

Abstract
The protein kinase C (PKC) family of serine–threonine kinases has been implicated in behavioral responses to opiates, but little is known about the individual PKC isozymes involved. Here, we show that mice lacking PKCε have increased sensitivity to the rewarding effects of morphine, revealed as the expression of place preference and intravenous self-administration at very low doses of morphine that do not evoke place preference or self-administration in wild-type mice. The PKCε null mice also show prolonged maintenance of morphine place preference in response to repeated testing when compared with wild-type mice. The supraspinal analgesic effects of morphine are enhanced in PKCε null mice, and the development of tolerance to the spinal analgesic effects of morphine is delayed. The density of μ-opioid receptors and their coupling to G-proteins are normal. These studies identify PKCε as a key regulator of opiate sensitivity in mice.
Keywords: Analgesia, opioid, PKC, place preference, self-administration
Opiate drugs are important for the treatment of severe pain, but their usefulness is constrained by their significant abuse potential, with addiction to opiates being a major social and economic problem throughout the world. In the United States alone, the economic cost of heroin addiction is estimated at $22 billion a year (Mark et al. 2001), and the National Survey on Drug Use and Health (NSDUH) estimates that lifetime heroin users in the United States number 3.1 million (NSDUH 2004). An understanding of the molecular mechanisms by which opiates produce their euphoric and analgesic effects is thus vital to aid the design of novel analgesics with reduced abuse potential.
Protein kinase C (PKC) is a family of intracellular, phospholipid-dependent, serine–threonine kinases that transduce signals involving lipid second messengers (Nishizuka 2001; Olive & Messing 2004). The family is encoded by nine genes and is divided into three subclasses based on differences in structure and response to second messengers. Conventional PKCs (α, β, γ) are activated by calcium and diacylglycerol, novel PKCs (δ, ε, η, θ) are activated by diacylglycerol but not by calcium and atypical aPKCs (ζ and τ/λ) are insensitive to calcium and diacylglycerol but are modulated by other lipids. All isozymes except PKCθ are found in the brain, and PKCγ is found only in the central nervous system. Recent evidence indicates a role for the PKC family in behavioral responses to opiates. For example, the PKC inhibitor calphostin C prevents the development of morphine-induced place preference (Narita et al. 2001a), and chronic morphine treatment produces an increase in membrane-associated PKC activity in the limbic forebrain (Narita et al. 2002b).
Determining the role of individual PKC isozymes in mediating behavioral responses to morphine is difficult to achieve pharmacologically, as currently available inhibitors such as Ro 31-8220 and GF 109203x do not exhibit specificity for individual PKC isoforms (Wilkinson et al. 1993), and have even been shown to affect kinases other than PKC, such as glycogen synthase kinase-3 (Hers et al. 1999), mitogen-activated protein kinase-activated protein (MAPKAP) kinase 1β and p70 S6 kinase (Alessi 1997). Ro 31-8220 has even been shown to directly inhibit sodium channels (Lingameneni et al. 2000). Other PKC inhibitors such as calphostin C also have little specificity for individual PKC isoforms and affect targets other than PKC, including Ras guanyl nucleotide releasing protein (RasGRP) (Lorenzo et al. 2000), phospholipase D (Sciorra et al. 2001) and L-type calcium channels (Hartzell & Rinderknecht 1996). Thus, we chose a genetic approach by studying behavioral responses to morphine in mice lacking the epsilon isoform of PKC. A similar approach revealed that mice lacking PKCγ show reduced place preference for morphine (Narita et al. 2001a) but enhanced morphine analgesia (Narita et al. 2001b).
There is evidence that PKCε is activated by opiates. Treatment of neurons cultured from 6-day-old chicken embryos with the μ-opioid receptor agonist morphiceptin increases PKCε activity as measured by kinase and membrane translocation assays (Mangoura & Dawson 1993). Also, treatment of the neuronal cell line SH-SY5Y with 1 μM D-Ala2, Nme-Phe4, G1yol5-enkephalin (DAMGO) for 4 h causes translocation of PKCε from cytosol to membrane fractions, whereas more prolonged treatment for 48 h causes an almost complete disappearance of PKCε immunoreactivity from these cells (Kramer & Simon 1999). DAMGO (1 μM)-stimulated PKCε translocation has also been observed after 1 h in SH-SY5Y cells (Mandyam et al. 2003). These results suggest that PKCε is a downstream mediator of μ-opioid receptor stimulation, raising the possibility that PKCε modulates behavioral responses to opiates.
Here, we investigated whether PKCε null mice show altered responses to opiates using models of both addictive behavior and analgesia. We report that these mice are more sensitive to both the analgesic and the rewarding effects of morphine and acquire intravenous morphine self-administration at a training dose that fails to induce self-administration in wild-type mice. These results identify PKCε as an important modulator of behavioral responses to morphine.
Materials and methods
Mice
The PKCε null mice were generated by homologous recombination as described previously (Khasar et al. 1999) and back-crossed onto the C57BL/6J background for more than 10 generations. To generate mice for experiments, we crossed C57BL/6J PKCε+/− mice with 129/S4 mice to generate F1 hybrid PKCε+/− mice. These were intercrossed to generate F2 hybrid littermates for experiments. Mice were genotyped by polymerase chain reaction of tail biopsies (5- to 10-mm fragment) obtained from non-anesthetized, 10-day-old pups. Experimentally naive PKCε null and wild-type mice were housed in standard Plexiglas cages with food and water available ad libitum. The colony room was maintained on a 12-h light:12-h dark cycle with lights on at 0600 h. Mice were between the ages of 2 and 6 months at the time of testing. Animal care and handling procedures were in accordance with Ernest Gallo Clinic and Research Center Institutional Animal Care and Use Committee and National Institutes of Health guidelines.
