

Background: Calcium-sensing receptor (CaSR) plays a critical role in the regulation of epithelial ion transport.
Results: CaSR activators induce Ca2+ signaling and duodenal bicarbonate secretion (DBS).
Conclusion: CaSR triggers Ca2+-dependent DBS, likely through receptor-operated channels, intermediate conductance Ca2+-activated K+ channels, and the cystic fibrosis transmembrane conductance regulator.
Significance: Dietary CaSR activators may modulate the physiological process of DBS that is critical for duodenal mucosal protection.
Keywords: Bicarbonate, Calcium, Calcium Channel, Calcium Imaging, Epithelial Cell, G Protein-Coupled Receptor (GPCR), Calcium-sensing Receptor, Cytosolic Ca2+ Concentrations, Transepithelial HCO3− secretion, Receptor-Op
Abstract
Epithelial ion transport is mainly under the control of intracellular cAMP and Ca2+ signaling. Although the molecular mechanisms of cAMP-induced epithelial ion secretion are well defined, those induced by Ca2+ signaling remain poorly understood. Because calcium-sensing receptor (CaSR) activation results in an increase in cytosolic Ca2+ ([Ca2+]cyt) but a decrease in cAMP levels, it is a suitable receptor for elucidating the mechanisms of [Ca2+]cyt-mediated epithelial ion transport and duodenal bicarbonate secretion (DBS). CaSR proteins have been detected in mouse duodenal mucosae and human intestinal epithelial cells. Spermine and Gd3+, two CaSR activators, markedly stimulated DBS without altering duodenal short circuit currents in wild-type mice but did not affect DBS and duodenal short circuit currents in cystic fibrosis transmembrane conductance regulator (CFTR) knockout mice. Clotrimazole, a selective blocker of intermediate conductance Ca2+-activated K+ channels but not chromanol 293B, a selective blocker of cAMP-activated K+ channels (KCNQ1), significantly inhibited CaSR activator-induced DBS, which was similar in wild-type and KCNQ1 knockout mice. HCO3− fluxes across epithelial cells were activated by a CFTR activator, but blocked by a CFTR inhibitor. CaSR activators induced HCO3− fluxes, which were inhibited by a receptor-operated channel (ROC) blocker. Moreover, CaSR activators dose-dependently raised cellular [Ca2+]cyt, which was abolished in Ca2+-free solutions and inhibited markedly by selective CaSR antagonist calhex 231, and ROC blocker in both animal and human intestinal epithelial cells. Taken together, CaSR activation triggers Ca2+-dependent DBS, likely through the ROC, intermediate conductance Ca2+-activated K+ channels, and CFTR channels. This study not only reveals that [Ca2+]cyt signaling is critical to modulate DBS but also provides novel insights into the molecular mechanisms of CaSR-mediated Ca2+-induced DBS.
Introduction
Cytosolic free Ca2+ ([Ca2+]cyt)4 plays an essential role in a variety of mammalian cells through the regulation of many biological functions, including neurotransmitter release, muscle contraction, gene regulation, cell proliferation, and apoptosis (1). Therefore, dysregulation of [Ca2+]cyt homeostasis may result in pathological changes in many systems. Under physiological conditions, various mechanisms are controlling Ca2+ homeostasis in the human body, one of which is the calcium-sensing receptor (CaSR) (2). The CaSR is a plasma membrane protein initially cloned from bovine parathyroid cells. It is a member of the G protein-coupled receptor family and regulates the synthesis of parathyroid hormone in response to changes in serum Ca2+ concentrations (3–5).
CaSR activation elicits complex intracellular signaling events through the modulation of a wide range of intracellular mediators, including Gαq/11 proteins and phospholipase C (PLC). These, in turn, stimulate both inositol trisphosphate production and PKC activation, which increases [Ca2+]cyt (4, 5). Activation of the CaSR has been shown to increase [Ca2+]cyt in different types of mammalian cells, especially in parathyroid cells, epithelial cells, osteocytes, cardiomyocytes, and smooth muscle cells (4, 5). In addition, CaSR activation can stimulate Gαi proteins and phosphodiesterase, leading to a decrease in cyclic AMP and cyclic GMP levels (4, 5).
It has been demonstrated that the CaSR is expressed along the entire gastrointestinal tract and plays a critical role in normal gut physiology (6). Recent studies have been mainly performed on its functions in modulating gastrin and gastric acid secretion and intestinal fluid and electrolyte transports (6–9). Extracellular Ca2+ ([Ca2+]o) stimulates gastric acid and bicarbonate secretion in the guinea pig (10, 11), suggesting that gastric surface epithelial cells are capable of sensing changes in Ca2+ to modulate gastric secretion, likely through CaSR activation. Although it is well documented that the CaSR inhibits intestinal transepithelial Cl− secretion by blocking cyclic AMP signaling (7), little is known about the role of the CaSR in intestinal transepithelial HCO3− secretion, which is a critical factor in duodenal mucosal protection and mainly under the control of cyclic AMP and Ca2+ signaling. Although the physiological roles and molecular mechanisms of cyclic AMP-induced HCO3− secretion are relatively well defined, those induced by Ca2+ signaling remain poorly understood in most epithelia, especially in intestinal epithelia (12). Moreover, although it is known that Ca2+ signaling is critical for duodenal bicarbonate secretion (DBS), the molecular mechanisms controlling [Ca2+]cyt homeostasis in duodenal epithelial cells are poorly understood.
In our previous studies, we proposed that Ca2+ and cyclic AMP signaling may play different roles in the regulation of intestinal transepithelial HCO3− and Cl− secretion. We found that although cyclic AMP plays a major role in intestinal Cl− secretion, Ca2+ signaling may be critical for transepithelial HCO3− secretion (13). However, activation of most well defined receptors expressed in intestinal epithelial cells usually increase both [Ca2+]cyt and intracellular cyclic AMP levels, making it difficult to distinguish between [Ca2+]cyt- and cyclic AMP-regulated epithelial ion transports. Because CaSR activation results in an increase in [Ca2+]cyt but a decrease in intracellular cyclic AMP levels (3–5), we hypothesized that the CaSR is a suitable receptor system for further delineating the role of [Ca2+]cyt- and cAMP-mediated intestinal epithelial ion transports in general and HCO3− secretion in particular.
Therefore, in this study, we sought to investigate CaSR modulation of [Ca2+]cyt-mediated DBS and the underlying mechanisms. We found that CaSR activation triggers Ca2+-dependent duodenal transepithelial HCO3− secretion, likely through the receptor-operated channels (ROCs), the intermediate-conductance Ca2+-activated K+ channels (IKCa), and the cystic fibrosis transmembrane conductance regulator (CFTR) channels. This study not only reveals that [Ca2+]cyt signaling is critical to modulate DBS but also provides novel insights into the underlying molecular mechanisms of CaSR-induced Ca2+-dependent DBS.
EXPERIMENTAL PROCEDURES
Animal Study
The animal use protocol was approved by the University of California San Diego Committee on Investigations Involving Animal Subjects. All experiments were performed with adult Harlan C-57 black mice; homozygous CFTR knockout (CFTR−/−) mice and their wild-type littermates (CFTR+/+), which were established as described previously (13); and mice deficient in KCNQ1 (kcnq1−/−) and their wild-type littermates (kcnq1+/+), which were generated as described earlier (14).
