

ABSTRACT
The basal layer of the epidermis contains stem cells and transit amplifying cells that rapidly proliferate and differentiate further into the upper layers of the epidermis. A number of molecules have been identified as regulators of this process, including p63 (also known as tumor protein 63) and Notch1. However, little is known about the mechanisms that regulate the transitions from stem cell to proliferating or differentiating transit amplifying cell. Here, we demonstrate that epiprofin (Epfn, also known as Sp6) plays crucial distinct roles in these transition stages as a cell cycle regulator and a transcription factor. Epfn knockout mice have a thickened epidermis, in which p63-expressing basal cells form multiple layers owing to the accumulation of premature transit amplifying cells with reduced proliferation and a reduction in the number of differentiating keratinocytes expressing Notch1. We found that low levels of Epfn expression increased the proliferation of human immortalized keratinocyte (HaCaT) cells by increasing EGF responsiveness and superphosphorylation of Rb. By contrast, high levels of Epfn expression promoted cell cycle exit and differentiation, by reducing E2F transactivation and inducing Notch1 expression. Our findings identify multiple novel functions of Epfn in epidermal development.
KEY WORDS: Sp transcription factor, Skin development, Proliferation, Differentiation, Keratinocyte, Transit amplifying cell, Stem cell, p63, Notch
INTRODUCTION
Skin self-renews throughout life by constantly replenishing keratinocytes in the outermost layer of the epidermis in a process that involves balancing the production of stem cells and transit amplifying cells, which are the committed keratinocyte precursors (Jones and Watt, 1993). The epidermis arises from a single ectodermal layer and renews by highly coordinated regulatory programs controlling proliferation and differentiation. The epidermis consists of four distinct layers that are mostly composed of keratinocytes at different stages of maturation. The basal layer of the epidermis contains stem cells and transit amplifying cells. Transit amplifying cells divide a limited number of times (three to five) before commitment to terminal differentiation. Basal keratinocytes attach to the underlying basement membrane through integrins, such as α3β1 and α6β4. Other integrins, such as α2β1 and α5β1, required for collagen/laminin and fibronectin binding, respectively, are also involved in basal keratinocyte interaction with the basement membrane (Burgeson and Christiano, 1997). When transit amplifying cells eventually stop proliferating and begin to differentiate, the expression of these integrins is reduced, resulting in detachment of keratinocytes from the basement membrane and cell movement towards the upper layers of the epidermis (Fuchs, 2008).
Proliferation of transit amplifying cells is stimulated by the mitogenic epidermal growth factor (EGF) through the EGF receptor (EGFR)–ERK–MAPK signaling pathway. Although the EGFR is also expressed by stem cells, these cells generally respond poorly to EGF (Jensen and Watt, 2006; Powell et al., 2012; Wong et al., 2012). The difference in EGF responsiveness is due in part to the reduced levels of the Lrig proteins in transit amplifying cells. These proteins negatively regulate EGF-induced signaling by promoting the ubiquitylation and degradation of EGFR (Jensen and Watt, 2006; Laederich et al., 2004; Watt and Jensen, 2009). Expression of Lrig1 is essential for epidermal homeostasis; Lrig1-knockout mice show epidermal hyperplasia, and knockdown of Lrig1 expression in cultured human keratinocytes induces stem cell expansion (Jensen and Watt, 2006; Suzuki et al., 2002). Several cell cycle regulators have been implicated in skin morphogenesis. For example, retinoblastoma protein (Rb) and the associated pocket proteins (p107 and p130; also known as RBL1 and RBL2, respectively) regulate epidermal differentiation and maintenance (Ruiz et al., 2004).
Coordinated epidermal differentiation programs are regulated by a series of genes. Among them, Notch1 and the deltaN isoform of p63 (also known as tumor protein 63) are crucial regulators of epidermal keratinocyte development (Blanpain et al., 2006). The transcription factor p63 is necessary for the proper development of skin, teeth, limbs and craniofacial tissues. In the epidermis, p63 is required to maintain the population of stem cells and transit amplifying cells and to ensure transit amplifying cell commitment to the epidermal cell lineage (Koster et al., 2004; Mills et al., 1999; Pellegrini et al., 2001; Romano et al., 2012; Truong et al., 2006; Yang et al., 1999). A deficiency of p63 results in the absence of keratinocyte stratification and differentiation (Mills et al., 1999; Yang et al., 1999). Loss of Notch signaling also disrupts epidermal differentiation (Lowell et al., 2000; Rangarajan et al., 2001), as evidenced by hyperkeratosis, whereas overexpression of activated Notch1 in basal keratinocytes induces the loss of proliferative capacity and induction of premature differentiation (Blanpain et al., 2006; Lefort et al., 2007; Nguyen et al., 2006). The expression of Notch1 and p63 in the epidermis is mutually exclusive; Notch1 is primarily expressed by differentiating keratinocytes, whereas p63 expression occurs in the basal layer, which mainly contains stem cells and maturing transit amplifying cells (Nguyen et al., 2006). A negative regulatory loop involving p63 and Notch signaling controls transit amplifying cell commitment to keratinocyte differentiation. Thus, the balance between p63 and Notch1 expression and signaling is crucial for the development and controlled renewal of the epidermis. However, it is not clear how transit amplifying cell cycle exit is promoted and how Notch1 transcription is induced during the transition from proliferation to differentiation.
