

Abstract
The potential use of smallpox virus as a bioterror agent and the endemic presence of monkeypox virus in Africa underscores the need for better therapies for orthopoxvirus infections. The only existing clinical experience treating vaccinia and smallpox infections has been with Marboran, which suggested that antiviral therapies could be effective in treating and preventing smallpox infections, but this compound has not been pursued. Drugs that have been approved for other indications, like cidofovir, could be approved for the treatment of orthopoxvirus infections in a timely manner, and this compound has already been approved for emergency treatment of smallpox and complications from vaccination. Its lack of activity when given orally, however, limits its use in a major outbreak involving large numbers of people exposed to the virus. The discovery and development of new therapies can be achieved more rapidly by drawing on the experience and successes with other antiviral agents, particularly with the herpesviruses. This review will discuss the orthopoxvirus replication cycle in detail noting specific viral functions and their associated gene products that have the potential to serve as new targets for drug design and development. This discussion is designed to help investigators relate these targets to parallel functions and existing assays in other virus systems that have been used successfully in drug development. The rapid progress that has been achieved in recent years should yield new drugs for the treatment of these infections and might also reveal new strategies for antiviral therapy with other viruses.
Keywords: Orthopoxvirus, small pox antiviral, vaccinia, drugs, targets, replication
Background and Introduction
Since smallpox was eradicated worldwide in 1980, there has been little interest in developing therapies for variola virus, the causative agent of this disease. In recent years, however, there has been increasing concern that variola virus or the related monkeypox virus, may be a potential threat as a weapon of bioterrorism [1-6]. The only well-studied chemotherapeutic agents for treatment of smallpox in humans were the isatin-β-thiosemicarbazones (IBT) particularly the methyl derivative termed methisazone (Marboran), which were reported to have activity in animal models [7-9] and in human disease [10,11]. Efficacy was generally considered to be achieved only through prophylaxis and interest in these compounds dwindled over the years. Although there has been only a nominal interest during the past 30 years in developing therapies for smallpox, a few active compounds including interferon and interferon inducers [12], and a variety of nucleosides or nucleotides have been reported to have activity against vaccinia virus (VV) [12-19]. A few drugs approved for other indications, such as human immunodeficiency virus, hepatitis virus, or herpesvirus infections, which have also been reported to have some activity against either VV or cowpox viruses (CV) are summarized in Table 1 [20].
Table 1. Activity of Licensed Antiviral Drugs Against Vaccinia and Cowpox Viruses in HFF Cellsa.
| Compound | Cytotoxicity | Vaccinia Copenhagen | Cowpox Brighton | ||
|---|---|---|---|---|---|
| CC50 (μM)b | EC50(μM)c | SId | EC50(μM) | SI | |
| Marboran | >427 | 3.3 ± 3.2 | >130 | 14 ± 0.3 | >31 |
| Cidofovir | >317 | 31 ± 5.4 | >10 | 42 ± 5.4 | >7.5 |
| Idoxuridine | >260 | 6.0 ± 0.2 | >43 | 2.0 ± 0.2 | >130 |
| Trifluridine | >338 | 1.7 | >199 | 1.5 | >225 |
| Vidarabine | >351 | 12 | >29 | 45 | >8 |
| Fialuridine | >269 | 1.5 ± 0.05 | >179 | 0.2 ± 0.08 | >1345 |
| Adefovir dipivoxil | 117 | 5.1 ± 0.7 | 23 | 13 ± 8.8 | 9.0 |
adapted from Kern, 2003 [20].
cytotoxic dose.
effective dose given as the average of two or more assays ± the standard deviation.
selective index is the the CC50/ EC50.
Since there is little incentive for industry to spend hundreds of millions of dollars to develop a drug against a disease that currently does not exist, the emphasis has been on identifying antiviral agents that are already approved for other indications. One such compound, cidofovir (CDV), is approved for intravenous use in the treatment of cytomegalovirus (CMV) retinitis in HIV-infected patients. The drug is very active in tissue culture cells against all the orthopoxviruses that have been tested including VV, CV, monkeypox virus, and variola virus [13,15,17,20-23]. The activity of CDV against orthopoxviruses is of particular interest since the compound has been shown to be active in animal models infected with VV and CV [14,21,23-28]. Importantly, it has recently been reported to be highly effective in treating monkeys infected with variola virus or monkeypox virus [29]. Although CDV is a highly effective inhibitor of orthopoxvirus replication, it is absorbed poorly when administered orally [30], and lack of oral bioavailability is a major limitation to the use of this drug in a large-scale emergency situation such as a smallpox outbreak. Its toxicity and lack of oral activity provide a rationale for the discovery and development of new orally active chemotherapeutic agents for treatment and/or prevention of orthopoxvirus infections whether acquired in a natural setting or through bioterrorism activities.
There are numerous nucleoside or nucleotide analogs that have been reported to have antiviral activity against VV or CV [13]. As mentioned previously, CDV is the most promising candidate on this list and has been approved for emergency treatment of smallpox. Idoxuridine and trifluridine, which are both approved for topical treatment of herpes simplex virus keratitis do not have a sufficient toxicology database to support parenteral use. Vidarabine, the first parenteral therapy approved for serious herpesvirus infections, is not active orally and was not very active in murine models for VV or CV infections [29,31].
The results of in vitro efficacy studies also indicated that adefovir dipivoxil and fialuridine were active candidates [20]. Although fialuridine is very active in tissue culture, its lack of activity in mouse models of VV [31] and its previous toxicity history in treatment of hepatitis in humans probably precludes it as a serious candidate. Adefovir dipivoxil, on the other hand, appears to be a serious candidate in that it is very active against VV and CV replication in tissue culture, has good oral bioavailability [32], and has been approved for treatment of hepatitis B. This compound needs to be evaluated further against monkeypox virus and variola virus before its real potential is known.
Advances in technology and targeted drug design for diseases such as AIDS, hepatitis, and others have not been applied until recently to the development of effective agents for orthopoxviruses. Lessons learned from the development of drugs for other viral diseases, particularly the herpesviruses, can be applied directly to the search for new inhibitors of orthopoxvirus replication. Adaptation of existing assays and screening techniques to this problem requires establishing the relationship of orthopoxvirus functions and gene products to those in other viruses that are well characterized and for which, assays and known inhibitors exist. The aim of this review is to highlight the steps involved in the replication of these viruses and emphasize those specific viral functions that are essential for replication. Each step in the viral replication cycle is described in detail focusing on specific viral functions that are known targets or hold promise as targets for antiviral therapy. This discussion is intended to stimulate the development of antiviral therapies for orthopoxvirus infections by providing information that can help investigators apply their expertise in other viruses to the field. It is hoped that this information will lead to the development of specific inhibitors for critical replication targets that can eventually be developed as agents for treatment of orthopoxvirus infections.
Overview of Orthopoxvirus Replication
The orthopoxvirus genus consists of 10 viruses including variola virus, VV, CV, monkeypox virus, and camelpox virus [33]. The prototypic member of this genus is VV. This virus has a characteristic virion morphology common to all viruses in the genus, distinguished by its large size (270 × 350 nm) and rather rectangular appearance. Two major types of infectious virions are produced in cell culture; i) intracellular mature virions (IMV) found within infected cells that are the most abundant form and ii) extracellular enveloped virions (EEV) that are IMV surrounded by a secondary lipoprotein envelope and ultimately released from the cell into the external environment. While EEV have been implicated in dissemination in vivo, IMV are very stable and are thought to be responsible for transmission between hosts.
Orthopoxvirus genomes are linear, double stranded DNA with inverted tandem repeats of variable length at both ends. Genomic DNA terminates in hairpin structures, such that it would form one circular single stranded piece of DNA if it were completely denatured [34]. The complete genome of the prototypic Western Reserve (WR) strain of VV has been sequenced and is 1941711 base pairs in length (AY243312). The Copenhagen (M35027), Ankara (U94848), and Tian Tan (AF095689) strains have also been entirely sequenced and are largely colinear with WR, with the exception of some insertions and deletions that are likely the result of extensive passage. Most other orthopoxvirus genomes have also been sequenced in their entirety and more than one strain have been sequenced for variola virus, CV and camelpox virus (for a current listing, see http://www.poxvirus.org).
The Copenhagen strain of VV was the first strain of this virus to be sequenced and 263 potential genes were identified in the 191,636 base pair genome [35]. A more recent annotation of the WR strain by Esposito and colleagues, included a number genes not present in Copenhagen and annotated 206 nonredundant genes (AY243312). This set of genes includes all the viral proteins required for viral replication in vitro and also contains numerous genes that are likely required for in vivo replication including an array of immunomodulatory genes [36]. A recent analysis identified a set of 49 genes that are completely conserved between the insect and vertebrate poxvirus families and a total of 90 genes that are conserved among the chordate poxviruses and are thought to reflect a minimum complement of genes required for replication of poxviruses [37]. This set of genes likely contains a number of good targets for antiviral therapies since they are highly conserved and are likely required for essential functions in replication of all orthopoxviruses.