Ussing Chamber Experiments in Vitro
The proximal duodenum removed from mice was immediately placed in ice-cold iso-osmolar mannitol with indomethacin (10 μm) solution. The duodenal tissue was stripped of seromuscular layers and then mounted in the Ussing chambers (window area, 0.1 cm2). Experiments were performed under continuous short-circuited conditions (voltage current clamp, VCC 600, Physiologic Instruments, San Diego, CA), and luminal pH was maintained at 7.40 by the continuous infusion of 5 mm HCl under the automatic control of a pH-stat system (ETS 822, Radiometer America, Westlake, OH). Duodenal short circuit currents (Isc) and HCO3− secretion were measured simultaneously as described previously (15). The rate of luminal bicarbonate secretion is expressed as micromolar per square centimeter per hour. The Isc was measured in microamperes and converted into μEq per square centimeters per hour. After basal parameters were measured for a 30-min period, CaSR activators were added to both the mucosal and serosal sides of the Ussing chamber because the CaSR has been identified on both the apical and basolateral membranes of epithelial cells (7, 16). In some experiments, tissues were treated with inhibitors for 10 min after the baseline recording, followed by addition of CaSR activators. Electrophysiological parameters and bicarbonate secretion were recorded for a total of 90 min. During this experimental period, the vehicle did not significantly change Isc and HCO3− secretion, as shown in our previous control experiments (15). The mucosal solution used in Ussing chamber experiments contained the following: 140 mm Na+, 5.4 mm K+, 1.2 mm Ca2+, 1.2 mm Mg2+, 120 mm Cl−, 25 mm gluconate, and 10 mm mannitol. The serosal solution contained the following: 140 mm Na+, 5.4 mm K+, 1.2 mm Ca2+, 1.2 mm Mg2+, 120 mm Cl−, 25 mm HCO3−, 2.4 mm HPO42−, 2.4 mm H2PO42−, 10 mm glucose, and 0.01 mm indomethacin. The osmolalities for both solutions were ∼300 mosmol/kg of H2O.
Epithelial Cell Culture
As described previously (17, 18), SCBN, a duodenal epithelial cell line of canine origin (19), and Caco-2 and HEK-293 cells, human epithelial cell lines, were fed with fresh DMEM supplemented with 10% fetal bovine serum, l-glutamine, and streptomycin every 2–3 days. SW-480, a human colon cancer cell line, was fed with fresh L15 supplemented with 10% fetal bovine serum and streptomycin. After the cells had grown to confluence, they were replated onto 12-mm round coverslips (Warner Instruments Inc., Hamden, CT) and incubated for at least 24 h before use for [Ca2+]cyt and pHi measurements.
Measurement of [Ca2+]cyt in Epithelial Cells by Digital Ca2+ Imaging
[Ca2+]cyt levels in epithelial cells were measured by digital Ca2+ imaging as described previously (20). Cells grown on coverslips were loaded with 5 μm Fura-2/AM in physiological salt solution, described below, at room temperature (∼22 °C) for 50 min and then washed for 30 min. Thereafter, the coverslips with epithelial cells were mounted in a perfusion chamber on a Nikon microscope stage (Nikon Corp., Tokyo, Japan). The ratio of Fura-2/AM fluorescence with excitation at 340 or 380 nm (F340/380) was followed over time and captured using an intensified charge-coupled device camera (ICCD200) and a MetaFluor imaging system (Universal Imaging Corp., Downingtown, PA). The physiological salt solution used in digital Ca2+ measurement contained the following: 140 mm Na+, 5 mm K+, 2 mm Ca2+, 147 mm Cl2−, 10 mm Hepes, and 10 mm glucose (pH 7.4). For the Ca2+-free solution, Ca2+ was omitted, but 0.5 mm EGTA was added. The osmolality for all solutions was ∼300 mosmol/kg of H2O.
Measurement of HCO3− Fluxes in SCBN Cells
pHi measurements in SCBN cells were applied as described previously (21). Briefly, cells grown on coverslips were incubated with 2 μm 2′,7′-bis(2-carboxyethyl)-5-(and -6)carboxyfluorescein-AM in physiological salt solution, described above, for 30 min at room temperature and then washed for 30 min. The ratio of 2′,7′-bis(2-carboxyethyl)-5-(and -6)carboxyfluorescein fluorescence with excitation at 495 or 440 nm (F495/440) was captured using an intensified charge-coupled device camera and a MetaFluor imaging system. The NaCl/HCO3− solutions contained the following: 120 mm NaCl, 25 mm NaHCO3, 2.5 mm K2HPO4, 1 mm MgSO4, 1 mm CaCl2, and 10 mm glucose equilibrated with 5% CO2/95% O2 (pH 7.4). In Na+-free (Na+-free/HCO3−) solutions, Na+ was replaced with N-methyl-d-glucamine. In HCO3−-free solutions, NaHCO3 was replaced with NaCl (in NaCl/Hepes solution) or with N-methyl-d-glucamine (in Na+-free/Hepes solution). In experiments in which cells were acidified, 30 mm NH4C1 replaced an equal amount of N-methyl-d-glucamine. The ratio of the 2′,7′-bis(2-carboxyethyl)-5-(and -6)carboxyfluorescein fluorescence was calibrated in terms of pHi by incubating the cells in a high K+ solution (KCl replaced NaCl) and then permeabilizing the cells with 10 μm nigericin. Then the pH of the bathing solution was stepped between pH 6.3 and 7.4. The F495/440 was linear over this pH range. Cells were first perfused with either the NaCl/HCO3− or NaCl/Hepes solution in the chamber for 15 min to allow the pHi to stabilize. Then the cells were switched to Na+-free/HCO3− or Na+-free/Hepes for 5 min to remove Na+ from the cells. The cells were then treated with the NH4-containing solution for 5 min, and when the NH4-containing solution was removed, cells were acidified to pHi 6.0–6.5. Rates of pHi recovery after treatment with drugs were calculated by linear regression analysis between pH 6.0 and 6.5.
Western Blot Analysis
The specific anti-CaSR antibody used in this study is an affinity-purified monoclonal antibody raised against a synthetic peptide corresponding to the extracellular domain (residues 214–235) of the human CaSR (Labome, Princeton, NJ). Its cross-reactivity with rodents, specificity, and applications have been described previously (7, 22). A Western blot analysis of mouse duodenal mucosae and intestinal epithelial cells was applied as described previously (15). PVDF membranes (Millipore Corp., Billerica, MA) with resolved proteins were incubated with the anti-CaSR antibody or GAPDH (1:5000, Ambion, Austin, TX). After washing with PBS plus 1% Tween (PBST), the rabbit anti-mouse secondary antibody was applied to the membranes, which were treated with a chemiluminescent solution (Fivephoton Biochemicals, San Diego, CA) and then exposed to x-ray film. Densitometric analysis of the blots was performed with the use of an AlphaImager digital imaging system (Alpha Innotech, San Leandro, CA).
Immunohistochemistry
Immunohistochemistry was carried out as described previously (7). Briefly, the slides with duodenal tissues from C-57 mice or with intestinal epithelial cells were incubated with an anti-CaSR monoclonal antibody (1:100 dilution, Labome). The primary antibodies were detected with biotinylated goat anti-mouse IgG (Vector Laboratories, Burlingame, CA) secondary antibodies. Immunoreactivity was detected using a horseradish peroxidase (3′-,3′-diaminobenzidine) kit (BioGenex, San Francisco, CA) followed by counterstaining with hematoxylin, dehydration, and mounting. Slides were then examined with a Nikon Eclipse 800 Research microscope. To demonstrate the CaSR specificity of the antibody labeling, a control experiment was performed in which the primary antibody was omitted. All incubations were performed at room temperature.
Chemicals and Solutions
Spermine, U-73122, genistein, GdCl3, clotrimazole, and CFTRinh-172 were purchased from Sigma. 2-Aminoethoxydiphenyl borate (2-APB) and chromanol 293B were purchased from Tocris Bioscience (Ellisville, MO). Fura-2/AM and 2′,7′-bis(2-carboxyethyl)-5-(and -6)carboxyfluorescein were from Invitrogen. Anti-CaSR monoclonal antibody (catalog no. MA1–934, a mouse mAb) was from Labome. The other chemicals were obtained from Fisher Scientific (Santa Clara, CA).
Statistical Analysis
Results are expressed as mean ± S.E. Differences between means were considered to be statistically significant at p < 0.05 using Student's t test or one-way analysis of variance followed by Newman-Keuls post hoc test, as appropriate.