Conditioned place preference
Mice were trained in Med Associates (Georgia, VT, USA) Place Preference Chambers. These consisted of two test chambers, 16.8 × 12.7 × 12.7 cm, separated by a central access chamber, 7.2 × 12.7 × 12.7 cm. One test chamber was white with a grid floor, the other black with a rod floor. The central access chamber was gray with a smooth floor. Mice were given a 30-min habituation session, consisting of free access to all chambers, on the day immediately prior to commencement of training. Mice were trained by being placed into the central access chamber immediately following morphine or saline administration. The door to the appropriate side was remotely opened and then closed once the mouse had entered the test chamber. The morphine-paired compartment was assigned randomly across subjects. Mice were trained in four 30-min sessions. Mice were treated with morphine every other day, with saline treatment on intervening days. On the day immediately following the final training session, mice were tested by allowing them free access to all chambers for 30 min. A mouse was considered to have developed conditioned place preference for morphine if the time it spent in the drug-paired chamber was significantly greater than the time spent in the non-drug-paired side as determined by a Wilcoxon signed rank test where P < 0.05 (Newton et al. 2004). The same analysis was performed on data obtained during the habituation session performed prior to the first training day to detect any baseline preference for a particular chamber. None was found in either genotype (data not shown) and thus an unbiased place preference protocol was used. Naive mice were used for every morphine dose. Extinction of conditioned place preference was measured by repeating only the test procedure on the days indicated.
Hotplate assay
Mice were placed one at a time onto to a 52°C hotplate (Accusan Instruments, Columbus, OH, USA), and the time that elapsed until they licked or withdrew their paws, or jumped from the plate, was recorded by an examiner blind to their genotype. The mice were then given an injection of morphine [1–10 mg/kg intraperitoneal (i.p.) in saline] and retested 30 min later. Mice were not permitted to exceed 30 seconds of exposure to the hotplate to prevent prolonged painful stimulation. Data were analyzed to account for this artificial ceiling and were calculated as the percentage of the maximum possible effect (%MPE) by the following formula: 100 × [(drug response time – basal response time)/(30 seconds – basal response time)] = %MPE (Balmberg & Bannon 1999). Data were analyzed by two-way analysis of variance (ANOVA) with factors for dose and genotype, from 5 to 10 mice per morphine dose and 33 to 35 mice for baseline measurements.
Tail-flick assay
Mice were held in a towel and their tails lowered into a beaker of water held at 54°C. Latency to remove the tail from the hot water was recorded. Animals were then given an injection of morphine (1–10 mg/kg i.p.) and the latency was recorded again 30 min later. Mice were not permitted to exceed 30 seconds of exposure to hot water to prevent prolonged painful stimulation. Data were expressed as %MPE (see above), using 4–10 mice per morphine dose. Baseline values are reported from all 25 mice. To assess the development of tolerance to morphine analgesia, the same procedure was repeated daily, using 10 mg/kg morphine only, for 7 days, using 10 mice per genotype. For 2 days prior to all tail-flick experiments, mice were acclimatized to being held in the towel.
Intravenous self-administration surgery
Mice were placed under general anesthesia with an isoflurane vapor mixture and then surgically prepared with chronic silastic jugular catheter implants (Roberts et al. 1997). The catheters were made of silastic tubing (inner diameter 0.3 mm; outer diameter 0.64 mm; 7-mm length). The jugular catheter was passed subcutaneously to a polyethylene assembly mounted on the back of the animal. This assembly consisted of a guide cannula (C313G; Plastics One, Roanoke, VA, USA), which was bent at a right angle halfway down the cannula. Silastic tubing was made soft with a short solvent bath (Hemo-De, 1 min) and slipped over the cannula. After drying, the cannula-tubing junction was encased in dental cement with a piece of fabric mesh secured to the bottom. The mesh served to anchor the assembly subcutaneously. After shaving the areas for incision, and application of 70% alcohol and povidone/iodine solution, incisions were made in the mid-scapular region as well as anteromedial to the right forearm above the external right jugular vein. A catheter was passed subcutaneously from the dorsal incision to the ventral incision, and the silastic tubing was inserted into the jugular vein, and then tied gently with suture thread to the vein. The dorsal incision was closed, 30 μl of physiological saline was flushed through the catheter and the exit point was then capped with a Tygon stopper. Catheter patency was verified by administering 0.02 ml of sodium brevital (5 mg/ml; Monarch Pharmaceuticals, Bristol, TN, USA) on the day after surgery and after the self-administration sessions.
Self-administration training and testing
Experimental sessions were conducted in mouse conditioning chambers (ENV-307W, Med Associates, St Albans, VT, USA) located within ventilated sound attenuation chambers. Each chamber was equipped with two levers on one wall spaced 5.5 cm apart. A fixed ratio 1 (FR 1) schedule of reinforcement was used whereby responses on the active lever resulted in the activation of the infusion pump for 5 seconds to deliver 25 μl of solution from a 10-ml syringe. Immediately following morphine delivery, there was a 20-second time-out period to prevent overdose. A cue light immediately above the active lever came on at the time of infusion and remained on throughout the time-out period. Responses on the inactive lever and during the time-out period were recorded but had no programed consequences. Sessions lasted for 60 min. Intravenous drug infusions were delivered by a software-operated infusion pump (Med Associates, St Albans, VT, USA) placed outside the sound-attenuating box, through a liquid swivel and a syringe. Tygon microbore tubing connected the components of the infusion apparatus to each other and to the exit port of the catheter.
[3H]DAMGO autoradiography
Naive mice were euthanized with CO2 followed by decapitation, and their brains were rapidly removed and placed in isopentane at −50°C for 15 seconds. Brains were then stored at −80°C. Cryosections were thaw-mounted onto gelatin slides, dessicated and stored at −80°C. For autoradiography, slides containing precut sections were preincubated in 50 mM Tris–HCl, pH 7.4, at 25°C for 30 min, followed by incubation in 5 nM [3H]DAMGO (American Radiolabeled Chemicals Inc., St Louis, MO, USA) 50 Ci/mmol) in 50 mM Tris–HCl for 60 min at 27°C. Non-specific binding was assessed using adjacent sections incubated in the presence of 1 μM naloxone (Surratt et al. 1994). Slides were rinsed twice, 5 min each in ice-cold 50 mM Tris–HCl, pH 7.4, and then dipped in ice-cold distilled water. They were then dried overnight at 4°C and placed in an X-ray cassette with Tritium Microscales (Amersham-Pharmacia, Piscataway, NJ, USA) and exposed to Kodak MR film with an intensifying screen for 3–5 weeks at −80°C. Autoradiograms were scanned using a flat-bed scanner and analyzed using the program IMAGE j (http://rsb.info.nih.gov/ij). The amount of bound [3H]DAMGO was calculated from optical density values using the tritium standards. Genotypes were then compared by two-tailed, unpaired t-tests for each brain region.
