

Abstract
Background/objectives
Fasting dyslipidemia is commonly observed in insulin resistant states and mechanistically linked to hepatic overproduction of very low density lipoprotein (VLDL). Recently, the incretin hormone glucagon-like peptide-1 (GLP-1) has been implicated in ameliorating dyslipidemia associated with insulin resistance and reducing hepatic lipid stores. Given that hepatic VLDL production is a key determinant of circulating lipid levels, we investigated the role of both peripheral and central GLP-1 receptor (GLP-1R) agonism in regulation of VLDL production.
Methods
The fructose-fed Syrian golden hamster was employed as a model of diet-induced insulin resistance and VLDL overproduction. Hamsters were treated with the GLP-1R agonist exendin-4 by intraperitoneal (ip) injection for peripheral studies or by intracerebroventricular (ICV) administration into the 3rd ventricle for central studies. Peripheral studies were repeated in vagotomised hamsters.
Results
Short term (7–10 day) peripheral exendin-4 enhanced satiety and also prevented fructose-induced fasting dyslipidemia and hyperinsulinemia. These changes were accompanied by decreased fasting plasma glucose levels, reduced hepatic lipid content and decreased levels of VLDL-TG and -apoB100 in plasma. The observed changes in fasting dyslipidemia could be partially explained by reduced respiratory exchange ratio (RER) thereby indicating a switch in energy utilization from carbohydrate to lipid. Additionally, exendin-4 reduced mRNA markers associated with hepatic de novo lipogenesis and inflammation. Despite these observations, GLP-1R activity could not be detected in primary hamster hepatocytes, thus leading to the investigation of a potential brain–liver axis functioning to regulate lipid metabolism. Short term (4 day) central administration of exendin-4 decreased body weight and food consumption and further prevented fructose-induced hypertriglyceridemia. Additionally, the peripheral lipid-lowering effects of exendin-4 were negated in vagotomised hamsters implicating the involvement of parasympathetic signaling.
Conclusion
Exendin-4 prevents fructose-induced dyslipidemia and hepatic VLDL overproduction in insulin resistance through an indirect mechanism involving altered energy utilization, decreased hepatic lipid synthesis and also requires an intact parasympathetic signaling pathway.
Keywords: Glucagon-like peptide-1 (GLP-1), Very low density lipoprotein (VLDL), Fasting dyslipidemia, Insulin resistance, Hepatic steatosis, Incretin
Abbreviations: apoB100, apolipoproteinB100; FFA, free fatty acid; GLP-1, glucagon-like peptide-1; GLP-1R, GLP-1 receptor; ICV, intracerebroventricular; ip, intraperitoneal; RER, respiratory exchange ratio; T2D, type 2 diabetes; VLDL, very low density lipoprotein
1. Introduction
A central pathophysiological feature of type 2 diabetes (T2D) and the metabolic syndrome is fasting dyslipidemia that is characterized by enhanced very low density lipoprotein (VLDL) production, formation of atherogenic small dense LDL, and decreased HDL-cholesterol [1]. While insulin normally acts as a negative regulator of VLDL production, decreased hepatic insulin sensitivity in T2D results in the overproduction of these triglyceride (TG)-rich apolipoprotein B100 (apoB100) containing particles. This ultimately leads to the fasting hypertriglyceridemia that is associated with enhanced cardiovascular risk [1]. Thus, regulation of hepatic VLDL production in insulin resistance is important for controlling fasting dyslipidemia and reducing the formation of atherogenic particles.
The incretin hormone glucagon-like peptide-1 (GLP-1) has been recently implicated in decreasing postprandial dyslipidemia in rodent models and diabetic patients by reducing intestinal lipoprotein production [2–5]. Cardiovascular biomarkers in T2D patients were also shown to improve significantly after GLP-1 receptor (GLP-1R) agonism as indicated by reduced plasma TG, as well as reduced LDL-cholesterol and total cholesterol levels [6–8]. In regard to the liver specifically, GLP-1R agonism in high-fat fed mice has been shown to enhance hepatic β-oxidation thereby reducing lipid stores [9] and making GLP-1 a potential therapy in the treatment of hepatic steatosis [9–12]. While recent studies suggest that hepatic VLDL production can be modulated by GLP-1R agonism [13] other literature indicates the lack of a hepatic GLP-1R [14–18]. Nonetheless, the GLP-1R has been detected in hypothalamic nuclei [19] and central GLP-1R activation has been shown to regulate both hepatic glycogen storage [20] and peripheral lipid deposition [21].
Given that GLP-1 has been shown to improve cardiovascular biomarkers and modulate hepatic lipid metabolism, we examined the effects of both peripheral and central GLP-1R agonism in the regulation of fasting dyslipidemia and VLDL production. In the present study, we employ the fructose-fed Syrian golden hamster as a model of diet-induced insulin resistance and VLDL overproduction [22]. Additional studies were carried out in C57BL/6 mice to assess whether the changes in VLDL are unique to the hamster model. The peripheral and central effects of the GLP-1R agonists exendin-4 or liraglutide on fasting dyslipidemia, hepatic steatosis and VLDL production were examined. In the present report, we provide experimental evidence for a potential mechanism by which GLP-1R agonism indirectly regulates hepatic VLDL production and fasting dyslipidemia; namely through changes in hepatic and whole body energy balance and lipid metabolism which may occur through a parasympathetic signaling pathway.
2. Methods
2.1. Animals
Male Syrian golden hamsters (Mesocricetus auratus) weighing 110–140 g were purchased from Charles River (Montreal, QC, Canada), housed individually on a 12 h light–dark cycle and had access to food (chow) and water ad libitum. Blood was collected by retro-orbital bleeds after a 5 h fast prior to initiating diet experiments. After a 1 week acclimatization period, hamsters were given a fructose diet (60% fructose 20% casein; Dyets, Bethlehem, USA) to induce insulin resistance [22]. Pair-fed hamsters received the same daily amount of food (in grams) as measured in exendin-4 treated hamsters. In the intervention study, hamsters received twice daily intraperitoneal (ip) injections of vehicle (saline) or exendin-4 (20 μg/kg; Bachem, Torrance, CA, USA) during the last 7 days of a 14 day feeding. This study was repeated in hamsters that underwent either vagotomy or sham surgery. In the preventative study, hamsters started the fructose diet and peripheral exendin-4 treatment simultaneously for a total of 10 days. For the central studies, hamsters received once daily intracerebroventricular (ICV) administration of vehicle or exendin-4 (250 ng) into the 3rd ventricle of the brain for the final 4 days of a 14 day feeding. Hamster weights and food consumption were monitored every 1–3 days.
Male C57BL/6J mice weighing 30–35 g were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Mice were maintained on a 12 h light–dark cycle and had access to food (chow) and water ad libitum. After a 1 week acclimatization period, mice were fed a chow diet with 4% fructose water for 16 weeks and received daily ip injections of liraglutide (20 μg/kg; Bachem, Torrance, CA, USA) or vehicle during the last 4 weeks. All animal protocols were approved by the Animal Ethics Committee of the Hospital for Sick Children.
2.2. ICV cannula implantation
Hamsters were anesthetized using a ketamine xylazine mixture (80:10 mg/kg) given ip. A 31 gauge guide cannula was inserted into the third ventricle using flat skull coordinates (5 mm depth at the intersection of midline and bregma) and affixed using acrylic glue and dental cement. Hamsters were allowed to recover for 1 week and given Metacam (1 mg/kg) as a postoperative analgesic.
2.3. Subdiaphragmatic truncal vagotomy
An upper abdominal midline incision was made under isoflurane anesthesia and the left lobe of the liver was retracted laterally using saline-soaked gauze and cotton swabs. The anterior and posterior trunks of the vagus nerve on the subdiaphragmatic esophagus were identified using a microscope and exposed with the use of blunt dissection. Fine tip electrocautery and micro forceps was used for isolation of both trunks from the surrounding tissues and then cauterized cautiously before their bifurcations. The abdominal muscle wall layer and skin was repaired by sutures. In the sham surgery, transections were not preformed following exposing the vagal trunks.
2.4. Assessment of hepatic lipoprotein production
Hamsters underwent jugular vein catheterization as previously described in Ref. [3]. Hamsters were allowed to recover for 24 h and after a 5 h fast a baseline blood sample (400 μl) was collected. Hamsters then received vehicle or exendin-4 by ip (20 μg/kg) or ICV (250 ng) injections. Pluoronic F-127 (poloxamer; 20% in saline, 0.5 g/kg) or Triton-WR1339 (0.5 g/kg) was given 10 min after initial treatment by ip injection or iv infusion, respectively and blood was collected at 2, 4 and 6 h. Liver and adipose tissue were excised under isoflurane anesthesia for weight analysis.