RESULTS
Protein Expression of the CaSR in Mouse Duodenum Mucosal Tissues
To examine CaSR expression in mouse duodenum mucosa, both Western blot and immunohistochemistry analyses were performed. As shown in our Western blot analysis (Fig. 1), the antibody identified a significant band at ∼120–130 kDa in lysates of mouse duodenum mucosal tissues, indicating protein expression of the CaSR (8, 23). Fig. 2A shows typical villous crypt structures of mouse duodenum mucosa with H&E staining. Fig. 2B shows representative images of CaSR immunohistochemistry in duodenum mucosa. Intense CaSR immunoreactivity (brown) was noted on both apical and basolateral membranes of the villous and crypt epithelial cells (Fig. 2B, right panel). However, no specific signal for the CaSR was observed when CaSR primary antibody was omitted (Fig. 2C). Therefore, cellular distribution and location of the CaSR in mouse duodenum mucosa was detected by immunohistochemistry.
FIGURE 1.
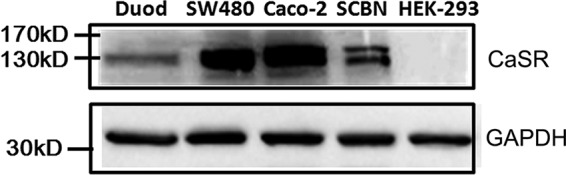
Protein expression of the CaSR in mouse duodenal mucosa and human epithelial cells. After mouse duodenal mucosal tissues (Duod), SCBN cells, SW-480 cells, and Caco-2 cells (human colonic epithelial cells), and HEK-293 cells (human epithelial cells used as a negative control) were lysed, Western blot analysis was performed to detect protein expression of the CaSR using a specific anti-CaSR monoclonal antibody. GAPDH was used as a loading control. These data are representative of three experiments with similar results.
FIGURE 2.

Immunohistochemistry on sections obtained from mouse duodenal mucosal tissues. A, H&E staining of mouse duodenal mucosa showing normal morphology and typical villous crypt structures. B, representative immunoreactivity of CaSR proteins (brown) in the villus and crypts of the duodenal mucosa at different magnifications. C, representative immunoreactivity without incubation of primary anti-CaSR antibody in the villus and crypts of duodenal mucosa as a negative control. Magnifications are ×100, ×200, and ×400 in the left, center, and right panels, respectively. These data are representative of at least three experiments with similar results.
Protein Expression of the CaSR in Intestinal Epithelial Cells
To examine CaSR expression in intestinal epithelial cells, both Western blot and immunohistochemistry analyses were performed on SCBN, SW-480, and Caco-2 cells, two human intestinal epithelial cell lines commonly used in the literature for physiological and pathological studies of intestinal ion transports. As shown by the Western blot analysis in Fig. 1, the antibody identified a strong band at ∼120–130 kDa in both SW480 and Caco-2 cells. However, the antibody identified one band of ∼120–130 kDa and another band of ∼140–150 kDa in the SCBN cell line, which is similar to previous reports (8, 23), indicating CaSR protein expression in duodenal epithelial cells. However, our Western blot results show that the expression of CaSR protein is severalfold higher in human intestinal epithelial cells than in mouse duodenum mucosal tissues (Fig. 1), suggesting a higher CaSR expression in pure epithelial cells than in mucosal tissues that contain various cell types. To rule out the possible nonspecific staining of the CaSR in the tissues and cell lines, we also used parental HEK-293 cells as negative controls. Indeed, the antibody did not detect any CaSR expression in these cells (Fig. 1).
Fig. 3 shows representative images of CaSR immunocytochemistry in these epithelial cells. Intense CaSR immunoreactivity was noted in intestinal epithelial SCBN, SW-480, and Caco-2 cells (Fig. 3, A, C, and E) but not in epithelial HEK-293 cells (Fig. 3G). No specific signal for the CaSR was observed when the CaSR primary antibody was omitted (Fig. 3, B, D, F, and H). Therefore, by immunocytochemistry, the CaSR was verified in intestinal epithelial cells, which is consistent with its presence in the epithelial cells of rat duodenum mucosa (24).
FIGURE 3.

Immunohistochemistry of CaSR proteins on intestinal epithelial cells. A and B, representative immunoreactivity of CaSR proteins with (A) or without (B) primary anti-CaSR antibody in SCBN cells. C and D, representative immunoreactivity of CaSR proteins with (C) or without (D) primary anti-CaSR antibody in human colonic epithelial SW-480 cells. E and F, representative immunoreactivity of CaSR proteins with (E) or without (F) primary anti-CaSR antibody in human colonic epithelial Caco-2 cells. G, representative immunoreactivity with primary anti-CaSR antibody in human epithelial HEK-293 cells as a negative control. Magnification is ×100 for all images. These data are representative of at least three experiments with similar results.
Role of the CaSR in Regulating Duodenal HCO3− Secretion and Isc
The CaSR has been functionally demonstrated along the entire gastrointestinal epithelium, where it plays an important role in the regulation of gastric acid and intestinal Cl− secretion. Therefore, in our initial studies, Ussing chamber experiments were conducted to test whether the CaSR is involved in duodenal mucosal ion transports, especially DBS. Because it is now evident that CFTR channels are essential for transepithelial HCO3− and Cl− secretion in most gastrointestinal epithelia (25, 26), both CFTR knockout and wild-type mice were used to test whether CaSR activation can modulate duodenal Isc and HCO3− secretion. After basal Isc and HCO3− secretion were recorded for 30 min, two commonly used CaSR activators, spermine (1 mm) and Gd3+ (0.5 mm), were added to both sides of the tissues because the CaSR is not restricted to one side of epithelial cells (7, 16). As shown in Fig. 4, A and B, in both CFTR knockout and wild-type mice, spermine and Gd3+ did not significantly affect duodenal basal Isc (p > 0.05, n = 6). The net peak HCO3− secretion, calculated as the difference between the baseline and the peak value at 10 min, was used to describe the CaSR-activated HCO3− secretion. As shown in Fig. 4, C and D, both spermine and Gd3+ markedly stimulated DBS in wild-type mice (p < 0.01, n = 6), which was inhibited significantly by U73122 (10 μm), a selective PLC inhibitor (p < 0.01, n = 6). However spermine and Gd3+ did not stimulate DBS in CFTR knockout mice (not significant, n = 6). Therefore, these data suggest that CaSR activation selectively stimulates DBS through the PLC pathway and CFTR channels.
FIGURE 4.

Effects of different CaSR activators on duodenal Isc and DBS in CFTR KO and WT mice. A, time course of duodenal Isc in wild type mice after addition of spermine (1 mm) or Gd3+ (0.5 mm) to Ussing chambers as indicated by the arrow. B, time course of duodenal Isc in CFTR knockout mice after addition of spermine or Gd3 to Ussing chambers as indicated by the arrow. C, spermine-stimulated net peak DBS in the presence or the absence of U73122 (10 μm) in wild-type mice and in CFTR knockout mice. D, Gd3+-stimulated net peak DBS in the presence or the absence of U73122 in wild-type mice and in CFTR knockout mice. Values are expressed as mean ± S.E. for five to six experiments. **, p < 0.01 versus each activator-stimulated net peak DBS in WT mice.
Involvement of ROC and IKCa in CaSR-mediated HCO3− Secretion
It is well known that cation channels are expressed in intestinal epithelia and that Ca2+ signaling is critical to modulate epithelial ion transport, likely through the activation of intermediate Ca2+-activated K+ channels (IKCa or KCNN4) (15). We further examined whether ROC and IKCa are involved in CaSR-mediated DBS. As shown in Fig. 5A, chromanol 293B (10 μm), a selective blocker of cAMP-activated K+ channels (KCNQ1) (27–29), did not significantly affect spermine-induced net peak DBS (not significant, n = 6). However, clotrimazole (30 μm), a selective blocker of IKCa (30, 31), markedly inhibited spermine-induced net peak DBS (p < 0.01, n = 6). 2-APB (100 μm), a commonly used blocker of ROC (32), also markedly inhibited spermine-induced net peak DBS (p < 0.01, n = 6) (Fig. 5A). Moreover, when spermine-induced net peak DBS were compared between KCNQ1 knockout and wild-type mice, no significant differences were found between these two types of mice (NS, n = 6) (Fig. 5B). Again, spermine (1 mm) did not significantly affect the basal duodenal Isc of both KCNQ1 knockout and wild-type mice (NS, n = 6) (Fig. 5C). Therefore, our data indicate that Ca2+ signaling, ROC, and IKCa, but not cAMP signaling and KCNQ1, are involved in CaSR-mediated DBS.
