

Smooth muscle plays important roles in vascular development. Study of smooth muscle differentiation of human embryonic stem cell–derived mesenchymal cells identifies olfactomedin 2 as a novel regulator. Olfactomedin 2 regulates smooth muscle gene transcription by empowering serum response factor binding to the CArG box in smooth muscle gene promoters.
Abstract
Transforming growth factor-β (TGF-β) plays an important role in smooth muscle (SM) differentiation, but the downstream target genes regulating the differentiation process remain largely unknown. In this study, we identified olfactomedin 2 (Olfm2) as a novel regulator mediating SM differentiation. Olfm2 was induced during TGF-β–induced SM differentiation of human embryonic stem cell–derived mesenchymal cells. Olfm2 knockdown suppressed TGF-β–induced expression of SM markers, including SM α-actin, SM22α, and SM myosin heavy chain, whereas Olfm2 overexpression promoted the SM marker expression. TGF-β induced Olfm2 nuclear accumulation, suggesting that Olfm2 may be involved in transcriptional activation of SM markers. Indeed, Olfm2 regulated SM marker expression and promoter activity in a serum response factor (SRF)/CArG box–dependent manner. Olfm2 physically interacted with SRF without affecting SRF-myocardin interaction. Olfm2-SRF interaction promoted the dissociation of SRF from HERP1, a transcriptional repressor. Olfm2 also inhibited HERP1 expression. Moreover, blockade of Olfm2 expression inhibited TGF-β–induced SRF binding to SM gene promoters in a chromatin setting, whereas overexpression of Olfm2 dose dependently enhanced SRF binding. These results demonstrate that Olfm2 mediates TGF-β–induced SM gene transcription by empowering SRF binding to CArG box in SM gene promoters.
INTRODUCTION
Vascular smooth muscle (SM) differentiation is an essential component of vascular development. The alterations in the differentiated state of the SM contribute to a variety of major cardiovascular diseases, including atherosclerosis, hypertension, restenosis, and vascular aneurysms (Schwartz, 1983, 1997; Kocher and Gabbiani, 1986; Glukhova et al., 1988; Aikawa et al., 1993; Libby, 2001; Owens et al., 2004). A hallmark of this SM phenotypic plasticity is the altered transcription of SM-specific differentiation genes, including SM α-actin (α-SMA), SM22α, and SM myosin heavy chain (SMMHC; Regan et al., 2000). Serum response factor (SRF), a widely expressed MCM1, Agamous, Deficiens, SRF–box transcription factor, binds as a homodimer to a DNA consensus sequence known as a CArG box (CC(A/T)6GG), which is found in the promoter regions of SM-specific genes. SRF-CArG association is required for transcriptional activation of SM genes (Miano, 2003). A large number of environmental cues, including growth factors/inhibitors, inflammatory mediators, and cell–cell and cell–matrix interactions, have been shown to regulate SM differentiation. Transforming growth factor-β (TGF-β) is among the most potent soluble growth factors that activate SM contractile gene expression in specified SM and non-SM types (Hirschi et al., 1998; Hu et al., 2003; Masszi et al., 2003; Chen and Lechleider, 2004; Sinha et al., 2004; Deaton et al., 2005; Tang et al., 2010).
SM originates from multiple sources during vascular development. Various in vitro model systems have been developed to mimic SM development in vivo, including C3H10T1/2 cells, neural crest cells, A404, embryoid body, and embryonic stem cells (Hirschi et al., 1998; Manabe and Owens, 2001; Chen and Lechleider, 2004; Compton et al., 2006; Xie et al., 2007; Han et al., 2010; Yamaguchi et al., 2011). We previously developed a novel in vitro model for TGF-β–induced SM differentiation from human embryonic stem cell–derived mesenchymal cells (hES-MCs; Boyd et al., 2009; Guo et al., 2013). The precise molecular mechanisms governing the SM differentiation process, however, remain largely unknown.
Olfactomedin 2 (Olfm2) belongs to the family of olfactomedin domain–containing proteins, which contains at least 13 members in mammals (Karavanich and Anholt, 1998). Olfm2 expression is developmentally regulated, and Olfm2 knockdown reduces eye size, inhibits optic nerve extension, and disrupts anterior CNS and head development, including neural crest cell–derived cartilaginous structures of the pharyngeal arches in zebrafish (Nakaya and Tomarev, 2007; Lee et al., 2008). Olfm2 is able to form heterodimers with other family members, such as Olfm1, Olfm3, and myocilin, in early eye development (Sultana et al., 2011). Effects of Olfm2 on eye development are likely associated with Pax6 signaling in developing zebrafish eyes (Lee et al., 2008). Pax6 is a master transcriptional factor for eye development and functions and has been shown to physically interact with TGF-β, contributing to maintaining the functional status of eyes (Lang, 2004; Shubham and Mishra, 2012). These data suggest a possible involvement of Olfm2 in the TGF-β signaling cascade during early eye development. In human, a R144Q substitution in Olfm2 protein is believed to be the disease-causing mutation in Japanese patients with open-angle glaucoma (Funayama et al., 2006). However, the role of Olfm2 in vascular development, especially in SM differentiation, has never been reported.
In an analysis of TGF-β–induced SM differentiation of hES-MCs, we found that Olfm2 was dramatically up-regulated by TGF-β. We thus hypothesize that Olfm2 may be one of the TGF-β downstream targets mediating SM differentiation. Indeed, we found that Olfm2 plays an important role in TGF-β–induced SM differentiation. Olfm2 was up-regulated along with the expression of SM markers in TGF-β–treated hES-MCs. TGF-β also induced the nuclear accumulation of Olfm2. Both gain- and loss-of-function studies indicated that Olfm2 was essential for the expression of SM markers. Olfm2 promoted SM differentiation in a SRF/CArG box-dependent manner. Olfm2 physically interacted with SRF to counteract the association between SRF and HERP1 (also known as Hrt2, Hey2, Hesr2, and CHF1), a transcriptional repressor of SM genes, which further enabled Olfm2 to enhance SRF binding to CArG box in SM marker promoter, leading to transcriptional activation of SM marker genes. Together our results identify Olfm2 as a novel regulator for SM differentiation.