GTP-γS binding
The coupling of μ-opioid receptors to G-protein activation was assessed by measuring the binding of radiolabeled GTY-γS to brain slices following incubation with the μ-receptor agonist DAMGO. Precut brain frozen sections were preincubated in TME buffer (100 mM NaCl, 3 mM MgCl2, 0.2 mM EGTA, 50 mM Tris–HCl, pH 7.4, and 1 U/ml adenosine deaminase) for 10 min at 25°C. Sections were then incubated in TME with 2 mM guanosine diphosphate (GDP) at 25°C for 15 min. Sections were subsequently incubated with TME containing 20 μM DAMGO, 2 mM GDP, 0.04 nM [35S]GTP-γS (Amersham-Pharmacia, >1000 Ci/mmol) and 0.2 mM dithiothreitol for 2 h at 25°C. Non-specific binding was assessed using adjacent sections incubated in the presence of 10 μM GTP-γS (Sigma, St Louis, MO, USA). Sections were washed twice in 50 mM Tris–HCl for 2 min at 5°C, then once in deionized water for 1 min at 5°C. Slides were dried overnight at 4°C under a fan and apposed to Kodak MR film for 72 h. Autoradiograms were scanned using a flat-bed scanner and analyzed using the program IMAGE j (http://rsb.info.nih.gov/ij). The percent stimulation was calculated by comparing [35S]GTP-γS binding in DAMGO-stimulated and unstimulated sections. Genotypes were then compared by two-tailed, unpaired t-tests for each brain region.
Results
PKCε null mice show increased sensitivity to morphine-induced conditioned place preference
The PKCε null and wild-type mice were trained in a conditioned place preference paradigm with a range of morphine doses. Both genotypes showed significant place preference when trained with either 5 mg/kg or 0.5 mg/kg morphine, but only PKCε null mice showed conditioned place preference at the very low training dose of 0.1 mg/kg morphine (Fig. 1a). We also tested the maintenance of place preference for morphine in mice trained with 5 mg/kg morphine. In wild-type animals, place preference was reduced 17 days after the original trial and was absent at 30 days after the original test. In contrast, PKCε null mice showed robust maintenance of place preference during the 30-day period (Fig. 1b). These results demonstrate that PKCε null mice are more sensitive to the rewarding properties of morphine and that they retain memory of morphine reward longer than wild-type littermates.
Figure 1. Morphine conditioned place preference is enhanced in PKCε null mice.
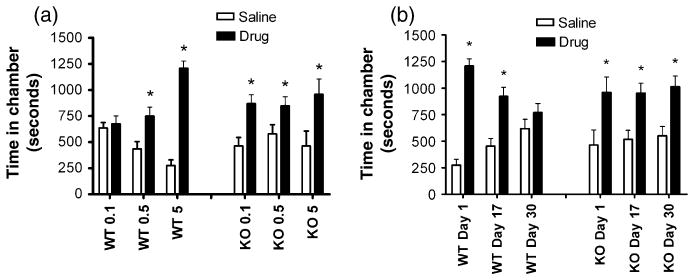
(a) PKCε null mice developed conditioned place preference to a sub-threshold dose of morphine (0.1 mg/kg i.p.) that did not cause place preference in wild-type mice. Both genotypes developed preference to 0.5 and 5 mg/kg morphine. (b) Place preference to 5 mg morphine is maintained for a 30-day period in PKCε null mice but extinguishes in wild-type mice. Results are mean ± standard error of the mean values from 10 to 11 mice of each genotype. * P < 0.05 compared with the saline-paired side by Wilcoxon signed rank test.
Operant morphine self-administration
Since PKCε null mice were more sensitive to the rewarding effects of morphine in the conditioned place preference paradigm, we wished to determine if they could acquire morphine self-administration at a low training dose (0.1 mg/kg). We trained mice to self-administer morphine intravenously over seven daily 60-min sessions on an FR 1 schedule. The PKCε null mice demonstrated a significant level of responding on the active lever compared with the inactive lever, whereas wild-type mice did not (Fig. 2a). A two-way ANOVA with factors for lever and genotype did not reveal an overall main effect of genotype (F(1,34) = 2.23, P = 0.1449) or lever (F(1,34) = 1.57, P = 0.2187) but did reveal a significant interaction between the two factors (F(1,34) = 4.319, P = 0.0453). Post hoc Bonferroni tests revealed a significant difference between levers for PKCε null mice only (P < 0.05). The administration dose was then reduced to 0.05 mg/kg, and the null mice reduced their responding such that there was no longer a significant difference between the active and inactive levers in either genotype by two-way ANOVA (Fig. 2b). In addition, PKCε null mice earned a greater number of infusions than wild-type mice during the 0.1-mg/kg morphine sessions (Fig. 2c); a two-way repeated measures ANOVA with a between-subjects factor for genotype and a repeated measure for trial showed an effect of genotype (F(1,204) = 8.751, P = 0.0088) and post hoc Bonferroni tests revealed a significant difference in trials 3 and 4 (P < 0.05). These data indicate that, at a low-infusion dose, PKCε null mice demonstrate morphine self-administration whereas wild-type mice do not. The overall response rates were quite low even for the PKCε null mice, possibly due to these animals being C57BL/6 × 129/S4 hybrids, since the 129 background has been associated with low levels of operant self-administration (Kuzmin & Johansson 2000).
Figure 2. Morphine self-administration is enhanced in PKCε null mice.

Mice were implanted with intravenous catheters delivering morphine on an FR 1 schedule for thirteen 60-min sessions (0.1 mg/kg per infusion for seven sessions, 0.05 mg/kg for six sessions). PKCε null mice showed significantly more responding on the active vs. the inactive lever for 0.1 mg/kg infusions, whereas wild-type mice did not show preferential responding on either lever (a). This preference for the active lever was abolished when the infusion dose was reduced to 0.05 mg/kg (b). In addition, the total number of rewards earned by PKCε null mice was greater than that earned by wild-type mice (c). *P < 0.05 compared with wild-type mice at that dose by post hoc Bonferroni tests.
