

Abstract
Upon antigen/allergen recognition, epidermal Langerhans’ cells (LC) are mobilized and migrate to the local lymph node where they play a major role in initiating or regulating immune responses. It had been proposed that all chemical allergens induce LC migration via common cytokine signals delivered by TNF-α and IL-1β. Here the dependence of LC migration on TNF-α following treatment of mice with various chemical allergens has been investigated. It was found that under standard conditions the allergens oxazolone, paraphenylene diamine, and trimellitic anhydride, in addition to the skin irritant sodium lauryl sulfate, were unable to trigger LC mobilization in the absence of TNF-α signalling. In contrast, two members of the dinitrohalobenezene family (2,4-dinitrochlorobenzene [DNCB] and 2,4-dinitrofluorobenzene [DNFB]) promoted LC migration independently of TNF-R2 (the sole TNF-α receptor expressed by LC) and TNF-α although the presence of IL-1β was still required. However, increasing doses of oxazolone overcame the requirement of TNF-α for LC mobilization, whereas lower doses of DNCB were still able to induce LC migration in a TNF-α-independent manner. These novel findings demonstrate unexpected heterogeneity among chemical allergens and furthermore that LC can be induced to migrate from the epidermis via different mechanisms that are either dependent or independent of TNF-α. Although the exact mechanisms with regard to the signals that activate LC have yet to be elucidated, these differences may translate into functional speciation that will likely impact on the extent and quality of allergic sensitization.
Keywords: chemical allergens, IL-1β, Langerhans’ cell, TNF-α
Introduction
Epidermal Langerhans’ cells (LC) are the outermost immune sentinels of the skin.1,2 The view initially was that LC were predominantly immunostimulatory cells and critical cellular vectors of cutaneous immune responses.3,4 Such conclusions were drawn primarily from studies of contact hypersensitivity in mice, but there soon emerged indications that the conventional LC paradigm might not provide the complete picture of the role of LC in skin sensitization.5 More recently there has developed an appreciation that LC are not the only, and possibly not even the most important, antigen presenting cells in the acquisition of skin sensitization. It is now apparent that dermal dendritic cells (DC) frequently play a decisive role in the acquisition of skin sensitization.6 Although in certain circumstances LC may be required for the optimal development of skin sensitization in mice,7 there is now persuasive evidence that these cells have, as possibly their primary function, an immunoregulatory role in the skin immune system. It has been proposed that LC are committed to immune regulation and the induction and maintenance of tolerance,8 and it has been shown that LC orchestrate and contain cutaneous immune responses to parasites (such as Leishmania),9 bacteria 10 and contact allergens.11–13 There may exist several mechanisms through which LC are able to regulate cutaneous immune responses, including via the induction of T regulatory cell responses in draining lymph nodes.9,10,14 The implication is that, irrespective of whether LC are primed to deliver immunostimulatory or counter-regulatory signals, their mobilization and migration from the skin to regional lymph nodes is an important element in the orchestration of cutaneous immune responses to chemical allergens and other antigens.
We and others have demonstrated previously that LC migration in mice in response to contact allergens is induced by proinflammatory cytokines.15 Upon activation by cytokines LC migrate from the epidermis towards CXCL12 expressed in the dermis via expression of CXCR4 on LC, and subsequently to the draining lymph node via up-regulation of CCR7 expression.16,17 It was found that in mice and in humans local administration of either IL-1β or TNF-α was able to induce LC mobilization.18–21 Moreover, in a series of experiments conducted largely using 4-ethoxymethylene-2-phenyloxazol-5-one (oxazolone), a potent skin sensitizing chemical, it was shown that the mobilization of LC following local encounter with contact allergen was dependent upon the availability of both TNF-α and IL-1β acting downstream of IL-18.19,22,23 Based upon these observations it had been proposed that the initiation of LC activation following topical exposure to a contact allergen requires the receipt by LC of two independent signals delivered by IL-1β (acting through IL-1RI) and TNF-α, (acting exclusively through TNF-R2; the sole receptor for TNF-α displayed by LC),24 and that these cytokines together provoke the changes required for LC mobilization and migration, including down-regulation of E-cadherin and increased expression of CCR7.25 The assumption has been that this paradigm holds true for all contact allergens, and that in the absence of either cytokine LC mobilization will not be initiated.
In the investigations described here, we have challenged this assumption, and questioned specifically whether the availability of TNF-α is a universal requirement for LC mobilization in response to contact allergens. The results of these investigations illustrate that there is not a mandatory requirement for TNF-α for the initiation of LC migration during skin sensitization.
Materials and methods
Animals
BALB/c strain mice were obtained from Harlan Olac (Bicester, UK). Mice deficient in TNF-R2 (TNF-R2-/- strain mice) 26 were a kind gift from J.J. Peschon, Department of Molecular Immunology, Immunex Corp., Seattle, USA. As the TNF-R2−/− strain mice were on a C57BL6 background, C57BL6 strain mice (Harlan Olac) were used as controls in these experiments and are referred to throughout as TNF-R2+/+. Animals were 8-12 weeks old, with the exception of the experiment presented in Fig. 3biii, where animals were 20 weeks old. Mice were housed on sterilized wood bedding with materials provided for environmental enrichment. Food (Special Diet Services, Witham, UK) and water were available ad libitum. The ambient temperature was maintained at 21 ± 2°C and relative humidity was 55 ± 10% with a 12 hr light/dark cycle. All procedures were approved by the UK Home Office and carried out in compliance with the UK Animals (Scientific Procedures) Act, 1986 under a Home Office granted Project Licence.
Figure 3.