We previously identified epiprofin (Epfn, also known as Sp6) based on mRNA analysis of tooth germs and demonstrated its expression in certain ectodermal tissues such as teeth, skin, hair follicles and limb buds (Nakamura et al., 2004). Epfn is a member of the Sp family of transcription factors, and it contains a transactivation/suppressor domain at the N-terminus and three C2H2 zinc finger motifs at the C-terminus for DNA binding (Suske et al., 2005). Epfn knockout (Epfn−/−) mice display striking phenotypic features, including defects in tooth morphology, supernumerary tooth formation, digit fusion, skin abnormalities and hairlessness (Hertveldt et al., 2008; Ibarretxe et al., 2012; Nakamura et al., 2008; Talamillo et al., 2010). In addition to excess teeth, Epfn−/− mice display severe enamel hypoplasia due to defects in dental epithelial differentiation into enamel-matrix-secreting ameloblasts (Nakamura et al., 2008). In the absence of Epfn, the inner dental epithelium loses its characteristic ability for rapid proliferation, leading to severe enamel hypoplasia (Nakamura et al., 2008). Therefore, Epfn might be a multifunctional regulator of both proliferation and differentiation, depending on the developmental stage.
In this study, we demonstrate that Epfn−/− mice develop hyperplastic epidermis and hyperkeratosis of the skin. In the Epfn−/− epidermis, p63-expressing basal keratinocytes form multiple cell layers and Notch1 expression is reduced. We found that, in the absence of Epfn, premature transit amplifying (pre-TA) cells with reduced proliferation activity accumulate and show partial commitment to differentiation. In cell culture, Epfn promotes proliferation of keratinocyte progenitors at low levels of Epfn expression, whereas high Epfn expression promotes cell cycle exit and induces differentiation through inducing Notch1. Our findings reveal that Epfn is a novel regulator of epidermal development.
RESULTS
Defects in skin development and in morphogenesis in Epfn−/− mice
Homozygous epiprofin-knockout (Epfn−/−) mice are smaller than normal and display defects in both skin development and hair formation (Fig. 1Ab), as well as the tooth dysmorphogenesis described previously (Hertveldt et al., 2008; Nakamura et al., 2008). By contrast, heterozygous littermates (Epfn+/−) show no obvious phenotype (Fig. 1Aa). Knockout mice are hairless over most of their bodies and have short, thin, wrinkled whiskers (Fig. 1Ac,d). There are no apparent histological abnormalities in the epidermal and pelage hair follicles of Epfn−/− mice until approximately embryonic day 16.5 (Fig. 1Ba,b). After this stage, however, abnormalities in the epidermis and hair follicles become progressively more severe. After postnatal day 7 (P7), Epfn−/− epidermal layers are significantly thicker than those of heterozygotes, owing to hyperkeratosis and hypercellularity (Fig. 1Bc–h).
Fig. 1.

Defects of skin and hair follicle development in Epfn−/− mice. (A) Defective skin, hair follicle and whisker formation (arrowheads) in 3-month-old Epfn+/− (a,c) and Epfn−/− (b,d) mice. (B) Defective skin morphogenesis and hair follicle formation. Histological analysis of developing skin and skin morphogenesis at E16.5 (a,b), P3 (c,d), P7 (e,f) and P14 (g,h) of Epfn+/− (a,c,e,g) and Epfn−/− (b,d,f,h) mice. Scale bars: 100 µm. (C) Expression of epidermal keratinocyte differentiation marker proteins in P10 Epfn+/− (a,c,e,g) and Epfn−/− (b,d,f,h) mice. The expression patterns of K10 (green; a–h), K5 (red; a,b), involucrin (Inv; red; c,d), filaggrin (Fil; red; e,f) and loricrin (Lor; red; g,h) are shown. Hoechst 33258 (blue) was used to stain DNA (a–h). Scale bars: 100 µm. (D) The expression of p63 (green) and Notch1 (red) (a,b) in P13 mice, and Hes1 (green; c,d) in P10 mice. In P10 mice, DNA was stained with DAPI (blue). The white dotted lines indicate the basement membrane of the epidermis. Also shown is Epfn expression (DAB) (e,f) in 3-month-old mice. Scale bars: 100 µm (a,b,e,f), 50 µm (c,d). (E) Ratios of cells immunopositive for p63, Notch1, Hes1 and Epfn in the epidermis of Epfn+/− and Epfn−/− mice. Data show the mean+s.e.m. (three independent experiments).