Mice were fasted for 5 h and a baseline blood sample (100 μl) was collected from the tail vein into lithium heparin-coated tubes. Mice then received a final ip injection of liraglutide (20 μg/kg) or vehicle. Poloxamer (0.5 mg/kg) was given 10 min after initial treatment by ip injection and blood (100 μl) was collected at 2 and 4 h by tail bleeds and at 6 h by cardiac puncture. Livers were excised under isoflurane anesthesia, embedded in OCT buffer and flash frozen in liquid nitrogen for histological analysis.
2.5. Lipoprotein isolation and fractionation
To separate the plasma, blood samples were centrifuged for 10 min at 4 °C and 6000 rpm. Plasma (100 μl) was overlayed with 1 ml of KBr (1.006 g/ml) and centrifuged for 5 h at 40,000 rpm at 4 °C (TLA-45 rotor; Beckman Coulter, ON, Canada). The VLDL fraction (Sf 20–400) was collected as the top 200 μl of the tube volume. To separate various lipoprotein fractions, baseline plasma samples from each treatment group were pooled together and FPLC analysis was carried out as previously described in Ref. [3].
2.6. Plasma lipid, insulin and glucose measurements
Plasma TG, cholesterol, glucose, AST and ALT were measured using Ortho Clinical Diagnostics V950 (VITROS, Buckinghamshire, UK) enzymatic-based colorimetric assay or enzymatic-based multiple-point rate assay according to the manufacturer's protocol. Plasma FFA, VLDL-TG and VLDL-cholesterol were determined by an enzymatic-based colorimetric assay (Randox, Crumlin, UK) and plasma insulin levels were measured by ELISA (Mercodia, Uppsala, Sweden) according to the manufacturer's instructions. ApoB100 immunoblotting was performed as previously described in Ref. [3].
To perform glucose tolerance tests, mice were fasted for 16 h and glucose (2 g/kg) was administered ip. Glucose levels were measured by tail vein sampling with a portable glucometer (Roche Diagnostics).
2.7. Metabolic cage studies
An additional set of hamsters underwent the intervention study described above, were placed into an 8-cage Comprehensive Lab Animal Monitoring System (CLAMS; Columbus Instruments, Columbus, OH, USA) on the 12th day, and given a minimum of 24 h to acclimatize. Respiratory exchange ratio (RER), CO2 production, O2 consumption, heat production and locomoter activity were monitored every 18 min during a 48 h period at room temperature. Cages were opened and calculations were stopped for 2 h between 8:30 and 10:30 am and for 1 h between 4:30 and 5:30 pm daily for replenishing food, measuring body weight and performing injections. On the final day, hamsters were removed from the cages, given a final ip injection of exendin-4 (20 μg/kg) or saline and subsequently fasted for 5 h. Hamsters were then anesthetized using isoflurane and liver tissue was excised and flash frozen in liquid nitrogen and stored at −80 °C.
2.8. Hepatocyte isolation and cAMP quantification
Primary hamster hepatocytes were isolated as previously described in Ref. [23] and cultured in DMEM with 10% FBS. Cells were plated in 24 well plates at a density of either 0.5 or 1 million cells/well and maintained at 37 °C and 5% CO2. Cell viability was determined by trypan blue exclusion assay.
Primary hamster hepatocytes were pre-treated with IBMX (500 μM) for 30 min prior to an additional 30 min treatment with exendin-4 (50 nM), forskolin (20 μM) or vehicle. Cells were lysed and prepared for a cAMP ELISA according to manufacturer's protocol (Applied Biosystems, California, USA). cAMP assay measurements were obtained using a luminometer (Envision, PerkinElmer, Turku Finland).
2.9. Lipid extraction and analysis
Primary hepatocytes were treated with or without 0.4 mM oleic acid for 16 h and subsequently treated with exendin-4 (1 nM, 20 nM, 50 nM and 100 nM) for 6 h in the presence of 20% BSA and maintained at 37 °C and 5% CO2. Lipids were extracted using a hexane:isopropanol (3:2) solvent mixture and TG was quantified using an enzymatic-based colorimetric assay (Randox).
Lipids were extracted from liver tissue as previously described in Ref. [24]. For histological lipid analysis, frozen liver tissue was sectioned and stained with Oil Red O.
2.10. Polymerase chain reaction (PCR)
Flash frozen tissues were crushed in liquid nitrogen using a mortar and pestle. Total RNA was extracted using Qiagen RNeasy Mini Kit according to manufacturer's instructions. cDNA synthesis and quantitative RT-PCR were carried out as previously described in Ref. [25]. Primers are shown in Table S1.
2.11. Statistical analyses
All results are presented as mean ± SEM. Statistical comparisons were performed using two-tailed Student's unpaired t-test, Mann–Whitney U test, linear regression, one-way ANOVA or two-way ANOVA with the Bonferroni post hoc test. All tests were performed using GraphPad Prism software (San Diego, CA, USA).
3. Results
3.1. Peripheral GLP-1R agonism enhances satiety and reduces body weight
To understand the effect of peripheral GLP-1R stimulation in insulin resistant states, hamsters were put on a 60% fructose diet to induce insulin resistance and VLDL overproduction [22]. Insulin resistance was assumed based on increased dyslipidemia previously shown to occur within this hamster model in insulin resistant states [22]. In an intervention study, fructose-fed hamsters received ip exendin-4 administration for the final 7 days of a 14 day feeding (Figure 1A). Consistent with a known role to enhance satiety, short-term peripheral exendin-4 rapidly reduced food consumption (Figure 1B). This led to a gradual and significant reduction in body mass (Figure 1B,C) despite no significant changes in liver or fat pad weight (Figure S1A,B). Plasma AST and ALT levels were unchanged following exendin-4 treatment (Figure S1C,D). To confirm the above effects, and to assess whether or not exendin-4 could prevent the onset of fructose-induced dyslipidemia, a preventative study was carried out Figure 1D) in which hamsters received both exendin-4 and the fructose diet simultaneously for 10 days. In the preventive study, exendin-4 similarly decreased both food consumption and body mass (Figure 1E,F), however, the satiety effect normalized by the 10th day leading to a plateau in body weight loss. Despite recovered satiety, exendin-4 prevented the diet-induced increase in both fat pad and liver weight (Figure S2A,B) which was further associated with a trend towards decreased hepatic TG and cholesterol levels (Figure S2C,D). No changes in plasma AST and ALT levels were observed (Figure S2E,F).
Figure 1.
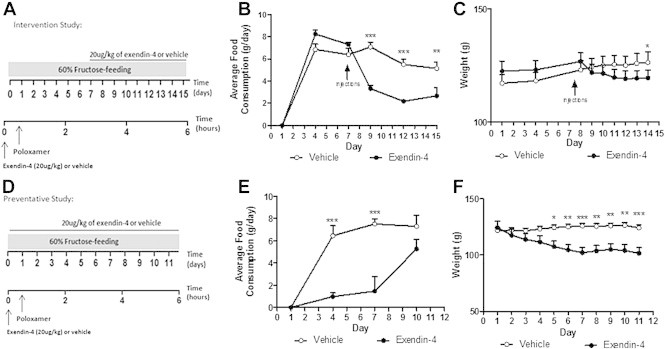
Short term peripheral exendin-4 decreases food consumption and body weight. Fructose-fed hamsters received vehicle or exendin-4 (20 μg/kg) for 7 (A–C; Intervention study) or 10 days (D–F; Preventative study). Fasting plasma was collected following poloxamer (0.5 g/kg). Average daily food consumption (B,E) and body weight (C,F) during intervention (B,C) and preventative (D,F) studies. Intervention n = 5–7; Preventative n = 6; *p < 0.05, **p < 0.01, ***p < 0.001 as analyzed by two-way ANOVA with Bonferroni post hoc test.