The VV replication cycle presents many novel targets for the development of antiviral drugs (see Fig. (1). Briefly, both IMVs and EEVs bind to and enter cells with release of cores into the cytoplasm. The virus cores uncoat and early gene expression occurs using constituent proteins packaged within the virion. Expression of early genes leads to synthesis of intermediate gene transcription factors and the initiation of viral DNA synthesis, which allows the expression of intermediate genes to proceed. Intermediate gene products include a set of late transcription factors that subsequently promote late transcription. Late structural proteins and enzymes are required for the assembly of virions, which occurs in electron dense areas of the cytoplasm and are also required to recruit early transcription factors destined to be packaged into maturing virions. Nascent genomic DNA in the form of very large concatameric intermediates is then resolved into unit length genomes and packaged into immature virions. Proteolytic events drive the maturation of immature particles into IMVs and a subset of these undergo a secondary envelopment where additional glycoproteins are acquired. Newly formed viruses are transported to the cell surface where they egress to form EEVs.
Fig. (1).
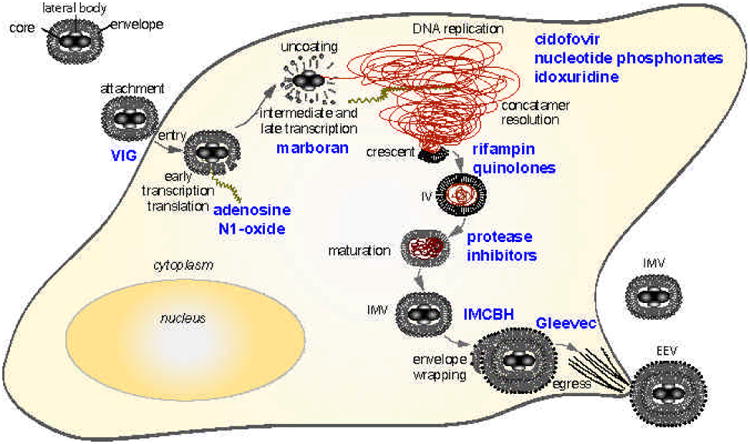
Inhibitors of the orthopoxvirus replication cycle. Virions enter the cell and express early, intermediate and late gene products that facilitate the replication of the virus. Distinct steps of the replication cycle are shown in black, and inhibitors that block replication at these stages are shown in blue. RNA transcripts are brown and nascent viral DNA is shown as red.
Below, each step of the replication process will be discussed in detail, noting the viral proteins that are known to participate in these functions. Following a summary of each phase of the replication cycle will be a description of inhibitors that are known to block viral replication at that particular stage, noting the molecular targets of the inhibitors if they are known (Table 2). Potential targets will also be discussed at the end of each section, concentrating on essential gene products insofar as they are known. In addition, the gene products discussed in each step of the orthopoxvirus life cycle are summarized in Table 3.
Table 2. Inhibitors with Defined Targets in VV.
| inhibitor | target | protein ID | function | reference |
|---|---|---|---|---|
| hydroxyurea | F4L (VACWR043) a | AAO89322.1 | ribonucleotide reductase small subunit | [151] |
| aphidicolin, araC, PAA | E9L (VACWR065) a | AAO89344.1 | DNA polymerase | [91,92] |
| hydroxyurea | I4L (VACWR073) c | AAO89352.1 | ribonucleotide reductase large subunit | [151] |
| IBT | G2R (VACWR080) a | AAO89359.1 | late transcription elongation factor | [73] |
| IBT | J3R (VACWR095) a, b | AAO89374.1 | poly(A) polymerase small subunit VP39; cap methyltransferase, and transcription elongation factor | [71] |
| rifampicin | D13L (VACWR118) a | AAO89397.1 | rifampicin resistance membrane protein; associates with inner surface immature virus membrane | [128] |
| IMCBH | F13L (VACWR052) a | AAO89331.1 | palmytilated EEV membrane protein; phospholipase motif, required for IEV formation | [132] |
| IBT | A24R (VACWR144) a | AAO89423.1 | DNA-dependent RNA polymerase subunit RPO132 | [70] |
| VIG | B5R (VACWR187) a | AAO89466.1 | EEV type-I membrane glycoprotein; | [45] |
| Ofloxacin enrofloxacin novobiocin | H6R (VACWR104) c | AAO89383.1 | topoisomerase I | [103,106] |
| TTP-6171 | I7L (VACWR076) | AAO89355.1 | protease | [130] |
mapped by drug resistance
drug dependence
identified in enzymatic assay
Table 3. Potential Targets for the Development of Drugs to Treat Orthopoxvirus Infections.
| Function | Gene Product | Comments |
|---|---|---|
| DNA processing and packaging | (A18R) | DNA helicase |
| (A22R) | Holliday junction resolvase | |
| (A32L) | ATPase; DNA packaging | |
| (A50R) | DNA ligase | |
| (H6R) | topoisomerase type IB | |
| (I6L) | telomere binding protein | |
| DNA replication | (A20R) | viral DNA polymerase processivity factor |
| (B1R) | serine-threonine kinase; essential for DNA replication | |
| (D4R) | uracil-DNA glycosylase; essential for DNA replication | |
| (D5R) | NTPase interacts with A20R; essential for DNA replication | |
| (E9L) | DNA polymerase | |
| (H5R) | morphogenesis-related; substrate of B1R kinase; | |
| (I3L) | ssDNA-binding phosphoprotein | |
| viral enzymes | (A48R) | thymidylate kinase |
| (A57R) | guanylate kinase | |
| (F4L, I4L) | ribonucleotide reductase small and large subunits | |
| (H1L) | tyr/ser protein phosphatase | |
| (J2R) | thymidine kinase | |
| entry and uncoating | (A28L) | unknown |
| (A33R) | EEV membrane phosphoglycoprotein; actin tail formation | |
| (H2R) | unknown | |
| (A17L) | IMV membrane protein | |
| (L1R) | IMV membrane protein; target of neutralizing antibody | |
| (B5R) | EEV type-I membrane glycoprotein | |
| morphogenesis | (A13L, A14L, A17L, A30L) | essential IMV membrane proteins |
| (D13L) | rifampicin resistance membrane protein | |
| (F10L) | essential serine-threonine kinase; morphogenesis | |
| (G7L) | virion structural protein | |
| (I7L,G1L) | essential viral proteinases | |
| (J1R) | virion protein; required for morphogenesis | |
| (H3L) | IMV morphogenesis involved in maturation (VP55) | |
| (L1R) | IMV membrane protein; target of neutralizing antibody; | |
| transcription and mRNA processing | (A24R, A29L, A5R, D7R, E4L, G5.5R, J4R, J6R) | RNA polymerase |
| (A18R) | DNA helicase; effects postreplicative viral transcription | |
| (D11L) | ATPase, nucleoside triphosphate phosphohydrolase-I | |
| (D12L, D1R) | small and large subunits of mRNA capping enzyme | |
| (E1L, J3L) | poly(A) polymerase catalytic subunits VP55 and V39 | |
| (G2R) | late transcription elongation factor | |
| (H4L) | RNA polymerase associated protein (RAP94) | |
| EEV formation | (A27L) | IMV surface protein, attachment, and microtubule transport |
| (A36R) | IEV transmembrane phosphoprotein; actin tail formation | |
| (B5R) | EEV membrane glycoprotein; membrane wrapping | |
| (F13L) | EEV membrane protein; required for membrane-wrapping | |
| (A34R) | EEV glycoprotein; adherence and actin tail formation |
Virus Entry and Uncoating
The binding of IMV and EEV to cells is poorly understood and the cell receptors for virus binding have not yet been described [38]. Neutralizing epitopes, however, on both IMV and EEV have been defined and may provide some insight as to the viral proteins involved in this process. An antibody specific for the IMV membrane protein A17L, prevents virus penetration [39] and a second IMV protein, L1R, was also shown to be a target of neutralizing antibodies [40]. Of the six described proteins specific for the EEV outer envelope, only vaccination with B5R or A33R were shown to be protective in an animal model and resulted in the production of neutralizing antibodies [41]. Following the binding of virus particles to the cell membrane, virus cores enter the cytoplasm in a process that is ill defined, but is not necessarily fusion mediated since newly delivered virus cores have been reported to retain membranous structures at least transiently [42]. The uncoating process renders viral DNA within virus cores susceptible to deoxyribonuclease [43]. This process is presumed to be mediated by a viral function since it does not occur in the presence of either RNA or protein synthesis inhibitors and a viral protease may be involved since specific proteolytic events are apparent [42]. Recently, conditional lethal mutations in A28L demonstrated directly that this gene was required specifically for penetration of the virus, while virus binding was unaffected [44]. Since additional defects were observed in syncytium formation, it appears that A28L might mediate membrane fusion events and suggests also that they may play an important role in entry. The complex events resulting in the entry of the virus cores in the cytoplasm likely involves the participation of other viral proteins that have not been characterized.