RESULTS
Olfm2 was induced during TGF-β–induced SM differentiation
TGF-β is an important determinant for SM lineage. To determine the role of Olfm2 in TGF-β–induced SM differentiation, we examined the expression of Olfm2 and SM markers in TGF-β–treated hES-MCs. As shown in Figure 1, Olfm2 expression markedly increased soon after TGF-β induction, and TGF-β induced Olfm2 expression in a time-dependent manner, with an 8.4-fold increase after 48-h treatment (Figure 1, A and B). Of importance, Olfm2 induction occurred earlier than expression of SM markers α-SMA, SM22α, and SMMHC (Figure 1, A–E, and Supplemental Figure S1). Moreover, Olfm2 expression was observed in smooth muscle of human aorta tissue (Figure 1F). These data suggest that Olfm2 is involved in TGF-β–induced SM differentiation.
FIGURE 1:

TGF-β–induced Olfm2 expression while promoting SM differentiation in hES-MCs. (A) TGF-β–induced Olfm2 expression along with the differentiation of hES-MCs. Serum-starved hES-MCs were treated with vehicle (−) or TGF-β (+; 1 ng/ml) for the time indicated. Western blotting was performed to examine the expression of Olfm2 and SM marker proteins. (B–E) Quantification of Olfm2 and SM marker expression by normalizing to α-tubulin shown in A. *p < 0.01 compared with vehicle-treated group (0 h; n = 3). (F) Olfm2 was expressed in human aortic smooth muscle. Human aorta was paraffin embedded and sectioned and stained with IgG or Olfm2 antibody as indicated. Olfm2 was shown to predominantly express in the nuclei of vascular smooth muscles.
Olfm2 was required for TGF-β–induced SM differentiation
To determine whether Olfm2 plays a role in SM differentiation, we tested whether Olfm2 is required for TGF-β–induced SM marker expression. As shown in Figure 2, A–E, knockdown of Olfm2 by an adenoviral vector expressing Olfm2 short hairpin RNA (shRNA; adenovirus [Ad]-shOlfm2) significantly attenuated TGF-β–induced SM marker expression, indicating that Olfm2 is essential for TGF-β–induced SM differentiation. To further determine the role of Olfm2 in SM differentiation, we tested whether Olfm2 alone can induce SM marker expression. As shown in Figure 2, F–J, ectopic expression of Olfm2 in serum-starved hES-MCs induced 3.2-, 2.3-, and 3.6-fold increases in α-SMA, SM22α, and SMMHC expression, respectively. These results demonstrate that Olfm2 can promote SM marker gene expression.
FIGURE 2:

Olfm2 was required for TGF-β–induced SM differentiation of hES-MCs. (A) Olfm2 knockdown suppressed TGF-β–induced SM differentiation. hES-MCs were transduced with Ad–green fluorescent protein (GFP) or Ad-shOlfm2, followed by vehicle (−) or TGF-β (+; 1 ng/ml) treatment for 48 h. Western blotting was performed to detect Olfm2 and SM marker expression. (B–E) Quantification of the protein expression shown in A. The protein expression was normalized to α-tubulin. *p < 0.01 compared with Ad-GFP–transduced group with vehicle treatment. #p < 0.01 compared with Ad-GFP–transduced group with TGF-β treatment (n = 3). (F) Olfm2 expression induced SM differentiation. hES-MCs were transfected with control or Olfm2 plasmid, followed by serum starvation for 24 h. Cell lysates were collected for Western blot analysis of the proteins indicated. (G–J) Quantification of the protein expression shown in F by normalizing to α-tubulin. *p < 0.01 compared with control plasmid–transfected group (n = 3).
TGF-β induced Olfm2 nuclear accumulation
To investigate how Olfm2 regulates TGF-β–induced SM differentiation, we first detected the subcellular location of Olfm2. Olfm2 immunostaining showed that Olfm2 was located in both cytoplasm and nuclei of TGF-β–untreated hES-MCs. TGF-β stimulation appeared to cause a majority of Olfm2 to accumulate in the nuclei (Figure 3A), suggesting that Olfm2 may serve as a nuclear factor to regulate transcriptional activation of SM markers. To confirm the Olfm2 nuclear location, we fractionated hES-MC protein extracts into nuclear and cytoplasmic portions for Olfm2 Western blotting analysis. As shown in Figure 3, B and C, before TGF-β treatment, only 38% of Olfm2 was located in the nuclei. TGF-β induction, however, increased the nuclear Olfm2 to 81% of the total protein, consistent with the immunostaining results in Figure 3A. These results indicate that TGF-β induces Olfm2 translocation to nuclei of hES-MCs.
FIGURE 3:
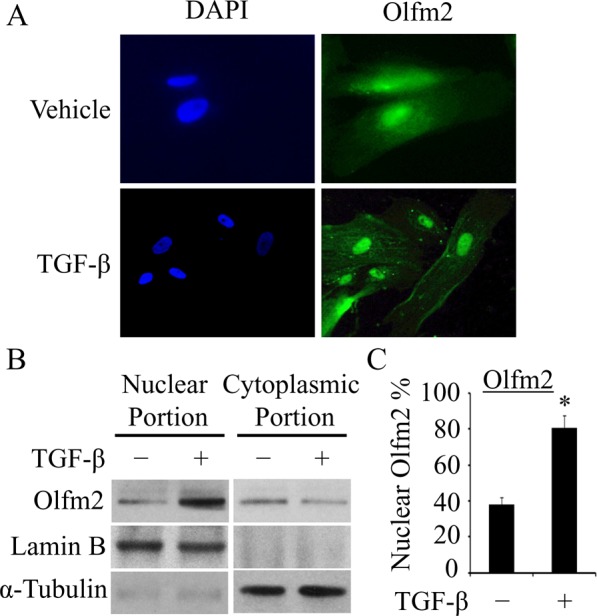
TGF-β induced Olfm2 nuclear accumulation. (A) TGF-β promoted the nuclear localization of Olfm2 in hES-MCs. Serum-starved hES-MCs were treated with vehicle or TGF-β (1 ng/ml) for 24 h. Immunostaining was performed to detect Olfm2 localization. 4′,6-Diamidino-2-phenylindole was used to stain nuclei. (B) TGF-β enhanced nuclear Olfm2 level. Cell lysates of hES-MCs treated with vehicle or TGF-β (24 h) were fractionated into nuclear and cytoplasmic portions and subjected to Western blot. Lamin B and α-tubulin were used as loading controls for the nuclear and cytoplasmic fractions, respectively. (C) Quantification of nuclear Olfm2 protein shown in B. *p < 0.01 compared with vehicle-treated group (n = 3).
