

Abstract
Protein kinase inhibitors are an important class of therapeutics. In addition, selective kinase inhibitors can often reveal unexpected biological insights, augmenting genetic approaches and playing a decisive role in preclinical target validation studies. Nevertheless, developing protein kinase inhibitors with sufficient selectivity and pharmacodynamic potency presents significant challenges. Targeting noncatalytic cysteines with covalent inhibitors is a powerful approach to address both challenges simultaneously. Here, we describe our efforts to design irreversible and reversible electrophilic inhibitors with varying degrees of kinase selectivity. Highly selective covalent inhibitors have been used to elucidate the roles of p90 ribosomal protein S6 kinases (RSK) in animal models of atherosclerosis and diabetes. By contrast, semi-promiscuous covalent inhibitors have revealed new therapeutic targets in disease-causing parasites and have shown utility as chemoproteomic probes for interrogating kinase occupancy in living cells.
1. Introduction
Protein phosphorylation by kinases regulates nearly every aspect of cellular physiology (Manning, Whyte, Martinez & Hunter, 2002). Because protein kinases are misregulated in many diseases, they have been hotly pursued as therapeutic targets for drug discovery. Out of 518 human protein kinases, only a small fraction have been targeted with selective inhibitors. By virtue of their high structural homology, particularly in the ATP binding site targeted by most small-molecule inhibitors, selective inhibition of distinct protein kinases remains a major challenge.
Covalent targeting of poorly conserved, noncatalytic cysteine residues with electrophilic kinase inhibitors has emerged as a powerful strategy for increasing potency and selectivity (Barf & Kaptein, 2012). Structural bioinformatics analysis of the human kinome has revealed ~200 kinase domains that have a solvent-exposed cysteine within striking distance of the ATP binding site (Liu, Sabnis, Zhao, Zhang, Buhrlage, Jones, et al., 2013; Leproult, Barluenga, Moras, Wurtz & Winssinger, 2011). In spite of the potential for improved selectivity, covalent inhibitors are typically avoided by the pharmaceutical industry out of fear of idiosyncratic toxicity resulting from drug-protein adducts (Uetrecht, 2008; Evans, Watt, Nicoll-Griffith & Baillie, 2004). Nevertheless, covalent drugs are making a comeback, especially in the context of molecularly targeted therapies for cancer (Singh, Petter, Baillie & Whitty, 2011).
An early example of a cysteine-targeted kinase inhibitor is 2’-thioadenosine, a proof-of-concept compound designed to form a disulfide bond with a poorly conserved cysteine (Cys797) in the EGFR kinase domain (Singh, Dobrusin, Fry, Haske, Whitty & McNamara, 1997). Subsequently, derivatization of an EGFR-selective quinazoline scaffold with an acrylamide electrophile resulted in an irreversible inhibitor with enhanced antitumor activity (Fry, Bridges, Denny, Doherty, Greis, Hicks, et al., 1998). The success of this approach is evident in the recent FDA approval of two irreversible cysteine-targeted kinase inhibitors. Afatinib, a quinazoline-based EGFR inhibitor (Solca, Dahl, Zoephal, Bader, Sanderson, Klein, et al., 2012), is approved for advanced non-small cell lung cancer, and ibrutinib, a Bruton's tyrosine kinase (BTK) inhibitor based on a pyrazolopyrimidine scaffold (Pan, Scheerens, Li, Schultz, Sprengeler, Burrill, et al., 2007), is approved for mantle cell lymphoma and chronic lymphocytic leukemia. Both compounds use an acrylamide electrophile to irreversibly target a cysteine shared by EGFR and BTK.
In this chapter, we describe our chemistry-focused approach to the design of electrophilic kinase inhibitors. Case studies illustrate strategies for targeting noncatalytic cysteines as well as the catalytic lysine in protein kinases. We discuss our recent discovery of reversible covalent kinase inhibitors and discuss a novel method for targeting noncatalytic cysteines with reversible electrophilic fragments. In addition to designing selective inhibitors, we have also developed covalent probes that are ‘semi-promiscuous’, targeting a relatively common cysteine or the conserved catalytic lysine. We have used these probes to identify new therapeutic kinase targets and quantify kinase target engagement in living cells by other inhibitors, including ponatinib, a leukemia drug recently associated with life-threatening side effects.
2. Design of irreversible cysteine-targeted kinase inhibitors
Identification of a suitable kinase target and a noncovalent recognition scaffold are primary considerations when embarking on a covalent inhibitor design project. If the goal is to obtain highly selective inhibitors, the kinase should have a solvent-exposed cysteine near a “druggable” pocket, typically within ~10 Å of the ATP binding site. Ideally, this cysteine should be found in only a handful of other human kinases. The design process usually starts with the identification of a noncovalent kinase-recognition scaffold that (1) inhibits the desired target with at least micromolar potency (e.g., IC50 <100 μM), and (2) can be modified to place an electrophilic moiety in close proximity to the targeted cysteine. Although a co-crystal structure of the scaffold bound to the kinase target is ideal for guiding the placement of electrophilic substituents, it is not essential; in a pinch, docking to a homology model may suffice. With the appropriate choice of electrophile, it is possible to transform a recognition scaffold with weak affinity and poor selectivity into a selective covalent inhibitor with potency in the low nanomolar to picomolar range.
Because IC50 values for irreversible inhibitors usually depend on the incubation time and ATP concentration, we hold these parameters constant to compare potencies across a series of compounds (e.g., 200 μM ATP, 30 min incubation). If desired, a real-time assay format (e.g., ADP Quest, DiscoveRX) can be used to measure the kinetics of kinase inhibition (Ki and kinact). To increase the stringency of the biochemical kinase assay and our ability to differentiate between highly thiol-reactive inhibitors, we often include 10 mM glutathione as a physiologically relevant competing nucleophile.
2.1 Irreversible covalent Inhibitors of RSK1/2/4
The p90 ribosomal protein S6 kinases comprise four closely related paralogs (RSK1-4) activated downstream of the Ras-MAPK pathway (Hauge & Frödin, 2006; Romeo, Zhang & Roux, 2012). RSKs contain two kinase domains, an AGC-family N-terminal kinase domain (NTD) and a CAMK-family C-terminal kinase domain (CTD) connected by a short linker with several regulatory phosphorylation sites. Following phosphorylation of Thr577 (human RSK2 numbering) by ERK, the activated CTD autophosphorylates Ser386 within the linker segment (Fisher & Blenis, 1996, Dalby, Morrice, Caudwell, Avruch & Cohen, 1998). This serves as a docking site for PDK1, which phosphorylates the NTD activation loop, leading to full activation of the NTD and downstream signaling (Frödin, Jensen, Merienne & Gammeltoft, 2000). RSKs have been shown to phosphorylate dozens of proteins involved in diverse cellular processes, including the sodium/hydrogen exchanger NHE1 (Takahashi, Abe, Gallis, Aebersold, Spring, Krebs, et al., 1999; Cuello, Snabaitis, Cohen, Taunton & Avikran, 2007), the translation initiation factor eIF4B (Shahbazian, Roux, Mieulet, Cohen, Raught, Taunton, et al., 2006), the tumor suppressor kinase LKB1 (Sapkota, Cummings, Newell, Armstrong, Bain, Frödin, et al. 2007; Doehn, Hauge, Frank, Jensen, Duda, Nielsen, et al., 2009), and the transcription factor c-Fos (David, Mehic, Bakiri, Schilling, Mandic, Priemel, et al., 2005). Many if not most phosphorylation sites attributed to RSK have also been linked to other kinases (e.g., kinases downstream of PI3K and p38 MAPK), depending on the cellular context. RSK hyperactivity has been implicated in tumor cell invasion (Doehn, et al., 2009; Kang, Elf, Lythgoe, Hitosugi, Taunton, Zhou, et al., 2010; Smolen, Zhang, Zubrowski, Edelman, Luo, Yu, et al., 2010), as well as endothelial dysfunction and atherosclerosis (Le, Heo, Takei, Lee, Woo, Chang, et al., 2013).