3.2. Peripheral GLP-1R stimulation prevents the onset of fructose-induced dyslipidemia
Our laboratory has previously demonstrated a role for exendin-4 in the regulation of postprandial dyslipidemia [3]. To determine whether exendin-4 plays a similar role in fasting dyslipidemia, we examined changes in fasting plasma lipids, glucose and insulin levels prior to and following the intervention study. As expected, the fructose diet significantly elevated both fasting plasma TG and cholesterol levels despite any changes in plasma glucose (Figure 2A–C). Peripheral GLP-1R stimulation prevented fructose-induced hypertriglyceridemia (Figure 2A) and hypercholesterolemia (Figure 2B) and reduced basal levels of fasting plasma glucose (Figure 2C; Figure S3A). Strikingly similar results were obtained following the preventative study (Figure 2D–F; Figure S3C), confirming the ability of peripheral GLP-1R agonism to prevent diet-induced fasting dyslipidemia. Reduction in fasting lipid levels were also accompanied by significantly decreased fructose-induced hyperinsulinemia (Figure S3B).
Figure 2.
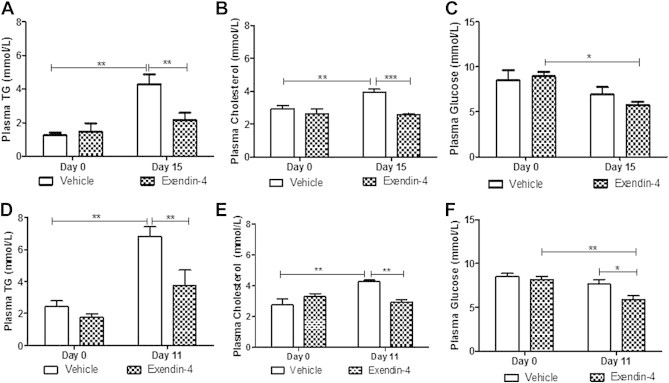
Peripheral exendin-4 prevents fructose-induced dyslipidemia. Plasma from vehicle or exendin-4 hamsters on Day 0 and after the intervention (Day 15; A–C) or preventative study (Day 11; D–F). (A,D) Plasma triglyceride (TG), (B,E) cholesterol and (C,F) glucose. Intervention n = 5–7; Preventative n = 6; *p < 0.05, **p < 0.01, ***p < 0.001 as analyzed by two-way ANOVA with Bonferroni post hoc test.
3.3. Exendin-4 reduces hepatic lipoprotein production and lipidation in insulin resistant states
Based on the observation that exendin-4 prevented the onset of fructose-induced dyslipidemia, we next sought to determine whether these effects were due to modulation of VLDL by using poloxamer to block lipoprotein clearance and allow for the rate of production to be assessed. In the intervention study, exendin-4 significantly reduced VLDL-associated TG, cholesterol and apoB100 accumulation (Figure 3A–C). The preventative study showed reduced VLDL-cholesterol and apoB100 following exendin-4 treatment (Figure 3E–F) with no significant changes in VLDL-TG (Figure 3D). Similar reductions were observed for plasma TG, cholesterol and apoB100 levels following both the intervention and preventative study (Figure S4).
Figure 3.
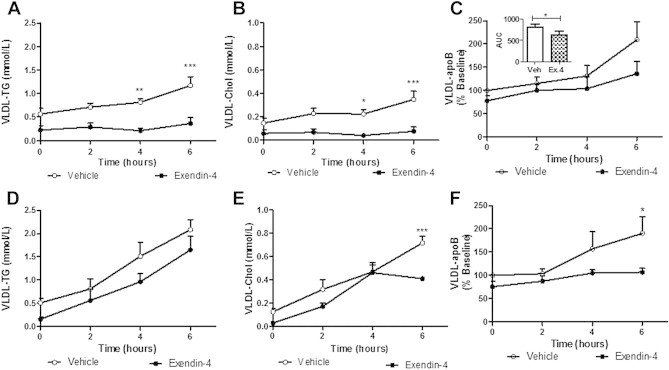
Peripheral exendin-4 decreases VLDL associated-TG, -cholesterol and apoB100 accumulation. VLDL isolated from vehicle or exendin-4 hamsters in the intervention (A–C) or preventative (D–F) study. (A,D) VLDL-triglyceride, (B,E) cholesterol, (C,F) and apoB100 accumulation with area under the curve (AUC) for VLDL-apoB100 (C). Intervention n = 5–7; Preventative n = 6; *p < 0.05, **p < 0.01, ***p < 0.001 as analyzed by two-way ANOVA with Bonferroni post hoc test. AUC analyzed by Mann–Whitney U test.
A linear regression analysis was carried out to determine whether the above changes were due to decreased rate of production or due to differences at baseline. For the intervention study, slopes were significantly decreased in exendin-4 treated hamsters for plasma-TG, VLDL-TG and VLDL-cholesterol (Table S2A) thereby suggesting reduced lipidation of VLDL particles. For the preventative study, slopes were significantly different for VLDL-apoB100 levels (Table S2B) suggesting reduced particle production following exendin-4 administration.
3.4. The ability of peripheral GLP-1R agonism to prevent fasting dyslipidemia is independent of changes in food intake
GLP-1R agonism enhanced satiety and significantly reduced food consumption in hamsters in both the intervention and preventative studies (Figure 1). To determine whether the changes in fasting lipid levels were independent of changes in food consumption, an intervention study was repeated under pair-fed conditions. While the fructose diet significantly enhanced both fasting plasma TG and FFA levels in food-restricted controls, this effect was negated by exendin-4 administration (Figure 4A,C). Despite no changes in fasting plasma cholesterol (Figure 4B), exendin-4 significantly reduced fasting VLDL-TG, VLDL-cholesterol and LDL-cholesterol levels (Figure 4D,E) in comparison to pair-fed controls. These data suggest that exendin-4-mediated decreases in fasting dyslipidemia and VLDL are independent of changes in food consumption.
Figure 4.
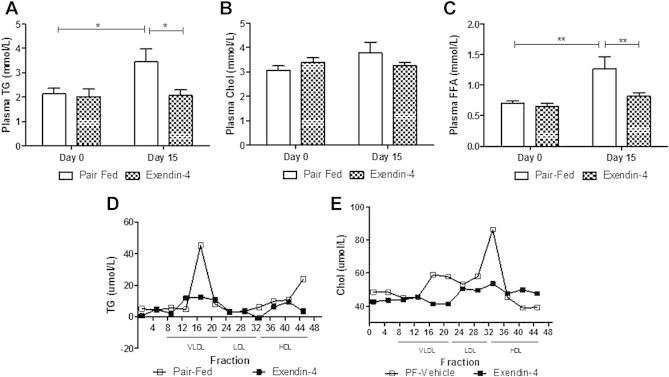
Exendin-4 decreases dyslipidemia and VLDL–TG independent of changes in food intake. Fasting plasma from pair-fed or exendin-4 hamsters on Day 0 or following treatment (Day 15). (A) Plasma triglyceride (TG), (B) cholesterol and (C) FFA levels. FPLC (D) TG and (E) cholesterol. n = 10–11. (D,E). *p < 0.05, **p < 0.01, ***p < 0.001 as analyzed by two-way ANOVA with Bonferroni post hoc test.
3.5. The GLP-1R mediated amelioration in dyslipidemia and hepatic steatosis is not unique to hamsters and is also observed in a hyperlipidemic mouse model
To confirm that the hyperlipidemic effects of GLP-1R agonism were not unique to the hamster model, a chronic intervention study was carried out in liraglutide-treated mice receiving 4% fructose water and a chow diet for 16 weeks (Figure S5A). Peripheral liraglutide enhanced glucose tolerance after 3 weeks of treatment (Figure 5A) with no changes in weight (Figure S5B) or food consumption (data not shown). Liraglutide completely blunted fasting plasma TG accumulation (Figure 5B) and reduced VLDL-TG (Figure 5C) and VLDL-cholesterol levels (p = 0.051; Figure S5C). Additionally, liraglutide significantly reduced both hepatic TG and cholesterol accumulation (Figure 5D,E) which was visually confirmed by decreased oil red O staining (Figure 5F). Thus, peripheral GLP-1R agonism decreased fasting dyslipidemia, VLDL-lipidation and hepatic steatosis in a diet-induced hyperlipidemic mouse model despite any changes in weight or food consumption.
Figure 5.