Olfm2-induced SM marker expression depended on SRF/CArG
Although Olfm2 is a nuclear factor likely involving the SM gene transcription, analysis of Olfm2 protein structure using DNA-Binder software revealed that Olfm2 lacks the DNA-binding domain, suggesting that Olfm2 may serve as a coactivator for a key transcription factor that regulates SM differentiation. SRF is a key nuclear transcription factor regulating the expression of most SM marker genes by binding to highly conserved CArG box present within nearly all SM-specific promoters (Miano, 2003; Mack, 2011). Because Olfm2 is critical for SM marker expression, we sought to determine whether Olfm2 can induce SM marker expression in the absence of SRF. As shown in Figure 4, A–E, knockdown of SRF by shRNA blocked Olfm2-induced SM marker expression, suggesting that SRF was essential for Olfm2-induced SM differentiation. Moreover, CArG box mutations significantly inhibited Olfm2 induction of α-SMA and SM22α promoter activity (Figure 4, F and G), suggesting that CArG box is indispensable for Olfm2 function. These data demonstrate that Olfm2 mediates SM differentiation in a SRF/CArG-dependent manner.
FIGURE 4:

Olfm2 induced SM marker expression in an SRF/CArG-dependent manner. (A) Knockdown of SRF blocked Olfm2-induced SM marker expression. hES-MCs were cotransfected with control (–) or Olfm2 plasmid and control (shCtrl) or SRF shRNA (shSRF) plasmid as indicated, followed by serum starvation for 24 h. Western blotting was performed to detect the expression of proteins indicated. (B–E) Quantification of protein expression shown in A by normalizing to α-tubulin. *p < 0.01 compared with shCtrl-transfected group without Olfm2. #p < 0.01 compared with Olfm2-transfected group with shCtrl (n = 3). (F, G) CArG box mutation diminished the SM marker promoter activity activated by Olfm2. (F) α-SMA promoter (from −2.6 to +2.8 kb) constructs with wild-type (WT) or mutant CArG box either in the first intron (CArGm intron) or in the promoter region (CArGm (A+B)) were cotransfected with the control (Ctrl) or Olfm2 plasmid into hES-MCs for 48 h. (G) SM22α promoter constructs with wild-type (WT) or mutant CArG box in the two SRF-binding sites (CArGmnear or CArGmfar) were cotransfected with the control or Olfm2 plasmid into hES-MCs for 48 h. Luciferase assay was performed. Fold induction of promoter activity was calculated relative to the WT promoter cotransfected with the control plasmid (set as 1). *p < 0.01 compared with control plasmid–transfected WT promoter. #p < 0.01 compared with Olfm2 plasmid–transfected WT promoter (n = 3).
Olfm2 physically interacted with SRF
Because Olfm2 function in SM differentiation relies on SRF/CArG, we sought to determine whether Olfm2 interacts with SRF. A coimmunoprecipitation (CoIP) assay using endogenous proteins extracted from hES-MCs treated with vehicle or TGF-β showed that Olfm2 was coimmunoprecipitated with SRF (Figure 5A) and SRF was coimmunoprecipitated with Olfm2 (Figure 5C), demonstrating that Olfm2 physically interacted with SRF in hES-MCs. Their interaction was significantly enhanced by TGF-β, which was likely attributable to the increased expression of Olfm2 by TGF-β (Figure 5, A–D).
FIGURE 5:

Olfm2 physically interacted with SRF. (A, C) CoIP with endogenous proteins indicated that Olfm2 physically interacted with SRF. Serum-starved hES-MCs were treated with vehicle (–) or TGF-β (+; 1 ng/ml) for 24 h. Cell lysates were immunoprecipitated with normal IgG, SRF (A), or Olfm2 (C) antibody. The immunoprecipitates were blotted (IB) with Olfm2 (A) or SRF (C) antibody. The interaction between Olfm2 and SRF was enhanced by TGF-β induction. (B, D) Quantifications of Olfm2 immunoprecipitated with SRF (B) and of SRF immunoprecipitated with Olfm2 (D) as shown in A and C, respectively. The proteins coimmunoprecipitated were normalized to α-tubulin. *p < 0.01 compared with IgG-immunoprecipitated group. #p < 0.01 compared with SRF-immunoprecipitated (B) and Olfm2- immunoprecipitated (D) groups with vehicle treatment (n = 3).
Olfm2 did not affect SRF–myocardin interaction
Myocardin (Myocd) is a transcriptional coactivator of SRF, and Myocd-SRF interaction is crucial for the differentiation of SM lineage (Wang et al., 2001, 2002). Therefore we tested whether Olfm2 affects SRF and Myocd expression as well as their interaction. We found that the ectopic expression of Olfm2 had no effect on mRNA expression of either SRF or Myocd (Figure 6, A–C). To determine whether Olfm2 affects the interaction between SRF and Myocd, we transfected hES-MCs with FLAG-tagged Myocd expression plasmid and performed CoIP assays with endogenous SRF. As shown in Figure 6, D and E, Olfm2 did not alter the interaction between SRF and Myocd. These data suggest that mechanisms other than modulation of SRF/Myocd binding are involved in Olfm2-mediated SM differentiation.
FIGURE 6:

Olfm2 did not affect the expression of SRF, Myocd, and their interaction. (A–C) Olfm2 did not alter SRF and Myocd mRNA expression. hES-MCs were transfected with control (Ctrl) or Olfm2 plasmid, followed by serum starvation for 24 h. qPCR was performed to detect the mRNA expression of Olfm2, SRF, and Myocd as indicated. (D) Olfm2 did not affect SRF-Myocd interaction. hES-MCs were cotransfected with control (−; 20 μg) or Olfm2 plasmid (+; 10–20 μg) and FLAG-Myocd plasmid (Myocd, 20 μg) in 10-cm dishes. Cell lysates were immunoprecipitated with normal IgG or SRF antibody. The immunoprecipitates were blotted (IB) with FLAG, Olfm2, or SRF antibody. (E) Quantification of Myocd protein coimmunoprecipitated with SRF by normalizing to Myocd input level shown in D. N.S., not significant.
Olfm2 blocked HERP1 expression and its binding to SRF
HERP1 is a transcriptional repressor abundantly expressed in the developing vascular system (Nakagawa et al., 2000; Iso et al., 2001a, b). HERP1 has been shown to physically associate with SRF and inhibit SM differentiation by interfering with SRF binding to CArG box (Doi et al., 2005). Because both HERP1 and Olfm2 interact with SRF with opposite effects on SM differentiation, we hypothesize that TGF-β induction of Olfm2 promotes SM differentiation through inhibition of HERP1 expression or HERP1 binding to SRF. Indeed, TGF-β induced time-dependent suppression of HERP1 but up-regulated Olfm2 expression (Figure 7, A and B). Of importance, Olfm2 overexpression decreased HERP1 mRNA expression (Figure 7C), whereas knockdown of Olfm2 effectively attenuated TGF-β–induced blockade of HERP1 expression (Figure 7, D and E). These data suggest that Olfm2 mediated suppression of HERP1 by TGF-β. To determine whether Olfm2 affects HERP1 interaction with SRF, we performed CoIP assay in cells where Olfm2 expression was blocked. We found that TGF-β treatment inhibited HERP1-SRF interaction. However, knockdown of Olfm2 restored the HERP1 binding to SRF that was suppressed by TGF-β, indicating that Olfm2 mediated the TGF-β–induced dissociation of HERP1 from SRF (Figure 7, F and G). These data demonstrate that Olfm2 regulates TGF-β–induced SM differentiation by removing the inhibitory effect of HERP1, that is, releasing SRF from HERP1.