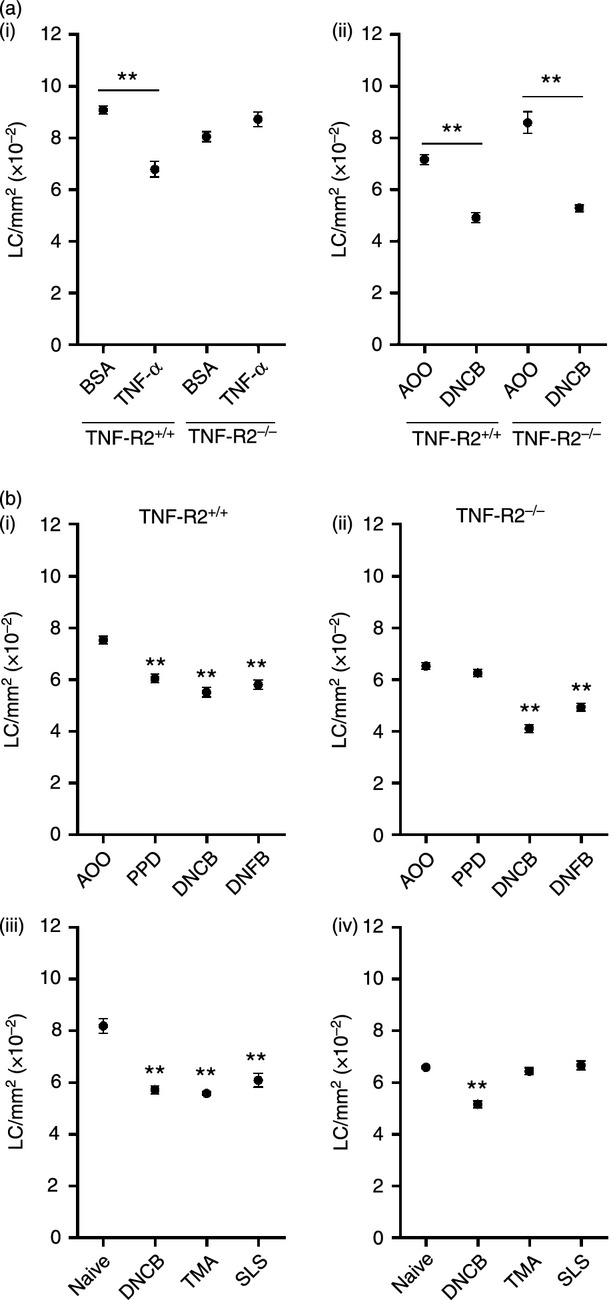
LC migration in TNF-R2−/− strain mice: not all allergens are impaired. TNF-R2+/+ and TNF-R2−/− strain mice received (ai) intradermal injections of TNF-α (or BSA control) or (aii) topical application of 1% DNCB (or AOO vehicle control) to the dorsum of both ears. TNF-R2+/+ (bi) and TNF-R2−/− (bii) strain mice received topical applications of 1% PPD, 0·25% DNFB, 1% DNCB or AOO vehicle control alone. TNF-R2+/+ (biii) and TNF-R2−/− (biv) strain mice received topical application of 1% DNCB, 25% TMA or 10% SLS to the dorsum of both ears, naïve control mice remained untreated. After 4 hr (or 17 hr for TMA) LC frequency was assessed as described in the legend to Fig. 1 (LC/mm2 per group ± SEM; each graph represents one individual experiment, n = 4–8 ears). The significance of differences between the treated and control groups was determined by a one-way anova followed by Tukey's post-test (**P < 0·01).
Genotyping TNF-R2−/− mice
Mice were genotyped, using mRNA derived from a tail snip, to confirm TNF-R2 deficiency. Two sets of primers (Sigma, Poole, UK) were utilized: forward CCACCGCTGCCCCTATGGCG and reverse TAGCACAGCACATCTGAGCC (a region in the second exon of the TNF-R2 gene that is disrupted in TNF-R2−/− but intact in wild type mice) (http://blast.ncbi.nlm.nih.gov/).
Chemicals and exposure
Mice received 25 μl of 0·5% or 3% oxazolone, 0·25% or 1% 2,4-dinitrochlorobenzene (DNCB), 0·25% 2,4-dinitrofluorobenzne (DNFB), 25% trimellitic anhydride (TMA), 1% paraphenylendiamine (PPD) in 4 : 1 acetone:olive oil (AOO); or 25% sodium lauryl sulfate (SLS) in dimethylformamide (all from Sigma) to the dorsum of both ears. Treatment with oxazolone, DNCB, DNFB, PPD and SLS was for 4 hr for LC enumeration, analysis of IL-1β expression and epidermal cell suspensions or 6 hr for TNF-α and IL-1β secretion. Treatment with TMA was for 17 hr, as compared with contact allergens, the mobilization of LC in response to TMA has slower kinetics and significant LC migration is not observed until 17 hr after exposure.27 Control mice were either treated with AOO alone or remained untreated (naïve). After treatment animals were sacrificed and ears were excised.
Cytokine treatment
Mice received 30 μl intradermal injections of recombinant murine TNF-α (50 ng) or IL-1β (50 ng or 300 ng) (R&D Systems, Abingdon, UK) formulated in 0·1% bovine serum albumin/phosphate buffered saline (BSA/PBS; or BSA/PBS as vehicle control) to both ear pinnae using a fine gauge hypodermic needle. Four hours after injection animals were sacrificed and ears were excised for LC enumeration.
Exposure to lactoferrin
Lactoferrin (LF) from human milk (Sigma) was reconstituted in PBS and added to aqueous cream BP (Boots, Nottingham, UK) (0·5 μg and 5 μg). Cream containing LF or cream alone (30 μl) was applied to the dorsum of ears of mice. Application of LF was for 2 hr prior to treatment with either oxazolone (0·5% or 3%), DNCB (1%) or IL-1β (50 or 300 ng).
Antibody administration
Mice received 30 μl intradermal injections of affinity purified goat anti-mouse anti-IL-1β (3 μg; R&D Systems) or rabbit anti-mouse anti-TNF-α (10 μg; PeproTech, London, UK) antibody. Normal goat serum (NGS) or normal rabbit serum (NRS) at a 1 in 5 dilution in PBS were used as controls for anti-IL-1β and anti-TNF-α antibody, respectively. Two hours following antibody administration mice were treated with either oxazolone or DNCB.
Preparation and analysis of epidermal cell suspensions
Ears were removed 4 hr following treatment with chemical and were split with the aid of forceps into dorsal and ventral halves. Dorsal halves were incubated with 0·25% trypsin (Life Technologies, Paisley, UK) in Hanks’ Balanced Salt Solution (HBSS; Life Technologies) for 20 min at 37°. Epidermal sheets were removed and washed thoroughly in 10% fetal calf serum (FCS)/RPMI (Life Technologies). A single cell suspension was prepared by gentle mechanical disaggregation through 200-mesh stainless steel gauze, washed twice in RPMI containing 20% FCS and resuspended in 10% FCS/RPMI. Epidermal cells (5 × 105) were incubated on ice for 30–45 min with anti-mouse I-Ad/I-Ed (rat IgG2a, clone 2G9, BD Pharmingen, San Diego, CA) diluted to 2 μg/ml in 5% FCS/PBS followed by allophycoerythrin-labelled goat anti-rat IgG (BD Pharmingen). Apoptotic cells were identified following staining for annexin V- fluorescein isothiocyanate (FITC)/propidium iodide (PI) according to the manufacturer's instructions (R&D Systems). Cells were washed and resuspended in 1% FCS/0·05% azide/PBS for analysis of 105 cells using a FACSCalibur flow cytometer (BD Biosciences, Oxford, UK).