We used a sequence alignment of the human kinome (Buzko & Shokat, 2002) to identify poorly conserved, noncatalytic cysteines in the ATP binding site. Structure-guided analysis of this alignment revealed 11 kinases (Figure 1A), including the CTDs of RSK1-4, with a cysteine projecting down from the “ceiling” of the ATP pocket, near the C-terminal end of the glycine-rich loop. Motivating our studies at the time, few useful inhibitors were known for any of these kinases. We were also motivated by the distance and geometry constraints imposed by this cysteine, which lies deeper in the pocket than the EGFR cysteine. These constraints forced us to consider alternatives to the acrylamide electrophiles used to target EGFR. Of the 11 kinases with this particular cysteine, we noticed that only RSK1/2/4 (but not RSK3) have a threonine in the “gatekeeper” position (Figure 1A), which in principle could open up an extended hydrophobic pocket that is less accessible in kinases with larger gatekeepers (Liu, Bishop, Witucki, Kraybill, Shimizu, Tsien, et al., 1999). Although we lacked knowledge of any RSK CTD inhibitors or crystal structures, our goal was to design an inhibitor that simultaneously exploited both selectivity filters found uniquely in RSK1/2/4, the Cys at the end of the Gly-rich loop and the Thr gatekeeper (Cohen, Zhang, Shokat & Taunton, 2005).
Figure 1.

Structural bioinformatics-based design of cysteine-targeted RSK inhibitors. (a) Sequence alignment reveals two selectivity filters unique to RSK1/2/4: a noncatalytic Cys and a Thr gatekeeper. (b) Multi-kinase inhibitor PP1 bound to the Src-family kinase HCK (PDB code: 1QCF), with N2 of PP1 proximal to Val284, corresponding to Cys436 in RSK2. (c) Chemical structure of PP1 and the irreversible RSK inhibitor, FMK.
A co-crystal structure of the pyrazolopyrimidine PP1 bound to HCK (Schindler, Sicheri, Pico, Gazit, Levitszki & Kuriyan, 1999), a SRC-family kinase with a Thr gatekeeper but otherwise distantly related to RSK, inspired the design of a fluoromethylketone inhibitor, prosaically named FMK (Figure 1B and 1C). We predicted that Cys436 in the RSK2 CTD would occupy a similar space as Val284 in HCK and would be within striking distance of a fluoromethylketone appended to C6 of a PP1-like pyrrolopyrimidine scaffold. A hydroxypropyl substituent at N7, predicted to be solvent accessible, would enable derivatization with fluorophores and affinity tags. Although fluoromethylketones have been used to target highly reactive catalytic cysteines in Cys proteases, their ability to covalently target noncatalytic cysteines had not been explored extensively.
FMK irreversibly inhibits RSK2 in biochemical (IC50 ~15 nM) and cellular assays (EC50 ~300 nM), and both CTD selectivity filters are required for potent inhibition (Cohen, et al., 2005). Mutation of either Cys436 to Val or the gatekeeper Thr493 to Met confers resistance (biochemical IC50 >3 μM). Given that it has a similar kinase-recognition scaffold as PP1, which reversibly inhibits SRC and many other tyrosine kinases, we were surprised to find that FMK was extremely selective when tested against a large panel of kinases (unpublished results in collaboration with Eric Johnson, AbbVie). Out of 150 kinases tested, FMK inhibited only five with an IC50 below 5 μM; only two kinases had sub-micromolar IC50s (S6K1 ~0.5 μM and PTK5 ~0.7 μM). Contrary to a published report (Bain, Plater, Elliot, Shpiro, Hastie, Mclauchlan, et al., 2007), FMK failed to inhibit LCK or SRC (IC50 >5 μM), whereas PP1 potently inhibits both kinases. The enhanced selectivity of FMK relative to PP1 suggests that the fluoromethylketone sterically clashes with most kinases that would otherwise accommodate the pyrrolopyrimidine scaffold.
We employed a similar strategy to target the structurally analogous cysteine in NEK2, a cell cycle-regulated kinase that localizes to centrosomes and is overexpressed in many cancers. A co-crystal structure of the oxindole SU11652 (related to the kidney cancer drug, sunitinib) bound to NEK2 suggested that the C5 position could project electrophiles to engage Cys22. Whereas the oxindole scaffold (including SU11652 and sunitinib) inhibited NEK2 weakly (IC50 >5 μM), appending a propynamide electrophile to C5 led to an irreversible NEK2 inhibitor that does not cross-react with PLK1, an essential cell cycle kinase with an equivalent cysteine (Henise & Taunton, 2011). Again, mutation of the key cysteine in NEK2 conferred resistance.
Applications of irreversible covalent kinase inhibitors
An advantage of irreversible (or slowly reversible; see below) covalent inhibitors that target a noncatalytic cysteine is the relative ease with which a cellular phenotype can be attributed to inactivation of the desired kinase, as opposed to inhibiting myriad off-target kinases. An inhibitor washout experiment is the simplest method: following acute treatment with a saturating amount of the inhibitor (e.g., 5 μM FMK, 30-60 min incubation at 37°C), cells are washed extensively (3 x 5 min with compound-free media) and the biological readout is assessed (e.g., RSK autophosphorylation in response to growth factor stimulation). Cellular phenotypes that persist after inhibitor washout may derive from irreversible inactivation of the desired kinase, whereas phenotypes that disappear after washout are likely due to reversible inhibition of irrelevant targets. Drug washout experiments may be confounded by rapid re-synthesis of the kinase, giving a false-negative result; depending on the re-synthesis rate, this is more likely to be an issue if the phenotype is scored many hours after the washout step (e.g., cell proliferation). In addition, some inhibitors are not easily washed out and may accumulate in cells at high concentrations. To control for this potential artifact and provide additional evidence linking a cellular phenotype to covalent targeting, we recommend synthesizing an isosteric version of the inhibitor that is not electrophilic. With FMK, for example, we synthesized the corresponding methyl ketone lacking the fluorine (unpublished results). With the NEK2 inhibitor, we added a methyl group to the electrophilic carbon of the propynamide (Henise & Taunton, 2011). The resulting “negative control” compounds are inactive vs. RSK and NEK2, but they have nearly identical physical properties and are similar in their ability to reversibly engage kinases.
