between waves 2 and 3 (the mode ages of which span these developmental time periods), in comparison to the phenotypic correlations between waves 1 and 2; and 3 and 4. This, again, reflects the possible changes in sleep that occur during the transition from childhood to adolescence.
Third, genetic factors contributed to ratings of “clinically significant insomnia” at all waves, from 33%, 38%, 14%, and 24% in waves 1, 2, 3, and 4, respectively. The genetic estimates are in line with estimates we would expect in adults21 in our sample at wave 1 and wave 2; yet are somewhat lower in our sample at later waves. This highlights the greater importance of the nonshared environment during adolescence in comparison with that in younger children, in whom a host of environmental and social changes are likely to take place, which may consequently interfere with sleep. Interestingly, our results highlight the contribution of nonadditive genetic effects at waves 1, 2, and 4, providing us with insight into the possible genetic mechanisms at play. However, the greater within-pair correlations in MZ twins compared with those of DZ twins could suggest an alternative explanation. Such a pattern of results could suggest the presence of sibling interaction, where one twin's behavior affects that of the co-twin, rather than nonadditive genetic effects.32,44 This seems plausible in the context of sleep, as the sleep behavior of one twin may similarly influence that of the co-twin if twins share a bedroom. Indeed, in the current sample, MZ twins were significantly more likely than DZ twins to share a bedroom with their co-twin in young childhood as well as at the time of assessment. That said, evidence of sibling interaction also requires greater variance in DZ twins as in comparison with MZ twins for the phenotypes of interest (i.e., insomnia). In the current sample, variances for MZ twins were comparable to those for DZ twins at each wave (unreported, but available upon request from the first author), making the pattern of results more consistent with an interpretation based on nonadditive genetic effects rather than sibling interaction. Although there is statistical support for a nonadditive component, our sample size has limited power to resolve the reduction in DZ correlations because of nonadditive effects from that caused by the effects of sibling interaction.
Fourth, evaluation of the multivariate model indicated that genetic factors influencing insomnia at wave 1 contribute to the maintenance of insomnia through adolescence. This is consistent with a study demonstrating that the stability of sleep difficulties from age 8 to 10 y was to the result of shared genetic effects.26 Additionally, new genetic influences come into play at wave 2, which further contribute to the maintenance of insomnia through adolescence. In contrast, no new genetic influences come into play at waves 3 and 4. It is likely that genes controlling the sleep-wake system are implicated in insomnia (such as those controlling the regulation of the sleep-wake switch, including the activity of acetylcholine, glutamate, gamma-aminobutyric acid (GABA), and the monoamines). Indeed, molecular genetic studies in adults have demonstrated associations between several of such genes and insomnia or poor sleep quality, including the serotonin transporter gene (5HTTLPR),45–48 monoamine oxidase-A,49 and GABA,50 among others.51 Other candidates may be genes implicated in the control of the circadian clock. Indeed, a polymorphism of the CLOCK 3111T/C polymorphism has been associated with insomnia in a sample of patients with major depression,52 although results are mixed.45,53 However, most molecular genetic studies focus on variation in normal sleep characteristics, or are speculative studies on sleep phenotypes in Drosophila, rather than focusing on clinically significant insomnia. Although this handful of studies provide clues as to the likely genes involved, further studies specifically focusing on insomnia populations are required. Furthermore, molecular genetic studies in childhood and adolescence will allow us to determine whether the same genetic pathways are involved in symptoms during these developmental periods as in adulthood.
The stability of genetic effects from wave 2 through wave 4 implies that the same set of genetic factors may contribute to insomnia over this time period. Studies spanning adolescence and adulthood are now required to chart the stability of genetic effects over longer time frames. This will enable us to determine whether insomnia in early childhood, adolescence, and adulthood stem from the same genetic pathways, and whether they are, genetically speaking, similar phenotypes.