Each of the proteins discussed above could be targeted in some manner to neutralize virus or to inhibit binding to cellular receptors. Indeed, antibodies specific for B5R in vaccinia immune globulin (VIG) appear to play the dominant role in the neutralizing activity against EEV [45]. It is possible that humanized monoclonal antibodies to any of these proteins could be produced and used to limit viral replication in vivo by neutralizing infectious virus. The development of such a therapy however, might face many hurdles given that antibodies are both expensive and lack oral bioavailability. Since a specific, albeit unidentified, viral protease appears to be required for the uncoating process, a protease inhibitor might be identified that could be used to treat viral infections through the specific inhibition of proteolytic events thought to be involved in this process. A28L is a new and excellent drug target since a fusion inhibitor could be developed as has been done for HIV [46]. The continued identification of viral proteins required for both virus entry and fusion will provide new and promising targets in the future.
Transcriptional Regulation and MRNA Processing
Gene expression in VV is regulated as a temporal cascade that is predominantly controlled at the transcriptional level [47]. The RNA polymerase and all transcription factors required for early stage transcription are packaged in virions [48]. This multisubunit enzyme is composed of at least 8 viral proteins that are expressed beginning at the early stage of viral infection [49]. An associated protein, RAP94 (H4L), is synthesized during the late stage of viral replication and is required for the packaging of the complex in virions [50], and together with two subunits of the viral early transcription factor (A7L and D6R) can confer specificity for early promoters [51]. Intermediate gene expression does not occur until the commencement of viral DNA synthesis and may reflect an inability of newly synthesized transcriptional proteins to access genomic DNA [52]. Three transcription factors, A1L, A2L, and G8R, are expressed at intermediate times and are required for the transcription of late viral promoters. Processing of VV transcripts is facilitated by a number of additional viral proteins that both cap and polyadenylate nascent viral transcripts. The capping enzyme is a heterodimer encoded by D1R and D12L that catalyzes each of the steps required to place a 7-methylguanosine cap on the 5′ end of viral mRNAs [53] and its biochemical activity has been well characterized [54]. A subsequent methylation of the penultimate nucleotide of the mRNA is catalyzed by an mRNA (nucleoside-2′-)-methyltransferase encoded by the viral J3R gene [55]. J3R also interacts with the RNA polymerase [56], and participates in the polyadenylation of viral transcripts as a heterodimer with the other subunit encoded by E1L [57].
It is possible that an inhibitor of the VV RNA polymerase could be developed, but no selective inhibitors have been described and thus it may be difficult to identify nontoxic molecules. The specificity of the VV capping and polyadenylation enzymes could also be exploited for the development of new antiviral agents, although it is possible that cellular enzymes could partially complement the function of viral enzymes involved in this process. Ribavirin triphosphate is a substrate for D1R and the capping enzyme, and can be covalently linked to the 5′ end of mRNAs instead of 7-methyl-guanylate [58]. The ribavirin derived cap structure confers stability on the mRNA, but adversely affects the ability of the transcripts to be translated and may result in the inefficient expression of viral genes. It is unclear if the inhibition observed with ribavirin in cell culture [59] is the result of this activity or the inhibition of IMP dehydrogenase, however it does not appear to be effective in an animal model [60]. It also appears that the translation of VV mRNA molecules can be specifically blocked with small molecule inhibitors. Adenosine N1-oxide is a selective inhibitor of VV replication that specifically blocks the translation of viral mRNA without affecting the translation of cellular messages [61]. Specific viral proteins affected by this compound have not been identified, but it holds promise as new approach for inhibiting viral replication.
Transcriptional Termination
The termination of transcription in orthopoxviruses exhibits some unique features [62] and is of particular interest because of its relationship to the mechanism of action of IBT and Marboran. Early transcripts are terminated prematurely in response to cis-acting sequences near the 3′ end of early transcriptional units in a process that requires both the capping enzyme [63] and a DNA dependent ATPase encoded by D11L [64]. Intermediate and late stage transcription differs in that the RNA polymerase reads through the early termination signals producing long transcripts with heterogeneous 3′ ends [65]. The A18R gene encodes an essential DNA helicase [66] and viruses with mutations in this gene yield aberrantly long transcripts, suggesting that it is an important transcript release factor [67]. Viruses with mutations in G2R are also defective and produce intermediate and late transcripts that are terminated prematurely, are uncharacteristically homogeneous, and under express large viral genes [68].
IBT and Marboran exhibit good antiviral activity against orthopoxviruses both in vitro and in vivo [7]. Despite their well characterized antiviral activity and limited clinical experience in humans [69], the mechanism of action of these compounds is complex and remains unclear. A large body of evidence suggests that IBT acts at the level of transcription and drug resistant mutations map to the 132 kilodalton (kDa) subunit of the RNA polymerase (A24R) [70] as well as J3R [71]. Interestingly, the deletion of the essential G2R gene can be complemented by minimizing premature termination either by deleting A18R [72], or through the use of IBT to minimize early termination [73]. Moreover, some viruses with mutations in J3R replicate poorly and can also be complemented through the addition of IBT [71]. While the molecular defects that occur in the presence of IBT have been characterized, it is not immediately apparent why this would have such an adverse affect on the replication of the virus. The presence of double stranded RNA resulting from aberrant termination may induce 2′-5′ oligoadenylate synthetase and the activation of RNaseL, thus inducing host defensive mechanisms [74]. Additional investigations with IBT are required to better understand its mechanism of action and the development of similar less toxic analogs might also lead to new therapies.
DNA Replication
The synthesis of VV DNA in general, and the DNA polymerase in particular is of paramount importance in the development of antiviral therapies because it is the target of many effective drugs against this and other viruses [13,20]. Viral DNA synthesis initiates after early transcription events within virus cores that uncoat and give rise to virus replication centers [75]. An origin of replication that resembles those seen in other viruses has not been described in VV; instead nicking events occur in the extrahelical bases of the terminal hairpins which results in the initiation of DNA synthesis in a process that is thought to involve I1L, I6L, and K4L [76]. A large number of viral proteins are likely required for the processive DNA synthesis that occurs during the replication process. Thus far, genetic studies with temperature sensitive (ts) mutants of VV have identified five complementation groups that are required for DNA synthesis and include the E9L DNA polymerase [77], D5R nucleoside triphosphatase [78], B1R protein kinase [79], D4R uracil DNA glycosylase [80,81], and the A20R polymerase processivity factor [82,83]. The DNA replication complex forms through the direct physical interaction of the molecular constituents and yields a large functional enzyme complex. The E9L DNA polymerase is a 1006 amino acid (aa) protein with a migration rate of 116 kDa that possesses both DNA polymerase and a proofreading activity within a single catalytically active polypeptide [82]. A20R is a 426 aa protein with an apparent migration rate of 48 kDa and appears to be a central molecule in the replication complex. This protein copurifies and coimmunoprecipitates with the DNA polymerase and greatly increases its processivity [84]. In addition to binding the DNA polymerase, this molecule appears to be important in recruiting other molecules to the replication complex and has been shown to physically associate with D4R, H5R, and D5R [85]. The D4R gene encodes a 218 aa protein with a migration rate of 25 kDa and possesses uracil DNA glycosylase activity [86]. Perhaps the most intriguing observation regarding this protein, is that point mutations which abrogate its enzymatic activity do not appear to impact DNA synthesis, viral replication, or yield of progeny virus [87]. Clearly, D4R has acquired an essential function that is unrelated to its enzymatic activity and may be related to its presence in the replication complex. The H5R gene encodes a 203 aa phosphoprotein that has been shown to be a transcription factor for late genes [88]. Its function in the replication complex is currently unclear, however B1R is known to associate with and phosphorylate this protein [89]. D5R encodes a 785 aa protein with a migration rate of 90 kDa. Viruses with ts mutations in D5R exhibit a fast stop phenotype suggesting that this molecule is part of the DNA replication complex [78] and it also may play a role in recombination [90].
The DNA polymerase has been identified as the target for many of the antiviral drugs with activity against orthopoxviruses [13,20]. Aphidicolin resistant mutants generated in the laboratory acquired a threonine or valine substitution at aa 498 that also conferred hypersensitivity to cytosine arabinoside (araC) [91]. Resistance to phosphonoacetic acid is conferred by any of three mutations; cysteine 356 to tyrosine, glycine 372 to aspartic acid, or glycine 380 to serine. Resistance to AraC requires any of the three mutations above with the addition of a phenylalanine 171 to tyrosine mutation [92]. Although CDV drug resistant mutants have been described, the mutations that confer resistance have not been mapped or sequenced [93].