The most convincing way to demonstrate that a cellular phenotype results from covalent inactivation of the desired kinase is to “rescue” the phenotype by reconstituting cells with a resistant allele of the kinase. Typically, the key Cys is mutated to Val, Ala, or Ser. In many cases, it is possible to confer resistance to the electrophilic inhibitor by expressing the mutant kinase in cells that also express the endogenous wild-type kinase; we have used this approach with our RSK and NEK2 inhibitors (Doehn, et al., 2009; Henise & Taunton, 2011). However, in certain cases, it may be difficult to achieve optimal expression levels of the transgene, or the inhibitor-bound endogenous kinase may act as a “dominant negative” and interfere with the function of the ectopically expressed mutant kinase. In such cases, it may be necessary to use cells in which the endogenous kinase has been genetically deleted or knocked down. Using zinc finger nuclease technology, Frödin and colleagues engineered FMK resistance by introducing a C436V mutation into the endogenous RSK2 (also known as RPS6KA3) gene (Chen, Pruett-Miller, Huang, Gjoka, Duda, Taunton, et al., 2011). Advances in CRISPR/Cas9 genome editing technology will likely make this process much easier (Friedland, Tzur, Esvelt, Colaiácovo, Church & Calarco, 2013; Wang, Yang, Shivalila, Dawlaty, Cheng, Zhang, et al., 2013).
2.2 Fluorescent and alkyne-tagged probes to quantify proteome-wide selectivity and RSK occupancy in vivo
To identify the intracellular targets of FMK and quantify RSK occupancy as a function of inhibitor concentration, we required tagged derivatives. We designed the following two probes for different applications (Cohen, Hadjivassiliou & Taunton, 2007): (1) FMK-PA, a more potent, clickable derivative for assessing proteome-wide selectivity, and (2) FMK-BODIPY, a cell-permeable fluorescent probe for convenient quantitation of RSK occupancy (Figure 2). Based on the proposed binding mode of FMK, we expected the hydroxypropyl group to be solvent exposed and able to accommodate various tags.
Figure 2.

FMK derivatives used to the elucidate pathophysiological roles of RSK. (a) Chemical structures of FMK-PA and FMK-BODIPY. (b) Kinome-wide selectivity of FMK-MEA, screened at 1 μM vs. 443 kinases (DiscoverX). Only RSK1 and RSK4 CTDs were bound >50% relative to DMSO controls. (c) Using FMK-BODIPY as a probe, RSK1/2 occupancy was assessed in cardiac tissue lysates derived from mice treated with the indicated dose of FMK-MEA. To visualize FMK-BODIPY-labeled RSK1 and RSK2, each kinase was separately immunoprecipitated with isoform-specific antibodies. A dose-dependent reduction in fluorescence indicates RSK occupancy by FMK-MEA. Figure adapted with permission from Lippincott Williams and Wilkins/Wolters Kluwer Health: Circulation, Le, et al., copyright 2013.
Substitution of the primary alcohol of FMK with propargylamine (via the di-Boc-protected mesylate) provided FMK-PA, amenable to bioorthogonal Cu(I)-mediated azide-alkyne cycloaddition, or click chemistry. FMK-PA was more potent than FMK in cellular assays, showing maximal inhibition of RSK autophosphorylation at 100 nM. After treating cells with FMK-PA, click chemistry with rhodamine-azide was performed on cell lysates. Only two rhodamine-labeled bands, corresponding to endogenous RSK1 and RSK2, were detected by SDS-PAGE and in-gel fluorescence scanning (Cohen, et al., 2007). Thus, FMK-PA appears to exhibit extraordinary proteome-wide selectivity, discriminating between Cys436 in RSK1/2 and thousands of off-target cysteines.
We also synthesized the fluorescent derivative FMK-BODIPY, which albeit less potent than FMK-PA, is cell permeable and reasonably selective for RSK1/2 when added to intact cells (3-5 μM in serum-free media) or cell lysates (Cohen, et al. 2007). FMK-BODIPY is more convenient than FMK-PA for quantifying RSK1/2 occupancy by other RSK inhibitors, as there is no requirement for click chemistry (Serafimova, Pufall, Krishnan, Duda, Cohen, Maglathlin, et al., 2012). Moreover, for reasons that aren't clear, detecting RSK1/2 with FMK-PA and click chemistry has proven difficult in certain contexts (e.g., lysates derived from mouse cardiac tissue). When assessing the pharmacodynamic potency of new RSK inhibitors in cells and mice, our “go-to” assay is to quantify RSK1/2 assay using FMK-BODIPY as the probe.
To elucidate the roles of RSK CTD activity in mouse disease models, we sought an FMK derivative with improved aqueous solubility. Based on the increased potency and solubility of FMK-PA, we synthesized the related methoxyethylamine derivative FMK-MEA (Figure 2B). When screened against a 443-kinase panel at concentration of 1 μM (DiscoveRX), FMK-MEA interacted with only two kinases, RSK1 and RSK4 CTD (Figure 2B; RSK2 CTD was not tested). This level of selectivity is rarely achieved with noncovalent kinase inhibitors. The bis-tosylate salt of FMK-MEA dissolves in saline at >25 mg/mL, enabling convenient dosing by intraperitoneal injection or oral gavage (Cmax ~600 nM in mice after 30 mg/kg oral dose, 38% bioavailability; unpublished results from Albany Molecular Research Inc.).
RSK activity is elevated in endothelial cells and cardiomyocytes in diabetic animals and has been implicated in heart disease exacerbated by diabetes. In a recent study, FMK-MEA was found to reduce endothelial dysfunction in two diabetic mouse models and was efficacious in an aggressive atherosclerosis model. We used FMK-BODIPY to demonstrate a correlation between RSK1/2 occupancy and a dose-dependent decrease in endothelial dysfunction readouts in mice treated with FMK-MEA (Le, et al., 2013). Although FMK-BODIPY-labeled RSK1/2 is easily detected in lysates derived from mouse lung, liver, and brain tissue, the signal to background is low in heart lysates, necessitating immunoprecipitation with anti-RSK1/2 antibodies (Figure 2C). A protocol for determining RSK1/2 occupancy in mouse cardiac tissue lysates using FMK-BODIPY is described below. The protocol can be used with fluorescent occupancy probes directed against other targets that are difficult to detect by gel electrophoresis of whole-cell lysates, as long as specific antibodies are available for the immunoprecipitation step. Relative to streptavidin affinity purification with biotinylated probes followed by Western blot detection, this protocol is simpler and easier to implement.
Assessing RSK1/2 occupancy after dosing mice with FMK-MEA
Administer FMK-MEA (or other RSK CTD inhibitors) or vehicle (2-4 mice per arm) by intraperitoneal injection or oral gavage; after 2-4 hours, sacrifice the mice according to a protocol pre-approved by your Institutional Animal Care and Use Committee. Harvest the desired tissues and freeze them immediately in liquid nitrogen. A time course (e.g., 6/12/24 hours after dosing) can be used to assess drug-target residence time and/or the rate of RSK re-synthesis.
While the tissue is still slightly frozen, mince it into small pieces with a clean razor blade.