Preparation of epidermal sheets and LC enumeration
Ears were removed and the dorsal halves were incubated for 1·5 hr in 0·02 mm ethylenediaminetetraacetic acid (EDTA) in PBS at 37°. The epidermis was removed, washed and fixed in acetone at −20° for 20 min. Epidermal sheets were stained for LC with 5 μg/ml MHC Class II (FITC rat anti-mouse IA-IE 2G9 [BD Biosciences]) diluted in 0·1% BSA/PBS for 1 hr. Epidermal sheets were mounted onto slides using Vectashield (Vector Laboratories Ltd, Peterborough, UK) and sealed with nail varnish. The frequencies of stained cells were assessed by fluorescence microscopy using an eyepiece with a calibrated grid (0·32 × 0·213) at ×40 magnification. Samples were blinded and for each sheet 10 consecutive fields were counted in the central portion of each ear. Data are expressed as the mean number of LC per mm2 per group.
Analysis of TNF-α and IL-1β production
Ears were removed and the dorsal halves were isolated, as described previously.27 Dorsal halves were floated on 250 μl 10% FCS/RPMI for 16 hr at 37°, 5% CO2. Supernatants were harvested and analyzed for TNF-α and IL-1β using a TNF-α and IL-1β DuoSet ELISA, respectively (R&D Systems) according to the manufacturer's instructions. The lower limit of accurate detection of the ELISA was approximately 30 pg/ml and 15 pg/ml for TNF-α and IL-1β, respectively.
Quantitative PCR
Ears were collected and immediately immersed in RNAlater (Life Technologies, Paisley, UK) for a minimum of 12 hr. Subsequently tissue was chopped with a scalpel into approximately 1 mm2 pieces and transferred into lysis buffer and homogenized with a T 10 basic ULTRA-TURRAX homogenizer (IKA, Staufen, Germany) until the tissue was dispersed. Subsequently mRNA was extracted using a Purelink extraction kit and cDNA was made using a High Capacity RNA-to-cDNA kit (both from Life Technologies) according to manufacturer's instructions. cDNA was used in quantitative real time PCR reactions using TaqMan primers. Expression of IL-1β was measured relative to the housekeeping gene hypoxanthine phosphoribosyltransferase 1 (HPRT). Results are presented as fold change in gene expression relative to untreated (naive) animals.
Statistical analyses
Statistical analyses were performed using graphpad prism 6 software (GraphPad Software Inc., La Jolla, CA). Data were analyzed using either a Student's t test or a one way anova followed by Tukey's post hoc test. Significance values are indicated as *P < 0·05, **P < 0·01, ***P < 0·001.
Results
In initial experiments the role of TNF-α in LC migration provoked by two chemically unrelated but potent allergens (oxazolone and DNCB) was compared using BALB/c strain mice and neutralizing anti-TNF-α antibody. Concentrations of oxazolone (0·5%) and DNCB (1%) were selected based on previous experience of skin sensitization in mice. The initial concentrations chosen were those that have been shown previously, when applied topically to the ears of BALB/c strain mice, to stimulate the migration of the proportion of LC that are able to rapidly leave the skin (20–30%), and to induce vigorous proliferative responses in the lymph nodes draining the site of exposure.27–29 Topical exposure to both allergens resulted in a significant loss of epidermal LC, approximately 19% and 22% reductions in LC frequency for oxazolone and DNCB, respectively (Fig. 1a). This is consistent with the proportion of LC (20–30%) that appears to be available for rapid mobilization in both murine and human skin.25 Prior administration of neutralizing anti-TNF-α antibody completely abrogated LC migration induced by oxazolone, but was without effect on DNCB-induced LC mobilization. Furthermore, very striking differences in LC morphology were noted between the allergens. Cells treated with oxazolone exhibited little change in morphology, whereas in the presence of neutralizing anti-TNF-α antibody the cell bodies appeared larger and the dendrites more brightly staining (Fig. 1e). Treatment with DNCB (regardless of the presence of anti-TNF-α antibody) caused an enlargement of cell bodies and appearance of a distinct honeycomb pattern of dendrites (Fig. 1f,g).
Figure 1.
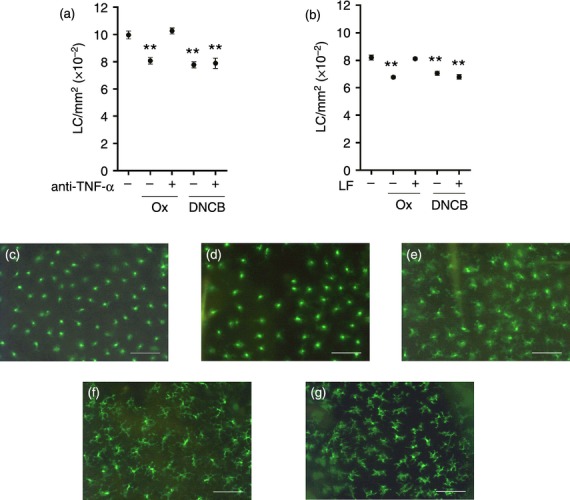
Anti-TNF-α antibody and lactoferrin inhibit oxazolone-, but not DNCB-induced LC migration. Mice (BALB/c) received (a) intradermal injections of anti-TNF-α antibody (or normal rabbit serum [NRS] control) or (b) topical application of human LF in aqueous cream (0·5 μg) (or aqueous cream alone control) to the dorsum of both ears. After 2 hr, mice received a topical application of 0·5% oxazolone (Ox) or 1% DNCB at the same site. Naïve control mice remained untreated. After a further 4 hr epidermal sheets were prepared and stained for LC using FITC-conjugated MHC class II antibody. (a) and (b) LC frequency expressed as mean LC/mm2 per group ± SEM (Each graph represents one individual experiment, n = 6 ears). The significance of differences between treated groups and naïve controls was analyzed by a one-way anova followed by Tukey's post-test (**P < 0·01). Representative micrographs of epidermal sheets from (c) naïve (untreated), (d) NRS/Ox, (e) anti-TNF-α/Ox, (f) NRS/DNCB, and (g) anti-TNF-α/DNCB; scale bar = 50 μm.
Given the differential dependence of the two allergens on TNF-α for LC migration indicated by neutralizing antibody administration, an alternative method for inhibiting cutaneous expression of this cytokine was employed. It has been demonstrated previously that topical application of human lactoferrin (LF) inhibits allergen (diphenylcyprone; oxazolone) and IL-1β-induced LC mobilization in both man and mouse as a consequence of the down-regulation of TNF-α.21,30–32 Prior topical administration of LF was without effect on DNCB-induced LC migration, whereas the oxazolone-induced reduction in LC frequency was almost completely abrogated (only 1% migration) under these conditions (Fig. 1b).