In addition to understanding the genetic mechanisms involved, the current study allows us to make inferences about the role of the environment. In univariate models, nonshared environmental influences accounted for the majority of variance in insomnia. In the multivariate model, only time-specific nonshared environmental influences were significant (with the exception of a small amount of overlapping nonshared environmental factors between waves 3 and 4), suggesting little overlap in the environmental influences contributing to insomnia. This finding suggests that environmental factors have only a transient effect on sleep, rather than contributing to sleep over time. This is in line with Spielman's “3P” model of insomnia, which proposes that “precipitating” factors (which may include environmental factors such as stressful life events) act as a trigger for the onset of insomnia in individuals with a predisposition to insomnia (such as genetic vulnerability); yet the maintenance of insomnia is influenced by distinct “perpetuating” factors after the precipitating factor has been surpassed.54
Despite these findings, this study has several limitations. First, although a strength of this study, these results reflect a sequential longitudinal cohort that contains individuals spanning the ages of 8 to 18 y at all waves; therefore, the results must be interpreted in terms of changes across time, rather than specifically focusing on discrete age groups. However, in each of the waves, particular ages were more common, and the sample mostly represents children and adolescents aged 8, 10, 14, and 15 y across the four waves. Although it would be theoretically possible to perform analyses based on discrete age groups irrespective of wave, the small sample size in the latter age groups would provide limited power to meaningfully report on age-related changes in the etiological influences. Second, the data are subjective in nature rather than measures of objective sleep difficulties. That said, insomnia is considered a subjective complaint, as clinical diagnosis is based purely on subjective measures,9 and it is often the case that individuals with insomnia exhibit no objectively recorded sleep deficit despite the subjective dissatisfaction with sleep quality or quantity.55 Accordingly, measuring insomnia by subjective methods appears most appropriate. Third, and on a related note, it is possible that our insomnia measures are confounded by traits that are typically associated with insomnia, such as depression and neuroticism.56,57 This would mean that our estimates of heritability, rather than purely reflecting insomnia, may to some extent reflect an underlying mood or personality disorder. In order to address these potential confounds, we additionally examined point biserial correlations between each of our insomnia variables and depression (measured using the Mood and Feelings Questionnaire [MFQ]58) and neuroticism (measured using the Emotionality, Activity, Sociability, and Impulsivity Temperament Survey [EASI-III]59) at waves 1–3 (as data from the MFQ and EASI were only available at theses waves; analyses available upon request from the first author). Although all of these correlations (with the exception of two) were significant, all were small (ranging between r = 0.05–0.26), suggesting minimal overlap between our insomnia variables, depression, and neuroticism. Accordingly, we can be confident that our estimates of heritability reflect sources of variance attributable to insomnia, to some extent independent of these potential confounds (we acknowledge that the best method to control for these potential confounds would be to regress out the effects of depression/neuroticism from our insomnia variables and examine the resulting change in A, C, and E; however, because these data were available only from waves 1–3 of the study, we are unable to treat the data equally across the four waves). Fourth, the current analyses are based on self-report responses from the CAPA interview rather than parent-reported symptoms. Although the accuracy of self-report in young children could be questioned, one study demonstrated that children as young as 8 y are able to report on their own symptoms.60 Other studies in young children largely focus on parent-reported symptoms, and so comparison with these studies should take this point into consideration. However, studies specifically comparing parent- and child-reported symptoms typically find that parents underestimate sleep disturbances in their children.61 Indeed, a previous paper reporting on insomnia symptoms from wave 1 of the current sample also demonstrate this pattern.30 Similarly, a study comparing adolescent- and parent-reported sleep patterns with actigraphy over the course of 1 w demonstrated little concordance between raters.62 Adolescents were more accurate at reporting on their sleep than were their parents. The general trend for parents to become progressively more inaccurate at reporting on their offsprings' sleep is likely because of their lack of awareness of the childrens' nighttime behavior. The current analyses may be the best representation of the sleep of these individuals. Finally, the small sample size in later waves meant that it was not possible to examine sex differences in the etiology of insomnia over time. Given that insomnia is typically more prevalent in females63 (a pattern that was also mirrored in the current data), it is possible that different mechanisms are at play between the sexes. Further investigation of sex specific genetic effects is warranted.
In conclusion, these findings contribute to our knowledge of the prevalence of insomnia symptoms and factors influencing insomnia in middle/late childhood through to adolescence. Insomnia symptoms were more prevalent in younger children, decreasing to estimates akin to those typically observed in adults, by adolescence. “Clinically significant insomnia” (as rated by clinicians) was moderately heritable at all waves, and is in line with heritability observed in adulthood in younger children, but somewhat lower during adolescence. At all waves the remaining source of variance was the nonshared environment, with no influence of family-wide (shared environmental) factors. Genetic influences on “clinically significant insomnia” showed a substantial degree of stability from wave 1 through wave 4, with new genetic factors coming into play at wave 2. Molecular genetic studies of childhood and adolescent insomnia are now required in order to determine the mechanism through which insomnia manifests and is maintained through these developmental periods. Such knowledge will provide us with clues as to biological mechanisms involved, and could facilitate the development of pharmaceutical treatments to target these pathways.