PKCε null mice show delayed development of analgesic tolerance by tail-flick and enhanced morphine analgesia on the hotplate
Having established that PKCε null mice show enhanced sensitivity to morphine conditioned place preference, we wished to investigate whether sensitivity was altered for morphine-induced analgesia. We used two separate measures of analgesia, the tail flick, which measures predominantly spinal responses, and the hotplate, which measures predominantly supraspinal responses (Javed et al. 2004; South & Smith 1998; Vylicky 1984). In the tail-flick assay, there was no genotypic difference in baseline responding (Fig. 3a) or in morphine dose response (Fig. 3b). However, when assessing the development of tolerance to morphine analgesia in the tail-flick assay using daily repeated testing after 10 mg/kg morphine, PKCε null mice showed delayed development of tolerance (Fig. 3c). A two-way repeated measures ANOVA with a between-subjects factor for genotype and a repeated measure for time showed an effect of genotype (F(1,95) = 5.53, P = 0.032) and day (F(6,95) = 43.06, P < 0.001) with an interaction between these factors (F(6,95) = 2.719, P = 0.018). Post hoc Bonferroni tests showed a significant difference (P < 0.001) in %MPE between wild-type and PKCε null mice on day 3. Baseline latency increased during repeated testing, but this did not differ between genotypes (Fig. 3a), and a two-way repeated measures ANOVA with factors for genotype and trial showed a significant effect of trial (F(6,96) = 14.84, P < 0.0001) but no effect of genotype (F(1,96) = 0.17, P = 0.6889) and no interaction between genotype and trial (F(6,96) = 1.08, P = 0.3794).
Figure 3. Acute supraspinal opioid analgesia is enhanced in PKCε null mice, whereas acute spinal analgesia is normal but the development of chronic tolerance is delayed.

Morphine (1–10 mg/kg i.p.) effects on spinal nociception were quantified 30 min after injection by measuring the tail-flick latency using a 56°C water bath. (a) There was no genotypic difference in baseline latency (trial 1) or in the latencies measured before each injection of 10 mg/kg morphine during the tolerance experiment (trials 2–7) increased similarly over time in both genotypes. (b) There was also no genotypic difference in morphine dose response in naive non-tolerant mice. (c) The development of analgesic tolerance was assessed by repeated daily testing 30 min after a 10-mg/kg dose of morphine. PKCε null mice showed delayed development of tolerance compared with wild-type mice. *P < 0.05 compared with wild-type mice on day 3 by post hoc Bonferroni tests. Morphine effects on supraspinal nociception were quantified by measuring paw withdrawal latency in mice 30 min after morphine (1–10 mg/kg i.p.) injection using a hotplate set at 52°C. (d) There was no difference in baseline latency. (e) PKCε null mice showed significantly greater withdrawal latency after morphine. *P < 0.05 compared with wild-type mice at 10 mg/kg morphine by post hoc Bonferroni tests.
To determine sensitivity to morphine in an assay of supra-spinal nociception, we performed a hotplate assay. Again, there was no difference in baseline response (Fig. 3d), but the analgesic response to morphine was greater in PKCε null mice (Fig. 3e). A two-way ANOVA showed significant main effects of genotype (F(1,60) = 6.78, P = 0.012) and dose (F(3,60) = 22.23, P < 0.001) with a difference between the genotypes at 10 mg/kg demonstrated by post hoc Tukey tests. Tolerance to morphine was not measured on the hotplate as repeated testing causes a reduction in response latency times even in drug-free animals, and genotypic differences in this habituation may affect the results (Gebhart et al. 1971; Plone et al. 1996).
PKCε null mice show normal μ-opioid receptor density and activation
Since PKCε null mice showed enhanced sensitivity to the rewarding and analgesic properties of morphine, we considered whether this was associated with increased μ-opioid receptor density or enhanced receptor activation of G-proteins. Receptor density was assessed using [3H]DAMGO autoradiography and receptor activation of G-protein signaling by [35S]GTP-γS binding after DAMGO stimulation. We found no differences between PKCε null and wild-type mice in either receptor density or G-protein activation in hippocampus, striatum or thalamus (Fig. 4a,b).
Figure 4. μ-Opioid receptor density and activation are unchanged in PKCε null mice.
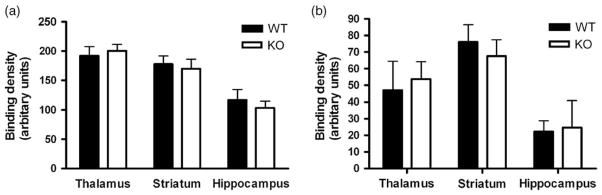
(a) μ-Opioid receptor density measured by [3H]DAMGO autoradiography was unchanged in PKCε null mice when compared with wild-type mice (P > 0.05, two-tailed t-test). (b) μ-Opioid receptor activation, measured by [35S]GTP-γS autoradiography after DAMGO stimulation, was unchanged in PKCε null mice compared with wild-type mice (P > 0.05, two-tailed t-test).
Discussion
Our results demonstrate a clear role for PKCε in regulating morphine reward and analgesia. PKCε null mice develop conditioned place preference to a very low training dose of morphine (0.01 mg/kg) that does not cause place preference in wild-type mice and maintain a robust morphine place preference over a 30-day drug-free period during which time place preference is extinguished in wild-type mice. Morphine analgesia is enhanced in PKCε null mice and intravenous morphine self-administration behavior is demonstrated at a low training dose that does not induce self-administration in wild-type mice. The differences in place conditioning between wild-type and PKCε null mice are not due to altered ability to learn the task, as both genotypes displayed conditioned place preference of similar magnitude when administered 5 mg/kg morphine. Therefore, our findings indicate that PKCε is an important regulator of morphine’s behavioral effects in animals and that pharmacological inhibition of PKCε may enhance responses to opioid drugs.