It might be argued that the reduction in LC frequency recorded in all strains of mice following topical exposure to dinitrobenzenes, regardless of the availability of TNF-α, is not a result of migration but is due to the cytotoxicity of these chemicals. Thus, the loss of LC is due to cell death rather than migration. The effect of DNCB on epidermal cell viability was therefore assessed as a function of annexin V/PI staining to determine the level of apoptosis/necrosis. Epidermal cell suspensions were prepared 4 hr following exposure of mice to 1% DNCB or AOO (the time point at which LC migration was monitored) and cells were stained for MHC class II to identify LC and with annexin V/PI. Three colour flow cytometric analysis of epidermal cell suspensions revealed that DNCB treatment was without impact on the frequency of viable LC (∼70%). Furthermore, the proportions of cells entering early apoptosis (annexin V+/PI−), late apoptosis (annexin V+/PI+) or necrosis (annexin V−/PI+), were very similar for cells isolated from AOO- or DNCB-treated animals (Fig. 2).
Figure 2.
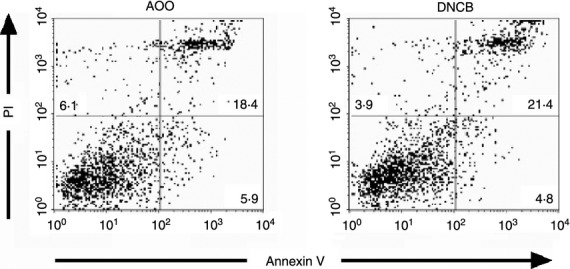
Treatment with DNCB does not induce higher levels of apoptosis or necrosis in LC. Groups of BALB/c strain mice (n = 2) were exposed topically on the dorsum of both ears to 25 μl of 1% DNCB in AOO or to AOO alone. Ears were removed 4 hr later and epidermal cell suspensions prepared for flow cytometric analysis of MHC class II, annexin V and PI expression. Cells were gated for MHC class II expression and analyzed for annexin V/PI staining. Results of representative experiments are shown. The percentage of MHC class II+ cells found in each quadrant is displayed.
An alternative explanation for the apparent lack of requirement for TNF-α in DNCB-stimulated LC migration is that DNCB provokes more vigorous and sustained expression of this cytokine than does oxazolone, and thus the methods employed (administration of anti-cytokine antibody and LF) are insufficient to neutralize the effects. The role of TNF-α in response to DNCB was therefore explored in knockout mice that are deficient in TNF-R2, the only TNF-α receptor expressed by LC.24 Intradermal administration of TNF-α stimulated significant LC mobilization in wild type mice (25%), whereas this cytokine was without effect in the TNF-R2−/− mice (Fig. 3ai). However, a very marked reduction in LC frequency was recorded in both strains following topical treatment with the contact allergen DNCB (31% and 38%; Fig. 3 aii), confirming the lack of dependence of this allergen on TNF-α for the induction of LC migration. This same strain of knockout mice was used to explore the requirement for TNF-α for LC mobilization induced by other stimuli. As oxazolone does not induce robust LC migration in C57 strain mice at the same concentrations used in BALB/c strain mice, other contact allergens (DNFB and PPD) were used in addition to the respiratory allergen TMA and the irritant SLS (Fig. 3b). Concentrations of allergen were selected on the basis of previous experience of doses that in each case provokes relatively vigorous lymph node activation with respect to proliferative responses.27,33,34 The irritant SLS does not provoke vigorous lymph node cell proliferative responses as this is a property of allergens, however, the dose selected has been shown previously to stimulate the rapid mobilization of LC in BALB/c strain mice.35 All stimuli caused the migration of LC with a similar level of vigour in wild type mice (between 20% and 30%) whereas only DNCB and its homologue DNFB were able to induce significant LC mobilization (37% and 24%) in the TNF-R2−/− mice.
For oxazolone-induced LC migration, it has been shown that at least two cytokine signals are required being provided by TNF-α and IL-1β.19,22,23 In subsequent experiments the dependence of DNCB-stimulated LC mobilization upon IL-1β was addressed using neutralizing anti-cytokine antibody (Fig. 4). The loss of epidermal LC was significantly impaired by pre-treatment with neutralizing anti-IL-1β antibody (24% to 3% and 26% to 12% migration recorded for oxazolone and DNCB, respectively). Interestingly, the lack of cutaneous IL-1β also had a differential impact on morphology of those LC that failed to migrate: for oxazolone-treated mice, the absence of IL-1β resulted in a more activated appearance, whereas for DNCB- treated mice, LC exhibited a less activated phenotype.
Figure 4.
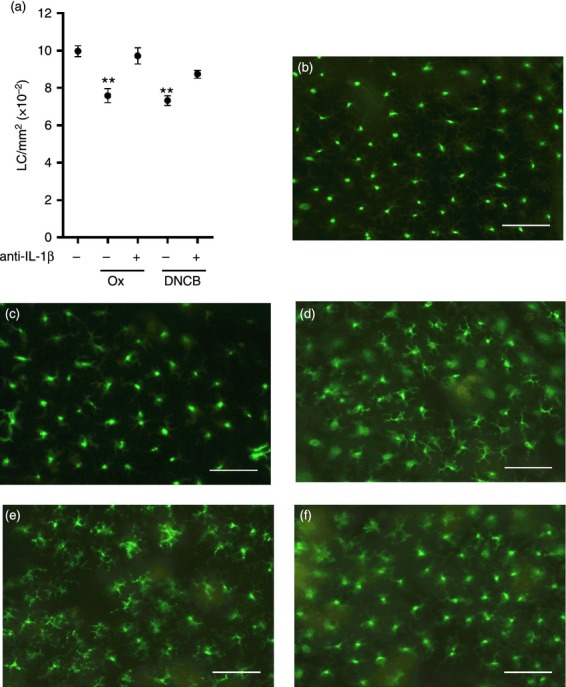
Allergen-induced LC migration is dependent upon IL-1β. BALB/c strain mice received intradermal injections of anti-IL-1β antibody (or normal goat serum [NGS] control) to the dorsum of both ears. After 2 hr, mice received a topical application of 0·5% Ox or 1% DNCB to the same site. Naïve control mice remained untreated. After 4 hr LC frequency was assessed as described in the legend to Figure 1 (mean LC/mm2 per group ± SEM; one individual experiment, n = 6 ears [a]). The significance of differences between treated groups and naïve controls was analyzed by a one-way anova followed by Tukey's post-test (**P < 0·01). Representative micrographs of epidermal sheets from (b) naïve, (c) NGS/Ox (d) anti-IL-1β/Ox, (e) NGS/DNCB and (f) anti-IL-1β/DNCB, scale bar = 50 μm.