FIGURE 5.
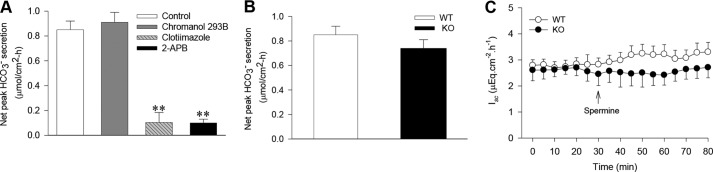
Effects of spermine and ion channel blockers on duodenal Isc and DBS in KCNQ1 KO and WT mice. A, spermine-stimulated net peak DBS in the presence or the absence of chromanol 293B (10 μm), clotrimazole (30 μm), or 2-APB (100 μm) in wild-type mice. B, comparison of spermine-stimulated net peak DBS in KCNQ1 knockout and wild-type mice. C, comparison of the time course of duodenal Isc in KCNQ1 knockout and wild-type mice after addition of spermine to Ussing chambers as indicated by the arrow. Spermine was used at 1 mm in A–C. Values are expressed as mean ± S.E. for six experiments. **, p < 0.01 versus the control (spermine-stimulated net peak DBS in wild-type mice).
CaSR Activation-induced HCO3− Fluxes across SCBN Cells
Because expression and function of CFTR channels has been well established in SCBN cells (18), they are commonly used for studies of small intestinal epithelial ion transports (18, 33, 34). We first tested the role of CFTR in HCO3− fluxes in SCBN cells. To this end, cells were treated with NH4Cl in Na+-free/HCO3− solution that caused the pHi first to increase (because of the entry of the weak base NH3) and then to decrease when the NH4 was washed from the bath. The cells remained acidic in the Na+-free/HCO3− solution, in which the pHi was kept relatively stable but recovered when cells were returned to NaCl/HCO3− solution (Fig. 6A), likely because of the operation of Na+/H+ exchange and other Na+- and HCO3−-dependent pHi regulators. To test for the ability of HCO3− to permeate though CFTR, genistein (50 μm), a commonly used CFTR activator (35), was added to cells that were acidified in Na+-free/HCO3− solution. We observed that pHi quickly recovered, and further recovery occurred after adding back NaCl/HCO3− solution (Fig. 6B). To examine whether genistein indeed activates HCO3− fluxes through the CFTR, cells were pretreated with CFTRinh-173 (10 μm), a commonly used CFTR blocker (35), which reversed genistein-induced pHi recovery in Na+-free/HCO3− solution (Fig. 6D). To test whether the genistein-activated, Na+-independent recovery of pHi was HCO3−-dependent, these experiments were also repeated in Na+-free and HCO3−-free Hepes-buffered solutions in which the acidified cells responded to genistein with only a slight effect on pHi, but a sustained pHi recovery occurred when Na+ was present in the Hepes solution (data not shown). Together, these findings are consistent with genistein-regulated HCO3− fluxes through the CFTR in the presence of extracellular HCO3−.
FIGURE 6.

Effects of different CaSR modulators and ion channel blockers on HCO3− fluxes in SCBN cells. A, control time course of pHi changes induced by NH4Cl in Na+-free/HCO3− solution. Treatment of cells with 30 mm NH4Cl in the solution caused the pHi first to increase and then to decrease when the NH4Cl was washed out. The cells remained acidic, and the pHi was relatively stable in Na+-free/HCO3− solution, but the pHi began to recover when the cells were returned to NaCl/HCO3− solution (Na+). B, genistein-induced HCO3− fluxes through CFTR channels. The time course of pHi changes in SCBN cells was similar to the control in A. However, after the NH4Cl was washed out, genistein (Gen, 50 μm) was added to the cells acidified in Na+-free/HCO3− solution, and the pHi began to recover, but further recovery occurred after adding back NaCl/HCO solution (Na+). C, spermine-induced HCO3− fluxes through CFTR channels. The time course of pHi changes was similar to B, but spermine (Sper, 1 mm) was added to the cells acidified in Na+-free/HCO3− solution. D, summary data showing the effects of different CaSR modulators and ion channel blockers on HCO3− fluxes in SCBN cells. CFTRinh-173 (10 μm), 2-APB (100 μm), and Gd3+ (0.5 mm) were applied to the experiments. Values are expressed as mean ± S.E. of 40–50 cells for each group. **, p < 0.01 versus control (Con) in A; ##, p < 0.01 versus their corresponding activators in B and C.
To test for the role of the CaSR in modulating HCO3− fluxes through the CFTR, similar experiments were performed with spermine (1 mm), which was added to cells acidified in Na+-free/HCO3− solution. As shown in Fig. 6C, spermine, like genistein, induced pHii recovery, which was reversed by 2-APB (100 μm). Similarly, Gd3+ (0.5 mm) induced pHi recovery in Na+-free/HCO3− solution (Fig. 6D). However, spermine and Gd3+ did not induce a significant pHi recovery in Na+-free, Hepes-buffered solutions (data not shown). These data from single cell studies are in agreement with those from a duodenal tissue study, indicating that ROC and CFTR channels are involved in CaSR-mediated transepithelial HCO3− secretion.
CaSR Activation Induces Ca2+ Signaling in Epithelial Cells
It is well documented that CaSR activation inhibits the intracellular cyclic AMP pathway in intestinal epithelial cells. However, little is known about Ca2+ signaling downstream of CaSR activation in these cells. In addition, although it is known that Ca2+ signaling is a critical regulator for DBS, so far Ca2+ signaling in duodenal epithelial cells is poorly understood. We therefore monitored [Ca2+]cyt changes in epithelial cells stimulated with different CaSR activators.
Following a short exposure to Ca2+-free solutions for 3 min, cells were superfused with different concentrations of [Ca2+]o (1.0–4.0 mm), which are close to the EC50 (∼2.0 mm) for CaSR activation (23). Although [Ca2+]o at 1.0 mm did not affect basal [Ca2+]cyt, significant increases were seen when [Ca2+]o increased to 4.0 mm (Fig. 7, A and B). Although [Ca2+]o is an endogenous CaSR activator, it may enter healthy cells through the store-operated Ca2+ entry pathway (32, 36) or may even directly leak into unhealthy cells. We also used the CaSR activator spermine and found that it dose-dependently increased [Ca2+]cyt in SCBN cells (Fig. 7, C and D). Moreover, both [Ca2+]o- and spermine-induced [Ca2+]cyt signaling was inhibited markedly by calhex 231 (3 μm), a selective CaSR antagonist (Fig. 7, E and F). These results provide direct evidence for the CaSR-mediated increase in [Ca2+]cyt in duodenal epithelial cells.
FIGURE 7.

Dose-dependent stimulation of [Ca2+]cyt by the CaSR agonist and inhibition by the CaSR antagonist in SCBN cells. After SCBN cells were loaded with Fura-2/AM, [Ca2+]cyt in the cells was measured by a digital Ca2+ imaging system. A, time course of [Ca2+]cyt changes induced by different concentrations of [Ca2+]o. B, summary data showing a dose-dependent peak [Ca2+]cyt response to [Ca2+]o stimulation. C, time course of [Ca2+]cyt changes induced by different concentrations of spermine in 2 mm [Ca2+]o-containing normal physiological solutions. D, summary data showing dose-dependent peak [Ca2+]cyt responses to spermine stimulation. E, time course of 4 mm [Ca2+]o-induced [Ca2+]cyt changes in the presence of calhex 231 (3 μm) in normal physiological solutions. F, summary data showing the inhibitory effect of calhex 231 on 4 mm [Ca2+]o- or 5 mm spermine-induced [Ca2+]cyt changes. Values are expressed as mean ± S.E. of 40–50 cells. *, p < 0.05; **, p < 0.01 versus the baselines before stimulation (Con) in B and D or versus the controls in the absence of calhex 231 in F.