Place each tissue sample into a 2 mL tube. Add 0.75 mL PBS supplemented with protease and phosphatase inhibitors (Roche). Note: buffers with stronger buffering capacity (e.g., 50 mM Tris, pH 8.0) may work better than PBS, depending on the target and the probe.
Prepare lysates using a mechanical rotor-stator style tissue homogenizer (Janke & Kunkel/IKA T25-Ultra-Turrax Homogenizer), keeping the tubes on ice. Begin by homogenizing for 30 seconds on low speed, allowing samples to rest for 30 seconds, and then homogenizing at high speed for 30 seconds.
Transfer the crude lysates to 1.5 mL ultracentrifuge tubes. Centrifuge samples at 4°C in a benchtop ultracentrifuge for 1 hour at 30,000 × g (Beckman Coulter Optima TLX Ultracentrifuge).
Transfer supernatants to new 1.5 mL tubes on ice and snap freeze in liquid nitrogen if desired.
Quantify protein concentration by Bradford assay, normalizing to the most dilute sample by adding cold PBS. Transfer 100 μL of the lysate to new tubes for labeling with the fluorescent occupancy probe, FMK-BODIPY.
Add FMK-BODIPY (5.25 μL of a 100 μM DMSO stock; 5 μM final concentration) and incubate for 1 hour at room temperature.
Remove 40 μL from each sample and add 10 μL of 5x Laemmli sample buffer (pre-IP lysate sample). Note: depending on the abundance of the target and the specificity and efficiency of fluorescent probe labeling, it may be possible to quantify target occupancy by SDS-PAGE analysis of the crude lysates, without the need for immunoprecipitation.
To the remaining 60 μL of the lysates treated with FMK-BODIPY, add 300 μL of cold PBS + 1% NP40. Keep samples on ice.
Add 10 μL of α-RSK1 and 10 μL of α-RSK2 antibodies (Santa Cruz Biotechnology, sc-231 and sc-9986). Place samples in a rotator at 4°C for 2-4 hours.
Add 30 μL of Protein G Dynabeads (Invitrogen 100-04D) to each tube, place in a rotator, and incubate overnight at 4°C. Note: shorter incubation times may suffice.
Place tubes in magnetic racks to separate the beads. Remove 40 μL and quench with 5x Laemmli sample buffer (post-IP lysate sample; Western blot analysis of pre- and post-IP lysate samples can be used to assess immunoprecipitation efficiency).
Discard the remaining supernatant and add 300 μL of cold PBS + 1% NP40 to wash. Place in a rotator for 5 minutes.
Place tubes in magnetic racks to separate beads, and discard the supernatant. Add 300 μL of cold PBS + 1% NP40, mix thoroughly, and transfer each sample to a new pre-chilled 1.5 mL Eppendorf tube. Place on a rotator at 4°C for 5 minutes.
Wash once more with 300 μL of cold PBS + 1% NP40 for 5 minutes, separate the beads with a magnetic rack, and discard the supernatant.
To elute the proteins from the beads, add 50 μL of 2x Laemmli sample buffer and 10 μL of freshly made 1 M DTT to the beads. Vortex briefly and heat for 1 minute at 90°C.
Resolve proteins by SDS-PAGE. Quantify BODIPY labeling of RSK1/2 on a Typhoon scanner (GE Healthcare). Transfer proteins from the gel to nitrocellulose to detect total RSK1/2 by Western blot (Figure 3C).
Figure 3.

Reversible targeting of noncatalytic cysteines with cyanoacrylamides. (a) Cyanoacrylamides, unlike acrylamides, form rapidly reversible adducts with thiols. (b) N-isopropyl cyanoacrylamide variant of FMK is a potent, selective, and reversible RSK inhibitor with slow dissociation kinetics. A co-crystal structure of a tert-butyl cyanoacrylate derivative bound to RSK2 shows a network of noncovalent interactions that cooperatively stabilize the covalent complex. Upon unfolding of RSK2, the covalent bond is rapidly reversed. Adapted with permission from Serafimova, et al., 2012, copyright Nature Publishing Group, 2012.
3. Targeting noncatalytic cysteines with reversible covalent inhibitors
Covalent irreversible inhibitors are typically avoided in drug discovery because of the potential for nonspecific alkylation, protein haptenization, and unpredictable toxicity (Uetrecht, 2008; Evans, et al., 2004). To minimize these concerns, electrophiles with attenuated reactivity, e.g. acrylamides, have been employed. However, the in vivo targeting specificity of acrylamide-based kinase inhibitors has not been established, and thus far, such drugs have only been developed to treat advanced cancer. As an alternative approach, we sought to target noncatalytic cysteines with reversible covalent inhibitors. In this case, reactions with off-target cysteines would be transient and ideally less likely to exert idiosyncratic toxic effects. By contrast, the cooperative formation of specific covalent and noncovalent interactions with the desired target would result in high-affinity binding and slow dissociation kinetics.
In this section, we describe our recent efforts to tune the reactivity of Michael acceptors for the design and discovery of reversible covalent inhibitors. Although our work has focused on kinases, this strategy should be applicable to other targets with druggable cysteines.
3.1 Reversible Michael acceptors for cysteine-targeting applications
To explore the possibility of trapping cysteine thiols with alternative electrophiles, we synthesized several Michael acceptors activated by one or two electron-withdrawing groups (EWGs) attached to the incipient α-carbanion (Serafimova, et al., 2012). As anticipated, α-cyanoacrylamides (with two EWGs) reacted with simple thiols like glutathione more rapidly than acrylamides or acrylonitriles. What was unexpected was that the thiol adducts could not be isolated and instantaneously reverted back to the cyanoacrylamides upon dilution. The faster thiol elimination rate relative to the corresponding acrylamide adduct makes sense given the increased carbon acidity of the cyanoacrylamide adduct. However, the magnitude of the adduct's kinetic instability at physiological pH was unanticipated. Based on these kinetic properties, we reasoned that cyanoacrylamides could be exploited to yield cysteine-targeted, reversible covalent inhibitors with increased selectivity relative to irreversible inhibitors. These concepts led to the discovery of ultra-selective cyanoacrylamide-based RSK inhibitors with picomolar affinity and slow off-rates, the structural basis of which was revealed by x-ray crystallography (Figure 3). Because covalent bond formation is under thermodynamic control and requires multiple specific noncovalent interactions to stabilize the covalent complex, the likelihood of forming spurious adducts with off-target proteins is reduced relative to irreversible electrophiles like fluoromethylketones and acrylamides.
3.2 Electrophilic fragment-based ligand discovery with cyanoacrylamides
The intrinsically reversible, yet energetically favorable nature of the cysteine/cyanoacrylamide reaction suggested that this ‘sweet spot’ in electrophile space could form the basis of a fragment screening library for cysteine-containing targets (Miller, Paavilainen, Krishnan, Serafimova & Taunton, 2013). The approach is conceptually related to the disulfide fragment tethering technology developed by Erlanson, Wells, and colleagues (Erlanson, Braisted, Raphael, Randal, Stroud, Gordon, et al., 2000), but with critical differences: (1) unlike most disulfides, cyanoacrylamides can be used in cells and animals, and (2) the amide group can serve as a medicinal chemistry handle for optimizing affinity and selectivity. In developing a cyanoacrylamide fragment screening approach, we exploited both of these advantages. We tested ten cyanoacrylamide fragments against three cysteine-containing kinases (RSK2, NEK2, and PLK1) under stringent conditions (10 mM glutathione and 0.1 mM ATP). Even with such a small screening set, we identified unique inhibitors for all three kinases with potencies in the low micromolar to submicromolar range. Structure-guided merging of two of the initial fragment hits led to the first pan-MSK/RSK CTD inhibitor, with subnanomolar affinity and >500-fold selectivity over NEK2 and PLK1 (Figure 4).