DISCLOSURE STATEMENT
This was not an industry supported study. This work was supported by the Mid-Atlantic Twin Registry and Award No. UL1TR000058 from the National Institutes of Health's National Center for Advancing Translational Science. We also acknowledge the support of the Virginia Retirement System and the US Department of Social Security. Data analyses were performed at Northumbria University, UK. Data was collected at the Virginia Commonwealth University School of Medicine, Richmond, VA. Dr. Gehrman has received research grant support from Merck. The other authors have indicated no financial conflicts of interest.
REFERENCES
- 1.Barclay NL, Gregory AM. Sleep in childhood and adolescence: agespecific sleep characteristics, common sleep disturbances and associated difficulties. In: Andersen SL, Pine DS, editors. The Neurobiology of Childhood. Berlin, Germany: Springer Berlin Heidelberg; 2013. [Google Scholar]
- 2.Camhi SL, Morgan WJ, Pernisco N, Quan SF. Factors affecting sleep disturbances in children and adolescents. Sleep Med. 2000;1:117–23. doi: 10.1016/s1389-9457(99)00005-2. [DOI] [PubMed] [Google Scholar]
- 3.Archbold KH, Pituch KJ, Panahi P, Chervin RD. Symptoms of sleep disturbances among children at two general pediatric clinics. J Pediatr. 2002;140:97–102. doi: 10.1067/mpd.2002.119990. [DOI] [PubMed] [Google Scholar]
- 4.Zhang JH, Li AM, Kong APS, Lai KYC, Tang NLS, Wing YK. A community-based study of insomnia in Hong Kong Chinese children: prevalence, risk factoes and familial aggregation. Sleep Med. 2009;10:1040–6. doi: 10.1016/j.sleep.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 5.Fricke-Oerkermann L, Pluck J, Schredl M, et al. Prevalence and course of sleep problems in childhood. Sleep. 2007;30:1371–7. doi: 10.1093/sleep/30.10.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson EO, Roth T, Schultz L, Breslau N. Epidemiology of DSM-IV insomnia in adolescence: lifetime prevalence, chronicity, and an emergent gender difference. Pediatrics. 2006;117:E247–E56. doi: 10.1542/peds.2004-2629. [DOI] [PubMed] [Google Scholar]
- 7.Liu XC, Uchiyama M, Okawa M, Kurita H. Prevalence and correlates of self-reported sleep problems among Chinese adolescents. Sleep. 2000;23:27–34. [PubMed] [Google Scholar]
- 8.Calhoun SL, Fernandez-Mendoza J, Vgontzas AN, Liao D, Bixler EO. Prevalence of insomnia symptoms in a general population sample of young children and preadolescents: gender effects. Sleep Med. 2014;15:91–5. doi: 10.1016/j.sleep.2013.08.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.American Psychiatric Association. 5th ed. Arlington, VA: American Psychiatric Publishing; 2013. Diagnostic and Statistical Manual of Mental Disorders. [Google Scholar]
- 10.Morin CM, Belanger L, LeBlanc M, et al. The natural history of insomnia. A population-based 3-year longitudinal study. Arch Intern Med. 2009;169:447–53. doi: 10.1001/archinternmed.2008.610. [DOI] [PubMed] [Google Scholar]
- 11.Patten CA, Choi WS, Gillin JC, Pierce JP. Depressive symptoms and cigarette smoking predict development and persistence of sleep problems in US adolescents. Pediatrics. 2000;106:E23. doi: 10.1542/peds.106.2.e23. [DOI] [PubMed] [Google Scholar]
- 12.Morrison DN, McGee R, Stanton WR. Sleep problems in adolescence. J Am Acad Child Adolesc Psychiatry. 1992;31:94–9. doi: 10.1097/00004583-199201000-00014. [DOI] [PubMed] [Google Scholar]
- 13.Jenni OG, Fuhrer HZ, Iglowstein I, Molinari L, Largo RH. A longitudinal study of bed sharing and sleep problems among Swiss children in the first 10 years of life. Pediatrics. 2005;115:233–40. doi: 10.1542/peds.2004-0815E. [DOI] [PubMed] [Google Scholar]