We subsequently examined possible defects in epidermal differentiation at P10 by double-immunofluorescent staining using antibodies against differentiation markers, including the keratins K5 and K10, as well as involucrin, filaggrin and loricrin (Fig. 1C). In control Epfn+/− littermates, K5 was expressed in the basal epidermal layer (Fig. 1Ca), whereas K10 was expressed primarily in the suprabasal layer of the epidermis (Fig. 1Ca,c,e,g). In Epfn−/− mice, the K5- and K10-positive cells formed multiple cell layers in which the K5-positive cells were located in a lower layer of the thickened epidermis as compared with the K10-positive cells (Fig. 1Cb). Involucrin, filaggrin and loricrin were expressed in differentiating keratinocyte layers in the control epidermis (Fig. 1Cc,e,g), but the expression of these genes was reduced in the Epfn−/− epidermis (Fig. 1Cd,f,h).
To address the molecular basis of the abnormal formation of multiple keratinocyte layers in the Epfn−/− epidermis, we analyzed the expression of p63 and Notch1, which regulate cell fate and keratinocyte differentiation in the epidermis (Fig. 1Da,b). In the control P13 epidermis of heterozygous mice, p63 was expressed in the basal layer and Notch1 expression was observed in differentiating keratinocytes, in agreement with previous reports (Fig. 1Da,b) (Kurata et al., 2004; Mills et al., 1999; Rangarajan et al., 2001). In the Epfn−/− epidermis, the p63-positive cells formed multiple layers (Fig. 1Db) and the number of p63-expressing cells was increased (Fig. 1E), whereas Notch1 expression was virtually undetectable in the epidermis (Fig. 1Db,E). Expression of Hes1, a transcription factor and downstream target of Notch, was also considerably lower in the Epfn−/− epidermis compared with that of the heterozygotes (Fig. 1Dc,d,E), indicating perturbed Notch signaling in the Epfn−/− epidermis. Finally, Epfn was expressed in basal layer keratinocytes and in differentiating keratinocytes in the epidermis during embryonic stages in the control epidermis but not in the Epfn−/− epidermis (Fig. 1De,f,E). A schematic diagram of the expression pattern for p63, Notch1 and Epfn in the epidermis is shown in supplementary material Fig. S1.
Reduced keratinocyte proliferation and apoptosis in the Epfn−/− epidermis
The epidermis of Epfn−/− mice exhibited multiple layers of K5- and p63-expressing basal cells (Fig. 1C), suggesting dysregulation of both cell proliferation and apoptosis. We examined proliferation in the Epfn−/− epidermis by immunostaining for proliferating cell nuclear antigen PCNA (a marker of late G1 and S phases) and Ki67, and by BrdU incorporation. Apoptosis was analyzed by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining (Fig. 2A,B). In the P7 Epfn+/− epidermis, the majority of the basal epidermal keratinocytes formed a single cell layer, and most of the cells were PCNA-positive (Fig. 2Aa,B). The number of PCNA-positive cells in the basal layer was significantly lower in the Epfn−/− epidermis, but the total number of cells exhibiting some PCNA immunoreactivity was higher in the Epfn−/− epidermis because of the long half-life of PCNA and the hypercellularity (Fig. 2Aa,b; Fig. 2B). Many Ki67-positive cells were detected in the basal layer of the P7 Epfn+/− epidermis, whereas the number of Ki67-positive cells was reduced in the Epfn−/− epidermis (Fig. 2Ac,d; Fig. 2B). Similarly, short-term incorporation of BrdU for 4 h to detect transit amplifying cells revealed that a significantly greater number of basal cells were proliferating in the control P7 Epfn+/− epidermis compared with the Epfn−/− epidermis (Fig. 2Ae,f; Fig. 2B). These results suggest that transit amplifying cell proliferation is inhibited in the Epfn−/− epidermis. However, these cells accumulate, resulting in hypercellularity. In addition, TUNEL staining analysis revealed that the number of apoptotic cells in P3 Epfn−/− epidermis was significantly reduced compared with that in the Epfn+/− epidermis (Fig. 2Ag,h,B). The lower rate of programmed cell death in the early postnatal mutant epidermis might have contributed to the observed increase in epidermal layer thickness and hypercellularity in Epfn−/− mice.
Fig. 2.

Slower keratinocyte proliferation, reduced apoptosis and dysregulation of Rb phosphorylation in the Epfn−/− epidermis. (A) PCNA (a,b), Ki67 (c,d) and BrdU (e,f) staining in the P7 epidermis, and TUNEL staining (g,h) in the P3 epidermis. Scale bars: 100 µm. (B) Relative numbers of PCNA-, Ki67-, BrdU- and TUNEL-positive cells in Epfn+/− and Epfn−/− skin. Data show the mean+s.e.m. (three independent experiments); **P<0.01. (C) Proliferation of primary keratinocytes from newborn Epfn+/− and Epfn−/− mice. Data show the mean+s.e.m. (three independent experiments); *P<0.05. (D) Flow cytometric analysis of cell cycle progression in primary keratinocytes from newborn Epfn+/− and Epfn−/− mice. (E) Expression of cell cycle regulators as revealed by western blotting. Reduced Rb phosphorylation (phos-pRb) and expression of cell cycle regulators in primary keratinocytes from newborn Epfn+/− and Epfn−/− mice. Upper arrowhead and asterisk, phos-Rb; lower arrowhead and asterisk, Rb protein.