FIGURE 7:

Olfm2 inhibited HERP1 expression and its binding to SRF. (A, B) TGF-β suppressed HERP1 mRNA expression. Serum-starved hES-MCs were treated with TGF-β for the time indicated. qPCR was performed to examine Olfm2 (A) and HERP1 (B) mRNA expression. The mRNA expression was normalized to cyclophilin. *p < 0.01 compared with the vehicle-treated group (0 h). (C) Olfm2 inhibited HERP1 mRNA expression. hES-MCs were transfected with control or Olfm2 plasmid, followed by serum starvation for 24 h. qPCR was performed to detect HERP1 expression. *p < 0.01 compared with the control plasmid–transfected group. (D, E) Knockdown of Olfm2 attenuated TGF-β-induced down-regulation of HERP1. hES-MCs were transduced with Ad-GFP or Ad-shOlfm2 (shOlfm2), followed by vehicle (–) or TGF-β (+) treatment for 24 h. qPCR was performed to detect Olfm2 (D) and HERP1 (E) mRNA expression. *p < 0.01 compared with Ad-GFP–transduced group with vehicle treatment. #p < 0.01 compared with Ad-GFP-transduced group with TGF-β treatment. (F) Olfm2 was essential for TGF-β–inhibited HERP1 binding to SRF. hES-MCs were transduced with Ad-GFP or shOlfm2, followed by vehicle or TGF-β treatment for 24 h. Cell lysates were immunoprecipitated with normal IgG or SRF antibody. The immunoprecipitates were blotted (IB) with HERP1, Olfm2, and SRF antibody as indicated. (G) Quantification of HERP1 protein immunoprecipitated with SRF by normalized to α-tubulin as shown in F. *p < 0.01 compared with Ad-GFP-transduced and SRF-immunoprecipitated group with vehicle treatment; #p < 0.01 compared with Ad-GFP–transduced and SRF-immunoprecipitated group with TGF-β treatment (n = 3).
Olfm2 was required for SRF binding to SM marker promoter
Because SRF binding to CArG box is crucial for SM gene transcription and Olfm2 mediated SRF release from HERP1, we sought to determine whether Olfm2 plays a role in SRF binding to SM marker gene promoter. Previous studies showed that SRF binding to CArG box is functionally important for SM22α promoter activity both in vitro and in vivo (Miano, 2003; McDonald et al., 2006). We thus performed chromatin immunoprecipitation (ChIP) assays to determine whether Olfm2 affects SRF binding to this CArG box. As shown in Figure 8A, SRF weakly bound to the CArG box of SM22α promoter in a basal state. TGF-β treatment significantly enhanced the binding (Figure 8, A and B), consistent with previous studies (Hautmann et al., 1999). Knockdown of Olfm2, however, significantly diminished TGF-β–enhanced SRF binding to the promoter (Figure 8, A and B), suggesting that Olfm2 is essential for SRF binding to SM marker promoter. Moreover, ectopic expression of Olfm2 in TGF-β–untreated cells dose dependently enhanced SRF binding to SM22α promoter (Figure 8, D and E). To determine whether this is a common mechanism for Olfm2 to regulate SRF targets in SM differentiation, we tested the Olfm2 effect on SRF binding to another SM promoter, SMMHC promoter, and observed similar results as with SM22α promoter (Figure 8, A–F). These data demonstrate that Olfm2 is a novel factor facilitating SRF binding to SM gene promoter, a key event for SM differentiation.
FIGURE 8:

Olfm2 facilitated and promoted SRF binding to the promoters of SMC marker genes in a chromatin setting. (A–C) Knockdown of Olfm2 blocked TGF-β-induced SRF binding to SM22α and SMMHC promoters. hES-MCs were transduced with Ad-GFP or shOlfm2, followed by vehicle or TGF-β treatment (1 ng/ml) as indicated for 24 h. ChIP assay were performed to detect binding of SRF to CArG box in the SM22α and SMMHC promoters as indicated. Representative semiquantitative PCR (A) and qPCR (B, C) results. *p < 0.01 compared with the vehicle-treated groups; #p < 0.01 compared with Ad-GFP–transduced group treated with TGF-β for each corresponding gene. (D–F) Olfm2 enhanced SRF binding to CArG box in the SM22α and SMMHC promoters. hES-MCs were transfected with control (+; 20 μg) or Olfm2 plasmid (10–20 μg) in 10-cm cell culture dishes. ChIP assay was performed to detect binding of SRF to CArG box in the SM22α and SMMHC promoters as indicated. Representative semiquantitative PCR (D) and qPCR (E, F) results. *p < 0.01 compared with the control plasmid–transfected group for each corresponding gene (n = 3).
DISCUSSION
SM differentiation is an important process during vasculogenesis and angiogenesis, by which multipotential progenitor cells in the developing organism acquire contractile SM characteristics (Owens, 1995; Owens et al., 2004). However, the precise molecular mechanisms controlling SM differentiation are not fully understood. It is well established that the vascular and nervous systems of vertebrates share many features, such as common signaling pathways and similar anatomy (Carmeliet, 2003; Melani and Weinstein, 2010; Tam and Watts, 2010), suggesting that common factors might regulate both vascular and nervous development. Olfm2 is a developmentally regulated gene implicated in the early development of the CNS (Nakaya and Tomarev, 2007; Lee et al., 2008). Our results suggest that Olfm2 also plays an important role in SM differentiation. Olfm2 is able to regulate TGF-β–induced SM differentiation of hES-MCs because knockdown of Olfm2 inhibits SM marker expression induced by TGF-β, whereas ectopic expression of Olfm2 induces SM markers expression, although to a much lesser extent than with TGF-β induction (Figure 2 vs. Figure 1), consistent with other factors, such as SRF and Myocd, also being required for TGF-β–induced SM differentiation.