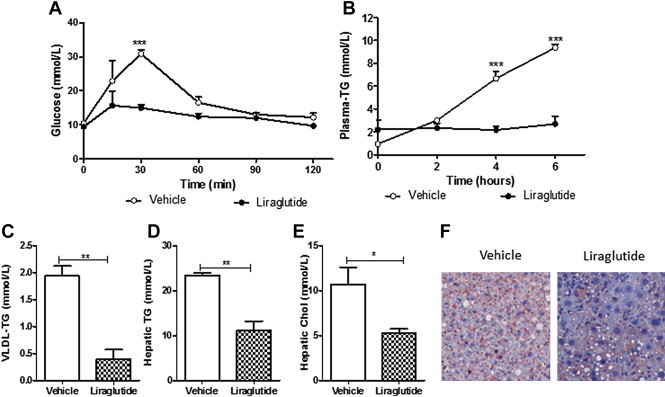
Liraglutide reduces dyslipidemia and hepatic steatosis in mice independent of changes in weight or food consumption. C57BL/6J mice receiving 4% fructose were treated with or liraglutide (20 μg/kg) for 4 weeks. Fasting plasma was collected following poloxamer (0.5 g/kg). (A) Glucose tolerance 3 weeks posttreatment. (B) Plasma triglyceride (TG) accumulation (C) VLDL-TG. (D,E) Hepatic TG and cholesterol. (F) Hepatic oil Red O staining. n = 3; *p < 0.05, **p < 0.01, ***p < 0.001. Analyzed by two-way ANOVA with Bonferroni post hoc test (A,B) or by Student's unpaired t-test (C–E).
3.6. Exendin-4 enhances whole body lipid utilization and regulates hepatic lipid homeostasis
To investigate whether changes in metabolic rate could account for the exendin-4 lipid lowering effects, hamsters were placed into metabolic cages during the intervention study. Despite a similar amount of weight loss between pair-fed and exendin-4 treated hamsters (Figure 6A), exendin-4 significantly reduced respiratory exchange ratio (RER) in comparison to both vehicle and pair-fed controls 3 h postinjection (Figure 6B; Figure S6A). This was accompanied by a decrease in CO2 production in comparison to vehicle (Figure 6C; Figure S6B). Interestingly, pair-fed and vehicle hamsters did not differ in RER and CO2 production indicating that exendin-4 causes a switch from carbohydrate to lipid as the main source of energy utilization independent of changes in satiety. No change in O2 consumption or heat production (Figure S6C,D) indicated no change in energy expenditure. Lastly, no changes in locomotor activity were observed (Figure S6E).
Figure 6.
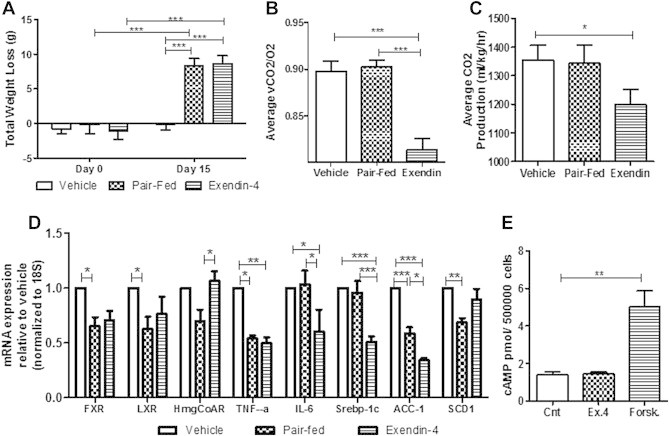
Exendin-4 increases lipid utilization and decreases hepatic de novo lipogenesis via an indirect mechanism. Metabolic cage analysis for vehicle, pair-fed and exendin-4 hamsters from the intervention study. (A) Body weight change. (B) RER and (C) CO2 averages 3 h postinjection. n = 10–11. Livers were excised for PCR analysis. (D) mRNA expression for hepatic inflammation and lipid homeostasis. n = 8. (E) cAMP levels of primary hamster hepatocytes treated ex vivo with vehicle, exendin-4 (100 nM) or forskolin (50 nM). n = 3. *p < 0.05, **p < 0.01, ***p < 0.001. Analyzed by two-way ANOVA with Bonferroni post hoc test (A) or by one way ANOVA (C–E).
To assess whether exendin-4 altered hepatic lipid metabolism, qRT-PCR analysis was performed. Both pair-fed and exendin-4 treated hamsters had significantly reduced fxr, lxr and TNF-α mRNA expression compared to vehicle (Figure 6D). Pair-feeding alone caused a reduction in mRNA expression of HMG-CoA reductase, the rate limiting enzyme for cholesterol production, and scd1, important in long chain FA synthesis (Figure 6D). Changes induced by exendin-4 in a food intake-independent manner included reduced hepatic IL-6 mRNA expression and markers for de novo lipogenesis including Srebp-1c and acc1 (Figure 6D). Therefore, while reductions in food intake may account for changes in long chain FA synthesis and improved cholesterol homeostasis, exendin-4 appears to play an important role in decreasing hepatic de novo lipogenesis.
3.7. GLP-1 indirectly targets the liver to modulate hepatic lipid and lipoprotein metabolism
The effects of exendin-4 on hepatic lipid metabolism and VLDL production raise the question of whether a hepatic GLP-1R exists. To test this, primary hamster hepatocytes were isolated and analyzed for active GLP-1R signaling via cAMP production. Ex vivo exendin-4 did not exhibit altered cAMP production (Figure 6E), did not alter cellular or secreted TG levels following oleic acid loading (Figure S7B,C), and did not change cell survival (data not shown). These data suggest the absence of a hepatic GLP-1R and indicate that indirect mechanisms account for the effect of GLP-1 on hepatic lipid and lipoprotein production.
3.8. Exendin-4 decreases fasting dyslipidemia via a parasympathetic signaling pathway
Recently central GLP-1R signaling has been implicated in regulating peripheral lipid deposition [21]. To assess the potential involvement of a central signaling pathway in the regulation of fasting dyslipidemia, fructose-fed hamsters received ICV administration of exendin-4 for 4 days (Figure 7A). Similar to the peripheral effects, central exendin-4 significantly reduced both body mass and food consumption (Figure 7B,C; Figure S8A,B) and prevented fructose-induced hypertriglyceridemia (Figure 7D) and hyperinsulinemia (Figure S8C). Central exendin-4 also caused a decrease in fasting VLDL associated-TG, -cholesterol and apoB100 and a simultaneous increase in LDL-TG, -cholesterol and -apoB100 as well as HDL-cholesterol (Figure 7E–G) This suggests that central exendin-4 results in the formation of smaller LDL-sized hepatic lipoprotein particles. Finally, no changes in plasma AST or ALT levels were observed (Figure S8D,E). Given the involvement of a central signaling pathway, we next investigated the potential involvement of parasympathetic signaling. A peripheral intervention study was repeated in vagotomized hamsters, however, a shorter 2 h time course was examined due to the more potent effect of triton infusion on lipid accumulation. Vagotomy alone induced weight loss (Figure 8A), decreased food consumption (Figure 8B) and non-significantly decreased fasting plasma TG levels (Figure 8C, figure; Day 0) in comparison to sham controls. Exendin-4 prevented weight gain, reduced food consumption and further prevented fructose-induced hypertriglyceridemia and hypercholesterolemia in sham but not vagotomized hamsters (Figure 8A–D). Exendin-4 also reduced VLDL production rate only in sham hamsters as indicated by reduced VLDL-associated TG, cholesterol, and apoB100 (Figure 8E,F; Figure S9A). Thus, the lipid-lowering effects of exendin-4 were negated in vagotomized hamsters suggesting the potential involvement of vagal signaling in the regulation of GLP-1 mediated reductions in fasting dyslipidemia. Lastly, no changes in plasma AST or ALT (Figure S9B,C).
Figure 7.
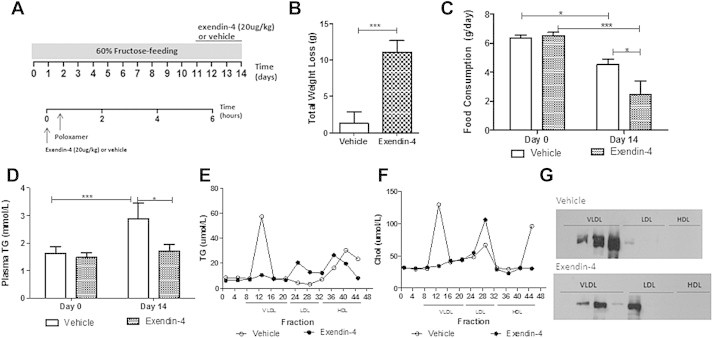
Central exendin-4 decreases weight and food consumption and decreases both fasting hypertriglyceridemia and VLDL-associated lipid and apoB100 levels. (A) Fructose-fed hamsters received intracerebroventricular (ICV) vehicle or exendin-4 (250 μg) and FPLC analysis was carried out. (B) Total change in weight. (C,D) Changes in food consumption and fasting plasma TG levels before (Day 0) and after (Day 14) the diet/treatment. (E–G) Triglyceride (TG), cholesterol and apoB100. n = 8; *p < 0.05, ***p < 0.001. Analyzed by two-way ANOVA with Bonferroni post hoc test (C,D) or by Student's unpaired t-test (B).