Results thus far indicate that loss of Epfn disrupts the normal balance of transit amplifying cell proliferation and differentiation that is necessary for proper skin morphogenesis. To examine the effects of Epfn on cell proliferation under controlled conditions, we used primary keratinocytes isolated from the epidermis of newborn Epfn+/− and Epfn−/− mice. There were significantly fewer cells in cultures derived from Epfn−/− mice compared with wild-type cultures after 4 days in serum-free and low-Ca2+ (differentiation-restricted) medium (KGM), suggesting that Epfn normally promotes keratinocyte proliferation (Fig. 2C). Flow cytometric analysis revealed that approximately half of the keratinocytes from the Epfn+/− epidermis were in the proliferating phases (G2/M and S), whereas the majority (∼70%) of the keratinocytes from the Epfn−/− epidermis were in the quiescent G0/G1 phase (Fig. 2D). Taken together, these results suggest that Epfn is essential for transit amplifying cell proliferation.
To determine the molecular pathways leading to cell cycle disruption in keratinocytes from Epfn−/− mice, we examined the expression and phosphorylation status of the cell cycle regulators Rb, p21 (also known as cyclin-dependent kinase inhibitor 1 or CDKN1A) and p107, and the cyclin-dependent kinases (CDKs) CDK4 and CDK6 (Fig. 2E). Cultured keratinocytes from the control Epfn+/− epidermis expressed both the phosphorylated form of Rb (phos-Rb) and the unphosphorylated form of Rb. The expression of phos-Rb and p21 was reduced in Epfn−/− keratinocytes, but the expression of p107 was not. CDK4 and CDK6 were expressed at similar levels in both cell types. These results suggest that Epfn promotes keratinocyte proliferation by regulating Rb phosphorylation and p21 expression (Fig. 2E).
Accumulation of premature transit-amplifying-cell-like keratinocytes in the Epfn−/− epidermis
The basal epidermis of Epfn−/− mice exhibited ectopic expression of keratins, and basal keratinocyte-like cells expressing K5 and p63 formed multiple cell layers (Fig. 1). Moreover, isolated keratinocytes from the Epfn−/− epidermis proliferated more slowly compared with keratinocytes derived from the Epfn+/− epidermis (Fig. 2C). To identify additional genes regulated by Epfn, we compared the expression of genes that are characteristic of normal stem cells and transit amplifying cells in Epfn−/− and Epfn+/+ primary keratinocytes using microarray analysis (Fig. 3A). In Epfn−/− keratinocytes, stem cell markers such as Krt15 (cytokeratin 15) and the Notch ligands Dll1 and Jag2 were significantly downregulated compared with their expression in wild-type cells, whereas other markers, such as Lrig3 and p63, were upregulated. The integrins α3 (Itga3), α6 (Itga6) and β1 (Itgb1), which are markers for both stem cells and transit amplifying cells, and Tfrc (transferrin receptor, also known as CD71), a marker of transit amplifying cells, were upregulated in Epfn−/− keratinocytes. However, Notch1 and Notch3, markers of differentiated keratinocytes, were downregulated in Epfn−/− keratinocytes, consistent with immunohistochemical observations using the antibodies against Notch1 and Hes1 (Fig. 1D,E). These differences in gene expression between Epfn+/+ and Epfn−/− keratinocytes were confirmed by quantitative PCR analysis using primer sets specific to individual genes (data not shown). Therefore, the premature transit-amplifying-like (pre-TA) cells that accumulated in the Epfn−/− epidermis have an immature phenotype, retaining certain stem cell marker genes while expressing only some genes specific to transit amplifying cells. Moreover, accumulated pre-TA cells in the Epfn−/− epidermis were not capable of rapid proliferation, which is a key characteristic of normal transit amplifying cells.
Fig. 3.