We observe enhanced responses to morphine in multiple behavioral measures of both reward and analgesia. Although addiction and analgesic tolerance both require chronic opiate exposure, it is generally believed that they are distinct biological phenomena (Hunt & Urch 2006). There are multiple possible substrates for PKCε that may modulate behavioral responses to opiates. Although PKC has been shown to directly phosphorylate the μ-opioid receptor (Zhang et al. 1996), our biochemical data, showing that μ-opioid receptor density and G-protein coupling are unchanged in PKCε null mice, suggest that PKCε acts downstream of the μ-opioid receptor in normal mice. Two of the principal downstream effectors of the μ-opioid receptor, via the release of G-protein βγ subunits, are G-protein-coupled inward rectifier K+ channels (GIRKs) (Bailey et al. 2004; Ikeda et al. 2002) and N-type calcium channels (Rusin & Moises 1995; Wu et al. 2004). Protein kinase C has been shown to phosphorylate and potentiate the function of GIRKs (Mao et al. 2004). There is also evidence that PKCε could modulate opiate responses via N-type calcium channels; βγ-mediated inhibition of N-type calcium channel function is antagonized by PKC activation (Cooper et al. 2000). It is likely that PKCε is the PKC that performs this function since PKCε forms a complex with the N-type calcium channel pore-forming α subunit through the enigma homolog 1 protein; moreover, a peptide inhibitor of PKCε inhibits both this interaction and phorbol ester enhancement of N-type channel function (Maeno-Hikichi et al. 2003). Studies showing stimulation of PKCε by μ-opioid-specific ligands (Kramer & Simon 1999; Mandyam et al. 2003) imply the existence of a negative feedback mechanism to counter μ-opioid inhibition of N-type calcium channels. These studies predict that, in mice lacking PKCε, this inhibitory feedback mechanism would be lost, allowing activation of μ-opioid receptors to result in more profound behavioral effects. Future studies utilizing pharmacological agents or double knockout mice may help determine whether PKCε modulates responses to opiates through actions at these candidate substrates.
The delayed extinction of morphine conditioned place preference is interesting as it represents an animal model of some of the behaviors displayed by human addicts who fail to inhibit their behavioral response to cues or contexts that are no longer rewarding. This deficit is thought to arise in part from impaired prefrontal cortex function (Jentsch & Taylor 1999; Kalivas & Volkow 2005). We are extending our studies to determine whether the extinction deficit shown here by PKCε null mice trained to express a conditioned place preference for morphine generalizes to a failure to extinguish Pavlovian conditioning or is specific for certain drugs.
Interestingly, our data indicate that acute responses to morphine in PKCε null mice are enhanced only in supraspinal systems, as the morphine dose–response curve and response magnitude were unaltered in a tail-flick assay, which examines spinal analgesia (South & Smith 1998; Vylicky 1984). However, we observed that the development of tolerance to the analgesic effects of morphine was delayed in the tail-flick assay. The molecular mechanisms by which repeated morphine exposure produces behavioral tolerance remain highly controversial (Bailey & Connor 2005), but numerous lines of evidence implicate a role for PKC. Behavioral studies demonstrate that co-administration of PKC inhibitors retards the development of spinal analgesic tolerance to morphine (Granados-Soto et al. 2000; Narita et al. 1994), and biochemical studies show that chronic morphine increases PKC activity in the spinal cord (Li & Roerig 1999). Our data suggest that PKCε may be an important isoform involved in these effects.
Although we have not assayed the pharmacokinetics of morphine in PKCε null mice, we do not see a difference in acute analgesic sensitivity in the tail-flick assay, suggesting that there is no baseline difference in morphine distribution or clearance. Since we did not measure morphine pharmacokinetics following repeated morphine administration, it is possible that the delayed development of analgesic tolerance we observed in PKCε null mice might be due to differences in pharmacokinetics during repeated morphine exposure. However, we think this is unlikely since it is generally believed that analgesic tolerance is not caused by a change in morphine pharmacokinetics following chronic drug administration, and this is supported by studies in rats (Mas et al. 2000).
The strategy of using knockout mice to determine the role of specific molecules in behavior has some limitations, such as the carry over of genes from the genetic background used to generate the embryonic stem (ES) cells (Wolfer et al. 2002). In the present study, the ES cells used to generate the null mutant were from the 129/S4 background (formerly 129/SvJae; see http://jaxmice.jax.org/info/bulletin/bulletin01.html). F1 hybrid mice were subsequently crossed into the C57BL/6 background (Khasar et al. 1999). Thus, the morphine phenotype of the PKCε null mice may be contributed to by genes from the 129/S4 background. However, it has been demonstrated that both 129/P3 and 129/SvJ mice have the opposite phenotype to PKCε null mice, with reduced sensitivity to morphine in measures of analgesia (129/P3) (Kest et al. 2002) and the development of morphine conditioned place preference (129/SvJ) (Dockstader & van der Kooy 2001). Although the mice used in the cited studies are not exactly the same as the 129 strain used here, the results suggest that the morphine phenotype of the PKCε null mice is not due to flanking genes carried over from the 129/S4 background. Additional considerations when using knockout mice that carry the null mutation from birth are that the phenotypes observed may result from indirect effects of gene deletion in other organs besides the brain, from developmental changes or from changes in the abundance of other gene products that compensate for the missing gene (Crawley 2000; Stephens et al. 2002). Site-specific injections of adult wild-type animals with a lentivirus encoding short hairpin RNA directed against PKCε may help address some of these issues in the future.
The increased response of PKCε null mice to morphine in the place-conditioning assay stands in contrast to the response of PKCγ null mice, which show no place preference for morphine (Narita et al. 2001b). This pattern resembles modulation of behavioral responses to ethanol by these PKC isozymes, where opposing roles for PKCε and PKCγ have been documented, with PKCε null mice being more sensitive to ethanol intoxication and voluntarily consuming less ethanol (Hodge et al. 1999), and PKCγ null mice showing the opposite phenotype (Harris et al. 1995). These behavioral phenotypes appear to correlate with opposing roles for these PKC isozymes in regulating the sensitivity of gamma-aminobutyric acid (GABAA) receptors to allosteric modulation by ethanol (Harris et al. 1995; Hodge et al. 1999; Proctor et al. 2003). Whether opposing actions at targets such as GABAA receptors mediate opposite effects of PKCε and PKCγ on behavioral responses to opiates remains to be investigated.