A number of drugs that have been approved for the treatment of herpesvirus infections are active against VV and CV in vitro (Table 1). Other compounds that have been in phase II/phase III clinical trials for these infections, are also active against orthopoxviruses [20]. It is interesting that all of the active compounds are either halogenated uracil analogs or phosphonate nucleotides. These results are consistent with a model in which the activity of nucleoside analogs against orthopoxviruses is not limited by their ability to inhibit the VV DNA polymerase, but rather by their ability to be phosphorylated by the thymidine kinase, J2R. Enzymatic characteristics of J2R have not been examined in detail, but the limited activity of nucleoside analogs against VV suggests that it exhibits a much narrower substrate specificity than the thymidine kinases encoded by herpesviruses. The good activity of nucleotide phosphonate analogs is also consistent with this model since the conversion to the monophosphate by J2R is not required. Clearly more research into the substrate specificity of J2R is required and could identify additional nucleosides that are highly active and nontoxic.
Concatamer Resolution and Packaging
DNA replication in orthopoxviruses rapidly produces significant quantities of viral DNA in the form of very large concatamers, that ultimately resolve into unit length genomes that are packaged within immature virions [94]. At least five viral gene products are thought to facilitate the resolution of unit length genomes from the tangle of concatameric intermediates consisting of approximately 10,000 copies of genomic DNA. The A22R gene encodes a specific Holliday junction resolvase that catalyzes the conversion of cruciform recombination intermediates into linear duplex products [95]. A 50 kDa dimeric deoxyribonuclease has been described and is thought to function in the resolution of concatemeric DNA intermediates, but the gene that encodes it has not been identified [96]. The A50R DNA ligase [97] and the DNA topoisomerase might also be expected to participate in this process [98]. H6R encodes a functional topoisomerase type IB that is packaged in virions and functions as part of the DNA replication complex [99]. A genetically engineered I6L ts mutant replicated DNA normally and correctly processed viral genomes, however, DNA failed to enter immature virions and proteolytic cleavage of the structural proteins was defective suggesting that it is required for the encapsidation of viral DNA [100].
Many of the processes that result in the formation of unit length genomes and their incorporation into IMVs could be targeted with small molecule inhibitors. The topoisomerase is a target of particular interest, since some quinolone antibiotics inhibit viral replication [101]. Recombinant viruses with deletions in the VV topoisomerase are viable, although they make smaller plaques in the absence of this enzymatic activity [102]. Nevertheless, a number of antibiotics [101,103] and a peptide inhibitor of this enzyme [104] significantly inhibit viral replication, arresting it early in morphogenesis. The quinolone antibiotic, ofloxacin inhibits the VV topoisomerase and has specific antiviral activity against VV in vitro [103]. Although relatively high concentrations of the compound were required in vitro, the drug was effective in reducing VV tail lesions in mice when the drug was given orally. Novobiocin has also been shown to inhibit the VV topoisomerase [105] and was reported to have weak activity in preventing tail lesions in mice infected with VV [103]. A recent study with fluoroquinolone antibiotics also demonstrated the specific inhibition of VV topoisomerase by enrofloxacin and suggested that related compounds with improved activity could be used as antiviral drugs [106]. The efficacy of topoisomerase inhibitors in mice suggests that they have the potential to provide therapeutic benefits in humans, notwithstanding the nonessential nature of this enzyme. Additional research is required to identify the most effective compounds and further evaluation of some of these inhibitors shown in Table 2 in animal models is warranted.
Specific inhibitors that prevent the cleavage and subsequent packaging of human cytomegalovirus DNA have been described [107,108]. It is possible that the specific cleavage and packaging of genomic DNA could be targeted in VV as well, and the telomere binding protein and proteins involved in the early stages of morphogenesis might be targets to consider [76].
Virion Morphogenesis and Release
Viral replication in the cytoplasm takes place in virus factories that are initially characterized by the presence of H5R, E5R, and the PKR inhibitor E3L [109]. The formation of virions occurs within the confines of these structures and initiates with the formation of crescents (Fig. (1)) containing the D13L spicule protein [110] and a membrane derived from the endoplasmic reticulum Golgi intermediate compartment [111]. D13L appears to localize to the concave surface of crescents and is thought to act as a scaffold to impose a rigid structure on the membrane involved [112]. The onset of morphogenesis is also dependent on the F10L protein kinase that phosphorylates A17L and A14L proteins and is required prior to the proteolytic cleavage of the A17L [113]. An assembly complex containing these proteins as well as A30, G7, and J1R is required for the formation of virions and also appears to increase the activity of the F10L kinase [114]. Immature virions containing genomic DNA undergo a condensation event during the maturational process and mature cores are formed as a result of proteolytic events catalyzed by the I7L protease [115]. Also required for the maturation of immature virions is the G1L metalloprotease [116], the myristylated L1R protein [117] and the A13L phosphoprotein [118].
After maturation, IMVs are transported out of the virus factory via microtubules [119] by a mechanism that requires A27L [120]. A subset of IMVs acquire a second envelope from either the early endosomes or the Golgi through a complex wrapping mechanism and are eventually released as EEVs [121]. This process is also dependent on A27L [120] as well as F13L [122] and B5R [123,124]. IMVs are actively transported to the cell surface on microtubules [125]. At the cell surface, the B5R membrane protein activates Src, which in turn phosphorylates A36R and promotes the formation of actin tails at the base of the virions [126]. The formation of these structures at the cell surface promotes cell to cell spread of the virus and is thought to be one of the factors contributing to the release of EEV [121].
This stage of the viral replication cycle is complex and has both proven and potential molecular targets. The antibiotic, rifampin inhibits the replication of vaccinia early in morphogenesis and blocks the formation of crescents [127]. Resistance to this drug maps to D13L [128] and treatment with the drug blocks the localization of D13L to virosomes [129]. It is possible that this drug or more effective analogs could be developed for the treatment of orthopoxvirus infections in vivo. The I7L protease is also clearly an important target for the development of VV inhibitors and experience with human immunodeficiency virus illustrates the tremendous potential of protease inhibitors. Recently a specific inhibitor of I7L, TTP-6171, was described and continued research in this arena will likely yield a number of candidate compounds for the treatment of orthopoxvirus infections [130].
N1-isonicotinoyl-N2-3-methyl-4-chlorobenzoylhydrazine (IMCBH) inhibits vaccinia replication at the stage of EEV formation [131]. Subsequent experiments with viruses resistant to the effects of this drug mapped the mutation to the major EEV envelope protein F13L [132]. In the presence of this compound, morphologically mature IMV appeared to accumulate in the cytoplasm [133], and the secondary membrane wrapping did not appear to take place [132]. It is possible that IMCBH, or another molecule with a similar activity could be used to inhibit VV replication in vivo, however, one report suggested that it was not protective in either mice or rabbits [131].
The egress of EEV could also be targeted through the inhibition of Src and related kinases which are required for actin based motility at the cell surface. Indeed, the inhibitor PP1 has been shown to inhibit the formation of actin tails [134], and the Src kinase inhibitor, Gleevec, has also been shown to inhibit the egress of EEV in a comet forming assay (Daniel Kalman, personal communication). While the notion that cellular kinases can be considered targets for antiviral therapy is controversial, many candidate drugs with this mechanism of action have been approved and are safe for use in humans such as Iressa, Rapamycin and Velcade [135]. Inhibitors such as IMCBH and Gleevec are capable of inhibiting the production of EEV although their activity can only be assessed in cell culture through comet assays. Nonetheless, the production of EEV is clearly important in vivo and it remains possible that the inhibition of EEV production may be sufficient to limit the spread of infection in vivo and prevent mortality [136].
Additional Viral Enzymes
VV encodes a number of viral proteins with well characterized enzymatic activity as well as proteins that share significant homology with known enzymes. Among them are essential viral gene products that are known to function in one or more of the stages in the replication cycle discussed above. Some viral enzymes have defined biochemical activity, yet it isn't clear how they function in viral replication so they will be discussed here without reference to the stage of viral replication where they are thought to function. In addition, a number of viral proteins share related biochemical functions and warrant discussion as a group noting their potential to serve as targets for antiviral chemotherapy and will be discussed in this context.