Of interest, TGF-β induces Olfm2 nuclear accumulation, making Olfm2 a nuclear factor involved in the transcriptional regulation of SM contractile genes. Analysis of Olfm2 protein sequence using cNLS Mapper Software reveals a nuclear localization signal, an 11–amino acid sequence of GYPKRSAGEAF, at the region of AA364-374. This sequence can be recognized and bound by importin-α. Therefore Olfm2 is most likely translocated into the nuclei of hES-MCs by TGF-β via an importin-α–mediated nuclear transport mechanism. Another possibility is that Olfm2 may form a complex with other signaling mediators, such as β-catenin or Notch intracellular domain. This complex may translocate into nucleus in a Smad3-dependent manner, which may be explored in the future.
Olfm2 function in SM marker expression/transcription is associated with SRF and CArG box because knockdown of SRF or CArG box mutation markedly inhibits Olfm2-induced SM marker gene expression and transcription. In fact, Olfm2 physically interacts with SRF, and this interaction is promoted by TGF-β. Of interest, Olfm2-SRF interaction does not affect Myocd-SRF interaction, suggesting that Myocd and Olfm2 may bind to different domains of the SRF protein. However, Olfm2 appears to be critical for SRF binding to CArG box in SM marker promoters, which is attributed to the different roles of Olfm2. First, Olfm2 affects SRF binding to SM marker promoter by releasing SRF from its binding to HERP1. HERP1 is a downstream target of Notch signaling (Nakagawa et al., 2000; Iso et al., 2001a, b). As a translational repressor, HERP1 inhibits Notch-mediated SM differentiation via a negative feedback mechanism. HERP1 also inhibits TGF-β–induced SM differentiation (King et al., 2006; Tang et al., 2008). Olfm2 blocks HERP1 interaction with SRF and thus makes SRF abundant for SM marker promoter. Olfm2 also inhibits HERP1 expression. Second, Olfm2 facilitates SRF binding to SM promoters, given that blockade of Olfm2 expression diminishes TGF-β–induced SRF binding to both SM22α and SMMHC promoters. Finally, Olfm2 may enhance the affinity of SRF binding to SM promoters, given that the enrichment of SRF-binding region in SM22α and SMMHC promoters is significantly increased in Olfm2-overexpressed cells.
Smad proteins are known to translocate to nuclei to regulate SM marker gene expression upon TGF-β induction. Smads appear to affect Olfm2 function by regulating its expression, given that knockdown of Smad3 completely and knockdown of Smad2 significantly block Olfm2 expression (Supplemental Figure S2). In addition, Smad3 inhibits Herp1 expression (Supplemental Figure S3), which is likely due to the induction of Olfm2. Of interest, both Smad3 and Olfm2 interact with SRF, but there is no detectable interaction between Olfm2 and Smad2 or Smad3 (unpublished data). One explanation is that Smad3 interacts with SRF in the early phase of SM lineage determination because Smad3 is phosphorylated and translocated into nuclei in a few minutes after TGF-β induction. Its phosphorylation/nuclear location declines after 1–2 h. Olfm2 interaction with SRF, however, occurs later, given that Olfm2 is not induced until after 1 h of TGF-β treatment. Therefore Olfm2 may interact with SRF at a slightly late phase to further promote SM marker gene transcription.
Identification of Olfm2 function in SM gene transcription provides novel insights into TGF-β–induced SM differentiation. In the undifferentiated hES-MC cells or the initial phase of SM differentiation, Olfm2 is expressed at a low level, and SRF interacts with Smad3 and binds weakly to CArG box due to a substantial binding of HERP1 to SRF, resulting in a repressed state or very low level of SM marker gene transcription. When Olfm2 is induced by TGF-β via Smads and translocated into nuclei, it binds to SRF, causing SRF dissociation from HERP1, and thus removes the inhibitory effect of HERP1. Furthermore, Olfm2 facilitates and enhances SRF binding to CArG box in SM marker promoters, leading to a strong induction of SM marker expression (Figure 9). The homeostatic balance between Olfm2 and HERP1 expression appears to be an important factor in determining whether SM-specific marker genes can be effectively induced by TGF-β after the initial phase of SM differentiation.
FIGURE 9:
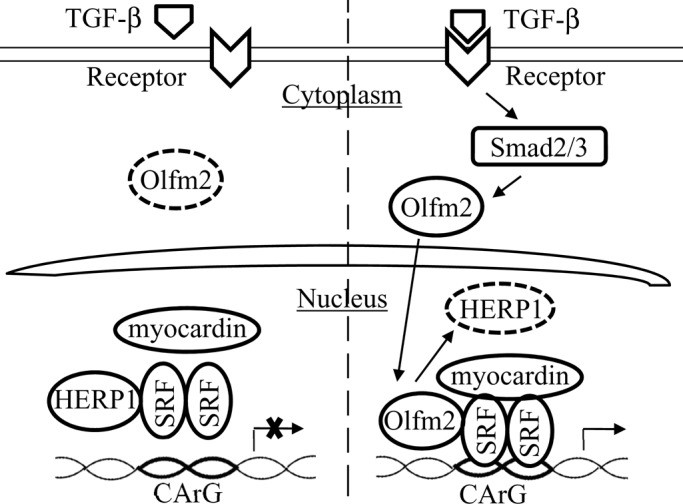
Schematic mechanism by which Olfm2 regulates SM marker gene transcription. On TGF-β stimulation, Olfm2 is induced by Smad2/3 and then translocated into nuclei of progenitor cells, where it binds to SRF, causing SRF dissociation from HERP1. Olfm2 then facilitates and even promotes SRF binding to CArG box in the SM gene promoter, resulting in increased transcriptional activation of SM markers.
MATERIALS AND METHODS
Cell culture and transfection
hES-MCs were maintained in αMEM (Fisher Scientific, Pittsburgh, PA) containing 10% mesenchymal stem cell–qualified fetal bovine serum (Hyclone) and 2 mM l-glutamine (Hyclone, Logan, UT). Cells were grown in serum-free medium for 16–24 h, followed by incubation with TGF-β (1 ng/ml) for various times as needed. For transfections, cells were cultured in 12- or 6-well plates or 6- or 10-cm dishes and transfected with different plasmids 24 h after plating with an 80–90% confluence using Lipofectamine LTX (Invitrogen, Grand Island, NY) according to the manufacturer's instruction.