Figure 4.

Cyanoacrylamide fragment screening identifies pan-MSK/RSK inhibitors. (a) Using a RSK2 kinase assay, we screened a panel of 10 cyanoacrylamide fragments (up to 300 μM) and discovered 1 and 2 as the most potent hits. We solved co-crystal structures of 1 and 2 bound RSK2 (PDB codes: 4JG6 and 4JG7). An overlay of both structures suggested the design of 3-aryl indazole variants. (b) Structure-guided optimization led to 3-aryl indazole 3 (RMM-46), which potently and selectively blocks MSK and RSK signaling in cells. Adapted with permission from Miller, R. M., Paavilainen, V. O., Krishnan, S., Serafimova, I. M. & Taunton, J. (2013). Electrophilic fragment-based design of reversible covalent kinase inhibitors. J. Am. Chem. Soc., 135, 5298 – 5301. Copyright 2013, American Chemical Society.
Assembling and screening a cyanoacrylamide fragment library
Cyanoacrylamides are easy to synthesize in one step via Knoevenagel condensation of 2-cyanoacetamide with an aldehyde fragment. Searching the ZINC database (Irwin, Sterling, Mysinger, Bolstad & Coleman, 2012) revealed over 12,000 commercially available aldehyde fragments (MW <250 Da). We typically mix aryl or heteroaryl aldehydes (0.3 mmol) with one equivalent each of 2-cyanoacetamide and piperidine in THF (~100 mM) for 24-48 hours. The pure cyanoacrylamide often precipitates in the reaction mixture and can be isolated by brief centrifugation through a 0.22 μm filter unit. For cyanoacrylamides that do not precipitate, we recommend preparative TLC as an efficient purification method. While cyanoacrylamides are often found in commercial small-molecule screening libraries, they tend to be larger (>300 Da) and have large hydrophobic substituents on the amide nitrogen. Such compounds may have lower solubility, which can lead to screening artifacts. Thus far, we have only used unsubstituted (primary) amides derived from 2-cyanoacetamide for initial fragment screens; we then incorporate amide substituents during the subsequent hit optimization phase. This latter modification step may be critical for certain cellular assays, as we have found that primary cyanoacrylamides are more prone to hydrolysis (via retro-Knoevenagel reaction to give the aldehyde and 2-cyanoacetamide) than secondary or tertiary cyanoacrylamides. Cyanoacrylate esters are even less stable.
Nearly all of the cyanoacrylamides that we have tested react reversibly with simple thiols like glutathione (GSH) and β-mercaptoethanol (BME). Monitoring the thermodynamics and kinetics of these reactions provides a simple approximation of an electrophile's intrinsic reactivity toward cysteine residues in unstructured regions of proteins. One way to compare the intrinsic reactivity of different cyanoacrylamides is to measure the equilibrium dissociation constant (Kd) by titrating GSH or BME in buffered solution. The reaction is conveniently monitored by UV-vis spectroscopy, as most β-aryl or -heteroaryl cyanoacrylamides have a strong absorption band (300-400 nm) that is disrupted upon thiol conjugate addition (Figure 5A). The Kd can be determined fitting the titration data to a one-site binding model using GraphPad Prism or similar software (Figure 5B). Reversibility can be demonstrated by diluting the thiol/cyanoacrylamide adduct into buffer that either lacks or contains excess thiol and monitoring recovery of the cyanoacrylamide UV absorption band (Figure 5C). For most thiol/cyanoacrylamide adducts, a new equilibrium is established within seconds upon dilution. Cyanoacrylamide/thiol equilibria can also be characterized by 1H NMR.
Figure 5.

Characterization of thiol conjugate addition reactions with cyanoacrylamides. (a) Reaction of cyanoacrylamide 1 with BME is accompanied by a decrease in the 340 nm absorption band. (b) The Kd is determined by fitting the titration data to a simple one-site binding model. (c) Dilution of the BME/cyanoacrylamide adduct into buffer lacking BME establishes a new equilibrium favoring the cyanoacrylamide.
Depending on the cyanoacrylamide, Kd values with GSH or BME typically range from 1–100 mM. Assuming millimolar GSH concentrations in cells, a cyanoacrylamide with a Kd of < 10 mM will be significantly bound to GSH, yet it will still be available to engage the desired target due to rapid β-elimination from GSH and dynamic equilibration. In our initial fragment screen, there was no correlation between kinase inhibition potency and BME reactivity as determined by Kd measurements (unpublished results). This further supports our conclusion that increased potency derives from specific noncovalent interactions involving the unique β-substituent of the cyanoacrylamide, in addition to the covalent bond formation. To expand the noncovalent recognition potential of the electrophile, we have begun to explore alternative Michael acceptors for electrophilic fragment screens and structure-based inhibitor design (Krishnan, Miller, Tian, Mullins, Jacobson & Taunton, 2014). Given the challenge of predicting whether a new electrophilic chemotype will undergo reversible thiol-addition reactions, we routinely measure the intrinsic reactivity and reversibility toward BME or GSH using the methods described above.
4. Semi-promiscuous covalent inhibitors as chemoproteomic probes
Quantifying drug/target engagement in living cells is a challenging problem for which new chemical methods are needed (Moellering & Cravatt, 2012). This issue is especially critical for kinase inhibitors, which may interact with any one of 500 structurally related ‘off-target’ kinases. High-throughput assays with purified proteins often fail to recapitulate the state of endogenous kinases in their native subcellular milieu, in which dynamic regulatory interactions modulate their affinity for ATP and inhibitors. Powerful technologies have been developed for quantifying interactions with hundreds of endogenous kinases, but these probes only work in dilute cell lysates (Patricelli, Nomanbhoy, Wu, Brown, Zhou, Zhang, et al., 2011; Bantscheff, Eberhard, Abraham, Bastuck, Boesche, Hobson, et al., 2007). We were therefore motivated to develop complementary probes for quantifying inhibitor/kinase engagement in living cells.
4.1 Identification of new therapeutic kinase targets with a semi-promiscuous inhibitor
We recently designed a chemoproteomic probe based on hypothemycin, an electrophilic polyketide natural product (Figure 6), for the purpose of identifying and prioritizing potential therapeutic targets in the disease-causing parasite, T. brucei (Nishino, Choy, Gushwa, Oses-Prieto, Koupparis, Burlingame, et al., 2013). Hypothemycin had previously been shown to covalently inactivate several human kinases bearing a cysteine adjacent to the conserved DXG motif, albeit with widely varying potency. In many eukaryotes, including humans and trypanosomes, such ‘CDXG kinases’ comprise 10-20% of the kinome. We found that hypothemycin potently kills T. brucei parasites. Twenty-one of the 182 protein kinases in T. brucei have a CDXG motif and were therefore strong candidates for mediating hypothemycin's trypanocidal effects.