Figure 8.
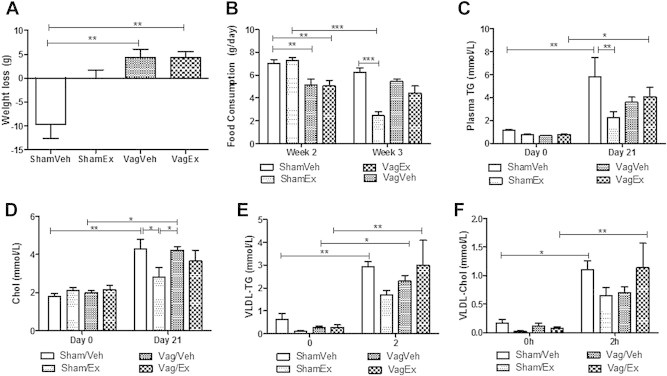
Exendin-4 requires vagal signaling to reduce fasting dyslipidemia. Hamsters underwent a subdiaphragmatic truncal vagotomy/sham surgery and allowed 1 week recovery followed by the intervention study. (A) Total weight loss (B) Changes in food consumption on fructose (week 2) and during treatment (week 3). (C) Changes in fasted plasma TG and (D) cholesterol levels before (Day 0) and after the study (Day 21). (E,F) Changes in VLDL-TG and VLDL-cholesterol up to 2 h posttriton. n = 3. *p < 0.05, **p < 0.01, ***p < 0.001. Analyzed by one way (A) or two-way ANOVA (B–F) with Bonferroni post hoc test.
4. Discussion
GLP-1R agonism has been suggested as a treatment for hepatic steatosis due to its ability to decrease hepatic lipid content [9–12]. Given that VLDL production is dependent on hepatic lipid availability, we investigated the effects of GLP-1R agonism on hepatic lipoprotein overproduction and fasting dyslipidemia associated with insulin resistance. Here we demonstrate that both short term peripheral and central GLP-1R stimulation induce weight loss, enhanced satiety and prevent fructose-induced dyslipidemia. The inability to confirm expression of a hepatic GLP-1R suggests an indirect mechanism. While vagotomy alone caused a modest reduction in fasting plasma TG, the exendin-4 mediated lipid lowering effects were negated in absence of vagal signaling. This suggests the involvement of a parasympathetic signaling pathway, however, the mechanism also appears to involve changes in whole body energy utilization.
Recently, Parlevliet et al. [13] have shown that 4 weeks of peripheral exendin-4 administration in high-fat fed apoE*3-leiden mice reduced VLDL production as indicated by decreased apoB levels 2 h posttreatment. We have confirmed these results in a non-transgenic hamster model showing more prolonged reductions in VLDL up to 6 h posttreatment. The hamster is a more ideal model given its similarity to humans in that the liver only produces apoB100-containing VLDL, thus allowing for determination of the origin (hepatic or intestinal) of the lipoprotein being examined. Thus, we were able to specifically show hepatic-derived apoB100 levels were reduced after exendin-4. While many studies have demonstrated the effects of chronic GLP-1R agonism on the liver [12,13,26], we show that the decreases in VLDL production occur within as little as 7 days of treatment indicating a short-term treatment regime may be effective. As a more novel aspect, we also investigated a neuronal mechanism and show that central exendin-4 induce similar reductions in both weight, food intake and fructose-induced fasting dyslipidemia in comparison to peripheral treatment.
Substantial evidence indicates that both peripheral and central GLP-1R are implicated in the regulation of satiety and weight loss (reviewed in Ref. [27]). While a decrease in dyslipidemia may be expected with weight loss, we show that the lipid lowering effects of exendin-4 (particularly TG) are independent of changes in satiety. This is in line with other studies showing that chronic peripheral GLP-1R agonism reduces hepatic lipid levels and enhances hepatic FA oxidation independent of any significant changes in weight [9]. In fact, decreases in cholesterol are more likely due to decreased weight loss, as mRNA markers for cholesterol homeostasis were equally decreased in pair-fed controls and there were no significant changes in plasma cholesterol levels between pair-fed and exendin-4 treated hamsters.
It is likely that the exendin-4 mediated decrease in hepatic de novo lipogenesis is a main contributor to decreased VLDL production since apoB100 particles are degraded in the absence of sufficient lipid availability [28]. This is supported by studies showing both GLP-1R agonism and raising endogenous levels of GLP-1 decrease hepatic lipid levels by enhancing β-oxidation and decreasing de novo lipogenesis [9,12–14,16,26,29,30]. The observed reduction in VLDL may further be explained by the exendin-4 mediated switch from burning carbohydrates to lipids as the main source of energy utilization [13], which we further show to be independent of changes in food consumption. This is supported by a clinical study in lean and obese patients showing that differences in RER reflected different rates of de novo lipogenesis [31]. Therefore the exendin-4 induced switch in metabolism may contribute to decreases observed in fasting dyslipidemia.
The presence of a hepatic GLP-1R remains controversial while some studies have detected hepatic GLP-1R mRNA or protein [9,10,32] and other studies are unable to detect a hepatic GLP-1R in similar animal models [14,16,33]. To complicate data interpretation, GLP-1R antibodies have been shown to provide a positive signal for the receptor even in GLP-1R knockout mice, indicating the likelihood for false positive detection [14]. Given that the GLP-1R shares high sequence homology with other genes in the liver, mRNA analysis may also provide false positive results. To avoid this, one study carried out PCR on the whole GLP-1R in mouse liver and indicated the lack of a hepatic GLP-1R [14]. Nonetheless, enhanced cAMP production was observed in rat hepatocytes treated with exendin-4 [12] and additional studies show decreased hepatic lipid and increased cell viability in human hepatocytes following exendin-4 administration [10,29]. We attempted to reproduce these results using similar conditions in primary hamster hepatocytes, however, our data cannot confirm the presence of a hepatic GLP-1R, which is in agreement with Flock et al. who were unable to detect any changes in cAMP levels after GLP-1 or exendin-4 exposure in primary mouse hepatocytes [16]. More recently, the development of a new transgenic mouse model to avoid antibody detection methods also indicated the lack of a hepatic GLP-1R [33]. Therefore, the exendin-4 mediated reduction in hepatic VLDL production and hepatic de novo lipogenesis are likely due to indirect mechanisms.
Given the lack of a hepatic GLP-1R, we next investigated the involvement of a central signaling pathway. The hypothalamus has previously been implicated in the regulation of hepatic VLDL production. Central administration of glucose into the 3rd ventricle inhibits VLDL secretion, while neuropeptide Y administered promotes VLDL production [34,35]. Interestingly, the hypothalamus, which lies adjacent to the 3rd ventricle, has a dense population of GLP-1Rs [19]. Our studies show that central exendin-4 into the 3rd ventricle prevents fructose-induced dyslipidemia. Given that GLP-1 and GLP-1R agonists can cross the blood brain barrier, the peripheral administration of exendin-4 had access to both peripheral and central receptors. Alternatively, in the ICV studies, we believe the observed effects to be solely central given that the dose of exendin-4 administered was likely too low to have peripheral effects. Our results that show a central effect of exendin-4 on fasting dyslipidemia are in agreement with Panjwani et al., who show that acute central exendin-4 administration significantly reduces hepatic TG secretion [14]. Central GLP-1 has also previously been shown to activate vagal nerve activity [36] however we are the first to show that the effects of GLP-1R agonism on fasting dyslipidemia and VLDL production require intact vagal signaling. While vagotomy alone caused modest but non-significant reductions in fasting plasma TG, the reductions did not match those induced by exendin-4 in sham hamsters. Given that exendin-4 administration was unable to further reduce dyslipidemia following vagotomy, this suggests that the indirect mechanism induced by exendin-4 requires an intact parasympathetic signaling pathway. It is possible that the mechanism involves an afferent-efferent signaling pathway since GLP-1Rs have been detected on visceral afferent nerve terminals in the hepatic portal bed [33,37]. This is supported by a study where an albumin-exendin-4 conjugate that was unable to cross the blood brain barrier was still able to activate a central circuit [38]. Additional studies are required to further develop and uncover the indirect mechanism of exendin-4 that mediates the effects on hepatic lipid and lipoprotein metabolism.