In contrast to their opposite responses in place-conditioning assays, both PKCε and PKCγ null mice show an enhanced analgesic response to morphine (Narita et al. 2001b) and delayed development of analgesic tolerance (Zeitz et al. 2001). For PKCγ null mice, unlike PKCε null mice, this may result from increased G-protein activation after μ-opioid receptor stimulation (Narita et al. 2002a). Protein kinase Cε is expressed throughout the central and peripheral nervous system (Choi et al. 2002) and is a key mediator of signal transduction within primary afferent nociceptors that promotes inflammatory hyperalgesia (Aley et al. 2000; Khasar et al. 1999). In contrast, PKCγ is not expressed in the peripheral nervous system, and appears instead to modulate nociception through actions in the dorsal horn of the spinal cord, where it is involved in reorganization of spinal cord circuitry following nerve injury (Martin et al. 1999, 2001). Given the different anatomical distribution of these two PKC isozymes and the distinct morphine phenotypes of mutant mice lacking them, it is likely that PKCε and PKCγ act through different mechanisms to enhance sensitivity to opiate analgesia. These studies demonstrate the importance of using isozyme-specific reagents to detect PKC isozyme-specific functions as commonly available pharmacological PKC inhibitors show little or no specificity for different PKC isozymes.
In summary, our data demonstrate that in mice lacking PKCε, the acute rewarding and central analgesic effects of morphine are enhanced, whereas acute spinal analgesic responses are unchanged. However, the development of tolerance to spinal analgesia is delayed, suggesting that PKCε operates through multiple mechanisms to regulate behavioral responses to morphine. Determining these mechanisms may aid in the development of novel analgesics with reduced abuse potential.
Acknowledgments
The authors thank Jackie Connolly and Christine Orr for technical assistance. This work was supported by United States Public Health Service Grants AA013588 (to R.O.M.) and AA014983 (to C.W.H.) and by funds provided by the State of California for medical research on alcohol and substance abuse through the University of California at San Francisco to R.O.M.
References
- Alessi DR. The protein kinase C inhibitors Ro 318220 and GF 109203X are equally potent inhibitors of MAPKAP kinase-1β (Rsk-2) and p70 S6 kinase. FEBS Lett. 1997;402:121–123. doi: 10.1016/s0014-5793(96)01510-4. [DOI] [PubMed] [Google Scholar]
- Aley KO, Messing RO, Mochly-Rosen D, Levine JD. Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci. 2000;20:4680–4685. doi: 10.1523/JNEUROSCI.20-12-04680.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Connor M. Opioids: cellular mechanisms of tolerance and physical dependence. Curr Opin Pharmacol. 2005;5:60–68. doi: 10.1016/j.coph.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Bailey CP, Kelly E, Henderson G. Protein kinase C activation enhances morphine-induced rapid desensitization of mu-opioid receptors in mature rat locus ceruleus neurons. Mol Pharmacol. 2004;66:1592–1598. doi: 10.1124/mol.104.004747. [DOI] [PubMed] [Google Scholar]
- Balmberg AB, Bannon AW. Models of nociception: hotplate, tail-flick, and formalin tests in rodents. In: Taylor GP, editor. Current Protocols in Neuroscience. Vol. 2. John Wiley and Sons; Hoboken, NJ: 1999. pp. 8.9.1–8.9.15. [DOI] [PubMed] [Google Scholar]
- Choi DS, Wang D, Dadgar J, Chang WS, Messing RO. Conditional rescue of protein kinase C epsilon regulates ethanol preference and hypnotic sensitivity in adult mice. J Neurosci. 2002;22:9905–9911. doi: 10.1523/JNEUROSCI.22-22-09905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper CB, Arnot MI, Feng ZP, Jarvis SE, Hamid J, Zamponi GW. Cross-talk between G-protein and protein kinase C modulation of N-type calcium channels is dependent on the G-protein beta subunit isoform. J Biol Chem. 2000;275:40777–40781. doi: 10.1074/jbc.C000673200. [DOI] [PubMed] [Google Scholar]
- Crawley JN. What’s Wrong With My Mouse? Wiley-Liss; New York: 2000. [Google Scholar]
- Dockstader CL, van der Kooy D. Mouse strain differences in opiate reward learning are explained by differences in anxiety, not reward or learning. J Neurosci. 2001;21:9077–9081. doi: 10.1523/JNEUROSCI.21-22-09077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebhart GF, Sherman AD, Mitchell CL. The influence of learning on morphine analgesia and tolerance development in rats tested on the hot plate. Psychopharmacologia. 1971;22:295–304. doi: 10.1007/BF00401791. [DOI] [PubMed] [Google Scholar]
- Granados-Soto V, Kalcheva I, Hua X, Newton A, Yaksh TL. Spinal PKC activity and expression: role in tolerance produced by continuous spinal morphine infusion. Pain. 2000;85:395–404. doi: 10.1016/S0304-3959(99)00281-X. [DOI] [PubMed] [Google Scholar]
- Harris RA, McQuilkin SJ, Paylor R, Tonegawa S, Wehner JM. Mutant mice lacking the γ isoform of protein kinase C show decreased behavioral actions of ethanol and altered function of γ-aminobutyrate type A receptors. Proc Natl Acad Sci U S A. 1995;92:3658–3662. doi: 10.1073/pnas.92.9.3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartzell HC, Rinderknecht A. Calphostin C, a widely used protein kinase C inhibitor, directly and potently blocks L-type Ca channels. Am J Physiol. 1996;270:C1293–C1299. doi: 10.1152/ajpcell.1996.270.5.C1293. [DOI] [PubMed] [Google Scholar]