- 14.Clarkson S, Williams S, Silva PA. Sleep in middle childhood--a longitudinal study of sleep problems in a large sample of Dunedin children aged 5-9 years. Aust Paediatr J. 1986;22:31–5. doi: 10.1111/j.1440-1754.1986.tb00179.x. [DOI] [PubMed] [Google Scholar]
- 15.Strauch I, Meier B, Kaiser F. Developmental aspects of sleep onset insomnia in adolescents. In: Koella WP, Rüther E, Schulz H, editors. Sleep ‘85. Stuttgart: Fischer; 1985. pp. 386–8. [Google Scholar]
- 16.Gregory AM, O'Connor TG. Sleep problems in childhood: a longitudinal study of developmental change and association with behavioral problems. J Am Acad Child Adolesc Psychiatry. 2002;41:964–71. doi: 10.1097/00004583-200208000-00015. [DOI] [PubMed] [Google Scholar]
- 17.Tononi G, Cirelli C. Sleep function and synaptic homeostasis. Sleep Med Rev. 2006;10:49–62. doi: 10.1016/j.smrv.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 18.Maquet P. The role of sleep in learning and memory. Science. 2001;294:1048–52. doi: 10.1126/science.1062856. [DOI] [PubMed] [Google Scholar]
- 19.Peirano PD, Algarin CR. Sleep in brain development. Biol Res. 2007;40:471–8. [PubMed] [Google Scholar]
- 20.Besedovsky L, Lange T, Born J. Sleep and immune function. Pflug Arch Eue J Phy. 2012;463:121–37. doi: 10.1007/s00424-011-1044-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barclay NL, Gregory AM. Quantitative genetic research on sleep: a review of normal sleep, sleep disturbances and associated emotional, behavioural, and health-related difficulties. Sleep Med Rev. 2013;17:29–40. doi: 10.1016/j.smrv.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 22.Van den Oord EJ, Verhulst FC, Boomsma DI. A genetic study of maternal and paternal ratings of problem behaviors in 3-year-old twins. J Abnorm Psychol. 1996;105:349–57. doi: 10.1037//0021-843x.105.3.349. [DOI] [PubMed] [Google Scholar]
- 23.Gregory AM, Eley TC, O'Connor GT, Plomin R. Etiologies of associations between childhood sleep and behavioral problems in a large twin sample. J Am Acad Child Adolesc Psychiatry. 2004;43:744–51. doi: 10.1097/01.chi/0000122798.47863.a5. [DOI] [PubMed] [Google Scholar]
- 24.Gregory AM. A genetic decomposition of the association between parasomnias and dyssomnias in 8-year-old twins. Arch Pediatr Adolesc Med. 2008;162:299–304. doi: 10.1001/archpedi.162.4.299. [DOI] [PubMed] [Google Scholar]
- 25.Gregory AM, Rijsdijk FV, Dahl RE, McGuffin P, Eley TC. Associations between sleep problems, anxiety, and depression in twins at 8 years of age. Pediatrics. 2006;118:1124–32. doi: 10.1542/peds.2005-3118. [DOI] [PubMed] [Google Scholar]
- 26.Gregory AM, Rijsdijk FV, Lau JYF, Dahl RE, Eley TC. The direction of longitudinal associations between sleep problems and depression symptoms: a study of twins aged 8 and 10 years. Sleep. 2009;32:189–99. doi: 10.1093/sleep/32.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barclay NL, Eley TC, Buysse DJ, Archer SN, Gregory AM. Diurnal preference and sleep quality: same genes? A study of young adult twins. Chronobiol Int. 2010;27:278–96. doi: 10.3109/07420521003663801. [DOI] [PubMed] [Google Scholar]
- 28.Barclay NL, Eley TC, Buysse DJ, Rijsdijk FV, Gregory AM. Genetic and environmental influences on different components of the ‘Pittsburgh Sleep Quality Index’ and their overlap. Sleep. 2010;33:659–68. doi: 10.1093/sleep/33.5.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moore M, Slane J, Mindell JA, Burt S, Klump KL. Genetic and environmental influences on sleep problems: a study of preadolescent and adolescent twins. Child Care Health Dev. 2011;37:638–41. doi: 10.1111/j.1365-2214.2011.01230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gehrman PR, Meltzer LJ, Moore M, Pack AI, Perlis ML, Eaves LJ. Heritability of insomnia symptoms in youth and their relationship to depression and anxiety. Sleep. 2011;34:1641–6. doi: 10.5665/sleep.1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knutson KL. The association between pubertal status and sleep duration and quality among a nationally representative sample of US adolescents. Am J Hum Biol. 2005;17:418–24. doi: 10.1002/ajhb.20405. [DOI] [PubMed] [Google Scholar]