Protein Kinases
B1R protein kinase (VPK1) is one of two essential protein kinases in VV with a migration rate of 34 kDa [137] and well characterized kinase activity [79]. The H5R product in VV is a natural substrate of this enzyme [138] and amino acids Thr-84 and Thr-85 were shown to be specifically phosphorylated [89]. Both H5R and B1R and are present in virosomes [138] and co-localize to punctate sites in the cytoplasm that are precursors to sites of viral DNA synthesis [139]. Mutants with ts defects that map to B1 fail to synthesize viral DNA [137], but it is unclear how this enzyme is involved in DNA synthesis or if it is related to the phosphorylation of H5R. Engineered mutations in H5R were hypothesized to be defective in DNA synthesis, but rather, exhibited a profound defect in morphogenesis reminiscent of those seen with F10 kinase mutants [140]. Recently identified human paralogs of B1R were also capable of complementing the defects observed in B1R ts mutants, but failed to phosphorylate H5 [141].
The F10L protein kinase (VPK2) is required for viral replication and is the major kinase encapsidated in virions. The product of this essential gene exhibits both autophosphorylation and transphosphorylation activity primarily on serine residues [142], but also appears to be involved in the phosphorylation of tyrosine residues on the A17 protein, which is one of the natural substrates for this kinase [143]. Mutant viruses with ts lesions in F10 appear to arrest at a very early stage in virion morphogenesis [144,145].
If specific inhibitors of B1R or F10L could be identified, they might be developed into antiviral drugs provided they have an adequate safety profile. One advantage of targeting protein kinases is that many classes of broadly active small molecule inhibitors have been described and strategies have been developed to confer specificity on existing lead compounds [146]. Thus, developing specific inhibitors of these kinases could start by identifying active drug classes and synthesizing analogs to increase the selectivity of these molecules by methods already developed. It might also be possible to use the protein targets of these kinases as a guide to design high affinity peptide substrates of these enzymes. These peptide inhibitors, could potentially serve as competitive inhibitors of the essential protein kinases.
Nucleoside and Nucleotide Kinases
Vaccinia encodes three proteins that have been shown, or hypothesized to possess nucleoside or nucleotide kinase activities. The thymidine kinase encoded by J2R is not required for viral replication, but is thought to have attenuating influence on viral replication in vivo [147]. Nevertheless, it is a valuable target for antiviral therapy in that it has the potential to confer selectivity on nucleoside analogs through selective phosphorylation in infected cells. Researchers have long used idoxuridine or bromodeoxyuridine to select for viruses that have mutations in this locus since the thymidine kinase preferentially phosphorylates the nucleoside, which in turn is phosphorylated to the level of the triphosphate and specifically inhibits DNA synthesis [148]. VV also expresses a thymidylate kinase (A48R) that has been shown to be enzymatically active [149]. Although this gene is not required for viral replication, it remains possible that this enzyme could preferentially activate nucleotide analogs, like CDV, and contribute substantially to the selectivity of this class of antiviral drugs. It is also possible that the a guanylate kinase homolog encoded by A57R could be exploited in a similar manner [35]. This gene is intact in two strains of CV, however each of the sequenced VV and variola virus isolates are missing the amino terminus of this protein including the ATP binding site and it is unclear if this is the result of passage or if this is a characteristic of both variola virus and VV.
Developing therapies that take advantage of the selectivity afforded by these nucleoside and nucleotide kinases will not likely be accomplished through screening strategies to identify compounds that are selectively phosphorylated by these kinases. Rather, nucleoside and nucleotide analogs that are active in antiviral assays and are nontoxic are likely to be phosphorylated by one or more of these kinases. Subsequent modifications of lead compounds will likely optimize activation by viral enzymes through the identification of the analogs with the greatest selective index.
Ribonucleotide Reductase
Vaccinia virus expresses F4L and I4L as a catalytically active heterodimeric ribonucleotide reductase in infected cells [150]. This enzyme converts ribonucleotides to the corresponding deoxyribonucleotides at the level of the diphosphate and is thought to provide deoxyribonucleoside triphosphates for DNA replication in the cytoplasm where these precursors do not exist in uninfected cells. The ribonucleotide reductase inhibitor, hydroxyurea, inhibits viral replication and drug resistant mutants have been shown to over express this enzyme [151]. While the large subunit of this enzyme is nonessential for viral replication [152] it is possible that specific inhibitors of this enzyme could affect replication of the virus in vivo.
H1L Protein Phosphatase
H1L encodes a 171 aa protein with a migration rate of approximately 20 kDa and exhibits phosphatase activity on substrates containing either phosphotyrosine or phosphothreonine and homologs are present in all orthopoxviruses [153-155]. H1L is apparently essential for viral replication since it has not been possible to generate recombinant viruses with deletions in this region [156]. Recombinant viruses with repressible expression of H1L are viable but exhibit reduced replication when H1 is repressed. Experiments with this virus demonstrated that F18 (DNA binding protein) is hyperphosphorylated in repressed virions and early transcription from these entities did not occur [157].
Summary and Comments
The preceding discussion of orthopoxvirus replication with regard to targets for antiviral chemotherapy, examined each of the stages of viral replication in detail and noted many viral proteins that perform critical functions. It is important at this stage to distinguish among existing drugs that could possibly be used to treat these infections, and known inhibitors of other viral proteins that have the potential to be developed as antivirals. Of the existing drugs with activity against these viruses (Table 1), CDV has the best characterized activity and has been approved for the emergency treatment of smallpox infections [20]. Orally bioavailable analogs of this compound also exhibit good activity and are currently under development [158]. It remains possible that approved drugs for other indications might exhibit antiviral activity against smallpox and some of these are under investigation.
A number of inhibitors of orthopoxvirus replication have been described and their mechanisms of action has been discussed herein. The molecular targets for many of these compounds have been formally ascribed to viral targets by genetic and biochemical means (Table 2). It is possible that derivatives of some of these inhibitors could be developed further as antiviral drugs, particularly those that have been approved for other indications and have already been shown to exhibit some activity in animal models (rifampin, ofloxacin, novobiocin). However, additional experiments are required to confirm their activity in animal models, and their activity also needs to be confirmed against variola virus and monkeypox virus.
Other than the DNA polymerase, the most promising targets likely fall among the genes required for viral replication. While this review has focused on essential genes, it is possible that inhibitors of other targets required for pathogenesis, such as CrmA and the complement control gene could also be considered as viable targets [36]. The most promising potential molecular targets have been discussed in detail above and are summarized in Table 3. While some of proteins, such as the protease are being screened at this writing, others remain unexploited until assays can be developed, or adapted from other systems. The material presented here was intended to help investigators relate experiences in other viral systems to similar aspects of orthopoxvirus replication. The application of technology and strategies used successfully for the development of therapies for other viruses, as well as current efforts underway should result in the discovery and development of new drugs for the treatment of these infections.
Acknowledgments
The authors thank Dick Moyer and Pete Turner (University of Florida, Gainesville, FL) for their helpful comments and critical reading of the manuscript.