- Hers I, Tavare JM, Denton RM. The protein kinase C inhibitors bisindolylmaleimide I (GF 109203x) and IX (Ro 31-8220) are potent inhibitors of glycogen synthase kinase-3 activity. FEBS Lett. 1999;460:433–436. doi: 10.1016/s0014-5793(99)01389-7. [DOI] [PubMed] [Google Scholar]
- Hodge CW, Mehmert KK, Kelley SP, McMahon T, Haywood A, Olive MF, Wang D, Sanchez-Perez AM, Messing RO. Supersensitivity to allosteric GABAA receptor modulators and alcohol in mice lacking PKCε. Nat Neurosci. 1999;2:997–1002. doi: 10.1038/14795. [DOI] [PubMed] [Google Scholar]
- Hunt SP, Urch CE. Pain, opiates and addiciton. In: McMahon SB, Koltzenburg M, editors. Wall and Melzacks Textbook of Pain. 5. Elsevier; London: 2006. pp. 349–359. [Google Scholar]
- Ikeda K, Kobayashi T, Kumanishi T, Yano R, Sora I, Niki H. Molecular mechanisms of analgesia induced by opioids and ethanol: is the GIRK channel one of the keys? Neurosci Res. 2002;44:121–131. doi: 10.1016/s0168-0102(02)00094-9. [DOI] [PubMed] [Google Scholar]
- Javed RR, Dewey WL, Smith PA, Smith FL. PKC and PKA inhibitors reverse tolerance to morphine-induced hypothermia and supraspinal analgesia in mice. Eur J Pharmacol. 2004;492:149–157. doi: 10.1016/j.ejphar.2004.03.061. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Taylor JR. Impulsivity resulting from frontostriatal dysfunction in drug abuse: implications for the control of behavior by reward-related stimuli. Psychopharmacology (Berl) 1999;146:373–390. doi: 10.1007/pl00005483. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Volkow ND. The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry. 2005;162:1403–1413. doi: 10.1176/appi.ajp.162.8.1403. [DOI] [PubMed] [Google Scholar]
- Kest B, Hopkins E, Palmese CA, Adler M, Mogil JS. Genetic variation in morphine analgesic tolerance: a survey of 11 inbred mouse strains. Pharmacol Biochem Behav. 2002;73:821–828. doi: 10.1016/s0091-3057(02)00908-5. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO. A novel nociceptor signaling pathway revealed in protein kinase C ε mutant mice. Neuron. 1999;24:253–260. doi: 10.1016/s0896-6273(00)80837-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer HK, Simon EJ. Role of protein kinase C (PKC) in agonist-induced mu-opioid receptor down-regulation: II. Activation and involvement of the alpha, epsilon, and zeta isoforms of PKC. J Neurochem. 1999;72:594–604. doi: 10.1046/j.1471-4159.1999.0720594.x. [DOI] [PubMed] [Google Scholar]
- Kuzmin A, Johansson B. Reinforcing and neurochemical effects of cocaine: differences among C57, DBA, and 129 mice. Pharmacol Biochem Behav. 2000;65:399–406. doi: 10.1016/s0091-3057(99)00211-7. [DOI] [PubMed] [Google Scholar]
- Li Y, Roerig SC. Alteration of spinal protein kinase C expression and kinetics in morphine, but not clonidine, tolerance. Biochem Pharmacol. 1999;58:493–501. doi: 10.1016/s0006-2952(99)00107-0. [DOI] [PubMed] [Google Scholar]
- Lingameneni R, Vysotskaya TN, Duch DS, Hemmings HC., Jr Inhibition of voltage-dependent sodium channels by Ro 31-8220, a ’specific’ protein kinase C inhibitor. FEBS Lett. 2000;473:265–268. doi: 10.1016/s0014-5793(00)01532-5. [DOI] [PubMed] [Google Scholar]
- Lorenzo PS, Beheshti M, Pettit GR, Stone JC, Blumberg PM. The guanine nucleotide exchange factor RasGRP is a high-affinity target for diacylglycerol and phorbol esters. Mol Pharmacol. 2000;57:840–846. [PubMed] [Google Scholar]
- Maeno-Hikichi Y, Chang S, Matsumura K, Lai M, Lin H, Nakagawa N, Kuroda S, Zhang JF. A PKC epsilon-ENH-channel complex specifically modulates N-type Ca2+ channels. Nat Neurosci. 2003;6:468–475. doi: 10.1038/nn1041. [DOI] [PubMed] [Google Scholar]
- Mandyam CD, Thakker DR, Standifer KM. Micro--opioid-induced desensitization of opioid receptor-like 1 and micro-opioid receptors: differential intracellular signaling determines receptor sensitivity. J Pharmacol Exp Ther. 2003;306:965–972. doi: 10.1124/jpet.103.051599. [DOI] [PubMed] [Google Scholar]
- Mangoura D, Dawson G. Opioid peptides activate phospholipase D and protein kinase C-e in chicken embryo neuron cultures. Neurobiology. 1993;90:2915–2919. doi: 10.1073/pnas.90.7.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Wang X, Chen F, Wang R, Rojas A, Shi Y, Piao H, Jiang C. Molecular basis for the inhibition of G protein-coupled inward rectifier K(+) channels by protein kinase C. Proc Natl Acad Sci U S A. 2004;101:1087–1092. doi: 10.1073/pnas.0304827101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark TL, Woody GE, Juday T, Kleber HD. The economic costs of heroin addiction in the United States. Drug Alcohol Depend. 2001;61:195–206. doi: 10.1016/s0376-8716(00)00162-9. [DOI] [PubMed] [Google Scholar]
- Martin WJ, Liu H, Wang H, Malmberg AB, Basbaum AI. Inflammation-induced up-regulation of protein kinase Cgamma immunoreactivity in rat spinal cord correlates with enhanced nociceptive processing. Neuroscience. 1999;88:1267–1274. doi: 10.1016/s0306-4522(98)00314-5. [DOI] [PubMed] [Google Scholar]
- Martin WJ, Malmberg AB, Basbaum AI. PKCgamma contributes to a subset of the NMDA-dependent spinal circuits that underlie injury-induced persistent pain. J Neurosci. 2001;21:5321–5327. doi: 10.1523/JNEUROSCI.21-14-05321.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mas M, Sabater E, Olaso MJ, Horga JF, Faura CC. Genetic variability in morphine sensitivity and tolerance between different strains of rats. Brain Res. 2000;866:109–115. doi: 10.1016/s0006-8993(00)02255-1. [DOI] [PubMed] [Google Scholar]
- Narita M, Feng Y, Makimura M, Hoskins B, Ho IK. A protein kinase inhibitor, H-7, inhibits the development of tolerance to opioid antinociception. Eur J Pharmacol. 1994;271:543–545. doi: 10.1016/0014-2999(94)90817-6. [DOI] [PubMed] [Google Scholar]