Figure 6.
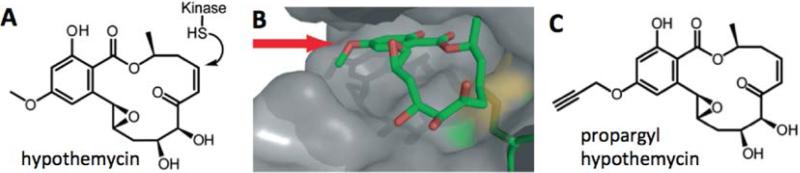
Semi-promiscuous occupancy probe for CDXG kinases. (a) Hypothemycin reacts with the cysteine in CDXG kinases via conjugate addition to the cis-enone. (b) Co-crystal structure of hypothemycin bound to ERK2 (PDB code: 3C9W) suggests the methyl ether as a derivatization site for an alkyne tag. (c) Chemical structure of propargyl-hypothemycin, prepared from desmethyl-hypothemycin and propargyl bromide.
To identify the relevant target(s), we synthesized a propargyl ether derivative of hypothemycin (guided by co-crystal structure) for quantitative chemoproteomic experiments. T. brucei cell lysates were pretreated with increasing concentrations of hypothemycin, followed by treatment with the propargyl-hypothemycin probe, click chemistry with biotin azide, streptavidin affinity purification, and mass spectrometry analysis. We used isobaric mass spectrometry tags (Ross, Huang, Marchese, Williamson, Parker, Hatten, et al., 2004) to quantify the relative amount of every protein identified in the streptavidin eluates as a function of increasing hypothemycin concentration. This analysis revealed that propargyl hypothemycin specifically labeled 11 CDXG kinases (without labeling any non-CDXG kinases), yet only three kinases were competed with nanomolar concentrations of hypothemycin. A similar quantitative occupancy assay in intact parasites revealed that only TbCLK1, a previously unstudied CLK-family kinase, was fully engaged by cytotoxic concentrations of hypothemycin. RNAi knockdown demonstrated that TbCLK1 (but not the closely related paralog, TbCLK2) is essential for T. brucei viability. Hence, this study led to the discovery of a new potential therapeutic target for African sleeping sickness, as well as a chemoproteomic probe for quantifying inhibitor occupancy of TbCLK1 and other CDXG kinases in living cells.
4.2 Targeting the catalytic lysine with covalent probes
Thus far, we have discussed the design of probes that target noncatalytic cysteines. To develop cellular occupancy probes that target a broader swath of the kinome than propargyl-hypothemycin, we turned to the conserved lysine, which coordinates the ADP leaving group during catalysis (e.g., K295 in human SRC, Figure 7A). While this lysine does not act as a catalytic nucleophile in the phospho-transfer step, its reactivity toward certain electrophiles may be enhanced by electrostatic interactions with conserved Asp and Glu residues; the latter interactions are observed in many but not all kinase structures. Targeting the catalytic Lys in kinases without targeting thousands of other lysines frequently found on protein surfaces presents a potential chemoselectivity challenge, and few Lys-targeted kinase probes have been reported that are suitable for interrogating kinase occupancy in cells. Nevertheless, ATP-biotin and ADP-biotin acylate lysine residues in kinases and other ATP-binding proteins in gel-filtered cell lysates and are useful as occupancy probes (Patricelli, et al., 2011).
Figure 7.

Design of a semi-promiscuous, lysine-targeted kinase probe. (a) Structure of SRC bound to AMP-PNP (PDB code: 2SRC), showing distances from the terminal phosphate to the catalytic Lys295 and nonconserved Cys277. (b) Design of a lysine-targeted sulfonyl fluoride probe, SF1, by merging FSBA and PP1. Adapted with permission from Gushwa. N. N., Kang, S., Chen, J. & Taunton, J. (2012). Selective targeting of distinct active site nucleophiles by irreversible SRC-family kinase inhibitors. J. Am. Chem. Soc., 134, 20214 – 20217. Copyright 2012, American Chemical Society.
Flurosulfonylbenzoyl adenosine (FSBA, Figure 7B) has been reported to react selectively with the catalytic Lys of several kinases (Kamps, Taylor & Sefton, 1984), although this probe is not sufficiently potent to be used in cells. Our strategy was to retain the sulfonyl fluoride electrophile of FSBA while increasing the intrinsic affinity of the recognition scaffold by incorporating a p-tolyl-substituted pyrrolopyrimidine, similar to the promiscuous tyrosine kinase inhibitor PP1. A propargyl group attached to the ribose 2'-OH fortuitously increased the potency of the sulfonyl fluoride probe SF1 toward all kinases tested (unpublished results). After brief treatment of intact human cells with SF1, click chemistry with biotin-azide and streptavidin affinity purification revealed efficient labeling of endogenous SRC-family kinases. By replacing the benzoyl sulfonyl fluoride of SF1 with a vinylsulfonate, we generated a second more selective probe that targets a proximal cysteine (Figure 7A) found in only 3 SRC-family kinases (e.g., SRC, but not LCK, LYN, HCK, or FYN), in addition to FGFR1-4 and a few other kinases (Gushwa, Kang, Chen & Taunton, 2012). Other groups have developed useful irreversible inhibitors that target this cysteine (Kwarcinski, Fox, Steffey, & Soellner, 2012; Zhou, Hur, McDermott, Dutt, Xian, Ficarro, et al., 2010).
Using SF1 to probe kinase occupancy in living cells, we discovered that the recently approved drug ponatinib potently engages the SRC-family kinase, LCK, while having no apparent effect on SRC or FYN. Given that all three kinases were previously shown to be sensitive to low nanomolar concentrations of ponatinib in biochemical assays, these results illustrate potential differences when measuring native kinase engagement in cells. In our initial study with SF1, Western blot detection with specific antibodies enabled quantitation of affinity-purified, probe-labeled kinases. Because antibodies against many human kinases are commercially available, this method provides a convenient alternative to instrument-intensive quantitation by mass spectrometry. Nevertheless, quantitation by Western blotting is limited in terms of the number of kinases that can be analyzed in a single experiment. In unpublished work, we have used SF1 in combination with quantitative mass spectrometry to identify a large set of T. brucei kinases, only a few of which are labeled by propargyl-hypothemycin and none of which are tyrosine kinases. Many of these kinases are essential for viability and hence, potential therapeutic targets. Given that little is known about their upstream activators or downstream substrates, quantitative occupancy assays with probes such as propargyl-hypothemycin and SF1 are among the best ways to demonstrate on-target cellular effects of new inhibitors. We anticipate that SF1 and related sulfonyl fluoride probes will have broad utility for kinase occupancy studies in human cells as well as disease-causing parasites.