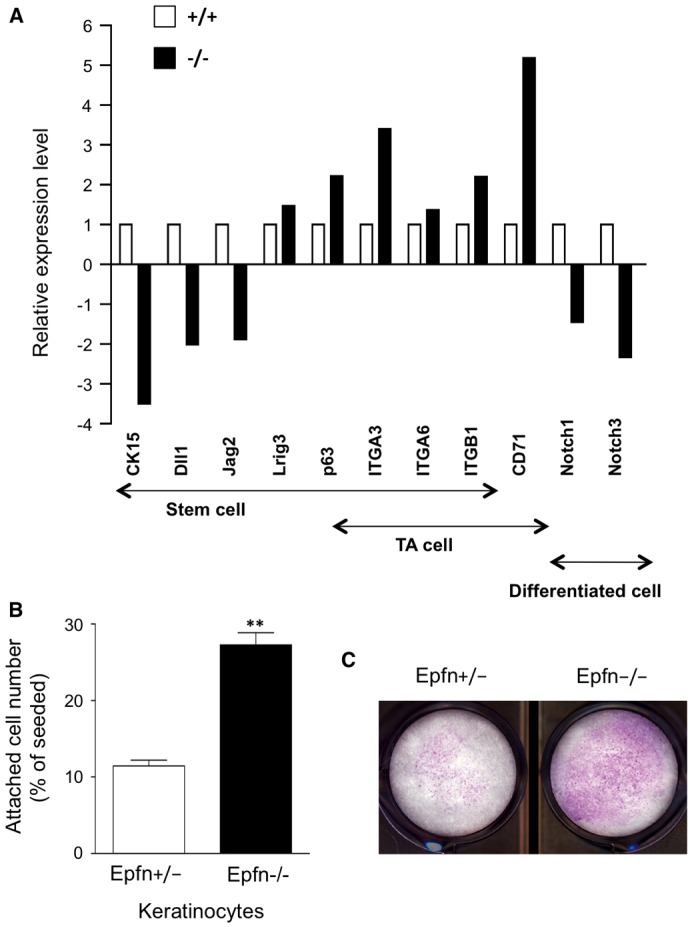
Characteristics of keratinocytes from the Epfn−/− epidermis. (A) Expression of various genes characteristic of stem cells, transit amplifying (TA) cells and mature keratinocytes in primary cultures derived from newborn Epfn+/+ and Epfn−/− mouse epidermis as measured by microarray analysis. (B) Cell attachment and (C) colony formation assays were performed using primary keratinocytes from newborn Epfn+/− and Epfn−/− mice. Data in B show the mean+s.e.m. (four independent experiments); **P<0.01.
Basal keratinocytes express integrins such as α3β1, α6β4 and α5β1 at the basal cell surface, and these act to anchor immature cells to the underlying basement membrane (Burgeson and Christiano, 1997). When transit amplifying cells differentiate, the expression of these integrins is reduced and the cells detach and migrate towards the surface layers (Fuchs, 2008). However, immunostaining of the basal Epfn−/− epidermis revealed integrin β6 expression over the entire peripheral cell surface (data not shown), consistent with an immature phenotype. In vitro, stem cells rapidly attach to fibronectin-coated wells within 15 min of incubation because of strong integrin expression (Jones and Watt, 1993). To characterize the functional effects of integrin expression by pre-TA cells in the Epfn−/− epidermis, we analyzed the attachment activity of keratinocytes from the Epfn−/− epidermis to fibronectin (Fig. 3B). Approximately 30% of the keratinocytes from the Epfn−/− epidermis attached to fibronectin-coated wells within 15 min of incubation, compared with only 10% of the keratinocytes from Epfn+/− mice. Colony-forming assays confirmed that the Epfn−/− keratinocytes formed more cell colonies than the Epfn+/− keratinocytes (Fig. 3C), indicating that these Epfn−/− keratinocytes retained certain stem-cell-like and immature cell properties.
Roles of Epfn in proliferation of HaCaT cells and keratinocytes
To address the mechanism of Epfn action in proliferation, we used the human epidermal keratinocyte cell line HaCaT, which proliferates in KGM but can be induced to differentiate in KGM containing a high Ca2+ concentration (Boukamp et al., 1988). HaCaT cells express Epfn at a low level in the proliferation medium (Fig. 4A, left two lanes; no exogenous Ca2+ addition). Elevating the Ca2+ levels in the medium by the addition of 0.7 mM or 1.2 mM Ca2+ increased Epfn expression as well as that of markers of keratinocyte differentiation, such as filaggrin (Fig. 4A, right four lanes). Transfection of a targeted Epfn short hairpin (sh)RNA into HaCaT cells reduced the endogenous RNA and protein levels of the target (Fig. 4Ba). Epfn shRNA significantly reduced HaCaT cell proliferation (Fig. 4Bb) in the low-Ca2+ medium, consistent with the results in the primary keratinocyte cultures from the Epfn−/− mice (Fig. 2C). We observed similar results using a different Epfn shRNA (data not shown).
Fig. 4.

Epfn expression in differentiated HaCaT cells and the role of Epfn in EGF-mediated cell proliferation. (A) Immunoblot analysis of Epfn and filaggrin expression in differentiating HaCaT cells cultured in a low-Ca2+ medium (KGM) with the addition of 0 mM, 0.7 mM or 1.2 mM Ca2+ for 4 days (4d) or 7 days (7d). (B) HaCaT cell proliferation. (a) Upper panel shows western blot analysis of endogenous Epfn expression in HaCaT cells transfected with control shRNA and Epfn shRNA. Lower panel shows densitometric analysis of the western blot assays using NIH ImageJ. (b) Proliferation of HaCaT cells transfected with control shRNA and Epfn shRNA. (C) EGF-mediated proliferation of HaCaT cells transfected with control shRNA and Epfn shRNA. (D) EGF-mediated proliferation of human epidermal keratinocytes transfected with control shRNA and Epfn shRNA. (E) EGF-mediated proliferation of keratinocytes from the wild-type (Epfn+/+) and Epfn−/− epidermis. Primary keratinocytes were cultured in KGM for 4 days with or without recombinant EGF (10 ng/ml). Quantitative data show the mean+s.e.m. (three independent experiments); *P<0.05; **P<0.01.