Preparation of shRNA adenoviral vector
shRNA target sequence for Olfm2 was GCA CGT CCA GTT ACG AGT ACA CGG ACG TG. Double-stranded DNA–coding Olfm2 shRNAs were cloned into pRNAT-H1.1/Adeno shuttle vector (Genscript, Piscataway, NJ). Adenovirus was packaged in 293 cells (Agilent Technologies, Santa Clara, CA) and purified by CsCl2 gradient ultracentrifugation as previously described (Wang et al., 2011). For adenoviral transduction, hES-MCs were transduced with 100–multiplicity of infection of control or shRNA adenovirus for 48–72 h.
Immunocytofluorescence staining
hES-MCs were cultured on sterile coverslips and treated with TGF-β. Cells were then fixed with 1% paraformaldehyde (PFA) for 5 min, followed by methanol for 10 min at −20°C for Olfm2 staining. Anti-Olfm2 antibody was used at 1:100 in blocking buffer consisting of 5% bovine serum albumin in phosphate-buffered saline (PBS). Cells were then incubated with fluorescein isothiocyanate–conjugated secondary antibodies (1:100). Stained cells were imaged with a Nikon Eclipse 90i microscope, and images were captured with a Nikon 12.7MP digital Sight DS-Ri1 color camera.
Quantitative reverse transcription-PCR
Total RNA from cultured cells was extracted using TRIzol Reagent (Invitrogen) according to the manufacturer's instructions. Reverse transcription was performed using an iScript cDNA Synthesis kit (Bio-Rad). Quantitative reverse transcription-PCR (qPCR) was performed in a Mx3005P qPCR machine using SYBR Green master mix (Agilent Technologies). Each sample was amplified in triplicate. Primers for the qPCR were as follows. Olfm2, 5′-GGG AGG AGG TGA GGA ATC TC-3′ (forward) and 5′-CCA TCG TGT CAG TCA TCC AG-3′ (reverse); Myocd, 5′-TGC ATG CTG CTG TAA AGT CC-3′ (forward) and 5′-TAG CTG AAT CGG TGT TGC TG-3′ (reverse); SRF, 5′-CCT ACC AGC TTC ACC CTC AT-3′ (forward) and 5′-CGG GCT AGG GTA CAT CAT GT-3′ (reverse); and HERP1, 5′-TAG AGA AAA GGC GTC GGG AT-3′ (forward) and 5′-GTG TGC GTC AAA GTA GCC TT-3′ (reverse).
Western blotting
hES-MCs were cultured in αMEM or treated with TGF-β. Cells were washed two times with cold PBS, followed by protein extraction using radioimmunoprecipitation assay buffer (50 mmol/l Tris-HCI, pH 7.4, 1% Triton X-100, 0.25% [wt/vol] sodium deoxycholate, 150 mmol/l NaCl, 1 mmol/l ethylene glycol tetraacetic acid, 0.1% SDS, protease inhibitors, and phosphatase inhibitors). Protein concentration was measured using BCA Protein Assay reagent (Thermo Scientific, Waltham, MA ). A 5- or 10-μg amount of the lysates was resolved by SDS–PAGE and transferred to polyvinylidene fluoride membranes (Bio-Rad, Hercules, CA). Membranes were blocked with 5% nonfat dry milk in Tris-buffered saline and Tween 20 and then incubated with primary antibodies in the blocking buffer for 1–2 h, followed by incubation with horseradish peroxidase–conjugated secondary antibody for 1 h (Sigma-Aldrich, St. Louis MO). Signal detection was performed with enhanced chemiluminescence (Millipore, Billerica, MA). Antibodies used were anti-Olfm2 (Abcam, Cambridge, MA), anti-SRF (Santa Cruz Biotechnology, Dallas, TX), anti–α-SMA (Abcam), anti-SM22α (Abcam), anti-SMMHC (Biomedical Technologies, Stoughton, MA), anti–α-tubulin (Cell Signaling, Danvers, MA), anti-FLAG (Sigma-Aldrich), and anti-HERP1 (Santa Cruz Biotechnology).
Coimmunoprecipitation assay and immunoblotting analysis
Cells were washed with ice-cold mild lysis buffer containing protease inhibitor mix (Thermo Scientific). The lysates were incubated with immunoglobulin G (IgG; negative control), SRF, or Olfm2 antibodies using CoIP kit (Thermo Scientific). The immunoprecipitates were pelleted, washed, and subjected to immunoblotting according to the manufacturer's instructions.
Promoter reporter luciferase assay
hES-MCs were cotransfected with α-SMA or SM22α promoter constructs and control or Olfm2 plasmids as described previously (Li et al., 2007; Shi and Chen, 2013), followed by serum starvation for 24 h. Then a luciferase assay was performed using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI). Independent experiments were repeated three times.
Chromatin immunoprecipitation assay
ChIP assays were performed using ChIP kit (Millipore). Growth-arrested hES-MCs were treated with TGF-β (1 ng/ml) for 24 h. Chromatin complexes were immunoprecipitated with 3 μg of SRF antibody or IgG (negative control) according to the manufacturer's instructions. Semiquantitative PCR and qPCR were performed to amplify human SM22α promoter region containing CArG box sequence using the primer set 5′-CCC TCT CTC CAG CCA GAC T-3′ (forward) and 5′-CCC CAG TGA CTC CAC ACA G-3′ (reverse; Tanaka et al., 2008).
Statistical analysis
All values are expressed as mean ± SE. Data were analyzed using analysis of variance with pairwise comparisons between groups. p < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants HL107526, HL119053, and HL123302 to S.Y.C.
Abbreviations used:
- Ad
adenovirus
- ChIP
chromatin immunoprecipitation assay
- CoIP
coimmunoprecipitation
- HERP1
Hrt2, Hey2, Hesr2, and CHF1
- hES-MC
human embryonic stem cell–derived mesenchymal cell
- Olfm2
olfactomedin 2
- qPCR
quantitative reverse transcription-PCR
- shRNA
short hairpin RNA
- SM
smooth muscle
- α-SMA
smooth muscle α-actin
- SMMHC
smooth muscle myosin heavy chain
- SRF
serum response factor
- TGF-β
transforming growth factor-β.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-08-1255) on October 8, 2014.