The CaSR is a member of the G protein-coupled receptor family, and its activation mobilizes different Ca2+ sources in different cell types (4, 5). Therefore, we sought to elucidate the mechanisms of [Ca2+]cyt mobilization by CaSR activation in SCBN cells. To test whether ROCs are involved in CaSR activation, cells were superfused with spermine (3 mm) in the presence or the absence of 2 mm [Ca2+]o. As shown in Fig. 8A, spermine induced a significant increase in [Ca2+]cyt in [Ca2+]0-containing solutions, but not in [Ca2+]0-free solution. We further examined CaSR activation-mediated [Ca2+]0 entry mechanisms in SCBN cells. Spermine (3–10 mm) significantly elevated [Ca2+]cyt in [Ca2+]0-containing solutions (Fig. 7, C and D). However, treatment with 2-APB (100 μm) (Fig. 8, C and D) or SKF96365 (10 μm) (Fig. 7, E and F), two commonly used ROC inhibitors (37), significantly inhibited spermine-induced [Ca2+]0 entry, indicating that the CaSR-mediated Ca2+ entry pathway in SCBN cells involves the ROC.
FIGURE 8.

Spermine-mediated [Ca2+]o entry through ROCs in SCBN cells. A, time course of [Ca2+]cyt changes induced by spermine (5 mm) in [Ca2+]o-free or 2 mm [Ca2+]o-containing normal physiological solutions. B, summary data showing peak [Ca2+]cyt responses to spermine in [Ca2+]o or 2 mm [Ca2+]o-containing normal physiological solutions. C, time course of [Ca2+]cyt changes induced by different concentrations of spermine (3 and 10 mm) in the presence of 2-APB (100 μm). D, summary data showing the effect of 2-APB on spermine-induced increase in [Ca2+]cyt. E, time course of [Ca2+]cyt changes induced by different concentrations of spermine (3 and 5 mm) in the presence of SKF96365 (SKF, 10 μm). It is noteworthy that the cells still responded to 10 μm ATP although the spermine-induced increase in [Ca2+]cyt was abolished by SKF96365. F, summary data showing the effect of SKF96365 on the spermine-induced increase in [Ca2+]cyt. Values are expressed as mean ± S.E. of 40–50 cells for each group. *, p < 0.05; **, p < 0.01 versus the baselines before stimulation of spermine (Con) in B or versus the controls in the absence of inhibitors in D and F.
The functional activity of the CaSR was also characterized in human intestinal epithelial cells. As shown in Fig. 9, [Ca2+]o dose-dependently increased [Ca2+]cyt in SW-480 and Caco-2 intestinal epithelial cells with CaSR expression (Fig. 9, A, B, and D) but not in HEK-293 cells without CaSR expression (Fig. 9, C and D), confirming functional expression of the CaSR in human intestinal epithelial cells. Moreover, in SW-480 cells, spermine did not alter [Ca2+]cyt in [Ca2+]0-free solutions but induced a marked [Ca2+]cyt rise in [Ca2+]0-containing solutions (Fig. 9, E and F), which was inhibited significantly by 2-APB (100 μm) (Fig. 9, G and H), further indicating an important role of the ROC in CaSR-mediated Ca2+ entry in human intestinal epithelial cells.
FIGURE 9.

Functional expression of the CaSR in human epithelial cells. A–C, time courses of [Ca2+]cyt changes induced by different concentrations of [Ca2+]o in SW-480, Caco-2, and HEK-293 cells. ATP (10 μm) was used as a positive control. D, summary data showing dose-dependent peak [Ca2+]cyt responses to [Ca2+]o stimulation (0, 1, and 3 mm) in human epithelial cells. E, time course of [Ca2+]cyt changes induced by spermine (5 mm) in [Ca2+]o-free or 2 mm [Ca2+]o-containing normal physiological solutions. F, summary data showing peak [Ca2+]cyt responses to spermine in [Ca2+]o or 2 mm [Ca2+]o-containing solutions. G, time course of [Ca2+]cyt changes induced by different concentrations of spermine (3 and 5 mm) in the presence or the absence of 2-APB (100 μm) in [Ca2+]o-containing solutions. H, summary data showing the effect of 2-APB on the spermine-induced increase in [Ca2+]cyt. Values are expressed as mean ± S.E. of 30–40 cells. *, p < 0.05; **, p < 0.01 versus the baselines before stimulation (Con) in A–C and E or versus the controls in the absence of 2-APB in H.
DISCUSSION
In this study, we demonstrated a novel role for the CaSR in controlling [Ca2+]cyt signaling in duodenal epithelial cells to regulate Ca2+-dependent DBS and advance our understanding of the molecular mechanisms underlying CaSR-mediated [Ca2+]cyt rise in these cells and Ca2+-dependent transepithelial HCO3− secretion.
The CaSR is a member of the pheromone class of G-protein-coupled receptors that is expressed in a variety of tissues throughout the body and has been identified to mediate a wide array of physiological effects (3–5). In the parathyroid gland, it is responsible for regulating body calcium homeostasis by modulating the levels of parathyroid hormone and calcium in the circulation (2, 38). Following the cloning of the CaSR from bovine parathyroid cells in 1993 (2), studies were conducted to determine the expression of the receptor. The CaSR has been shown to be expressed along the entire gastrointestinal tract, where it has many physiological roles, such as modulation of gastrin and gastric acid secretion, intestinal fluid, and ion transports by sensing the concentrations of electrolytes, amino acids, and polyamines (2, 9, 39). Although the CaSR has been cloned for two decades, only one previous study implicated its role in pancreatic HCO3− secretion (40), and another study suggested its possible involvement in l-glutamate-mediated DBS (41). So far, CaSR-mediated intestinal transepithelial HCO3− secretion and the underlying molecular mechanisms are largely unknown.
The DBS is critical to defend the vulnerable duodenal epithelium against various aggressive factors (42, 43). The importance of DBS in protecting duodenal mucosa has been confirmed in patients with duodenal ulcer whose acid-stimulated DBS is only 41% of that in healthy subjects (44). The DBS is impaired in the duodenal tissues from patients with cystic fibrosis, suggesting a pivotal role of the CFTR in mediating the DBS (45). Because the CaSR has been demonstrated to regulate gastric secretion and intestinal Cl− secretion, it is reasonable to infer that it may also modulate intestinal HCO3− secretion. We applied both CaSR agonists and antagonists in two models of duodenal mucosal tissues and intestinal epithelial cells and confirmed that CaSR activation indeed stimulates duodenal transepithelial HCO3− secretion, which is consistent with a previous observation that perfusion of Ca2+ and spermine increased DBS in anesthetized rats (41). However, that study did not further test whether Ca2+ and spermine stimulate the DBS through CaSR activation in the duodenum. Therefore, our study provides novel insights into the CaSR-mediated DBS.
Following our observation that CaSR activation induces DBS, we aimed to elucidate the underlying mechanisms, as established previously for pancreatic HCO3− secretion (40). We demonstrate that CaSR activation raises [Ca2+]cyt in SCBN, SW-480, and Caco-2 cells, likely by evoking [Ca2+]0 entry through the ROC. The SCBN cell model was used in this study because this cell line is the only well characterized nontransformed duodenal epithelial cell line (17, 18); because it expresses functional CFTR channels and has been used widely in the study of Ca2+-dependent Cl− secretion (18, 33); because it secretes HCO3−, as demonstrated by this study and others (18); and because it expresses CaSR protein. Our data indicate that CaSR activation can raise [Ca2+]cyt, which then opens the IKCa and stimulates HCO3− fluxes through the CFTR in duodenal epithelial cells. Because the physiological roles and molecular mechanisms of [Ca2+]cyt-induced HCO3− secretion remain poorly understood in most epithelia (12), this study focuses on CaSR-mediated Ca2+ signaling in intestinal epithelial cells. CaSR activity is demonstrated in canine duodenal epithelial SCBN cells and human intestinal epithelial SW-480 and Caco-2 cells with CaSR expression but not in human epithelial HEK-293 cells without CaSR expression. These data strongly support an important role of the CaSR in regulating the [Ca2+]cyt-dependent function in both human and animal duodenal epithelial cells.