In order to examine whether the requirement of TNF-α was dependent on the strength of the stimulus for migration, higher doses of oxazolone were used (Fig. 5). As demonstrated previously, prior application of LF inhibited LC mobilization in response to 0·5% oxazolone with little or no migration occurring (0% and 3·4% for Fig. 5a and b, respectively). In contrast, the higher concentration of oxazolone (3%) induced migration of LC from the epidermis in the absence of TNF-α. Thus, despite prior treatment of mice with LF, 3% oxazolone caused a 13·6% or 18·3% reduction in LC numbers, in the two independent experiments displayed in Fig. 5(a,b) in which TNF-α was neutralized with 0·5 μg or 5 μg of LF, respectively. Based on these data one hypothesis was that migration can be induced in the absence of TNF-α when there is available increased levels of IL-1β produced as a consequence of a stronger stimulus by chemical allergen. We have previously demonstrated that intradermal injection of 50 ng IL-1β is sufficient to induce LC mobilization, and furthermore, that this is dependent upon local TNF-α.19 We sought to determine, therefore, whether higher concentrations of IL-1β were capable of overcoming the requirement for TNF-α for the initiation of LC mobilization (Fig. 5c). Consistent with previous observations, intradermal injection of 50 ng IL-1β induced a significant loss of LC from the epidermis which was blocked by LF treatment. Intradermal injection of 300 ng IL-1β induced the same level of LC migration as did the lower dose (23·0% and 25·6% for 50 ng and 300 ng, respectively) but in this instance that migration was unaffected by prior exposure of mice to LF.
Figure 5.
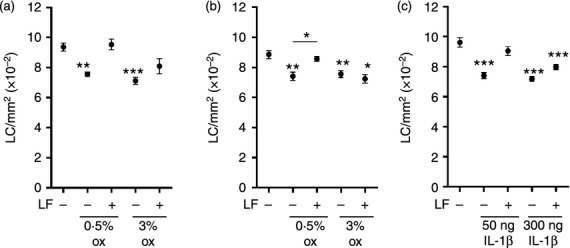
High doses of oxazolone and IL-1β induce LC migration independently of TNF-α. Mice (BALB/c) received topical application of human LF (0·5 μg [a, c] or 5 μg [b]) in aqueous cream (or aqueous cream alone) to the dorsum of both ears. After 2 hr, mice received a topical application of 0·5% or 3% oxazolone (Ox, [a, b]) or intradermal injection of 50 ng or 300 ng IL-β (c) at the same site. Naïve control mice remained untreated. After a further 4 hr epidermal sheets were prepared and stained for LC as described previously and LC frequency expressed as mean LC/mm2 per group ± SEM (each graph represents one individual experiment, n = 5–6 ears per group). The significance of differences between treated groups and naïve controls was analyzed by a one-way anova followed by Tukey's post-test (*P < 0·05, **P < 0·01, ***P < 0·001).
Given that high levels of IL-1β overcame the requirement for TNF-α for LC mobilization, allergen-induced cutaneous IL-1β expression and secretion were measured (Fig. 6). Following analysis of the kinetics of IL-1β gene expression (1 to 24 hr post allergen treatment; data not shown) a 4 hr time point was selected for analysis (Fig. 6a). Two concentrations of DNCB were used; the standard dose of 1% DNCB and a lower dose (0·25%) that induces equivalent LC migration to the standard dose in TNF-R2−/− strain mice (data not shown). Treatment with high dose oxazolone and DNCB each resulted in expression of IL-1β mRNA (∼25 fold increase). The lower doses of both allergens resulted in a more modest increase in IL-1β expression (5-10 fold increase). Baseline secretion of IL-1β protein by naïve explants was approximately 100 pg/ml (Fig. 6b). Treatment with high and low doses of DNCB resulted in significant increases (some 5 fold higher) in production of this cytokine, whereas exposure to oxazolone resulted in a dose dependent effect, with relatively modest (3 fold) induction at the lower dose (that failed to reach statistical significance) and more vigorous induction following stimulation with 3% oxazolone (9 fold increase). The secretion of TNF-α induced by exposure to low and high dose oxazolone and DNCB was also analyzed. Levels that were just above the lower limit of accurate detection (40–60 pg/ml) were recorded for this cytokine in tissue derived from control naïve mice. Treatment with allergen, however, was without impact on this baseline level of TNF-α production (data not shown).
Figure 6.
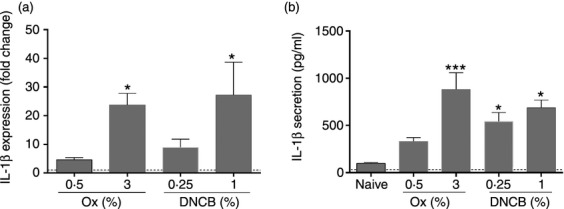
IL-1β secretion is dependent on allergen concentration. BALB/c strain mice received topical application of either oxazolone (0·5% or 3%) or DNCB (0·25% or 1%) to the dorsum of each ear. Naïve mice remained untreated. (a) After 4 hr, the ears were removed and homogenized and expression of IL-1β was measured relative to the housekeeping gene HPRT. Results are presented as fold change ± SEM in gene expression relative to untreated (naive) animals, which were used as a comparator and set to 1 (indicated by dotted line). Data are combined from 2 individual experiments, n = 4–8 ears per group. The significance of differences in the fold change between treatment groups and the 0·5% oxazolone group was analyzed using a one-way anova followed by Tukey's post-test, (*P < 0·05). (b) After 6 hr the ears were removed and the dorsal half was separated and floated on 250 μl RPMI media containing 10% FCS for 16 hr. The supernatants were harvested and analyzed for IL-1β by ELISA. Bars show the mean secreted IL-1β in pg/ml per group ± SEM, data are combined from 2 individual experiments, n = 4–6 ears per group. The significance of differences between the treatment groups and the naïve untreated control was analyzed using a one-way anova followed by Tukey's post-test, (*P < 0·05, ***P < 0·001).