- Narita M, Aoki T, Ozaki S, Yajima Y, Suzuki T. Involvement of protein kinase Cgamma isoform in morphine-induced reinforcing effects. Neuroscience. 2001a;103:309–314. doi: 10.1016/s0306-4522(00)00572-8. [DOI] [PubMed] [Google Scholar]
- Narita M, Mizoguchi H, Suzuki T, Dun NJ, Imai S, Yajima Y, Nagase H, Tseng LF. Enhanced mu-opioid responses in the spinal cord of mice lacking protein kinase Cgamma isoform. J Biol Chem. 2001b;276:15409–15414. doi: 10.1074/jbc.M009716200. [DOI] [PubMed] [Google Scholar]
- Narita M, Mizoguchi H, Khotib J, Suzuki M, Ozaki S, Yajima Y, Tseng LF, Suzuki T. Influence of a deletion of protein kinase C gamma isoform in the G-protein activation mediated through opioid receptor-like-1 and mu-opioid receptors in the mouse pons/medulla. Neurosci Lett. 2002a;331:5–8. doi: 10.1016/s0304-3940(02)00753-x. [DOI] [PubMed] [Google Scholar]
- Narita M, Mizuo K, Shibasaki M, Suzuki T. Up-regulation of the G(q/11alpha) protein and protein kinase C during the development of sensitization to morphine-induced hyperlocomotion. Neuroscience. 2002b;111:127–132. doi: 10.1016/s0306-4522(01)00515-2. [DOI] [PubMed] [Google Scholar]
- Newton PM, Orr CJ, Wallace MJ, Kim C, Shin HS, Messing RO. Deletion of N-type calcium channels alters ethanol reward and reduces ethanol consumption in mice. J Neurosci. 2004;24:9862–9869. doi: 10.1523/JNEUROSCI.3446-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizuka Y. The protein kinase C family and lipid mediators for transmembrane signaling and cell regulation. Alcohol Clin Exp Res. 2001;25:3S–7S. doi: 10.1097/00000374-200105051-00003. [DOI] [PubMed] [Google Scholar]
- NSDUH. Office of Applied Studies, Substance Abuse and Mental Health Services Adminsitration, US Department of Health and Human Services. 2004 http://oas.samhsa.gov/nsduh.htm.
- Olive MF, Messing RO. Protein kinase C isozymes and addiction. Mol Neurobiol. 2004;29:139–154. doi: 10.1385/mn:29:2:139. [DOI] [PubMed] [Google Scholar]
- Plone MA, Emerich DF, Lindner MD. Individual differences in the hotplate test and effects of habituation on sensitivity to morphine. Pain. 1996;66:265–270. doi: 10.1016/0304-3959(96)03048-5. [DOI] [PubMed] [Google Scholar]
- Proctor WR, Poelchen W, Bowers BJ, Wehner JM, Messing RO, Dunwiddie TV. Ethanol differentially enhances hippocampal GABA A receptor-mediated responses in protein kinase C gamma (PKC gamma) and PKC epsilon null mice. J Pharmacol Exp Ther. 2003;305:264–270. doi: 10.1124/jpet.102.045450. [DOI] [PubMed] [Google Scholar]
- Roberts AJ, Polis IY, Gold LH. Intravenous self-administration of heroin, cocaine, and the combination in Balb/c mice. Eur J Pharmacol. 1997;326:119–125. doi: 10.1016/s0014-2999(97)85405-2. [DOI] [PubMed] [Google Scholar]
- Rusin KI, Moises HC. μ-Opioid receptor activation reduces multiple components of high-threshold calcium current in rat sensory neurons. J Neurosci. 1995;15:4315–4327. doi: 10.1523/JNEUROSCI.15-06-04315.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciorra VA, Hammond SM, Morris AJ. Potent direct inhibition of mammalian phospholipase D isoenzymes by calphostin-c. Biochemistry. 2001;40:2640–2646. doi: 10.1021/bi002528m. [DOI] [PubMed] [Google Scholar]
- South SM, Smith MT. Apparent insensitivity of the hotplate latency test for detection of antinociception following intraperitoneal, intravenous or intracerebroventricular M6G administration to rats. J Pharmacol Exp Ther. 1998;286:1326–1332. [PubMed] [Google Scholar]
- Stephens DN, Mead AN, Ripley TL. Studying the neurobiology of stimulant and alcohol abuse and dependence in genetically manipulated mice. Behav Pharmacol. 2002;13:327–345. doi: 10.1097/00008877-200209000-00004. [DOI] [PubMed] [Google Scholar]
- Surratt CK, Johnson PS, Moriwaki A, Seidleck BK, Blaschak CJ, Wang JB, Uhl GR. -mu opiate receptor. Charged transmembrane domain amino acids are critical for agonist recognition and intrinsic activity. J Biol Chem. 1994;269:20548–20553. [PubMed] [Google Scholar]
- Vylicky L. Methods of testing pain mechanisms in animals. In: Wall P, Melzack R, editors. Textbook of Pain. 1. Churchill Livingstone; New York: 1984. pp. 178–185. [Google Scholar]
- Wilkinson SE, Parker PJ, Nixon JS. Isoenzyme specificity of bisindolylmaleimides, selective inhibitors of protein kinase C. Biochem J. 1993;294:335–337. doi: 10.1042/bj2940335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfer DP, Crusio WE, Lipp HP. Knockout mice: simple solutions to the problems of genetic background and flanking genes. Trends Neurosci. 2002;25:336–340. doi: 10.1016/s0166-2236(02)02192-6. [DOI] [PubMed] [Google Scholar]
- Wu ZZ, Chen SR, Pan HL. Differential sensitivity of N- and P/Q-type Ca2+ channel currents to a mu opioid in isolectin B4-positive and -negative dorsal root ganglion neurons. J Pharmacol Exp Ther. 2004;311:939–947. doi: 10.1124/jpet.104.073429. [DOI] [PubMed] [Google Scholar]
- Zeitz KP, Malmberg AB, Gilbert H, Basbaum AI. Reduced development of tolerance to the analgesic effects of morphine and clonidine in PKC gamma mutant mice. Pain. 2001;94:245–253. doi: 10.1016/S0304-3959(01)00353-0. [DOI] [PubMed] [Google Scholar]
- Zhang L, Yu Y, Mackin S, Weight FF, Uhl GR, Wang JB. Differential mu opiate receptor phosphorylation and desensitization induced by agonists and phorbol esters. J Biol Chem. 1996;271:11449–11454. doi: 10.1074/jbc.271.19.11449. [DOI] [PubMed] [Google Scholar]