REFERENCES
- Aikawa M, Sivam PN, Kuro-o M, Kimura K, Nakahara K, Takewaki S, Ueda M, Yamaguchi H, Yazaki Y, Periasamy M, et al. Human smooth muscle myosin heavy chain isoforms as molecular markers for vascular development and atherosclerosis. Circ Res. 1993;73:1000–1012. doi: 10.1161/01.res.73.6.1000. [DOI] [PubMed] [Google Scholar]
- Boyd NL, Robbins KR, Dhara SK, West FD, Stice SL. Human embryonic stem cell-derived mesoderm-like epithelium transitions to mesenchymal progenitor cells. Tissue Eng Part A. 2009;15:1897–1907. doi: 10.1089/ten.tea.2008.0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P. Blood vessels and nerves: common signals, pathways and diseases. Nat Rev Genet. 2003;4:710–720. doi: 10.1038/nrg1158. [DOI] [PubMed] [Google Scholar]
- Chen S, Lechleider RJ. Transforming growth factor-beta-induced differentiation of smooth muscle from a neural crest stem cell line. Circ Res. 2004;94:1195–1202. doi: 10.1161/01.RES.0000126897.41658.81. [DOI] [PubMed] [Google Scholar]
- Compton LA, Potash DA, Mundell NA, Barnett JV. Transforming growth factor-beta induces loss of epithelial character and smooth muscle cell differentiation in epicardial cells. Dev Dyn. 2006;235:82–93. doi: 10.1002/dvdy.20629. [DOI] [PubMed] [Google Scholar]
- Deaton RA, Su C, Valencia TG, Grant SR. Transforming growth factor-beta1-induced expression of smooth muscle marker genes involves activation of PKN and p38 MAPK. J Biol Chem. 2005;280:31172–31181. doi: 10.1074/jbc.M504774200. [DOI] [PubMed] [Google Scholar]
- Doi H, Iso T, Yamazaki M, Akiyama H, Kanai H, Sato H, Kawai-Kowase K, Tanaka T, Maeno T, Okamoto E, et al. HERP1 inhibits myocardin-induced vascular smooth muscle cell differentiation by interfering with SRF binding to CArG box. Arterioscler Thromb Vasc Biol. 2005;25:2328–2334. doi: 10.1161/01.ATV.0000185829.47163.32. [DOI] [PubMed] [Google Scholar]
- Funayama T, Mashima Y, Ohtake Y, Ishikawa K, Fuse N, Yasuda N, Fukuchi T, Murakami A, Hotta Y, Shimada N. SNPs and interaction analyses of noelin 2, myocilin, and optineurin genes in Japanese patients with open-angle glaucoma. Invest Ophthalmol Vis Sci. 2006;47:5368–5375. doi: 10.1167/iovs.06-0196. [DOI] [PubMed] [Google Scholar]
- Glukhova MA, Kabakov AE, Frid MG, Ornatsky OI, Belkin AM, Mukhin DN, Orekhov AN, Koteliansky VE, Smirnov VN. Modulation of human aorta smooth muscle cell phenotype: a study of muscle-specific variants of vinculin, caldesmon, and actin expression. Proc Natl Acad Sci USA. 1988;85:9542–9546. doi: 10.1073/pnas.85.24.9542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Stice SL, Boyd NL, Chen SY. A novel in vitro model system for smooth muscle differentiation from human embryonic stem cell-derived mesenchymal cells. Am J Physiol Cell Physiol. 2013;304:C289–C298. doi: 10.1152/ajpcell.00298.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Li N, Tian X, Kang J, Yan C, Qi Y. Endogenous transforming growth factor (TGF) beta1 promotes differentiation of smooth muscle cells from embryonic stem cells: stable plasmid-based siRNA silencing of TGF beta1 gene expression. J Physiol Sci. 2010;60:35–41. doi: 10.1007/s12576-009-0063-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hautmann MB, Adam PJ, Owens GK. Similarities and differences in smooth muscle alpha-actin induction by TGF-beta in smooth muscle versus non-smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999;19:2049–2058. doi: 10.1161/01.atv.19.9.2049. [DOI] [PubMed] [Google Scholar]
- Hirschi KK, Rohovsky SA, D’Amore PA. PDGF, TGF-beta, and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J Cell Biol. 1998;141:805–814. doi: 10.1083/jcb.141.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Wu Z, Phan SH. Smad3 mediates transforming growth factor-beta-induced alpha-smooth muscle actin expression. Am J Respir Cell Mol Biol. 2003;29:397–404. doi: 10.1165/rcmb.2003-0063OC. [DOI] [PubMed] [Google Scholar]
- Iso T, Sartorelli V, Chung G, Shichinohe T, Kedes L, Hamamori Y. HERP, a new primary target of Notch regulated by ligand binding. Mol Cell Biol. 2001a;21:6071–6079. doi: 10.1128/MCB.21.17.6071-6079.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iso T, Sartorelli V, Poizat C, Iezzi S, Wu HY, Chung G, Kedes L, Hamamori Y. HERP, a novel heterodimer partner of HES/E(spl) in Notch signaling. Mol Cell Biol. 2001b;21:6080–6089. doi: 10.1128/MCB.21.17.6080-6089.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karavanich C, Anholt RR. Evolution of olfactomedin. Structural constraints and conservation of primary sequence motifs. Ann NY Acad Sci. 1998;855:294–300. doi: 10.1111/j.1749-6632.1998.tb10585.x. [DOI] [PubMed] [Google Scholar]
- King IN, Kathiriya IS, Murakami M, Nakagawa M, Gardner KA, Srivastava D, Nakagawa O. Hrt and Hes negatively regulate Notch signaling through interactions with RBP-Jkappa. Biochem Biophys Res Commun. 2006;345:446–452. doi: 10.1016/j.bbrc.2006.04.097. [DOI] [PubMed] [Google Scholar]
- Kocher O, Gabbiani G. Cytoskeletal features of normal and atheromatous human arterial smooth muscle cells. Hum Pathol. 1986;17:875–880. doi: 10.1016/s0046-8177(86)80637-2. [DOI] [PubMed] [Google Scholar]
- Lang RA. Pathways regulating lens induction in the mouse. Int J Dev Biol. 2004;48:783–791. doi: 10.1387/ijdb.041903rl. [DOI] [PubMed] [Google Scholar]
- Lee JA, Anholt RR, Cole GJ. Olfactomedin-2 mediates development of the anterior central nervous system and head structures in zebrafish. Mech Dev. 2008;125:167–181. doi: 10.1016/j.mod.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Luo Z, Huang W, Lu Q, Wilcox CS, Jose PA, Chen S. Response gene to complement 32, a novel regulator for transforming growth factor-beta-induced smooth muscle differentiation of neural crest cells. J Biol Chem. 2007;282:10133–10137. doi: 10.1074/jbc.C600225200. [DOI] [PubMed] [Google Scholar]
- Libby P. Current concepts of the pathogenesis of the acute coronary syndromes. Circulation. 2001;104:365–372. doi: 10.1161/01.cir.104.3.365. [DOI] [PubMed] [Google Scholar]