In summary, we show that exendin-4 prevents fructose-induced dyslipidemia, VLDL overproduction and hepatic steatosis through an indirect mechanism that involves changes in whole body energy metabolism and decreased hepatic de novo lipogenesis. We further show that the exendin-4 mediated effects involve the CNS, particularly vagal signaling. Our data begin to uncover the indirect mechanism by which GLP-1 targets the liver and further suggest that GLP-1R agonism can be an effective approach in the treatment of metabolic dyslipidemia commonly observed in diabetic states.
Funding
This work was supported in part by an operating grant from the Heart and Stroke Foundation of Ontario (# 000220) to K.A. J.T. is supported by Banting and Best Diabetes Yow Kam-Yuen Graduate Scholarship in Diabetes Research, Ontario Graduate Scholarship and MITACS student scholarships.
Acknowledgments
The authors are grateful to ManKhun Chan (Department of Pediatric Laboratory Medicine, Division of Clinical Biochemistry, The Hospital for Sick Children, Toronto, ON, Canada) who assisted with plasma measurements.
Conflicts of interest
There are no conflicts of interest to declare.
Appendix A. Supplementary data
The following are the supplementary data related to this article:
Figure S1.
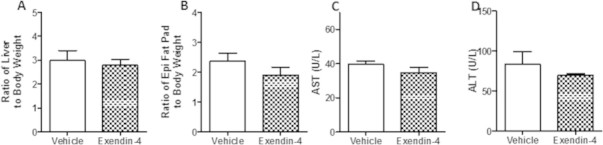
Peripheral exendin-4 does not alter liver or fat pad weight in an intervention treatment strategy. Hamsters underwent the intervention study and plasma and tissue was collected from vehicle and exendin-4 hamsters after a 5 h fast. (A) Liver and (B) epididymal fat pad weight. (C) Plasma AST and (D) ALT following a 5 h fast. n = 5–7. Analyzed by Student's unpaired t-test.
Figure S2.
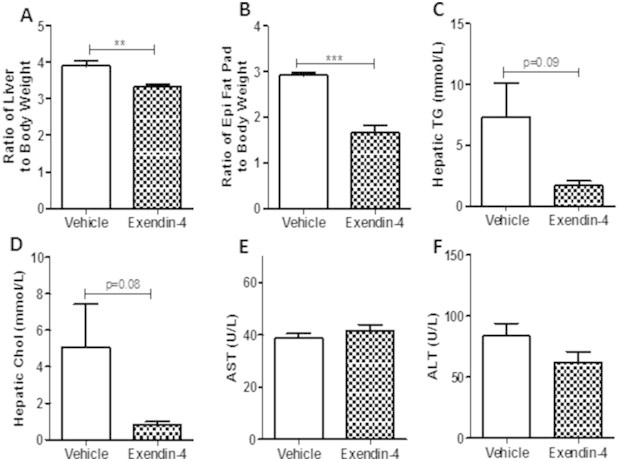
Peripheral exendin-4 decreases liver weight and lipid content in a preventative treatment strategy. Hamsters underwent the intervention study and plasma and tissue was collected from vehicle and exendin-4 hamsters after a 5 h fast. (A) Liver weight and (B) epididymal fat pad weight. (C) Hepatic triglyceride and (D) cholesterol content following the preventative study. (E) Plasma AST and (F) ALT levels following a 5 h fast. n = 6; **p < 0.01, ***p < 0.001 as analyzed by Student's unpaired t-test.
Figure S3.
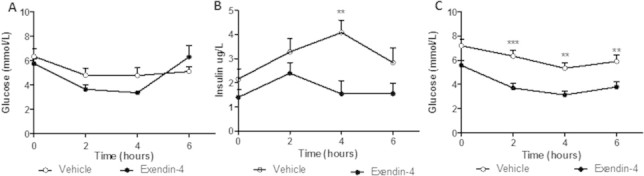
Peripheral exendin-4 decreases hyperinsulinemia and fasting plasma glucose. Following the intervention (A,B) or preventative study (C), vehicle and exendin-4 hamsters were fasted for 5 h and plasma was collected over 6 h. (A,C) Fasting plasma glucose and (B) insulin levels. Intervention n = 5–7; Preventative n = 6; **p < 0.01, ***p < 0.001 as analyzed by two-way ANOVA with Bonferroni post hoc test.
Figure S4.
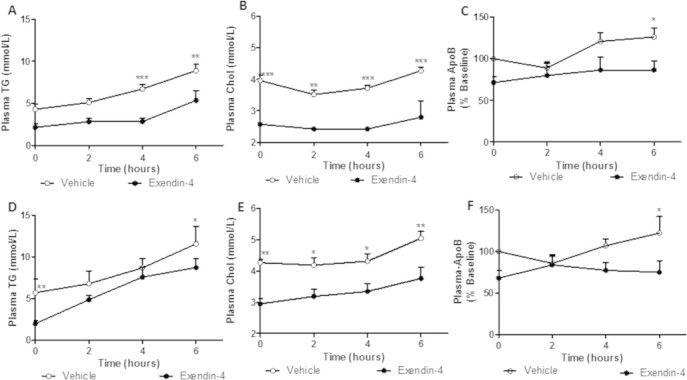
Peripheral exendin-4 decreases plasma TG, cholesterol and apoB100 accumulation. Following the intervention (A–C) or preventative (D–F) study, hamsters were fasted for 5 h and received an intraperitoneal (ip) injection of vehicle or exendin-4 (20 μg/kg) followed by an ip injection of poloxamer (0.5 g/kg). (A,D) Plasma triglyceride, (B,E) cholesterol, (C,F) and apoB100 accumulation. Intervention n = 5–7; Preventative n = 6; *p < 0.05, **p < 0.01, ***p < 0.001 as analyzed by two-way ANOVA with Bonferroni post hoc test.
Figure S5.
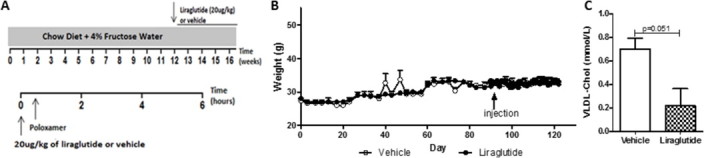
Liraglutide reduces VLDL-cholesterol without changes in weight. (A) C57BL/6J mice on a chow diet + 4% fructose water for 16 weeks were given a once daily intraperitoneal (ip) injection of vehicle or liraglutide (20 μg/kg) during the final 4 weeks. On the final day, an ip injection of poloxamer (0.5 g/kg) was administered following a 5 h fast. (B) Body weight was measured for the duration of the study (16 weeks). (C) VLDL-cholesterol levels 6 h after poloxamer. n = 3. Analyzed by two-way ANOVA with Bonferroni post hoc test (B) or by Student's unpaired t-test (C).
Figure S6.
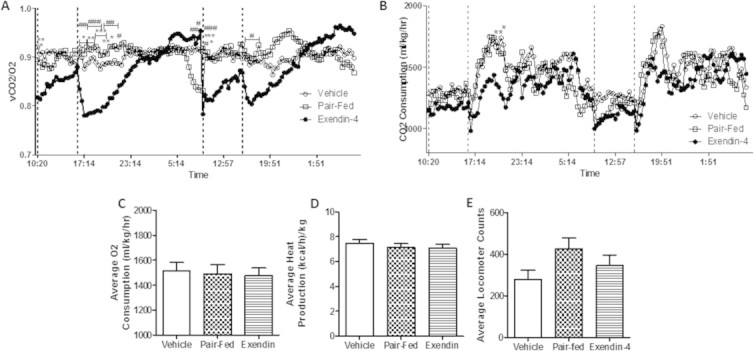
Exendin-4 acutely reduces RER and CO2 production. Vehicle, pair-fed and exendin-4 treated hamsters underwent the intervention study and placed into CLAMS metabolic cages for the final 48 h. Dashed lines indicate injection timepoints. (A) RER and (B) CO2 production of hamsters in the cages over time. (C) O2 consumption, (D) heat production and (E) total movement average 3 h postinjection. Dashed lines indicate injection time. n = 10–11. *p < 0.05, **p < 0.01, ***p < 0.001. *Significance between vehicle and exendin-4; #Significance between pair-fed and exendin-4. Analyzed by two-way ANOVA with Bonferroni post hoc test (A,B) or by one-way ANOVA (C–E).