Discussion
It is well-established that topical encounter with a contact allergen and the acquisition of skin sensitization is associated with the mobilization of epidermal LC and their migration away from the skin.25 The assumption originally was that a proportion of LC acquired and processed antigen and transported it from the skin to draining lymph nodes, via afferent lymphatics, for presentation to, and activation of, responsive T lymphocytes. There is now an appreciation, however, that LC are not exclusively immunostimulatory cells, and that they may have as their primary responsibility the maintenance of tolerance and regulation of cutaneous immune responses.8,13 Nevertheless, irrespective of their functional attributes and the roles they play in the acquisition and regulation of skin sensitization, it remains the case that in both mouse and man the mobilization of local LC and their migration from the skin is a hallmark of exposure to a contact allergen.25
The assumption has been that the activation and mobilization of LC induced via exposure to chemical allergens is mediated via cytokine and chemokine signalling. Previous investigations have suggested that contact allergen-induced mobilization is dependent upon the receipt by LC of at least 2 independent cytokine signals, delivered by TNF-α and IL-1β, acting downstream of IL-18.19,22,23 The view was that TNF-α and IL-1β (acting respectively through TNF-R2 and IL-1RI receptors expressed by LC) would together provoke the changes necessary for LC to free themselves from the surrounding epidermal tissue matrix and migrate from the skin, move across the basement membrane and into the dermis via CXCR4/CXCL12 interaction, and ultimately locate within the paracortical regions of draining lymph nodes by up regulating CCR7 expression.16,17,25,36
The data reported here challenge that assumption. We have shown that topical exposure of mice to DNCB (or to its homologue DNFB) induces LC mobilization through a mechanism that is independent of a requirement for TNF-α. The independence of DNCB (and DNFB)-induced LC mobilization of TNF-α appears to be the exception rather than the rule. Thus, at standard concentrations that induce equivalent levels of LC migration, it was found that other chemical allergens that are not dinitrohalobenzenes (oxazolone and PPD), a chemical respiratory allergen (TMA), and the non-sensitizing skin irritant SLS, all require TNF-α signalling for LC mobilization from the epidermis. Additionally, other stimuli that cause LC migration, such as UVB, have also been shown to be dependent upon TNF-α.37 Furthermore, in a murine skin explant model in which LC migrate spontaneously due to trauma-induced cytokine expression, in wild type mice there was a requirement for both TNF-α and IL-1β production, as revealed by neutralizing anti-cytokine antibodies.38
On this basis the as yet unproven implication is that the stimulation of LC mobilization by the majority of external stimuli (including chemical allergens, skin irritants and UVB) is typically dependent upon TNF-α. The intriguing question is, therefore, the basis for the absence of a requirement for TNF-α seen with the dinitrohalobenzenes. To that end it was relevant to consider whether an important variable with regard to TNF-α dependency is the strength of the signal delivered by chemical allergen at the skin surface. To explore this the dose-dependence of the requirement for TNF-α was examined with both oxazolone and DNCB. Consistent with that hypothesis it was found that a higher concentration of oxazolone than that used normally resulted in an equivalent level of LC mobilization, but without a requirement for TNF-α. Lower doses of DNCB still caused the TNF-α-independent migration of LC. The interpretation is that chemicals such as DNCB and DNFB that are naturally potent contact allergens and that deliver to the skin signals of a certain magnitude will provoke LC migration without the need for TNF-α. The corollary is that other chemical allergens that deliver different or less vigorous signals to the skin may require TNF-α unless increased concentrations are administered.
It is important to emphasize that the TNF-α-independent mobilization of LC by DNCB nevertheless required IL-1β. It has been assumed that IL-1β plays two roles in mobilizing LC; providing a stimulus for the production of TNF-α by keratinocytes, and also acting directly on LC themselves to provoke other changes required for migration.19,22,25 However, it has recently been shown that mice in which LC are unresponsive to IL-1 (LC lacking MyD88) migration in response to chemical allergen (the dinitrohalobenzene DNFB) is unaffected. The inference is that the direct interaction of IL-1 with LC may not be an essential pre-requisite for mobilization and that the mandatory activity of IL-1 is effected through some other cellular component, probably keratinocytes.39
We found here that intradermal exposure of mice to a higher concentration of IL-1β was also able to induce TNF-α-independent LC migration. On this basis, it could be argued that the signal elicited by certain contact allergens (such as DNCB), or by higher concentrations of other contact allergens (such as oxazolone), might be a function of more effective stimulation of local IL-1β production. However, the results reported here failed to disclose a clear relationship between the local production of IL-1β and TNF-α-independent LC migration. Collectively, these data suggest that although high local levels of IL-1β is sufficient to allow LC to proceed in the absence of TNF-α, there are other mechanisms through which this might be achieved. The nature of those mechanisms, and the pathway through which DNCB is able to elicit LC mobilization in the absence of TNF-α signalling, have yet to be defined.
It must be acknowledged that induced LC mobilization in the absence of TNF-α is not entirely without precedent. Thus, LC migration in mice infected with West Nile virus, a replicating, cutaneously acquired arbovirus, displays the same pattern of cytokine dependence to that observed for DNCB/DNFB; TNF-α independent and IL-1β dependent.40 Intradermal injection of virus resulted in a reduction in epidermal LC frequency in TNF-α knockout mice and following systemic administration of anti-TNF-α antibody to wild type mice, whereas antibody against IL-1β prevented virus-induced LC mobilization.40 The conclusion drawn is that there may exist a variety of stimuli that provide for the LC migration in the absence of TNF-α.
Irrespective of the pathways involved, it is possible that the nature of the cytokine signals that trigger LC mobilization have an impact on the tempo of migration/and or on the phenotypic and functional characteristics of trafficking LC. In this context it will be interesting to determine whether TNF-α-dependent and TNF-α-independent LC migration are associated with differential downstream consequences for lymph node activation and the quality and vigour of induced immune responses. Such is consistent with the observation that in certain circumstances LC, and other skin DC, have the potential to cause the selective stimulation of discrete functional subsets of T lymphocytes.41–43 There are a number of parallels with respect to LC migration in human and mouse. For example, the ability of TNF-α and IL-1β to induce LC migration in human skin, in addition to resting LC frequency decreasing with age, and that only 20–30% of LC possess the ability to migrate at any one time.44 Therefore it is likely that in humans also, LC migration, in certain circumstances, may be able to occur independently of TNF-α, and this would require further investigation.
Taken together the evidence reported here reveals that TNF-α is not always a mandatory requirement for the induction of LC mobilization and migration. Furthermore, there may be heterogeneity among chemical allergens with respect to a requirement for TNF-α in triggering the mobilization and migration of LC. The mechanistic relevance of such differences remains unclear. However, it is tempting to speculate that differences between chemical allergens with regard to the signals that activate LC may translate into important functional speciation – including possibly influences on the tempo of LC migration – that will in turn impact on the extent and quality of allergic sensitization.