- Mack CP. Signaling mechanisms that regulate smooth muscle cell differentiation. Arterioscler Thromb Vasc Biol. 2011;31:1495–1505. doi: 10.1161/ATVBAHA.110.221135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe I, Owens GK. Recruitment of serum response factor and hyperacetylation of histones at smooth muscle-specific regulatory regions during differentiation of a novel P19-derived in vitro smooth muscle differentiation system. Circ Res. 2001;88:1127–1134. doi: 10.1161/hh1101.091339. [DOI] [PubMed] [Google Scholar]
- Masszi A, Di Ciano C, Sirokmany G, Arthur WT, Rotstein OD, Wang J, McCulloch CA, Rosivall L, Mucsi I, Kapus A. Central role for Rho in TGF-beta1-induced alpha-smooth muscle actin expression during epithelial-mesenchymal transition. Am J Physiol Renal Physiol. 2003;284:F911–F924. doi: 10.1152/ajprenal.00183.2002. [DOI] [PubMed] [Google Scholar]
- McDonald OG, Wamhoff BR, Hoofnagle MH, Owens GK. Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo. J Clin Invest. 2006;116:36–48. doi: 10.1172/JCI26505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melani M, Weinstein BM. Common factors regulating patterning of the nervous and vascular systems. Annu Rev Cell Dev Biol. 2010;26:639–665. doi: 10.1146/annurev.cellbio.093008.093324. [DOI] [PubMed] [Google Scholar]
- Miano JM. Serum response factor: toggling between disparate programs of gene expression. J Mol Cell Cardiol. 2003;35:577–593. doi: 10.1016/s0022-2828(03)00110-x. [DOI] [PubMed] [Google Scholar]
- Nakagawa O, McFadden DG, Nakagawa M, Yanagisawa H, Hu T, Srivastava D, Olson EN. Members of the HRT family of basic helix-loop-helix proteins act as transcriptional repressors downstream of Notch signaling. Proc Natl Acad Sci USA. 2000;97:13655–13660. doi: 10.1073/pnas.250485597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaya N, Tomarev S. Expression patterns of alternative transcripts of the zebrafish olfactomedin 1 genes. Gene Expr Patterns. 2007;7:723–729. doi: 10.1016/j.modgep.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev. 1995;75:487–517. doi: 10.1152/physrev.1995.75.3.487. [DOI] [PubMed] [Google Scholar]
- Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- Regan CP, Adam PJ, Madsen CS, Owens GK. Molecular mechanisms of decreased smooth muscle differentiation marker expression after vascular injury. J Clin Invest. 2000;106:1139–1147. doi: 10.1172/JCI10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz SM. Cellular proliferation in atherosclerosis and hypertension. Proc Soc Exp Biol Med. 1983;173:1–13. doi: 10.3181/00379727-173-41601. [DOI] [PubMed] [Google Scholar]
- Schwartz SM. Smooth muscle migration in atherosclerosis and restenosis. J Clin Invest. 1997;100:S87–S89. [PubMed] [Google Scholar]
- Shi N, Chen SY. Cell division cycle 7 mediates transforming growth factor-beta-induced smooth muscle maturation through activation of myocardin gene transcription. J Biol Chem. 2013;288:34336–34342. doi: 10.1074/jbc.M113.498238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shubham K, Mishra R. Pax6 interacts with SPARC and TGF-beta in murine eyes. Mol Vis. 2012;18:951–956. [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Hoofnagle MH, Kingston PA, McCanna ME, Owens GK. Transforming growth factor-beta1 signaling contributes to development of smooth muscle cells from embryonic stem cells. Am J Physiol Cell Physiol. 2004;287:C1560–C1568. doi: 10.1152/ajpcell.00221.2004. [DOI] [PubMed] [Google Scholar]
- Sultana A, Nakaya N, Senatorov VV, Tomarev SI. Olfactomedin 2: expression in the eye and interaction with other olfactomedin domain-containing proteins. Invest Ophthalmol Vis Sci. 2011;52:2584–2592. doi: 10.1167/iovs.10-6356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam SJ, Watts RJ. Connecting vascular and nervous system development: angiogenesis and the blood-brain barrier. Annu Rev Neurosci. 2010;33:379–408. doi: 10.1146/annurev-neuro-060909-152829. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Sato H, Doi H, Yoshida CA, Shimizu T, Matsui H, Yamazaki M, Akiyama H, Kawai-Kowase K, Iso T, et al. Runx2 represses myocardin-mediated differentiation and facilitates osteogenic conversion of vascular smooth muscle cells. Mol Cell Biol. 2008;28:1147–1160. doi: 10.1128/MCB.01771-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Urs S, Boucher J, Bernaiche T, Venkatesh D, Spicer DB, Vary CP, Liaw L. Notch and transforming growth factor-beta (TGFbeta) signaling pathways cooperatively regulate vascular smooth muscle cell differentiation. J Biol Chem. 2010;285:17556–17563. doi: 10.1074/jbc.M109.076414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Urs S, Liaw L. Hairy-related transcription factors inhibit Notch-induced smooth muscle alpha-actin expression by interfering with Notch intracellular domain/CBF-1 complex interaction with the CBF-1-binding site. Circ Res. 2008;102:661–668. doi: 10.1161/CIRCRESAHA.107.165134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, Krieg PA, Olson EN. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell. 2001;105:851–862. doi: 10.1016/s0092-8674(01)00404-4. [DOI] [PubMed] [Google Scholar]
- Wang DZ, Li S, Hockemeyer D, Sutherland L, Wang Z, Schratt G, Richardson JA, Nordheim A, Olson EN. Potentiation of serum response factor activity by a family of myocardin-related transcription factors. Proc Natl Acad Sci USA. 2002;99:14855–14860. doi: 10.1073/pnas.222561499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JN, Shi N, Xie WB, Guo X, Chen SY. Response gene to complement 32 promotes vascular lesion formation through stimulation of smooth muscle cell proliferation and migration. Arterioscler Thromb Vasc Biol. 2011;31:e19–e26. doi: 10.1161/ATVBAHA.111.230706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie CQ, Zhang J, Villacorta L, Cui T, Huang H, Chen YE. A highly efficient method to differentiate smooth muscle cells from human embryonic stem cells. Arterioscler Thromb Vasc Biol. 2007;27:e311–e312. doi: 10.1161/ATVBAHA.107.154260. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S, Yamahara K, Homma K, Suzuki S, Fujii S, Morizane R, Monkawa T, Matsuzaki Y, Kangawa K, Itoh H. The role of microRNA-145 in human embryonic stem cell differentiation into vascular cells. Atherosclerosis. 2011;219:468–474. doi: 10.1016/j.atherosclerosis.2011.09.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.