Figure S7.
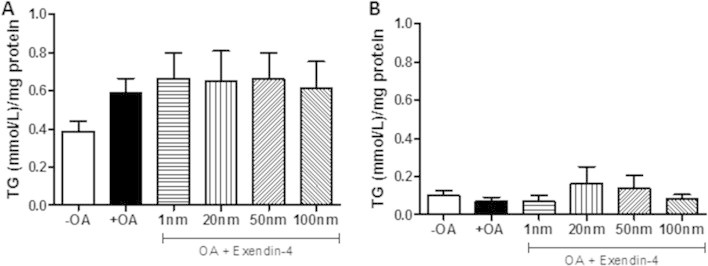
Primary hamster hepatocytes do not contain the GLP-1 receptor. Primary hamster hepatocytes were treated ex vivo with or without oleic acid (0.7 mM) for 16 h followed by 6 h of vehicle or exendin-4. (A) Cellular and (B) secreted TG levels in primary hamster hepatocytes. n = 3 in triplicate. Analyzed by two-way ANOVA with Bonferroni post hoc test.
Figure S8.
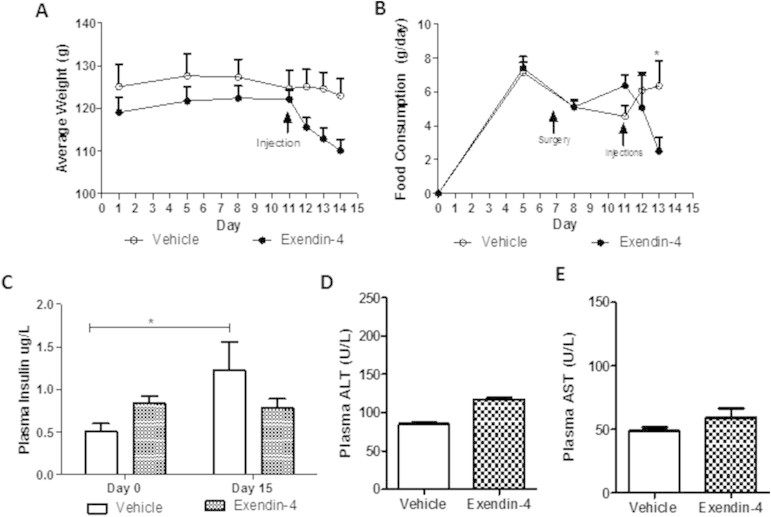
Central exendin-4 prevents fructose–induced hyperinsulinemia. (A,B) Daily weight and food consumption in hamsters that underwent the ICV study (C) Fasting plasma insulin levels before (day 0) and after (Day 14) the study. (D,E) Fasting plasma AST and ALT levels. n = 8. *p < 0.05, ***p < 0.001. (A–C) Analyzed by two-way ANOVA with Bonferroni post hoc test (A–C) or by unpaired Student's t-test (D,E).
Figure S9.
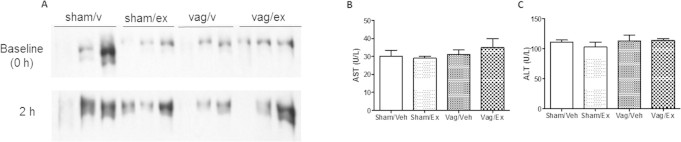
Vagotomy prevents exendin-4 mediated reductions in fasting VLDL-apoB100. (A) VLDL-ApoB100 blots in vagotomized or sham hamsters treated peripherally with vehicle or exendin-4. (B,C) Fasting plasma AST and ALT. n = 3.
References
- 1.Adeli K., Taghibiglou C., Van Iderstine S.C., Lewis G.F. Mechanisms of hepatic very low-density lipoprotein overproduction in insulin resistance. Trends in Cardiovascular Medicine. 2001;11(5):170–176. doi: 10.1016/s1050-1738(01)00084-6. [DOI] [PubMed] [Google Scholar]
- 2.Hein G., Baker C., Hsieh J., Farr S., Adeli K. GLP-1 and GLP-2 as yin and yang of intestinal lipoprotein production: evidence for predominance of GLP-2-stimulated postprandial lipemia in normal and insulin-resistant states. Diabetes. 2013;62(2) doi: 10.2337/db12-0202. PMID: 23028139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsieh J., Longuet C., Baker C.L., Qin B., Federico L.M., Drucker D.J. The glucagon-like peptide 1 receptor is essential for postprandial lipoprotein synthesis and secretion in hamsters and mice. Diabetologia. 2010;53(3):552–561. doi: 10.1007/s00125-009-1611-5. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz E.A., Koska J., Mullin M.P., Syoufi I., Schwenke D.C., Reaven P.D. Exenatide suppresses postprandial elevations in lipids and lipoproteins in individuals with impaired glucose tolerance and recent onset type 2 diabetes mellitus. Atherosclerosis. 2010;212(1):217–222. doi: 10.1016/j.atherosclerosis.2010.05.028. [DOI] [PubMed] [Google Scholar]
- 5.Farr S., Taher J., Adeli K. Glucagon-like peptide-1 as a key regulator of lipid and lipoprotein metabolism in fasting and postprandial states. Cardiovascular Hematological Disorders Drug Targets. 2014;14(2):126–136. doi: 10.2174/1871529x14666140505125300. [DOI] [PubMed] [Google Scholar]
- 6.Klonoff D.C., Buse J.B., Nielsen L.L., Guan X., Bowlus C.L., Holcombe J.H. Exenatide effects on diabetes, obesity, cardiovascular risk factors and hepatic biomarkers in patients with type 2 diabetes treated for at least 3 years. Current Medical Research and Opinion. 2008;24(1):275–286. doi: 10.1185/030079908x253870. [DOI] [PubMed] [Google Scholar]
- 7.Horton E.S., Silberman C., Davis K.L., Berria R. Weight loss, glycemic control, and changes in cardiovascular biomarkers in patients with type 2 diabetes receiving incretin therapies or insulin in a large cohort database. Diabetes Care. 2010;33(8):1759–1765. doi: 10.2337/dc09-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blonde L., Klein E.J., Han J., Zhang B., Mac S.M., Poon T.H. Interim analysis of the effects of exenatide treatment on A1C, weight and cardiovascular risk factors over 82 weeks in 314 overweight patients with type 2 diabetes. Diabetes, Obesity and Metabolism. 2006:436–447. doi: 10.1111/j.1463-1326.2006.00602.x. Sect. 8. [DOI] [PubMed] [Google Scholar]
- 9.Mells J.E., Fu P.P., Sharma S., Olson D., Cheng L., Handy J.A. Glp-1 analog, liraglutide, ameliorates hepatic steatosis and cardiac hypertrophy in C57BL/6J mice fed a western diet. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2012;302(2):G225–G235. doi: 10.1152/ajpgi.00274.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gupta N.A., Mells J., Dunham R.M., Grakoui A., Handy J., Saxena N.K. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology. 2010;51(5):1584–1592. doi: 10.1002/hep.23569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tushuizen M.E., Bunck M.C., Pouwels P.J., van Waesberghe J.H.T., Diamant M., Heine R.J. Incretin mimetics as a novel therapeutic option for hepatic steatosis. Liver International: Official Journal of the International Association for the Study of the Liver. 2006;26(8):1015–1017. doi: 10.1111/j.1478-3231.2006.01315.x. [DOI] [PubMed] [Google Scholar]
- 12.Ding X., Saxena N., Lin S., Gupta N., Anania F. Obesity pathogenesis; exendin-4, a GLP-1 receptor agonist, reverses hepatic steatosis in ob/ob mice. Obesity, Fitness and Wellness Week. 2006;43(1):173–181. doi: 10.1002/hep.21006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parlevliet E.T., Wang Y., Geerling J.J., Schröder-Van der Elst J.P., Picha K., O'Neil K. GLP-1 receptor activation inhibits VLDL production and reverses hepatic steatosis by decreasing hepatic lipogenesis in high-fat-fed APOE3-leiden mice. PloS One. 2012;7(11):e49152. doi: 10.1371/journal.pone.0049152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Panjwani N., Mulvihill E.E., Longuet C., Yusta B., Campbell J.E., Brown T.J. GLP-1 receptor activation indirectly reduces hepatic lipid accumulation but does not attenuate development of atherosclerosis in diabetic male ApoE(-/-) mice. Endocrinology. 2013;154(1):127. doi: 10.1210/en.2012-1937. [DOI] [PubMed] [Google Scholar]
- 15.Trevaskis J.L., Griffin P.S., Wittmer C., Neuschwander-Tetri B., Brunt E.M., Dolman C.S. Glucagon-like peptide-1 (GLP-1) receptor agonism improves metabolic, biochemical and histopathological indices of nonalcoholic steatohepatitis (NASH) in mice. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2012;302(8):G762–G772. doi: 10.1152/ajpgi.00476.2011. [DOI] [PubMed] [Google Scholar]
- 16.Flock G., Baggio L.L., Longuet C., Drucker D.J. Incretin receptors for glucagon-like peptide 1 and glucose-dependent insulinotropic polypeptide are essential for the sustained metabolic actions of vildagliptin in mice. Diabetes. 2007;56(12):3006–3013. doi: 10.2337/db07-0697. [DOI] [PubMed] [Google Scholar]
- 17.Dunphy J.L., Taylor R.G., Fuller P.J. Tissue distribution of rat glucagon receptor and GLP-1 receptor gene expression. Molecular and Cellular Endocrinology. 1998;141(1–2):179–186. doi: 10.1016/s0303-7207(98)00096-3. [DOI] [PubMed] [Google Scholar]