Although the CFTR has been thought to be principally activated by cyclic AMP, Ca2+ signaling can activate the CFTR or potentiate cyclic AMP-mediated CFTR activation (12). The [Ca2+]cyt elevation can stimulate mitochondrial ATP production, which is necessary for the process of epithelial HCO3− secretion (46). During the activation of CFTR, PKA uses ATP to phosphorylate and activate the R domain of CFTR (47). Therefore, the rise in [Ca2+]cyt can activate apical CFTR channels. It is also known that a rise in [Ca2+]cyt modulates the activities of Cl−/HCO3− exchangers, Na+/H+ exchangers, and Na+-HCO3− cotransport in epithelial cells (20, 48–51). The Ca2+-activated chloride channel has been suggested to contribute to HCO3− secretion in some epithelia (12). We reported previously that [Ca2+]cyt activates basolateral IKCa in murine duodenal epithelium to provide a driving force for HCO3− secretion (15). This study combining selective pharmacological inhibitors and knockout mice is in good agreement with other reports on the CaSR-Ca2+-IKCa pathway in the vascular system (52), further supporting our pervious notion that IKCa plays an essential role in Ca2+-mediated DBS (15). All of these actions of [Ca2+]cyt in epithelial cells may contribute to the molecular mechanisms underlying Ca2+-mediated transepithelial HCO3− secretion. However, our data demonstrate that CaSR-[Ca2+]cyt-IKCa-CFTR is a major pathway involved in CaSR-mediated DBS observed in this study (Fig. 10).
FIGURE 10.

Proposed model for CaSR-mediated Ca2+-dependent intestinal epithelial HCO3− secretion through activation of ROC-IKCa-CFTR channels. Dietary/nutrient CaSR modulators stimulate the plasma membrane CaSR of intestinal epithelial cells. CaSR activation elicits signaling events through modulation of intracellular mediators such as PLC, which induces [Ca2+]o entry through the ROCs. An increase in [Ca2+]cyt in epithelial cells activates the IKCa to lead to cell hyperpolarization, providing a driving force for transepithelial HCO3− secretion through the CFTR channels. Black arrows, simulation; red lines, inhibition; thick lines, physiological processes; thin lines, pharmacological activators or inhibitors or genetic knockout used for this study.
It is generally assumed that the regulatory mechanisms involved in intestinal epithelial HCO3− and Cl− secretion are similar, but this notion has never been fully studied and confirmed. Epithelial HCO3− and Cl− secretion is mainly under the control of cyclic AMP and Ca2+ signaling, which may interact and cross-talk to regulate epithelial ion transports (25, 42, 53, 54). Previous studies demonstrated that most well known secretagogues, such as forskolin, ACh, 5-HT, and PGE2, usually stimulate intestinal HCO3− and Cl− secretion in parallel (20, 42, 55, 56). It is not known, however, whether epithelial HCO3− and Cl− secretion has to occur in parallel and whether they are regulated by the same or different signaling/mechanisms. Notably, estrogen inhibits forskolin- and carbachol-induced rat colonic Cl− secretion (57). However, we revealed that estrogen stimulates DBS in humans and mice likely through Ca2+ signaling without altering basal duodenal Isc, an index primarily of epithelial Cl− secretion (13, 58, 59). Therefore, estrogen may play different roles in regulating intestinal HCO3− and Cl− secretion. These findings suggest that epithelial HCO3− and Cl− secretion may not be always triggered in parallel and they may not be regulated by the same signaling/mechanism. We therefore propose that different regulatory mechanisms may exist for intestinal HCO3− and Cl− secretion. Ca2+ signaling may play a key role in HCO3− secretion, but cyclic AMP may play a major role in Cl− secretion. Indeed, in this study, CaSR activation, resulting in an increase in [Ca2+]cyt but a decrease in intracellular cyclic AMP (4–6, 9), leads to a specific DBS without simultaneously altering duodenal Isc. Moreover, IKCa rather than cyclic AMP-activated K+ channels (KCNQ1) are found to be involved in CaSR-mediated DBS, indicating that a sole Ca2+ signaling in the absence of cyclic AMP can trigger the DBS. Therefore, this study not only confirms the pivotal role of [Ca2+]cyt as primary signaling in transepithelial HCO3− secretion but also further supports our notion that intestinal HCO3− and Cl− secretion can be triggered independently by different signaling/mechanisms (13).
What is the physiological relevance of this study? Food nutrients, such as dietary calcium, spermine, and l-amino acids, are CaSR activators that regulate gastric acid secretion, intestinal fluid, and ion transports. Here we confirmed a novel physiological role of these nutrients, namely DBS stimulation, and elucidated the underlying mechanisms. Because the DBS is critical for duodenal mucosal protection, these dietary CaSR modulators may also be involved in this physiological process through the [Ca2+]cyt-IKCa-CFTR cascade (Fig. 10). Because CaSR-mediated Ca2+ signaling can also stimulate Ca2+-activated chloride channel-dependent epithelial secretion that is independent of the CFTR, these CaSR modulators might be used to restore fluid secretion defects in cystic fibrosis disease (60). Moreover, understanding whether different cell signaling triggers distinct intestinal epithelial ion secretion is important for the development of better drugs that can specifically target either intestinal the HCO3− or Cl− secretion pathway. The medications that specifically trigger intestinal HCO3− secretion to protect gastrointestinal tract would not increase Cl− secretion, which might induce diarrhea. Also, the medications that specifically inhibit intestinal Cl− secretion to treat diarrhea would not reduce HCO3− secretion, which might induce gastrointestinal injury.
CONCLUSION
On the basis of this study, we conclude that dietary calcium and spermine could activate the CaSR in duodenal epithelial cells to specifically trigger Ca2+-dependent DBS that protects mucosa, CaSR activation-induced Ca2+ entry through ROC is critical to trigger DBS, and Ca2+ signaling regulates DBS, likely through activation of IKCa and CFTR channels. This study not only reveals that [Ca2+]cyt signaling is critical for CaSR-induced DBS but also provides novel insights into the molecular mechanisms of [Ca2+]cyt signaling-mediated transepithelial HCO3− secretion.
Acknowledgments
We thank Karl Pfeifer (National Institutes of Health) for KCNQ1 knockout mice.
This work was supported, in whole or in part, by National Institutes of Health Grant HL094728. This work was also supported by National Natural Science Foundation of China Grant 31371167 (to H. D.) and by the Department of Veterans Affairs (to V. V.).
- [Ca2+]cyt
- cytosolic Ca2+ concentration
- CaSR
- calcium-sensing receptor
- PLC
- phospholipase C
- [Ca2+]o
- extracellular Ca2+
- DBS
- duodenal bicarbonate secretion
- ROC
- receptor-operated channel
- IKCa
- intermediate conductance Ca2+-activated K+ channel(s)
- CFTR
- cystic fibrosis transmembrane conductance regulator
- Isc
- short circuit current
- 2-APB
- 2-aminoethoxydiphenyl borate
- 5-HT
- 5-hydroxytryptamine.