Abbreviations
- VV
Vaccinia virus
- CV
Cowpox virus
- CDV
Cidofovir
- IBT
Isatin-β-thiosemicarbazones
- IMV
Intracellular mature virion
- EEV
Extracellular enveloped virion
- VIG
Vaccinia immune globulin
- kDa
Kilodalton
- IMCBH
N1-isonicotinoyl-N2-3-methyl-4-chlorobenzoylhydrazine
- ts
Temperature sensitive
- aa
Amino acid
References
- 1.Breman JG, Henderson DA. N Engl J Med. 1998;339:556–559. doi: 10.1056/NEJM199808203390811. [DOI] [PubMed] [Google Scholar]
- 2.Breman JG, Henderson DA. N Engl J Med. 2002;346:1300–1308. doi: 10.1056/NEJMra020025. [DOI] [PubMed] [Google Scholar]
- 3.Henderson DA. Emerg Infect Dis. 1998;4:488–492. doi: 10.3201/eid0403.980340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heymann DL, Szczeniowski M, Esteves K. Br Med Bull. 1998;54:693–702. doi: 10.1093/oxfordjournals.bmb.a011720. [DOI] [PubMed] [Google Scholar]
- 5.Hutin YJ, Williams RJ, Malfait P, Pebody R, Loparev VN, Ropp SL, Rodriguez M, Knight JC, Tshioko FK, Khan AS, Szczeniowski MV, Esposito JJ. Emerg Infect Dis. 2001;7:434–438. doi: 10.3201/eid0703.010311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O'Toole T. Emerg Infect Dis. 1999;5:540–546. doi: 10.3201/eid0504.990416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bauer DJ. Br J Exp Pathol. 1955;36:105–114. [PMC free article] [PubMed] [Google Scholar]
- 8.Bauer DJ, Sheffield FW. Nature. 1959;184(Suppl. 19):1496–1497. doi: 10.1038/1841496b0. [DOI] [PubMed] [Google Scholar]
- 9.Boyle JJ, Haff RF, Stewart RC. Antimicrobial Agents Chemother. 1966;6:536–539. [PubMed] [Google Scholar]
- 10.Bauer DJ, St Vincent L, Kempe CH, Young PA, Downie AW. Am J Epidemiol. 1969;90:130–145. doi: 10.1093/oxfordjournals.aje.a121057. [DOI] [PubMed] [Google Scholar]
- 11.Bauer DJ, St vincent L, Kempe CH, Downie AW. Lancet. 1963;35:494–496. doi: 10.1016/s0140-6736(63)90230-7. [DOI] [PubMed] [Google Scholar]
- 12.De Clercq E, De Somer P. Appl Microbiol. 1968;16:1314–1319. doi: 10.1128/am.16.9.1314-1319.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Clercq E. Clin Microbiol Rev. 2001;14:382–397. doi: 10.1128/CMR.14.2.382-397.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Clercq E, Holy A, Rosenberg I. Antimicrob Agents Chemother. 1989;33:185–191. doi: 10.1128/aac.33.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Clercq E, Holy A, Rosenberg I, Sakuma T, Balzarini J, Maudgal PC. Nature. 1986;323:464–467. doi: 10.1038/323464a0. [DOI] [PubMed] [Google Scholar]
- 16.De Clercq E, Luczak M, Shugar D, Torrence PF, Waters JA, Witkop B. Proc Soc Exp Biol Med. 1976;151:487–490. doi: 10.3181/00379727-151-39241. [DOI] [PubMed] [Google Scholar]
- 17.De Clercq E, Sakuma T, Baba M, Pauwels R, Balzarini J, Rosenberg I, Holy A. Antiviral Res. 1987;8:261–272. doi: 10.1016/s0166-3542(87)80004-9. [DOI] [PubMed] [Google Scholar]
- 18.Neyts J, De Clercq E. Antimicrob Agents Chemother. 2001;45:84–87. doi: 10.1128/AAC.45.1.84-87.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naesens L, Snoeck R, Andrei G, Balzarini J, Neyts J, De Clercq E. Antiviral Chemistry and Chemotherapy. 1997;8:1–23. [Google Scholar]
- 20.Kern ER. Antiviral Res. 2003;57:35–40. doi: 10.1016/S0166-3542(02)00198-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baker RO, Bray M, Huggins JW. Antiviral Res. 2003;57:13–23. doi: 10.1016/S0166-3542(02)00196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kern ER, Hartline C, Harden E, Keith K, Rodriguez N, Beadle JR, Hostetler KY. Antimicrob Agents Chemother. 2002;46:991–995. doi: 10.1128/AAC.46.4.991-995.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neyts J, De Clercq E. J Med Virol. 1993;41:242–246. doi: 10.1002/jmv.1890410312. [DOI] [PubMed] [Google Scholar]
- 24.Bray M, Martinez M, Kefauver D, West M, Roy C. Antiviral Res. 2002;54:129–142. doi: 10.1016/S0166-3542(01)00220-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bray M, Martinez M, Smee DF, Kefauver D, Thompson E, Huggins JW. J Infect Dis. 2000;181:10–19. doi: 10.1086/315190. [DOI] [PubMed] [Google Scholar]
- 26.Smee DF, Bailey KW, Sidwell RW. Antivir Chem Chemother. 2001;12:71–76. doi: 10.1177/095632020101200105. [DOI] [PubMed] [Google Scholar]
- 27.Smee DF, Bailey KW, Wong M, Sidwell RW. Antiviral Res. 2000;47:171–177. doi: 10.1016/s0166-3542(00)00105-4. [DOI] [PubMed] [Google Scholar]
- 28.Quenelle DC, Collins DJ, Kern ER. Antimicrob Agents Chemother. 2003;47:3275–3280. doi: 10.1128/AAC.47.10.3275-3280.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huggins JW, Martinez MJ, Hartmann CJ, Hensley LE, Jackson DL, Refoeven DF, Kuksh DA, Lawson T, Miller DM, Mucker EA, Shamblin JD, Tate MK, Whitehour LA, Swiers SH, Jahrling PB. Antiviral Res. 2004;62:A57. [Google Scholar]
- 30.Cundy KC, Bidgood AM, Lynch G, Shaw JP, Griffin L, Lee WA. Drug Metab Dispos. 1996;24:745–752. [PubMed] [Google Scholar]
- 31.Smee DF, Sidwell RW. Nucleosides Nucleotides Nucleic Acids. 2004;23:375–383. doi: 10.1081/ncn-120028334. [DOI] [PubMed] [Google Scholar]
- 32.Barditch-Crovo P, Toole J, Hendrix CW, Cundy KC, Ebeling D, Jaffe HS, Lietman PS. J Infect Dis. 1997;176:406–413. doi: 10.1086/514057. [DOI] [PubMed] [Google Scholar]
- 33.Moss B. In: Fields' virology. Fields BN, Knipe DM, Howley PM, Griffin DE, editors. Lippincott Williams & Wilkins; Philadelphia: 2001. p. 3087. [Google Scholar]
- 34.Baroudy BM, Venkatesan S, Moss B. Cell. 1982;28:315–324. doi: 10.1016/0092-8674(82)90349-x. [DOI] [PubMed] [Google Scholar]
- 35.Goebel SJ, Johnson GP, Perkus ME, Davis SW, Winslow JP, Paoletti E. Virology. 1990;179:247–266. 517–263. doi: 10.1016/0042-6822(90)90294-2. [DOI] [PubMed] [Google Scholar]
- 36.Turner PC, Moyer RW. Virus Res. 2002;88:35–53. doi: 10.1016/s0168-1702(02)00119-3. [DOI] [PubMed] [Google Scholar]
- 37.Upton C, Slack S, Hunter AL, Ehlers A, Roper RL. J Virol. 2003;77:7590–7600. doi: 10.1128/JVI.77.13.7590-7600.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doms RW, Blumenthal R, Moss B. J Virol. 1990;64:4884–4892. doi: 10.1128/jvi.64.10.4884-4892.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ichihashi Y, Oie M. Virology. 1996;220:491–494. doi: 10.1006/viro.1996.0337. [DOI] [PubMed] [Google Scholar]
- 40.Wolffe EJ, Vijaya S, Moss B. Virology. 1995;211:53–63. doi: 10.1006/viro.1995.1378. [DOI] [PubMed] [Google Scholar]
- 41.Galmiche MC, Goenaga J, Wittek R, Rindisbacher L. Virology. 1999;254:71–80. doi: 10.1006/viro.1998.9516. [DOI] [PubMed] [Google Scholar]
- 42.Pedersen K, Snijder EJ, Schleich S, Roos N, Griffiths G, Locker JK. J Virol. 2000;74:3525–3536. doi: 10.1128/jvi.74.8.3525-3536.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pedley CB, Cooper RJ. J Gen Virol. 1987;68(Pt 4):1021–1028. doi: 10.1099/0022-1317-68-4-1021. [DOI] [PubMed] [Google Scholar]
- 44.Senkevich TG, Ward BM, Moss B. J Virol. 2004;78:2357–2366. doi: 10.1128/JVI.78.5.2357-2366.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bell E, Shamim M, Whitbeck JC, Sfyroera G, Lambris JD, Isaacs SN. Virology. 2004;325:425–431. doi: 10.1016/j.virol.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 46.Greenberg M, Cammack N, Salgo M, Smiley L. Rev Med Virol. 2004;14:321–337. doi: 10.1002/rmv.440. [DOI] [PubMed] [Google Scholar]
- 47.Broyles SS. J Gen Virol. 2003;84:2293–2303. doi: 10.1099/vir.0.18942-0. [DOI] [PubMed] [Google Scholar]
- 48.Munyon W, Paoletti E, Grace JT., Jr Proc Natl Acad Sci USA. 1967;58:2280–2287. doi: 10.1073/pnas.58.6.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baroudy BM, Moss B. J Biol Chem. 1980;255:4372–4380. [PubMed] [Google Scholar]