- 32.Eaves LJ, Silberg JL, Meyer JM, et al. Genetics and developmental psychopathology: 2. The main effects of genes and environment on behavioral problems in the Virginia Twin Study of Adolescent Behavioral Development. J Child Psychol Psyc. 1997;38:965–80. doi: 10.1111/j.1469-7610.1997.tb01614.x. [DOI] [PubMed] [Google Scholar]
- 33.Hewitt JK, Silberg JL, Rutter M, et al. Genetics and developmental psychopathology: 1. Phenotypic assessment in the Virginia Twin Study of Adolescent Behavioral Development. J Child Psychol Psychiatry. 1997;38:943–63. doi: 10.1111/j.1469-7610.1997.tb01613.x. [DOI] [PubMed] [Google Scholar]
- 34.Silberg JL, Rutter M, Tracy K, Maes HH, Eaves L. Etiological heterogeneity in the development of antisocial behavior: the Virginia Twin Study of Adolescent Behavioral Development and the Young Adult Follow-Up. Psychol Med. 2007;37:1193–202. doi: 10.1017/S0033291707000293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maes HH, Silberg JL, Neale MC, Eaves LJ. Genetic and cultural transmission of antisocial behavior: an extended twin parent model. Twin Res Hum Gen. 2007;10:136–50. doi: 10.1375/twin.10.1.136. [DOI] [PubMed] [Google Scholar]
- 36.Meyer JM, Silberg JL, Simonoff E, Kendler KS, Hewitt JK. The Virginia Twin-Family Study of Adolescent Behavioral Development: assessing sample biases in demographic correlates of psychopathology. Psychol Med. 1996;26:1119–33. doi: 10.1017/s0033291700035844. [DOI] [PubMed] [Google Scholar]
- 37.Simonoff E, Pickles A, Meyer JM, et al. The Virginia Twin Study of Adolescent Behavioral Development: influences of age, sex, and impairment on rates of disorder. Arch Gen Psychiatry. 1997;54:801–8. doi: 10.1001/archpsyc.1997.01830210039004. [DOI] [PubMed] [Google Scholar]
- 38.American Psychiatric Association. Text Revision. 3rd ed. Washington, DC: American Psychiatric Association; 1987. Diagnostic and Statistical Manual of Mental Disorders. [Google Scholar]
- 39.Neale MC. 4th ed. Richmond, VA: Virginia Commonwealth University Department of Psychiatry; 1997. Mx: Statistical Modeling. [Google Scholar]
- 40.Neale MC, Cardon LR. Dordrecht, Netherlands: Kluwer Academic Publishers; 1992. Methodology for Genetic Studies in Twins and Families. [Google Scholar]
- 41.Neale MC, Heath AC, Hewitt JK, Eaves LJ, Fulker DW. Fitting genetic models with LISREL: hypothesis testing. Behav Genet. 1989;19:37–49. doi: 10.1007/BF01065882. [DOI] [PubMed] [Google Scholar]
- 42.Laberge L, Petit D, Simard C, Vitaro F, Tremblay RE, Montplaisir J. Development of sleep patterns in early adolescence. J Sleep Res. 2001;10:59–67. doi: 10.1046/j.1365-2869.2001.00242.x. [DOI] [PubMed] [Google Scholar]
- 43.Ohayon MM, Lemoine P. Daytime consequences of insomnia complaints in the French general population. Encephale. 2004;30:222–7. doi: 10.1016/s0013-7006(04)95433-4. [DOI] [PubMed] [Google Scholar]
- 44.Rietveld MJ, Posthuma l D, Dolan CV, Boomsma DI. ADHD: sibling interaction or dominance: an evaluation of statistical power. Behav Genet. 2003;33:247–55. doi: 10.1023/a:1023490307170. [DOI] [PubMed] [Google Scholar]
- 45.Barclay NL, Eley TC, Mill J, et al. Sleep quality and diurnal preference in a sample of young adults: associations with 5HTTLPR, PER3 and CLOCK 3111. Am J Med Genet B Neuropsychiatr Genet. 2011;156:681–90. doi: 10.1002/ajmg.b.31210. [DOI] [PubMed] [Google Scholar]