One possible explanation for the reduced proliferation of Epfn−/− keratinocytes might be the reduction in EGF-mediated proliferation. The Lrig family of proteins promotes EGFR degradation and negatively regulates EGF-mediated stem cell proliferation, thereby maintaining a slow proliferation (Cai et al., 2009; Laederich et al., 2004; Muller et al., 2013; Rondahl et al., 2013). The microarray analysis showed upregulation of Lrig3 expression in Epfn−/− keratinocytes (Fig. 3A), suggesting that Epfn normally inhibits Lrig expression and that Epfn deficiency leads to a loss of the EGF-mediated proliferation of keratinocytes. We explored these possibilities by testing the EGF response in HaCaT cells transfected with control shRNA or Epfn shRNA (Fig. 4C). The HaCaT cells were incubated in the absence or the presence of EGF for 3 days and the cell proliferation was analyzed by colorimetric assays using the WST-8 Kit. EGF increased proliferation of the HaCaT cells transfected with control shRNA, whereas those transfected with Epfn shRNA did not respond to EGF (Fig. 4C). The expression level of Lrig1 was increased in the Epfn knockdown HaCaT cells (supplementary material Fig. S2). We confirmed the involvement of Epfn in EGF-mediated proliferation in primary human keratinocytes transfected with the same control shRNA and Epfn shRNA used for HaCaT cells (Fig. 4D). Two different Epfn shRNAs reduced the expression of endogenous Epfn in human keratinocytes (supplementary material Fig. S3). EGF treatment increased the proliferation of primary human keratinocytes, but it did not increase proliferation of the cells transfected with Epfn shRNA (Fig. 4D). Similarly, the primary mouse Epfn−/− keratinocytes from the Epfn−/− epidermis did not respond to EGF (Fig. 4E). These results suggest that Epfn promotes EGF-mediated proliferation of transit amplifying cells in part by suppressing Lrig expression.
Epfn promotes HaCaT cell proliferation by forming a complex with CDK4 and enhancing Rb phosphorylation
Next, we used HaCaT cells to examine whether Epfn promotes the proliferation of keratinocytes by interacting with cell cycle signaling molecules. We analyzed the dose-dependent effects of Epfn expression on HaCaT cell proliferation 2 days after transfection of HaCaT cells with a Halo-tagged Epfn expression vector under the control of CMV promoters containing various deletions (Fig. 5A). Western blotting using an anti-Epfn antibody revealed a gradual decrease in the expression of recombinant Halo-tagged Epfn with sequential deletions in the CMV promoter region (CMV, CMVd1; day3) (Fig. 5A). We found that low levels of Epfn expression (CMVd2, CMVd3) promoted more proliferation of HaCaT cells compared with that observed following high levels of Epfn expression (CMV, CMVd1) (Fig. 5B). These results suggest that low levels of Epfn expression are involved in promoting proliferation.
Fig. 5.

Epfn promotes HaCaT cell proliferation through enhancing Rb phosphorylation and Epfn–Cdk4 interaction. (A) Immunoblot analysis of Epfn expression in HaCaT cells transfected with various Halo-tagged Epfn expression vectors driven by either the full-length CMV promoter (CMV-Epfn) or versions of the CMV promoter containing deletions (CMVd1-Epfn, CMVd2-Epfn, CMVd3-Epfn). β-actin is shown as a loading control. (B) The relationship between Epfn expression levels and HaCaT cell proliferation. HaCaT cells were transfected with one of four Halo-tagged Epfn expression vectors. The proliferation was analyzed at various time-points. Data show the mean±s.e.m. (four independent experiments); **P<0.01. (C) Immunoblot analysis of cell cycle proteins. HaCaT cells were transfected with a His-tagged Epfn expression vector, and lysates were analyzed by western blotting using antibodies against the His tag, phos-Rb, E2F1 and β-actin (the loading control). (D) Co-immunoprecipitation assay of Epfn and Cdk4 (a) and an Rb phosphorylation assay (b) in HEK293 cells transfected with Mock or Epfn (Myc tag) expression vectors. IP, immunoprecipitated; IB, immunoblotted.