- 50.Zhang Y, Ahn BY, Moss B. J Virol. 1994;68:1360–1370. doi: 10.1128/jvi.68.3.1360-1370.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahn BY, Gershon PD, Moss B. J Biol Chem. 1994;269:7552–7557. [PubMed] [Google Scholar]
- 52.Keck JG, Baldick CJ, Jr, Moss B. Cell. 1990;61:801–809. doi: 10.1016/0092-8674(90)90190-p. [DOI] [PubMed] [Google Scholar]
- 53.Martin SA, Paoletti E, Moss B. J Biol Chem. 1975;250:9322–9329. [PubMed] [Google Scholar]
- 54.Shuman S. J Biol Chem. 1990;265:11960–11966. [PubMed] [Google Scholar]
- 55.Barbosa E, Moss B. J Biol Chem. 1978;253:7698–7702. [PubMed] [Google Scholar]
- 56.Mohamed MR, Latner DR, Condit RC, Niles EG. Virology. 2001;280:143–152. doi: 10.1006/viro.2000.0749. [DOI] [PubMed] [Google Scholar]
- 57.Gershon PD, Ahn BY, Garfield M, Moss B. Cell. 1991;66:1269–1278. doi: 10.1016/0092-8674(91)90048-4. [DOI] [PubMed] [Google Scholar]
- 58.Bougie I, Bisaillon M. J Biol Chem. 2004;279:22124–22130. doi: 10.1074/jbc.M400908200. [DOI] [PubMed] [Google Scholar]
- 59.Smee DF, Bray M, Huggins JW. Antivir Chem Chemother. 2001;12:327–335. doi: 10.1177/095632020101200602. [DOI] [PubMed] [Google Scholar]
- 60.Smee DF, Wong MH, Bailey KW, Beadle JR, Hostetler KY, Sidwell RW. Int J Antimicrob Agents. 2004;23:430–437. doi: 10.1016/j.ijantimicag.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 61.Kane EM, Shuman S. J Virol. 1995;69:6352–6358. doi: 10.1128/jvi.69.10.6352-6358.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Condit RC, Niles EG. Biochim Biophys Acta. 2002;1577:325–336. doi: 10.1016/s0167-4781(02)00461-x. [DOI] [PubMed] [Google Scholar]
- 63.Shuman S, Broyles SS, Moss B. J Biol Chem. 1987;262:12372–12380. [PubMed] [Google Scholar]
- 64.Shuman S, Moss B. J Biol Chem. 1988;263:6220–6225. [PubMed] [Google Scholar]
- 65.Mahr A, Roberts BE. J Virol. 1984;49:497–509. doi: 10.1128/jvi.49.2.497-509.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Simpson DA, Condit RC. J Virol. 1995;69:6131–6139. doi: 10.1128/jvi.69.10.6131-6139.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lackner CA, Condit RC. J Biol Chem. 2000;275:1485–1494. doi: 10.1074/jbc.275.2.1485. [DOI] [PubMed] [Google Scholar]
- 68.Black EP, Condit RC. J Virol. 1996;70:47–54. doi: 10.1128/jvi.70.1.47-54.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bray M, Wright ME. Clin Infect Dis. 2003;36:766–774. doi: 10.1086/374244. [DOI] [PubMed] [Google Scholar]
- 70.Condit RC, Easterly R, Pacha RF, Fathi Z, Meis R. J Virology. 1991;185:857–861. doi: 10.1016/0042-6822(91)90559-t. [DOI] [PubMed] [Google Scholar]
- 71.Latner DR, Xiang Y, Lewis JI, Condit J, Condit RC. Virology. 2000;269:345–355. doi: 10.1006/viro.2000.0243. [DOI] [PubMed] [Google Scholar]
- 72.Condit RC, Xiang Y, Lewis JI. Virology. 1996;220:10–19. doi: 10.1006/viro.1996.0280. [DOI] [PubMed] [Google Scholar]
- 73.Meis RJ, Condit RC. Virology. 1991;182:442–454. doi: 10.1016/0042-6822(91)90585-y. [DOI] [PubMed] [Google Scholar]
- 74.Bayliss CD, Condit RC. Virology. 1993;194:254–262. doi: 10.1006/viro.1993.1256. [DOI] [PubMed] [Google Scholar]
- 75.Beaud G. Biochimie. 1995;77:774–779. doi: 10.1016/0300-9084(96)88195-8. [DOI] [PubMed] [Google Scholar]
- 76.DeMasi J, Du S, Lennon D, Traktman P. J Virol. 2001;75:10090–10105. doi: 10.1128/JVI.75.21.10090-10105.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Traktman P, Sridhar P, Condit RC, Roberts BE. J Virol. 1984;49:125–131. doi: 10.1128/jvi.49.1.125-131.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Evans E, Traktman P. Chromosoma. 1992;102:S72–82. doi: 10.1007/BF02451789. [DOI] [PubMed] [Google Scholar]
- 79.Rempel RE, Traktman P. J Virol. 1992;66:4413–4426. doi: 10.1128/jvi.66.7.4413-4426.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Millns AK, Carpenter MS, DeLange AM. Virology. 1994;198:504–513. doi: 10.1006/viro.1994.1061. [DOI] [PubMed] [Google Scholar]
- 81.Stuart DT, Upton C, Higman MA, Niles EG, McFadden G. J Virol. 1993;67:2503–2512. doi: 10.1128/jvi.67.5.2503-2512.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McDonald WF, Traktman P. J Biol Chem. 1994;269:31190–31197. [PubMed] [Google Scholar]
- 83.Ishii K, Moss B. J Virol. 2001;75:1656–1663. doi: 10.1128/JVI.75.4.1656-1663.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Klemperer N, McDonald W, Boyle K, Unger B, Traktman P. J Virol. 2001;75:12298–12307. doi: 10.1128/JVI.75.24.12298-12307.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ishii K, Moss B. Virology. 2002;303:232–239. doi: 10.1006/viro.2002.1721. [DOI] [PubMed] [Google Scholar]
- 86.Upton C, Stuart DT, McFadden G. Proc Natl Acad Sci USA. 90:1993. 4518–4522. doi: 10.1073/pnas.90.10.4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.De Silva FS, Moss B. J Virol. 2003;77:159–166. doi: 10.1128/JVI.77.1.159-166.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kovacs GR, Moss B. J Virol. 1996;70:6796–6802. doi: 10.1128/jvi.70.10.6796-6802.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brown NG, Nick Morrice D, Beaud G, Hardie G, Leader DP. BMC Biochem. 2000;1:2. doi: 10.1186/1471-2091-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Roseman NA, Hruby DE. J Virol. 1987;61:1398–1406. doi: 10.1128/jvi.61.5.1398-1406.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Taddie JA, Traktman P. J Virol. 1991;65:869–879. doi: 10.1128/jvi.65.2.869-879.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Taddie JA, Traktman P. J Virol. 1993;67:4323–4336. doi: 10.1128/jvi.67.7.4323-4336.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Smee DF, Sidwell RW, Kefauver D, Bray M, Huggins JW. Antimicrob Agents Chemother. 2002;46:1329–1335. doi: 10.1128/AAC.46.5.1329-1335.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Moyer RW, Graves RL. Cell. 1981;27:391–401. doi: 10.1016/0092-8674(81)90422-0. [DOI] [PubMed] [Google Scholar]
- 95.Garcia AD, Moss B. J Virol. 2001;75:6460–6471. doi: 10.1128/JVI.75.14.6460-6471.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Merchlinsky M, Garon CF, Moss B. J Mol Biol. 1988;199:399–413. doi: 10.1016/0022-2836(88)90613-4. [DOI] [PubMed] [Google Scholar]
- 97.Smith GL, Chan YS, Kerr SM. Nucleic Acids Res. 1989;17:9051–9062. doi: 10.1093/nar/17.22.9051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Carpenter MS, DeLange AM. J Virol. 1991;65:4042–4050. doi: 10.1128/jvi.65.8.4042-4050.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shaffer R, Traktman P. J Biol Chem. 1987;262:9309–9315. [PubMed] [Google Scholar]
- 100.Grubisha O, Traktman P. J Virol. 2003;77:10929–10942. doi: 10.1128/JVI.77.20.10929-10942.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Richter S, Parolin C, Palumbo M, Palu G. Curr Drug Targets Infect Disord. 2004;4:111–116. doi: 10.2174/1568005043340920. [DOI] [PubMed] [Google Scholar]
- 102.Da Fonseca F, Moss B. Proc Natl Acad Sci USA. 2003;100:11291–11296. doi: 10.1073/pnas.1534874100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ikeda S, Yazawa M, Nishimura C. Antiviral Res. 1987;8:103–113. doi: 10.1016/0166-3542(87)90064-7. [DOI] [PubMed] [Google Scholar]
- 104.Klemm M, Cheng C, Cassell G, Shuman S, Segall AM. J Mol Biol. 2000;299:1203–1216. doi: 10.1006/jmbi.2000.3829. [DOI] [PubMed] [Google Scholar]
- 105.Sekiguchi J, Shuman S. Virology. 1997;235:129–137. doi: 10.1006/viro.1997.8684. [DOI] [PubMed] [Google Scholar]
- 106.Kamau E, Grove A. J Mol Biol. 2004;342:479–487. doi: 10.1016/j.jmb.2004.06.082. [DOI] [PubMed] [Google Scholar]