5. Conclusions and future directions
Understanding the intricate roles of protein kinases in cellular physiology requires the development of potent and selective small-molecule probes. Moreover, protein kinases remain an important and ever-expanding class of therapeutic targets for the treatment of cancer and other serious diseases. Although genetic gain- and loss-of-function studies can provide important clues about which kinases to target for a particular disease state, selective inhibitors are absolutely essential for early-stage target validation studies. Covalent targeting of noncatalytic cysteines has emerged as an efficient strategy for obtaining selective kinase inhibitors that exhibit prolonged target occupancy in vivo. Indeed, covalent kinase inhibitors often confer sustained inactivation of the desired target even when their pharmacokinetic properties are suboptimal. In this chapter, we have presented our laboratory's approach to the design of covalent probes and their application to the study of kinase targets for which there is minimal genetic validation in disease models. For example, our RSK1/2/4 inhibitor, FMK-MEA, has revealed a previously unknown role for RSK hyperactivity in atherosclerosis and endothelial dysfunction (Le, et al., 2013). Given that FMK-MEA targets the C-terminal kinase domain of three RSK paralogs, while sparing the N-terminal kinase domain, modeling its pharmacology with a purely genetic approach would be challenging.
The Protein Data Bank contains thousands of kinase/ligand co-crystal structures that reveal the precise location of potentially nucleophilic side chains relative to the bound ligands. For kinases and/or ligands whose structures have not been determined, the approximate locations of nucleophilic side chains (e.g., Cys or Lys) can be predicted from homology models and ligand docking. Knowledge of these distances and vectors – between a bound ligand and a noncatalytic Cys or catalytic Lys – should enable the design of either highly selective or semi-promiscuous covalent probes against many different kinases. One of our major goals for the future is to identify novel electrophilic functional groups with the optimal balance of reactivity and stability to target cysteine, lysine, and other potentially nucleophilic side chains in a selective, site-directed manner.
References
- Bain J, Plater L, Elliot M, Shpiro N, Hastie CJ, Mclauchlan H, Klevernic I, Arthur JSC, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem. J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, Mathieson T, Perrin J, Raida M, Rau C, Reader V, Sweetman G, Bauer A, Bouwmeester T, Hopf C, Kruse U, Neubauer G, Ramsden N, Rick J, Kuster B, Drewes G. Quantitative chemical proteomics revels mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 2007;25:1035–1044. doi: 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- Barf T, Kaptein A. Irreversible Protein Kinase Inhibitors: Balancing the Benefits and Risks. J. Med. Chem. 2012;55:6243–6262. doi: 10.1021/jm3003203. [DOI] [PubMed] [Google Scholar]
- Buzko O, Shokat KM. A kinase sequence database: sequence alignments and family assignment. Bioinformatics. 2002;18:1274. doi: 10.1093/bioinformatics/18.9.1274. [DOI] [PubMed] [Google Scholar]
- Chen F, Pruett-Miller SM, Huang Y, Gjoka M, Duda K, Taunton J, Collingwood TN, Frödin M, Davis GD. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat Methods. 2011;8:753–755. doi: 10.1038/nmeth.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MS, Hadjivassiliou H, Taunton J. A clickable inhibitor reveals context-dependent autoactivation of p90 RSK. Nat. Chem. Biol. 2007;3:156–160. doi: 10.1038/nchembio859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MS, Zhang C, Shokat KM, Taunton J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science. 2005;308:1318–1321. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuello F, Snabaitis AK, Cohen MS, Taunton J, Avkiran M. Evidence for direct regulation of myocardial Na+/H+ exchanger isoform 1 phosphorylation and activity by 90-kDa ribosomal S6 kinase (RSK): effects of the novel and specific RSK inhibitor fmk on responses to alpha1-adrenergic stimulation. Mol. Pharmacol. 2006;71:799–806. doi: 10.1124/mol.106.029900. [DOI] [PubMed] [Google Scholar]
- Dalby KN, Morrice N, Caudwell FB, Avruch J, Cohen P. Identification of regulatory phosphorylation sites in mitogen-activated protein kinase (MAPK)-activated protein kinase-1a/p90rsk that are inducible by MAPK. J. Biol. Chem. 1998;273:1496–1505. doi: 10.1074/jbc.273.3.1496. [DOI] [PubMed] [Google Scholar]
- David J, Mehic D, Bakiri L, Schilling AF, Mandic V, Priemel M, Idarraga MH, Reschke MO, Hoffman O, Amling M, Wagner EF. Essential role of RSK2 in c-Fos–dependent osteosarcoma development. J. Clin. Invest. 2005;115:664–672. doi: 10.1172/JCI22877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doehn U, Hauge C, Frank SR, Jensen CJ, Duda K, Nielsen JV, Cohen MS, Johansen JV, Winther BR, Lund LR, Winther O, Taunton J, Hansen SH, Frödin M. RSK Is a Principal Effector of the RAS-ERK Pathway for Eliciting a Coordinate Promotile/Invasive Gene Program and Phenotype in Epithelial Cells Mol. Cell. 2009:511–522. doi: 10.1016/j.molcel.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlanson DA, Braisted AC, Raphael DR, Randal M, Stroud RM, Gordon EM, Wells JA. Site-directed ligand discovery. Proc. Natl. Acad. Sci. 2000;97:9367–9372. doi: 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DC, Watt AP, Nicoll-Griffith DA, Baillie TA. Drug–Protein Adducts An Industry Perspective on Minimizing the Potential for Drug Bioactivation in Drug Discovery and Development. Chem. Res. Toxicol. 2004;17:3–16. doi: 10.1021/tx034170b. [DOI] [PubMed] [Google Scholar]
- Fisher TL, Blenis J. Evidence for two catalytically active kinase domains in pp90rsk. Mol. Cell. Biol. 1996;16:1212–1219. doi: 10.1128/mcb.16.3.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland AE, Tzur YB, Esvelt KM, Colaiácovo MP, Church GM, Calarco JA. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat Methods. 2013;10:741–743. doi: 10.1038/nmeth.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frödin M, Jensen CJ, Merienne K, Gammeltoft S. A phosphoserine-regulated docking site in the protein kinase RSK2 that recruits and activates PDK1. EMBO J. 2000;19:2924–2934. doi: 10.1093/emboj/19.12.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry DW, Bridges AJ, Denny WA, Doherty A, Greis KD, Hicks JL, Hook KE, Keller PR, Leopold WR, Loo JA, McNamara DJ, Nelson JM, Sherwood V, Smaill JB, Trump-Kallmeyer S, Dobrusin EM. Specific, irreversible inactivation of the epidermal growth factor receptor and erbB2, by a new class of tyrosine kinase inhibitor. Proc. Natl. Acad. Sci. 1998;95:12022–12027. doi: 10.1073/pnas.95.20.12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gushwa NN, Kang S, Chen J, Taunton J. Selective targeting of distinct active site nucleophiles by irreversible SRC-family kinase inhibitors. J. Am. Chem. Soc. 2012;134:20214–20217. doi: 10.1021/ja310659j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauge C, Frödin M. RSK and MSK in MAPK signaling. Journal of Cell Science. 2006;119:3021–3023. doi: 10.1242/jcs.02950. [DOI] [PubMed] [Google Scholar]