REFERENCES
- 1. Berridge M. J., Bootman M. D., Roderick H. L. (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529 [DOI] [PubMed] [Google Scholar]
- 2. Brown E. M., Gamba G., Riccardi D., Lombardi M., Butters R., Kifor O., Sun A., Hediger M. A., Lytton J., Hebert S. C. (1993) Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature 366, 575–580 [DOI] [PubMed] [Google Scholar]
- 3. Brown E. M., Vassilev P. M., Hebert S. C. (1995) Calcium ions as extracellular messengers. Cell 83, 679–682 [DOI] [PubMed] [Google Scholar]
- 4. Brown E. M., MacLeod R. J. (2001) Extracellular calcium sensing and extracellular calcium signaling. Physiol. Rev. 81, 239–297 [DOI] [PubMed] [Google Scholar]
- 5. Hofer A. M., Brown E. M. (2003) Extracellular calcium sensing and signalling. Nat. Rev. Mol. Cell Biol. 4, 530–538 [DOI] [PubMed] [Google Scholar]
- 6. Geibel J. P., Hebert S. C. (2009) The functions and roles of the extracellular Ca2+-sensing receptor along the gastrointestinal tract. Annu. Rev. Physiol. 71, 205–217 [DOI] [PubMed] [Google Scholar]
- 7. Cheng S. X. (2012) Calcium-sensing receptor inhibits secretagogue-induced electrolyte secretion by intestine via the enteric nervous system. Am. J. Physiol. Gastrointest. Liver Physiol. 303, G60–G70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheng S. X., Okuda M., Hall A. E., Geibel J. P., Hebert S. C. (2002) Expression of calcium-sensing receptor in rat colonic epithelium: evidence for modulation of fluid secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 283, G240–G250 [DOI] [PubMed] [Google Scholar]
- 9. Geibel J., Sritharan K., Geibel R., Geibel P., Persing J. S., Seeger A., Roepke T. K., Deichstetter M., Prinz C., Cheng S. X., Martin D., Hebert S. C. (2006) Calcium-sensing receptor abrogates secretagogue- induced increases in intestinal net fluid secretion by enhancing cyclic nucleotide destruction. Proc. Natl. Acad. Sci. U.S.A. 103, 9390–9397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Flemström G., Garner A. (1980) Stimulation of gastric acid and bicarbonate secretions by calcium in guinea pig stomach and amphibian isolated mucosa. Acta Physiol. Scand. 110, 419–426 [DOI] [PubMed] [Google Scholar]
- 11. Garner A., Flemström G. (1978) Gastric HCO3−-secretion in the guinea pig. Am. J. Physiol. 234, E535–E541 [DOI] [PubMed] [Google Scholar]
- 12. Jung J., Lee M. G. (2014) Role of calcium signaling in epithelial bicarbonate secretion. Cell Calcium 55, 376–384 [DOI] [PubMed] [Google Scholar]
- 13. Smith A., Contreras C., Ko K. H., Chow J., Dong X., Tuo B., Zhang H. H., Chen D. B., Dong H. (2008) Gender-specific protection of estrogen against gastric acid-induced duodenal injury: stimulation of duodenal mucosal bicarbonate secretion. Endocrinology 149, 4554–4566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Casimiro M. C., Knollmann B. C., Ebert S. N., Vary J. C., Jr., Greene A. E., Franz M. R., Grinberg A., Huang S. P., Pfeifer K. (2001) Targeted disruption of the Kcnq1 gene produces a mouse model of Jervell and Lange-Nielsen syndrome. Proc. Natl. Acad. Sci. U.S.A. 98, 2526–2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dong H., Smith A., Hovaida M., Chow J. Y. (2006) Role of Ca2+-activated K+ channels in duodenal mucosal ion transport and bicarbonate secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G1120–G1128 [DOI] [PubMed] [Google Scholar]
- 16. Sheinin Y., Kállay E., Wrba F., Kriwanek S., Peterlik M., Cross H. S. (2000) Immunocytochemical localization of the extracellular calcium-sensing receptor in normal and malignant human large intestinal mucosa. J. Histochem. Cytochem. 48, 595–602 [DOI] [PubMed] [Google Scholar]
- 17. Pang G., Buret A., O'Loughlin E., Smith A., Batey R., Clancy R. (1996) Immunologic, functional, and morphological characterization of three new human small intestinal epithelial cell lines. Gastroenterology 111, 8–18 [DOI] [PubMed] [Google Scholar]
- 18. Buresi M. C., Schleihauf E., Vergnolle N., Buret A., Wallace J. L., Hollenberg M. D., MacNaughton W. K. (2001) Protease-activated receptor-1 stimulates Ca2+-dependent Cl− secretion in human intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 281, G323–G332 [DOI] [PubMed] [Google Scholar]
- 19. Buret A., Lin Y. C. (2008) Genotypic characterization of an epithelial cell line for the study of parasite-epithelial interactions. J. Parasitol. 94, 545–548 [DOI] [PubMed] [Google Scholar]
- 20. Smith A. J., Chappell A. E., Buret A. G., Barrett K. E., Dong H. (2006) 5-Hydroxytryptamine contributes significantly to a reflex pathway by which the duodenal mucosa protects itself from gastric acid injury. FASEB J. 20, 2486–2495 [DOI] [PubMed] [Google Scholar]
- 21. Negulescu P. A., Machen T. E. (1990) Intracellular ion activities and membrane transport in parietal cells measured with fluorescent dyes. Methods Enzymol. 192, 38–81 [DOI] [PubMed] [Google Scholar]
- 22. Goebel S. U., Peghini P. L., Goldsmith P. K., Spiegel A. M., Gibril F., Raffeld M., Jensen R. T., Serrano J. (2000) Expression of the calcium-sensing receptor in gastrinomas. J. Clin. Endocrinol. Metab. 85, 4131–4137 [DOI] [PubMed] [Google Scholar]
- 23. Quinn S. J., Ye C. P., Diaz R., Kifor O., Bai M., Vassilev P., Brown E. (1997) The Ca2+-sensing receptor: a target for polyamines. Am. J. Physiol. 273, C1315–C1323 [DOI] [PubMed] [Google Scholar]
- 24. Chattopadhyay N., Cheng I., Rogers K., Riccardi D., Hall A., Diaz R., Hebert S. C., Soybel D. I., Brown E. M. (1998) Identification and localization of extracellular Ca2+-sensing receptor in rat intestine. Am. J. Physiol. 274, G122–G130 [DOI] [PubMed] [Google Scholar]
- 25. Seidler U., Blumenstein I., Kretz A., Viellard-Baron D., Rossmann H., Colledge W. H., Evans M., Ratcliff R., Gregor M. (1997) A functional CFTR protein is required for mouse intestinal cAMP-, cGMP- and Ca2+-dependent HCO3− secretion. J. Physiol. 505, 411–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Seidler U. E. (2013) Gastrointestinal HCO3− transport and epithelial protection in the gut: new techniques, transport pathways and regulatory pathways. Curr. Opin. Pharmacol. 13, 900–908 [DOI] [PubMed] [Google Scholar]
- 27. Jepps T. A., Greenwood I. A., Moffatt J. D., Sanders K. M., Ohya S. (2009) Molecular and functional characterization of Kv7 K+ channel in murine gastrointestinal smooth muscles. Am. J. Physiol. Gastrointest. Liver Physiol. 297, G107–G115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bett G. C., Morales M. J., Beahm D. L., Duffey M. E., Rasmusson R. L. (2006) Ancillary subunits and stimulation frequency determine the potency of chromanol 293B block of the KCNQ1 potassium channel. J. Physiol. 576, 755–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee W. K., Torchalski B., Roussa E., Thévenod F. (2008) Evidence for KCNQ1 K+ channel expression in rat zymogen granule membranes and involvement in cholecystokinin-induced pancreatic acinar secretion. Am. J. Physiol. Cell Physiol. 294, C879–C892 [DOI] [PubMed] [Google Scholar]
- 30. Rufo P. A., Merlin D., Riegler M., Ferguson-Maltzman M. H., Dickinson B. L., Brugnara C., Alper S. L., Lencer W. I. (1997) The antifungal antibiotic, clotrimazole, inhibits chloride secretion by human intestinal T84 cells via blockade of distinct basolateral K+ conductances: demonstration of efficacy in intact rabbit colon and in an in vivo mouse model of cholera. J. Clin. Invest. 100, 3111–3120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Devor D. C., Singh A. K., Gerlach A. C., Frizzell R. A., Bridges R. J. (1997) Inhibition of intestinal Cl− secretion by clotrimazole: direct effect on basolateral membrane K+ channels. Am. J. Physiol. 273, C531–C540 [DOI] [PubMed] [Google Scholar]