- 107.Buerger I, Reefschlaeger J, Bender W, Eckenberg P, Popp A, Weber O, Graeper S, Klenk HD, Ruebsamen-Waigmann H, Hallenberger S. J Virol. 2001;75:9077–9086. doi: 10.1128/JVI.75.19.9077-9086.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Underwood MR, Harvey RJ, Stanat SC, Hemphill ML, Miller T, Drach JC, Townsend LB, Biron KK. J Virol. 1998;72:717–725. doi: 10.1128/jvi.72.1.717-725.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Murcia-Nicolas A, Bolbach G, Blais JC, Beaud G. Virus Res. 1999;59:1–12. doi: 10.1016/s0168-1702(98)00114-2. [DOI] [PubMed] [Google Scholar]
- 110.Mohandas AR, Dales S. Virology. 1995;214:494–502. doi: 10.1006/viro.1995.0060. [DOI] [PubMed] [Google Scholar]
- 111.Sodeik B, Doms RW, Ericsson M, Hiller G, Machamer CE, van 't Hof W, van Meer G, Moss B, Griffiths G. J Cell Biol. 1993;121:521–541. doi: 10.1083/jcb.121.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sodeik B, Griffiths G, Ericsson M, Moss B, Doms RW. J Virol. 1994;68:1103–1114. doi: 10.1128/jvi.68.2.1103-1114.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Betakova T, Wolffe EJ, Moss B. J Virol. 1999;73:3534–3543. doi: 10.1128/jvi.73.5.3534-3543.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Szajner P, Jaffe H, Weisberg AS, Moss B. J Virol. 2003;77:3418–3429. doi: 10.1128/JVI.77.6.3418-3429.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Byrd CM, Bolken TC, Hruby DE. J Virol. 2002;76:8973–8976. doi: 10.1128/JVI.76.17.8973-8976.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ansarah-Sobrinho C, Moss B. J Virol. 2004;78:6855–6863. doi: 10.1128/JVI.78.13.6855-6863.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ravanello MP, Hruby DE. J Virol. 1994;68:6401–6410. doi: 10.1128/jvi.68.10.6401-6410.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Unger B, Traktman P. J Virol. 2004;78:8885–8901. doi: 10.1128/JVI.78.16.8885-8901.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ploubidou A, Moreau V, Ashman K, Reckmann I, Gonzalez C, Way M. Embo J. 2000;19:3932–3944. doi: 10.1093/emboj/19.15.3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sanderson CM, Hollinshead M, Smith GL. J Gen Virol. 2000;81:47–58. doi: 10.1099/0022-1317-81-1-47. [DOI] [PubMed] [Google Scholar]
- 121.Smith GL, Vanderplasschen A, Law M. J Gen Virol. 2002;83:2915–2931. doi: 10.1099/0022-1317-83-12-2915. [DOI] [PubMed] [Google Scholar]
- 122.Blasco R, Cole NB, Moss B. J Virol. 1991;65:4598–4608. doi: 10.1128/jvi.65.9.4598-4608.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Engelstad M, Smith GL. Virology. 1993;194:627–637. doi: 10.1006/viro.1993.1302. [DOI] [PubMed] [Google Scholar]
- 124.Wolffe EJ, Isaacs SN, Moss B. J Virol. 1993;67:4732–4741. doi: 10.1128/jvi.67.8.4732-4741.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.van Eijl H, Hollinshead M, Smith GL. Virology. 2000;271:26–36. doi: 10.1006/viro.2000.0260. [DOI] [PubMed] [Google Scholar]
- 126.Newsome TP, Scaplehorn N, Way M. Science. 2004;306:124–129. doi: 10.1126/science.1101509. [DOI] [PubMed] [Google Scholar]
- 127.Grimley PM, Rosenblum EN, Mims SJ, Moss B. J Virol. 1970;6:519–533. doi: 10.1128/jvi.6.4.519-533.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Baldick CJ, Jr, Moss B. Virology. 1987;156:138–145. doi: 10.1016/0042-6822(87)90444-2. [DOI] [PubMed] [Google Scholar]
- 129.Miner JN, Hruby DE. Virology. 1989;170:227–237. doi: 10.1016/0042-6822(89)90370-x. [DOI] [PubMed] [Google Scholar]
- 130.Byrd CM, Bolken TC, Mjalli AM, Arimilli MN, Andrews RC, Rothlein R, Andrea T, Rao M, Owens KL, Hruby DE. J Virol. 2004;78:12147–12156. doi: 10.1128/JVI.78.22.12147-12156.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kato N, Eggers HJ, Rolly H. J Exp Med. 1969;129:795–808. doi: 10.1084/jem.129.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Schmutz C, Payne LG, Gubser J, Wittek R. J Virol. 1991;65:3435–3442. doi: 10.1128/jvi.65.7.3435-3442.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hiller G, Eibl H, Weber K. J Virol. 1981;39:903–913. doi: 10.1128/jvi.39.3.903-913.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Frischknecht F, Moreau V, Rottger S, Gonfloni S, Reckmann I, Superti-Furga G, Way M. Nature. 1999;401:926–929. doi: 10.1038/44860. [DOI] [PubMed] [Google Scholar]
- 135.Schang LM. J Antimicrob Chemother. 2002;50:779–792. doi: 10.1093/jac/dkf227. [DOI] [PubMed] [Google Scholar]
- 136.Payne LG, Kristensson K. J Gen Virol. 1985;66(Pt 3):643–646. doi: 10.1099/0022-1317-66-3-643. [DOI] [PubMed] [Google Scholar]
- 137.Traktman P, Anderson MK, Rempel RE. J Biol Chem. 1989;264:21458–21461. [PubMed] [Google Scholar]
- 138.Beaud G, Beaud R, Leader DP. J Virol. 1995;69:1819–1826. doi: 10.1128/jvi.69.3.1819-1826.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Domi A, Beaud G. J Gen Virol. 2000;81:1231–1235. doi: 10.1099/0022-1317-81-5-1231. [DOI] [PubMed] [Google Scholar]
- 140.DeMasi J, Traktman P. J Virol. 2000;74:2393–2405. doi: 10.1128/jvi.74.5.2393-2405.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Boyle KA, Traktman P. J Virol. 2004;78:1992–2005. doi: 10.1128/JVI.78.4.1992-2005.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lin S, Broyles SS. Proc Natl Acad Sci USA. 1994;91:7653–7657. doi: 10.1073/pnas.91.16.7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Derrien M, Punjabi A, Khanna M, Grubisha O, Traktman P. J Virol. 1999;73:7287–7296. doi: 10.1128/jvi.73.9.7287-7296.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Traktman P, Caligiuri A, Jesty SA, Liu K, Sankar U. J Virol. 1995;69:6581–6587. doi: 10.1128/jvi.69.10.6581-6587.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wang S, Shuman S. J Virol. 1995;69:6376–6388. doi: 10.1128/jvi.69.10.6376-6388.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Noble ME, Endicott JA, Johnson LN. Science. 2004;303:1800–1805. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 147.Tartaglia J, Perkus ME, Taylor J, Norton EK, Audonnet JC, Cox WI, Davis SW, van der Hoeven J, Meignier B, Riviere M, et al. Virology. 1992;188:217–232. doi: 10.1016/0042-6822(92)90752-b. [DOI] [PubMed] [Google Scholar]
- 148.Byrd CM, Hruby DE. Methods Mol Biol. 2004;269:31–40. doi: 10.1385/1-59259-789-0:031. [DOI] [PubMed] [Google Scholar]
- 149.Hughes SJ, Johnston LH, de Carlos A, Smith GL. J Biol Chem. 1991;266:20103–20109. [PubMed] [Google Scholar]
- 150.Slabaugh MB, Johnson TL, Mathews CK. J Virol. 1984;52:507–514. doi: 10.1128/jvi.52.2.507-514.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Slabaugh MB, Mathews CK. J Virol. 1986;60:506–514. doi: 10.1128/jvi.60.2.506-514.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Child SJ, Palumbo GJ, Buller RM, Hruby DE. Virology. 1990;174:625–629. doi: 10.1016/0042-6822(90)90119-c. [DOI] [PubMed] [Google Scholar]
- 153.Rosel JL, Earl PL, Weir JP, Moss B. J Virol. 1986;60:436–449. doi: 10.1128/jvi.60.2.436-449.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hakes DJ, Martell KJ, Zhao WG, Massung RF, Esposito JJ, Dixon JE. Proc Natl Acad Sci USA. 1993;90:4017–4021. doi: 10.1073/pnas.90.9.4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Guan K, Hakes DJ, Wang Y, Park HD, Cooper TG, Dixon JE. Proc Natl Acad Sci USA. 1992;89:12175–12179. doi: 10.1073/pnas.89.24.12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Franke CA, Rice CM, Strauss JH, Hruby DE. Mol Cell Biol. 1985;5:1918–1924. doi: 10.1128/mcb.5.8.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Liu K, Lemon B, Traktman P. J Virol. 1995;69:7823–7834. doi: 10.1128/jvi.69.12.7823-7834.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Quenelle DC, Collins DJ, Wan WB, Beadle JR, Hostetler KY, Kern ER. Antimicrob Agents Chemother. 2004;48:404–412. doi: 10.1128/AAC.48.2.404-412.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]