- Henise J, Taunton J. Irreversible Nek2 kinase inhibitors with cellular activity. J. Med. Chem. 2011;54:4133–4146. doi: 10.1021/jm200222m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: a free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012;52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamps MP, Taylor SS, Sefton BM. Direct evidence that oncogenic tyrosine kinases and cyclic AMP-dependent protein kinase have homologous ATP-binding sites. Nature. 1984;310:589–592. doi: 10.1038/310589a0. [DOI] [PubMed] [Google Scholar]
- Kang S, Elf S, Lythgoe K, Hitosugi T, Taunton J, Zhou W, Muller S, Fan S, Sun S, Marcus AI, Gu T, Polakiewicz RD, Chen Z, Khuri FR, Shin DM, Chen J. p90 ribosomal S6 kinase 2 promotes invasion and metastasis of human head and neck squamous cell carcinoma cells. J. Clin. Invest. 2010;120:1165–1177. doi: 10.1172/JCI40582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan S, Miller RM, Tian B, Mullins RD, Jacobson MP, Taunton J. Design of reversible, cysteine-targeted Michael acceptors guided by kinetic and computational analysis. J. Am. Chem. Soc. 2014;136:12624–12630. doi: 10.1021/ja505194w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwarcinscki FE, Fox CC, Steffey ME, Soellner MB. Irreversible inhibitors of c-Src kinase that target a nonconserved cysteine. ACS Chem. Biol. 2012;7:1910–1971. doi: 10.1021/cb300337u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le NT, Heo KS, Takei Y, Lee H, Woo CH, Chang E, McClain C, Hurley C, Wang X, Li F, Xu H, Morrell C, Sullivan MA, Cohen MS, Serafimova IM, Taunton J, Fujiwara K, Abe J. A crucial role for p90RSK-mediated reduction of ERK5 transcriptional activity in endothelial dysfunction and atherosclerosis. Circulation. 2013;187:486–499. doi: 10.1161/CIRCULATIONAHA.112.116988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leproult E, Barluenga S, Moras D, Wurtz JM, Winssinger N. Cysteine mapping in conformationally distinct kinase nucleotide binding sites: application to the design of selective covalent inhibitors. J. Med. Chem. 2011;54:1347–1355. doi: 10.1021/jm101396q. [DOI] [PubMed] [Google Scholar]
- Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, Gray NS. Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol. 2013;20:146–159. doi: 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Bishop A, Witucki L, Kraybill B, Shimizu E, Tsien J, Ubersax J, Blethrow J, Morgan DO, Shokat KM. Structural Basis for selective inhibition of Src family kinases by PP1. Chem. Biol. 1999;6:671–678. doi: 10.1016/s1074-5521(99)80118-5. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Miller RM, Paavilainen VO, Krishnan S, Serafimova IM, Taunton J. Electrophilic fragment-based design of reversible covalent kinase inhibitors. J. Am. Chem. Soc. 2013;135:5298–5301. doi: 10.1021/ja401221b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moellering RE, Cravatt BF. How chemoproteomics can enable drug discovery and development. Chem. Biol. 2012;19:11–22. doi: 10.1016/j.chembiol.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino M, Choy JW, Gushwa NN, Oses-Prieto JA, Koupparis K, Burlingame AL, Renslo AR, McKerrow JH, Taunton J. Hypothemycin, a fungal natural product, indentifies therapeutic targets in Trypanosoma brucei. eLife. 2013;2:1–15. doi: 10.7554/eLife.00712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, Mendonca RV, Sweeney MD, Scott KC, Grothaus PG, Jeffery DA, Spoerke JM, Honigberg LA, Young PR, Dalrymple SA, Palmer JT. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem. 2007;2:58–61. doi: 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
- Patricelli MP, Nomanbhoy TK, Wu J, Brown H, Zhou D, Zhang J, Jagannathan S, Aban A, Okerberg E, Herring C, Nordin B, Weissig H, Yang Q, Lee JD, Gray NS, Kozarich JW. In situ kinase profiling reveals functionally relevant properties of native kinases. Chem. Biol. 2011;18:699–710. doi: 10.1016/j.chembiol.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo Y, Zhang X, Roux PP. Regulation and function of the RSK family of protein kinases. Biochem. J. 2012;441:553–569. doi: 10.1042/BJ20110289. [DOI] [PubMed] [Google Scholar]
- Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- Sapkota GP, Cummings L, Newell FS, Armstrong C, Bain J, Frödin M, Grauert M, Hoffmann M, Schnapp G, Steegmaier M, Cohen P, Alessi DR. BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem. J. 2007;401(I):29–38. doi: 10.1042/BJ20061088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler T, Sicheri F, Pico A, Gazit A, Levitzki A, Kuriyan J. Crystal structure of Hck in complex with a Src family-selective tyrosine kinase inhibitor. Mol. Cell. 1999;3:639–648. doi: 10.1016/s1097-2765(00)80357-3. [DOI] [PubMed] [Google Scholar]
- Serafimova IM, Pufall MA, Krishnan S, Duda K, Cohen MS, Maglathlin RL, McFarland JM, Miller RM, Frödin M, Taunton J. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol. 2012;8:471–476. doi: 10.1038/nchembio.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian D, Roux PP, Mieulet V, Cohen MS, Raught B, Taunton J, Hershey JW, Blenis J, Pende M, Sonenberg N. The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 2006;25:2781–2791. doi: 10.1038/sj.emboj.7601166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Dobrusin EM, Fry DW, Haske T, Whitty A, McNamara DJ. Structure-based design of a potent, selective, and irreversible inhibitor of the catalytic domain of the erbB receptor subfamily of protein tyrosine kinases. J. Med. Chem. 1997;40:1130–1135. doi: 10.1021/jm960380s. [DOI] [PubMed] [Google Scholar]
- Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Smolen GA, Zhang J, Zubrowski MJ, Edelman EJ, Luo B, Yu M, Ng LW, Scherber CM, Schott BJ, Ramaswamy S, Irimia D, Root DE, Haber DA. A genome-wide RNAi screen identifies multiple RSK-dependent regulators of cell migration. Genes & Dev. 2010;24:2654–2665. doi: 10.1101/gad.1989110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, Kraemer O, Himmelsbach F, Haaksma E, Adolf GR. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J. Pharmacol. Exp. Ther. 2012;343:342–350. doi: 10.1124/jpet.112.197756. [DOI] [PubMed] [Google Scholar]
- Takahashi E, Abe J, Gallis B, Aebersold R, Spring DJ, Krebs EG, Berk BC. p90(RSK) is a serum-stimulated Na+/H+ exchanger isoform-1 kinase. Regulatory phosphorylation of serine 703 of Na+/H+ exchanger isoform-1. J. Biol. Chem. 1999;274:20206–20214. doi: 10.1074/jbc.274.29.20206. [DOI] [PubMed] [Google Scholar]
- Uetrecht J. Idiosyncratic drug reactions: past, present, and future. Chem. Res. Toxicol. 2008;21:84–92. doi: 10.1021/tx700186p. [DOI] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Hur W, McDermott U, Dutt A, Xian W, Ficarro SB, Zhang J, Sharma SV, Brugge J, Meyerson M, Settleman J, Gray NS. A structure-guided approach to creating covalent FGFR inhibitors. Chem. Biol. 2010;17:285–295. doi: 10.1016/j.chembiol.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]