Acknowledgments
We would like to thank Kieran T Mellody for excellent technical assistance.
Glossary
- AOO
acetone: olive oil
- BSA
bovine serum albumin
- DC
dendritic cell
- DNCB,
2,4-dinitrochlorobenzene
- EDTA
ethylenediaminetetraacetic acid
- DNFB
2,4-dinitrofluorobenzene
- FCS
fetal calf serum
- FITC
fluorescein isothiocyanate
- HPRT
hypoxanthine phosphoribosyltransferase 1
- LF
lactoferrin
- LC
Langerhans’ cell
- NGS
normal goat serum
- NRS
normal rabbit serum
- oxazolone
4-ethoxymethylene-2-phenyloxazol-5-one
- PPD
paraphenylene diamine
- SDS
sodium lauryl sulfate
- TMA
trimellitic anhydride
Disclosures
The authors declare no financial or commercial conflict of interest.
References
- 1.Rowden G, Lewis MG, Sullivan AK. Ia antigen expression on human epidermal Langerhans cells. Nature. 1977;268:247–8. doi: 10.1038/268247a0. [DOI] [PubMed] [Google Scholar]
- 2.Katz SI, Tamaki K, Sachs DH. Epidermal Langerhans cells are derived from cells originating in the bone marrow. Nature. 1979;282:324–6. doi: 10.1038/282324a0. [DOI] [PubMed] [Google Scholar]
- 3.Toews GB, Bergstresser PR, Streilein JW. Epidermal Langerhans cell density determines whether contact hypersensitivity or unresponsiveness follows skin painting with DNFB. J Immunol. 1980;124:445–53. [PubMed] [Google Scholar]
- 4.Streilein JW, Bergstresser PR. Langerhans cell function dictates induction of contact hypersensitivity or unresponsiveness to DNFB in Syrian hamsters. J Invest Dermatol. 1981;77:272–7. doi: 10.1111/1523-1747.ep12482453. [DOI] [PubMed] [Google Scholar]
- 5.Streilein JW. Antigen-presenting cells in the induction of contact hypersensitivity in mice: evidence that Langerhans cells are sufficient but not required. J Invest Dermatol. 1989;93:443–8. doi: 10.1111/1523-1747.ep12284018. [DOI] [PubMed] [Google Scholar]
- 6.Bursch LS, Wang L, Igyarto B, et al. Identification of a novel population of Langerin+ dendritic cells. J Exp Med. 2007;204:3147–56. doi: 10.1084/jem.20071966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bennett CL, Noordegraaf M, Martina CA, et al. Langerhans cells are required for efficient presentation of topically applied hapten to T cells. J Immunol. 2007;179:6830–5. doi: 10.4049/jimmunol.179.10.6830. [DOI] [PubMed] [Google Scholar]
- 8.Shklovskaya E, O'Sullivan B, Ng LG, et al. Langerhans cells are precommitted to immune tolerance induction. Proc Natl Acad Sci USA. 2011;108:18049–54. doi: 10.1073/pnas.1110076108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kautz-Neu K, Noordegraaf M, Dinges S, et al. Langerhans cells are negative regulators of the anti-Leishmania response. J Exp Med. 2011;208:885–91. doi: 10.1084/jem.20102318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van der Aar AMG, Picavet DI, Muller FJ, et al. Langerhans cells favor skin flora tolerance through limited presentation of bacterial antigens and induction of regulatory T cells. J Invest Dermatol. 2013;133:1240–9. doi: 10.1038/jid.2012.500. [DOI] [PubMed] [Google Scholar]
- 11.Kaplan DH, Jenison MC, Saeland S, et al. Epidermal Langerhans cell-deficient mice develop enhanced contact hypersensitivity. Immunity. 2005;23:611–20. doi: 10.1016/j.immuni.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 12.Yoshiki R, Kabashima K, Sugita K, et al. IL-10-producing Langerhans cells and regulatory T cells are responsible for depressed contact hypersensitivity in grafted skin. J Invest Dermatol. 2009;129:705–13. doi: 10.1038/jid.2008.304. [DOI] [PubMed] [Google Scholar]
- 13.Bobr A, Olvera-Gomez I, Igyarto BZ, et al. Acute ablation of Langerhans cells enhances skin immune responses. J Immunol. 2010;185:4724–8. doi: 10.4049/jimmunol.1001802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gomez de Aguero M, Vocanson M, Hacini-Rechinel F, et al. Langerhans cells protect from allergic contact dermatitis in mice by tolerizing CD8+ T cells and activating Foxp3+ T cells. J Clin Invest. 2012;122:1700–11. doi: 10.1172/JCI59725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kimber I, Dearman RJ, Cumberbatch M, et al. Langerhans cells and chemical allergy. Curr Opin Immunol. 1998;10:614–9. doi: 10.1016/s0952-7915(98)80078-2. [DOI] [PubMed] [Google Scholar]
- 16.Ouwehand K, Santegoets SJ, Bruynzeel DP, et al. CXCL12 is essential for migration of activated Langerhans cells from epidermis to dermis. Eur J Immunol. 2008;38:3050–9. doi: 10.1002/eji.200838384. [DOI] [PubMed] [Google Scholar]
- 17.Saeki H, Moore AM, Brown MJ, Hwang ST. Cutting edge: secondary lymphoid-tissue chemokine (SLC) and CC chemokine receptor 7 (CCR7) participate in the emigration pathway of mature dendritic cells from the skin to regional lymph nodes. J Immunol. 1999;162:2472–5. [PubMed] [Google Scholar]
- 18.Cumberbatch M, Kimber I. Dermal tumour necrosis factor-α induces dendritic cell migration to draining lymph nodes and possibly provides one stimulus for Langerhans cell migration. Immunology. 1992;75:257–63. [PMC free article] [PubMed] [Google Scholar]
- 19.Cumberbatch M, Dearman RJ, Kimber I. Langerhans cells require signals from both tumour necrosis factor-α and interleukin-1β for migration. Immunology. 1997;92:388–95. doi: 10.1046/j.1365-2567.1997.00360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cumberbatch M, Griffiths CEM, Tucker SC, et al. Tumour necrosis factor-α induced Langerhans cell migration in humans. Br J Dermatol. 1999;141:192–200. doi: 10.1046/j.1365-2133.1999.02964.x. [DOI] [PubMed] [Google Scholar]
- 21.Cumberbatch M, Bhushan M, Dearman RJ, et al. IL-1β-induced Langerhans cell migration and TNF-α production in human skin: regulation by lactoferrin. Clin Exp Immunol. 2003;132:352–9. doi: 10.1046/j.1365-2249.2003.02146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cumberbatch M, Kimber I. Tumour necrosis factor-α is required for accumulation of dendritic cells in draining lymph nodes and for optimal skin sensitisation. Immunology. 1995;84:31–5. [PMC free article] [PubMed] [Google Scholar]