To identify the molecular mechanism of Epfn-mediated cell proliferation, we examined the expression of cell cycle regulators in HaCaT cells transfected with a His-tagged Epfn expression vector (Fig. 5C). Epfn expression increased the phosphorylation of Rb (Fig. 5C) but had no effect on Rb protein levels (data not shown) and did not affect the expression of E2F1, a transcription factor that binds to Rb in a cell-cycle-dependent manner and is a crucial regulator of cell cycle control. Epfn-induced Rb phosphorylation is consistent with decreased amounts of phos-Rb in Epfn−/− keratinocytes (Fig. 2E). Rb phosphorylation by the cyclin-D–CDK complex from the G1 to S phase of the cell cycle results in the release of active E2F1 from the E2F1–Rb complex. Consequently, E2F1 activates the transcription of genes that drive cell cycle progression. Because Epfn promoted Rb phosphorylation in HaCaT cells (Fig. 5C), we speculated that Epfn might also interact with the cyclin-D–CDK complex. As CDK4 is a crucial regulator of Rb phosphorylation, we assessed possible interactions between Epfn and CDK4 by co-immunoprecipitation assays (Fig. 5Da). HEK293T cells were co-transfected with Myc-tagged Epfn and CDK4 expression vectors, and the extracts were immunoprecipitated with an anti-Myc antibody and analyzed by western blotting using an anti-CDK4 antibody. The CDK4 antibody recognized the protein complex pulled down by anti-Myc, strongly suggesting that Epfn directly or indirectly complexes with CDK4 (Fig. 5Da). Moreover, the isolated Epfn–CDK4 complex strongly enhanced Rb phosphorylation in vitro (Fig. 5Db). These results suggest that Epfn complexes with CDK4 and that this Epfn–CDK4 complex promotes cell proliferation by enhancing the phosphorylation of Rb (schematic in Fig. 7B).
Fig. 7.

Schematic diagram of Epfn roles in keratinocyte proliferation and differentiation. (A) Epfn promotes rapid transit amplifying (TA) cell proliferation. In the absence of Epfn, pre-TA cells accumulate in the epidermis. Pre-TA cells are committed to differentiation but retain some stem cell characteristics, such as slow proliferation in part due to elevated Lrig expression. As Epfn expression increases, Epfn promotes cell cycle exit and activates Notch1 expression, triggering differentiation. Epfn-induced Notch expression suppresses p63 expression. In addition, Epfn might directly inhibit p63 expression. Epfn regulates the p63 and Notch signaling pathways that are essential for epidermal development, maintenance and renewal. KO, knockout. (B) Multiple functions of Epfn during keratinocyte proliferation and differentiation. In transit amplifying cells, Epfn enhances Rb phosphorylation by interacting with CDK4 to promote proliferation. As Epfn expression increases, Epfn binds to E2F, which inhibits cell progression and promotes cell cycle exit by inhibiting E2F transactivation activity. Epfn also promotes keratinocyte differentiation by inducing Notch1 transcription.
Epfn promotes cell cycle exit and differentiation
Transit amplifying cells rapidly proliferate, but only for a limited number of divisions before cell cycle exit for terminal differentiation. The factor involved in controlling this transition is not clear. Epfn is continuously expressed in basal and differentiating keratinocytes in the epidermis, and in the Epfn−/− epidermis, keratinocyte differentiation is reduced. In cell culture, Epfn expression was induced during HaCaT cell differentiation in the high-Ca2+ medium (Fig. 4A). Therefore, Epfn might play a pivotal role in promoting cell cycle exit and triggering differentiation processes. To address this hypothesis, we first examined the ability of Epfn to promote the differentiation of HaCaT cells in low-Ca2+ medium. We found that Epfn overexpression in HaCaT cells increased the mRNA expression of Notch1, K10 and involucrin compared with that of HaCaT cells transfected with a control vector (Fig. 6A). These results suggest that overexpression of Epfn promotes the expression of keratinocyte differentiation markers.
Fig. 6.

Epfn promotes HaCaT cell differentiation through regulating Notch1. (A) Expression of keratinocyte marker genes as revealed by qPCR in HaCaT cells transfected with either Mock or Epfn expression vectors. (B) Co-immunoprecipitation assay of Epfn and E2F1 in HaCaT cells transfected with E2F1 (HA tag) and Epfn (VSV tag) expression vectors. IP, immunoprecipitated; IB, immunoblotted. (C) E2F activity assay in HaCaT cells transfected with Mock and Epfn expression vectors. (D) RBP-Jk reporter activity in HaCaT cells transfected with either Mock or Epfn expression vectors. Quantitative data in A,C,D show the mean+s.e.m. (three independent experiments); **P<0.01. (E) ChIP analysis of the Notch1 promoter using the anti-VSV antibody in HaCaT cells transfected with either Mock or Epfn (VSV tag) expression vectors. Upper panel, in silico prediction of potential Sp transcription factor binding sites (Sp sites A and B) in the human Notch1 promoter. Lower panel, nuclear extracts from either VSV–Epfn- or Mock-transfected HaCaT cells were used for ChIP analysis with primer sets for Sp sites A and B. ab, antibody.