- 32. Parekh A. B., Putney J. W., Jr. (2005) Store-operated calcium channels. Physiol. Rev. 85, 757–810 [DOI] [PubMed] [Google Scholar]
- 33. Buresi M. C., Buret A. G., Hollenberg M. D., MacNaughton W. K. (2002) Activation of proteinase-activated receptor 1 stimulates epithelial chloride secretion through a unique MAP kinase- and cyclo-oxygenase-dependent pathway. FASEB J. 16, 1515–1525 [DOI] [PubMed] [Google Scholar]
- 34. Buresi M. C., Vergnolle N., Sharkey K. A., Keenan C. M., Andrade-Gordon P., Cirino G., Cirillo D., Hollenberg M. D., MacNaughton W. K. (2005) Activation of proteinase-activated receptor-1 inhibits neurally evoked chloride secretion in the mouse colon in vitro. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G337–345 [DOI] [PubMed] [Google Scholar]
- 35. Tuo B., Wen G., Seidler U. (2009) Differential activation of the HCO3− conductance through the cystic fibrosis transmembrane conductance regulator anion channel by genistein and forskolin in murine duodenum. Br. J. Pharmacol. 158, 1313–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Putney J. W., Jr. (2007) Recent breakthroughs in the molecular mechanism of capacitative calcium entry (with thoughts on how we got here). Cell Calcium 42, 103–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Villalba N., Stankevicius E., Garcia-Sacristán A., Simonsen U., Prieto D. (2007) Contribution of both Ca2+ entry and Ca2+ sensitization to the α1-adrenergic vasoconstriction of rat penile small arteries. Am. J. Physiol. Heart Circ. Physiol. 292, H1157–H1169 [DOI] [PubMed] [Google Scholar]
- 38. Hebert S. C., Brown E. M. (1995) The extracellular calcium receptor. Curr. Opin. Cell Biol. 7, 484–492 [DOI] [PubMed] [Google Scholar]
- 39. Riccardi D., Park J., Lee W. S., Gamba G., Brown E. M., Hebert S. C. (1995) Cloning and functional expression of a rat kidney extracellular calcium/polyvalent cation-sensing receptor. Proc. Natl. Acad. Sci. U.S.A. 92, 131–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bruce J. I., Yang X., Ferguson C. J., Elliott A. C., Steward M. C., Case R. M., Riccardi D. (1999) Molecular and functional identification of a Ca2+ (polyvalent cation)-sensing receptor in rat pancreas. J. Biol. Chem. 274, 20561–20568 [DOI] [PubMed] [Google Scholar]
- 41. Akiba Y., Watanabe C., Mizumori M., Kaunitz J. D. (2009) Luminal l-glutamate enhances duodenal mucosal defense mechanisms via multiple glutamate receptors in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 297, G781–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Flemström G., Isenberg J. I. (2001) Gastroduodenal mucosal alkaline secretion and mucosal protection. News Physiol. Sci. 16, 23–28 [DOI] [PubMed] [Google Scholar]
- 43. Allen A., Flemström G. (2005) Gastroduodenal mucus bicarbonate barrier: protection against acid and pepsin. Am. J. Physiol. Cell Physiol. 288, C1–C19 [DOI] [PubMed] [Google Scholar]
- 44. Isenberg J. I., Selling J. A., Hogan D. L., Koss M. A. (1987) Impaired proximal duodenal mucosal bicarbonate secretion in patients with duodenal ulcer. N. Engl. J. Med. 316, 374–379 [DOI] [PubMed] [Google Scholar]
- 45. Pratha V. S., Hogan D. L., Martensson B. A., Bernard J., Zhou R., Isenberg J. I. (2000) Identification of transport abnormalities in duodenal mucosa and duodenal enterocytes from patients with cystic fibrosis. Gastroenterology 118, 1051–1060 [DOI] [PubMed] [Google Scholar]
- 46. Maléth J., Hegyi P. (2014) Calcium signaling in pancreatic ductal epithelial cells: an old friend and a nasty enemy. Cell Calcium 55, 337–345 [DOI] [PubMed] [Google Scholar]
- 47. Chappe V., Irvine T., Liao J., Evagelidis A., Hanrahan J. W. (2005) Phosphorylation of CFTR by PKA promotes binding of the regulatory domain. EMBO J. 24, 2730–2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Namkung W., Lee J. A., Ahn W., Han W., Kwon S. W., Ahn D. S., Kim K. H., Lee M. G. (2003) Ca2+ activates cystic fibrosis transmembrane conductance regulator- and Cl− -dependent HCO3− transport in pancreatic duct cells. J. Biol. Chem. 278, 200–207 [DOI] [PubMed] [Google Scholar]
- 49. Cohen M. E., Reinlib L., Watson A. J., Gorelick F., Rys-Sikora K., Tse M., Rood R. P., Czernik A. J., Sharp G. W., Donowitz M. (1990) Rabbit ileal villus cell brush border Na+/H+ exchange is regulated by Ca2+/calmodulin-dependent protein kinase II, a brush border membrane protein. Proc. Natl. Acad. Sci. U.S.A. 87, 8990–8994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cohen M. E., Wesolek J., McCullen J., Rys-Sikora K., Pandol S., Rood R. P., Sharp G. W., Donowitz M. (1991) Carbachol- and elevated Ca2+-induced translocation of functionally active protein kinase C to the brush border of rabbit ileal Na+ absorbing cells. J. Clin. Invest. 88, 855–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bachmann O., Reichelt D., Tuo B., Manns M. P., Seidler U. (2006) Carbachol increases Na+- HCO3− cotransport activity in murine colonic crypts in a M3-, Ca2+/calmodulin-, and PKC-dependent manner. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G650–G657 [DOI] [PubMed] [Google Scholar]
- 52. Weston A. H., Absi M., Ward D. T., Ohanian J., Dodd R. H., Dauban P., Petrel C., Ruat M., Edwards G. (2005) Evidence in favor of a calcium-sensing receptor in arterial endothelial cells: studies with calindol and Calhex 231. Circ. Res. 97, 391–398 [DOI] [PubMed] [Google Scholar]
- 53. Flemstrom G., Garner A. (1982) Gastroduodenal HCO3− transport: characteristics and proposed role in acidity regulation and mucosal protection. Am. J. Physiol. 242, G183–G193 [DOI] [PubMed] [Google Scholar]
- 54. Lee R. J., Foskett J. K. (2010) cAMP-activated Ca2+ signaling is required for CFTR-mediated serous cell fluid secretion in porcine and human airways. J. Clin. Invest. 120, 3137–3148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tuo B. G., Sellers Z., Paulus P., Barrett K. E., Isenberg J. I. (2004) 5-HT induces duodenal mucosal bicarbonate secretion via cAMP- and Ca2+-dependent signaling pathways and 5-HT4 receptors in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 286, G444–G451 [DOI] [PubMed] [Google Scholar]
- 56. Dong H., Sellers Z. M., Smith A., Chow J. Y., Barrett K. E. (2005) Na+/Ca2+ exchange regulates Ca2+-dependent duodenal mucosal ion transport and HCO3− secretion in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G457–G465 [DOI] [PubMed] [Google Scholar]
- 57. Condliffe S. B., Doolan C. M., Harvey B. J. (2001) 17β-oestradiol acutely regulates Cl− secretion in rat distal colonic epithelium. J. Physiol. 530, 47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Barrett K. E., Smitham J., Traynor-Kaplan A., Uribe J. M. (1998) Inhibition of Ca2+-dependent Cl− secretion in T84 cells: membrane target(s) of inhibition is agonist specific. Am. J. Physiol. 274, C958–C965 [DOI] [PubMed] [Google Scholar]
- 59. Barrett K. E., Keely S. J. (2000) Chloride secretion by the intestinal epithelium: molecular basis and regulatory aspects. Annu. Rev. Physiol. 62, 535–572 [DOI] [PubMed] [Google Scholar]
- 60. Lee R. J., Foskett J. K. (2014) Ca signaling and fluid secretion by secretory cells of the airway epithelium. Cell Calcium 55, 325–336 [DOI] [PubMed] [Google Scholar]