- 18.Aviv V., Meivar-Levy I., Rachmut I.H., Rubinek T., Mor E., Ferber S. Exendin-4 promotes liver cell proliferation and enhances the PDX-1-induced liver to pancreas transdifferentiation process. Journal of Biological Chemistry. 2009;284(48):33509–33520. doi: 10.1074/jbc.M109.017608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larsen P.J., Tang-Christensen M., Holst J.J., Ørskov C. Distribution of glucagon-like peptide-1 and other preproglucagon-derived peptides in the rat hypothalamus and brainstem. Neuroscience. 1997;77(1):257–270. doi: 10.1016/s0306-4522(96)00434-4. [DOI] [PubMed] [Google Scholar]
- 20.Knauf C., Cani P.D., Perrin C., Iglesias M.A., Maury J.F., Bernard E. Brain glucagon-like peptide-1 increases insulin secretion and muscle insulin resistance to favor hepatic glycogen storage. Journal of Clinical Investigation. 2005;115(12):3554–3563. doi: 10.1172/JCI25764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nogueiras R., Perez-Tilve D., Veyrat-Durebex C., Morgan D.A., Varela L., Haynes W.G. Direct control of peripheral lipid deposition by CNS GLP-1 receptor signaling is mediated by the sympathetic nervous system and blunted in diet-induced obesity. Journal of Neuroscience. 2009;29(18):5916. doi: 10.1523/JNEUROSCI.5977-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taghibiglou C., Carpentier A., Van Iderstine S.C., Chen B., Rudy D., Aiton A. Mechanisms of hepatic very low density lipoprotein overproduction in insulin resistance. evidence for enhanced lipoprotein assembly, reduced intracellular ApoB degradation, and increased microsomal triglyceride transfer protein in a fructose-fed hamster model. Journal of Biological Chemistry. 2000;275(12):8416–8425. doi: 10.1074/jbc.275.12.8416. [DOI] [PubMed] [Google Scholar]
- 23.Taghibiglou C., Rudy D., Van Iderstine S.C., Aiton A., Cavallo D., Cheung R. Intracellular mechanisms regulating apoB-containing lipoprotein assembly and secretion in primary hamster hepatocytes. Journal of Lipid Research. 2000;41(4):499. [PubMed] [Google Scholar]
- 24.Basciano H., Miller A.E., Naples M., Baker C., Kohen R., Xu E. Metabolic effects of dietary cholesterol in an animal model of insulin resistance and hepatic steatosis. American Journal of Physiology. Endocrinology and Metabolism. 2009;297(2):E462–E473. doi: 10.1152/ajpendo.90764.2008. [DOI] [PubMed] [Google Scholar]
- 25.Yue J.T.Y., Mighiu P.I., Naples M., Adeli K., Lam T.K.T. Glycine normalizes hepatic triglyceride-rich VLDL secretion by triggering the CNS in high-fat fed rats. Circulation Research. 2012;110(10):1345–1354. doi: 10.1161/CIRCRESAHA.112.268276. [DOI] [PubMed] [Google Scholar]
- 26.Lee J., Hong S., Chae S.W., Kim D.H., Choi J.H., Bae J.C. Exendin-4 improves steatohepatitis by increasing Sirt1 expression in high-fat diet-induced obese C57BL/6J mice. PloS One. 2012;7(2):e31394. doi: 10.1371/journal.pone.0031394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams D.L. Minireview: finding the sweet spot: peripheral versus central glucagon-like peptide 1 action in feeding and glucose homeostasis. Endocrinology. 2009;150(7):2997–3001. doi: 10.1210/en.2009-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yao Z., Tran K., McLeod R.S. Intracellular degradation of newly synthesized apolipoprotein B. Journal of Lipid Research. 1997;38(10):1937. [PubMed] [Google Scholar]
- 29.Sharma S., Mells J.E., Fu P.P., Saxena N.K., Anania F.A. GLP-1 analogs reduce hepatocyte steatosis and improve survival by enhancing the unfolded protein response and promoting macroautophagy. PLOS One. 2011;6(9) doi: 10.1371/journal.pone.0025269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kern M., Klöting N., Niessen H.G., Thomas L., Stiller D., Mark M. Linagliptin improves insulin sensitivity and hepatic steatosis in diet-induced obesity. PloS One. 2012;7(6):e38744. doi: 10.1371/journal.pone.0038744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minehira K., Vega N., Vidal H., Acheson K., Tappy L. Effect of carbohydrate overfeeding on whole body macronutrient metabolism and expression of lipogenic enzymes in adipose tissue of lean and overweight humans. International Journal of Obesity. 2004;28(10):1291–1298. doi: 10.1038/sj.ijo.0802760. [DOI] [PubMed] [Google Scholar]
- 32.Svegliati-Baroni G., Saccomanno S., Rychlicki C., Agostinelli L., De Minicis S., Candelaresi C. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced bya high-fat diet in nonalcoholic steatohepatitis. Liver International. 2011;31(9):1285–1297. doi: 10.1111/j.1478-3231.2011.02462.x. [DOI] [PubMed] [Google Scholar]
- 33.Richards P., Parker H.E., Adriaenssens A.E., Hodgson J.M., Cork S.C., Trapp S. Identification and characterization of GLP-1 receptor-expressing cells using a new transgenic mouse model. Diabetes. 2014;63(4):1224. doi: 10.2337/db13-1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bruinstroop E., Pei L., Ackermans M.T., Foppen E., Borgers A.J., Kwakkel J. Hypothalamic neuropeptide Y (NPY) controls hepatic VLDL-triglyceride secretion in rats via the sympathetic nervous system. Diabetes. 2012;61(5):1043. doi: 10.2337/db11-1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lam T.K.T., Gutierrez-Juarez R., Pocai A., Bhanot S., Tso P., Schwartz G.J. Brain glucose metabolism controls the hepatic secretion of triglyceride-rich lipoproteins. Nature medicine. 2007;13(2):171–180. doi: 10.1038/nm1540. [DOI] [PubMed] [Google Scholar]
- 36.Cabou C., Campistron G., Marsollier N., Leloup C., Cruciani-Guglielmacci C., Pénicaud L. Brain glucagon-like peptide-1 regulates arterial blood flow, heart rate, and insulin sensitivity. Diabetes. 2008;57(10):2577–2587. doi: 10.2337/db08-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vahl T.P., Tauchi M., Durler T.S., Elfers E.E., Fernandes T.M., Bitner R.D. Glucagon-like peptide-1 (GLP-1) receptors expressed on nerve terminals in the portal vein mediate the effects of endogenous GLP-1 on glucose tolerance in rats. Endocrinology. 2007;148(10):4965–4973. doi: 10.1210/en.2006-0153. [DOI] [PubMed] [Google Scholar]
- 38.Baggio L.L., Huang Q., Cao X., Drucker D.J. An albumin-exendin-4 conjugate engages central and peripheral circuits regulating murine energy and glucose homeostasis. Gastroenterology. 2008;134(4):1137–1147. doi: 10.1053/j.gastro.2008.01.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.