- 23.Antonopoulos C, Cumberbatch M, Mee JB, et al. IL-18 is a key proximal mediator of contact hypersensitivity and allergen-induced Langerhans cell migration in murine epidermis. J Leukoc Biol. 2008;83:361–7. doi: 10.1189/jlb.0604352. [DOI] [PubMed] [Google Scholar]
- 24.Wang B, Fujisawa H, Zhuang L, et al. Depressed Langerhans cell migration and reduced contact hypersensitivity response in mice lacking TNF receptor p75. J Immunol. 1997;159:6148–55. [PubMed] [Google Scholar]
- 25.Kimber I, Cumberbatch M, Dearman RJ, et al. Cytokines and chemokines in the initiation and regulation of epidermal Langerhans cell mobilization. Br J Dermatol. 2000;142:401–12. doi: 10.1046/j.1365-2133.2000.03349.x. [DOI] [PubMed] [Google Scholar]
- 26.Peschon JJ, Torrance DS, Stocking KL, et al. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol. 1998;160:943–52. [PubMed] [Google Scholar]
- 27.Cumberbatch M, Clelland K, Dearman RJ, Kimber I. Impact of cutaneous IL-10 on resident epidermal Langerhans’ cells and the development of polarized immune responses. J Immunol. 2005;175:43–50. doi: 10.4049/jimmunol.175.1.43. [DOI] [PubMed] [Google Scholar]
- 28.Cumberbatch M, Dearman RJ, Kimber I. Influence of ageing on Langerhans cell migration in mice: identification of a putative deficiency of epidermal interleukin-1beta. Immunology. 2002;105:466–77. doi: 10.1046/j.1365-2567.2002.01381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hilton J, Dearman RJ, Basketter DA, Kimber I. Identification of chemical respiratory allergens: dose-response relationships in the mouse IgE test. Toxicol Mech Methods. 1995;5:51–60. [Google Scholar]
- 30.Cumberbatch M, Dearman RJ, Uribe-Luna S, et al. Regulation of epidermal Langerhans cell migration by lactoferrin. Immunology. 2000;100:21–8. doi: 10.1046/j.1365-2567.2000.00014.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Griffiths CE, Cumberbatch M, Tucker SC, et al. Exogenous topical lactoferrin inhibits allergen-induced Langerhans cell migration and cutaneous inflammation in humans. Br J Dermatol. 2001;144:715–25. doi: 10.1046/j.1365-2133.2001.04125.x. [DOI] [PubMed] [Google Scholar]
- 32.Almond RJ, Flanagan BF, Antonopoulos A, et al. Differential immunogenicity and allergenicity of native and recombinant human lactoferrins: Role of glycosylation. Eur J Immunol. 2013;43:170–81. doi: 10.1002/eji.201142345. [DOI] [PubMed] [Google Scholar]
- 33.Basketter DA, Dearman RJ, Hilton J, Kimber I. Dinitrohalobenzenes: evaluation of relative skin sensitization potential using the local lymph node assay. Contact Dermatitis. 1997;36:97–100. doi: 10.1111/j.1600-0536.1997.tb00421.x. [DOI] [PubMed] [Google Scholar]
- 34.Warbrick EV, Dearman RJ, Lea LJ, Basketter DA, Kimber I. Local lymph node assay responses to paraphenylenediamine: intra- and inter-laboratory evaluations. J Appl Toxicol. 1999;19:255–60. doi: 10.1002/(sici)1099-1263(199907/08)19:4<255::aid-jat573>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 35.Cumberbatch M, Dearman RJ, Groves RW, Antonopoulos C, Kimber I. Differential regulation of epidermal langerhans cell migration by interleukins (IL)-1alpha and IL-1beta during irritant- and allergen-induced cutaneous immune responses. Toxicol Appl Pharmacol. 2002;182(2):126–35. doi: 10.1006/taap.2002.9442. [DOI] [PubMed] [Google Scholar]
- 36.Kripke ML, Munn CG, Jeevan A, Tang JM, Bucana C. Evidence that cutaneous antigen-presenting cells migrate to regional lymph nodes during contact sensitization. J Immunol. 1990;145:2833–8. [PubMed] [Google Scholar]
- 37.Duthie MS, Kimber I, Dearman RJ, Norval M. Differential effects of UVA1 and UVB radiation on Langerhans cell migration in mice. J Photochem Photobiol, B. 2000;57:123–31. doi: 10.1016/s1011-1344(00)00087-7. [DOI] [PubMed] [Google Scholar]
- 38.Stoitzner P, Zanella M, Ortner U, et al. Migration of Langerhans cells and dermal dendritic cells in skin organ cultures: augmentation by TNF-alpha and IL-1beta. J Leukoc Biol. 1999;66:462–70. [PubMed] [Google Scholar]
- 39.Haley K, Igyártó BZ, Ortner D, Bobr A, Kashem S, Schenten D, Kaplan DH. Langerhans cells require MyD88-dependent signals for Candida albicans response but not for contact hypersensitivity or migration. J Immunol. 2012;188:4334–9. doi: 10.4049/jimmunol.1102759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Byrne SN, Halliday GM, Johnston LJ, et al. Interleukin-1beta but not tumor necrosis factor is involved in West Nile virus-induced Langerhans cell migration from the skin in C57BL/6 mice. J Invest Dermatol. 2001;117:702–9. doi: 10.1046/j.0022-202x.2001.01454.x. [DOI] [PubMed] [Google Scholar]
- 41.Klechevsky E, Morita R, Liu M, et al. Functional specializations of human epidermal Langerhans cells and CD14+ dermal dendritic cells. Immunity. 2008;29:497–510. doi: 10.1016/j.immuni.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Igyarto BZ, Haley K, Ortner D, et al. Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper cell responses. Immunity. 2011;35:260–72. doi: 10.1016/j.immuni.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seneschal J, Clark RA, Gahad A, et al. Human epidermal Langerhans cells maintain immune homeostasis in skin by activating skin resident regulatory T cells. Immunity. 2012;36:1–12. doi: 10.1016/j.immuni.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Griffiths CE, Dearman RJ, Cumberbatch M, Kimber I. Cytokines and Langerhans cell mobilisation in mouse and man. Cytokine. 2005;32:67–70. doi: 10.1016/j.cyto.2005.07.011. [DOI] [PubMed] [Google Scholar]