- 46.Brummett BH, Krystal AD, Ashley-Koch A, et al. Sleep quality varies as a function of 5-HTTLPR genotype and stress. Psychosom Med. 2007;69:621–4. doi: 10.1097/PSY.0b013e31814b8de6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deuschle M, Schredl M, Schilling C, et al. Association between a serotonin transporter length polymorphism and primary insomnia. Sleep. 2010;33:343–7. doi: 10.1093/sleep/33.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perlis RH, Mischoulon D, Smoller JW, et al. Serotonin transporter polymorphisms and adverse effects with fluoxetine treatment. Biol Psychiatry. 2003;54:879–83. doi: 10.1016/s0006-3223(03)00424-4. [DOI] [PubMed] [Google Scholar]
- 49.Brummett BH, Krystal AD, Siegler IC, et al. Associations of a regulatory polymorphism of monoamine oxidase-A gene promoter (MAOA-uVNTR) with symptoms of depression and sleep quality. Psychosom Med. 2007;69:396–401. doi: 10.1097/PSY.0b013e31806d040b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buhr A, Bianchi MT, Baur R, et al. Functional characterization of the new human GABA(A) receptor mutation beta3(R192H) Hum Genet. 2002;111:154–60. doi: 10.1007/s00439-002-0766-7. [DOI] [PubMed] [Google Scholar]
- 51.Gehrman PR, Byrne E, Gillespie NA, Martin NG. Genetics of Insomnia. Sleep Med Clin. 2011;6:191–202. [Google Scholar]
- 52.Serretti A, Benedetti F, Mandelli L, et al. Genetic dissection of psychopathological symptoms: insomnia in mood disorders and CLOCK gene polymorphism. Am J Med Genet B Neuropsychiatr Genet. 2003;121B:35–8. doi: 10.1002/ajmg.b.20053. [DOI] [PubMed] [Google Scholar]
- 53.Serretti A, Gaspar-Barba E, Calati R, et al. 3111T/C Clock gene polymorphism is not associated with sleep disturbances in untreated depressed patients. Chronobiol Int. 2010;27:265–77. doi: 10.3109/07420521003663785. [DOI] [PubMed] [Google Scholar]
- 54.Spielman AJ, Caruso L, Glovinsky PB. A behavioral perspective on insomnia treatment. Psychiat Clin North Am. 1987;10:541–53. [PubMed] [Google Scholar]
- 55.Edinger JD, Fins AI. The distribution and clinical-significance of sleep time misperceptions among insomniacs. Sleep. 1995;18:232–9. doi: 10.1093/sleep/18.4.232. [DOI] [PubMed] [Google Scholar]
- 56.van de Laar M, Verbeek I, Pevernagie D, Aldenkamp A, Overeem S. The role of personality traits in insomnia. Sleep Med Rev. 2010;14:61–8. doi: 10.1016/j.smrv.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 57.Buysse DJ, Angst J, Gamma A, Ajdacic V, Eich D, Rossler W. Prevalence, course, and comorbidity of insomnia and depression in young adults. Sleep. 2008;31:473–80. doi: 10.1093/sleep/31.4.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Angold A, Costello EJ, Pickles A, Winder F. London: Medical Research Council Child Psychiatry Unit; 1987. The Development of a Questionnaire for use in Epidemiological Studies of Depression in Children and Adolescents. [Google Scholar]
- 59.Buss AH, Plomin R. New York: Wiley; 1975. A Temperament Theory of Personality Development. [Google Scholar]
- 60.Merrell KW, McClun LA, Kempf KKG, Lund J. Using self-report assessment to identify children with internalizing problems: validity of the internalizing symptoms scale for children. J Psychoeduc Assess. 2002;20:223–39. [Google Scholar]
- 61.Gregory AM, Rijsdijk FV, Eley TC. A twin-study of sleep difficulties in school-aged children. Child Dev. 2006;77:1668–79. doi: 10.1111/j.1467-8624.2006.00966.x. [DOI] [PubMed] [Google Scholar]
- 62.Short MA, Gradisar M, Lack LC, Wright HR, Chatburn A. Estimating adolescent sleep patterns: parent reports versus adolescent self-report surveys, sleep diaries, and actigraphy. Nat Sci Sleep. 2013;5:23–6. doi: 10.2147/NSS.S38369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang B, Wing YK. Sex differences in insomnia: a meta-analysis. Sleep. 2006;29:85–93. doi: 10.1093/sleep/29.1.85. [DOI] [PubMed] [Google Scholar]