E2F transcription factors activate genes that promote cell cycle progression, and therefore they can induce cell cycle re-entry. In addition, E2F1 forms a heterocomplex with Sp family transcription factors (Lin et al., 1996; Rotheneder et al., 1999). Therefore, we examined the interactions between Epfn and E2F1 by co-transfection of HaCaT cells with the VSV-G-tagged Epfn and HA-tagged E2F1 expression vectors. The extracts were immunoprecipitated with an anti-HA antibody and western blotted using anti-E2F1 and anti-VSV antibodies. We found that Epfn coprecipitated with E2F1 (Fig. 6B), suggesting that Epfn forms a protein complex with E2F1. We subsequently examined E2F transactivation activity using an E2F reporter construct in HaCaT cells transfected with the E2F1 and Epfn expression vectors (Fig. 6C). Epfn and E2F1 expression reduced E2F-mediated transcriptional activity, suggesting that Epfn inhibited E2F1 activity and cell cycle progression by sequestering the protein within an Epfn–E2F1 complex. Complex formation between Epfn and E2F1 when Epfn expression is elevated might explain why maturing transit amplifying cells exit the cell cycle after only a limited number of cell divisions (Fig. 7).
Epfn promotes Notch1 transcription in HaCaT cell differentiation
Notch1 promotes keratinocyte differentiation (Rangarajan et al., 2001). It is therefore likely that Epfn overexpression promotes HaCaT cell differentiation through Notch1 activation. To examine whether or not Epfn induces Notch signaling, we used an RBP-Jk (also known as RBPJ) reporter construct, because RBP-Jk is a DNA-binding factor and interacts with Notch as a canonical Notch target protein (de la Pompa et al., 1997). We found that Epfn transfection significantly increased RBP-Jk reporter activity (Fig. 6D), suggesting that the Epfn-mediated increase in Notch1 mRNA led to the activation of Notch1 signaling. We further examined Epfn-induced Notch1 transcription by assaying Epfn binding to the Notch1 promoter using chromatin immunoprecipitation (ChIP) assays. The human Notch1 promoter contains two distinct putative Sp-binding regions (Sp site A at −3.0 kb and Sp site B at −50 bp), both of which have multiple potential Sp-binding motifs upstream of the Notch1 gene transcriptional start site (Fig. 6E, upper panel). ChIP analysis revealed that Epfn bound to Sp site A, but not to Sp site B (site B) (Fig. 6E, lower panel) or to other promoter regions (data not shown). Immunoprecipitation of nuclear extracts using a rabbit IgG antibody as a negative control resulted in no genomic amplification. These results suggest that Epfn overexpression promotes Notch1 transcription and concomitant activation of Notch-dependent gene transcription cascades that are necessary for the differentiation of transit amplifying cells into keratinocytes (Fig. 7).
DISCUSSION
Our in vivo and cell culture results revealed that Epfn is essential for epidermal keratinocyte development by promoting both proliferation and differentiation of transit amplifying cells through distinct mechanisms (Fig. 7). In the Epfn−/− epidermis, p63 was expressed in multiple cell layers (Fig. 1Da,b) rather than in just a single basal layer of the normal epidermis. These p63-expressing basal-layer-like cells lost their rapid proliferation activity, which is a characteristic of transit amplifying cells. In addition, the expression of Notch1 and other differentiation marker genes was severely inhibited (Fig. 1Db,E; supplementary material Fig. S1).
Significant reductions in the number of BrdU-positive cells were observed, and weak PCNA-positive cells were found in the multi-cell layers, likely corresponding to p63-positive cells in the multi-cell layer in the Epfn−/− epidermis (Fig. 2A,B). In addition, primary keratinocytes from Epfn−/− epidermis showed reduced proliferation activity (Fig. 2C) and accumulated in the G1/G0 phase (Fig. 2D). Knockdown of endogenous Epfn reduced the proliferation of HaCaT cells (Fig. 4B). These results suggest that Epfn is necessary for rapid proliferation of transit amplifying cells. The proliferation of HaCaT cells by forced expression of Epfn at a low level further supports the positive regulation of keratinocyte proliferation by Epfn.
Rb binds to and inactivates members of the E2F transcription factor family that normally serve to promote cell cycle progression to S phase. Phosphorylation of Rb by CDKs releases E2F from the inactive E2F–Rb complex, allowing E2Fs to activate the genes that are necessary for cell cycle progression (Hanahan, 2000; Harbour et al., 1999). Epfn deficiency reduces Rb phosphorylation in keratinocytes (Fig. 2E) and dental epithelial cells (Nakamura et al., 2008), whereas Epfn enhances Rb phosphorylation by forming a complex with CDK4, leading to the proliferation of HaCaT cells (Fig. 5C,D). Our data demonstrate the novel mechanism by which Epfn regulates the G1/S transition and is therefore involved in the rapid phase of transit amplifying cell proliferation. The Rb proteins also participate in cell lineage specification during adipogenesis, cardiogenesis, hematopoiesis and myogenesis (Bergh et al., 1999; Classon et al., 2000; Li et al., 2000; Papadimou et al., 2005). In myogenesis, Rb and Sp1 (the most ubiquitously expressed Sp family protein) interact with tissue-specific transcription factors to regulate cell fate and muscle cell differentiation, suggesting that phosphorylation of Rb family proteins by the Sp family proteins might be a common mechanism for controlling progenitor proliferation (Guo et al., 2003). However, it is not clear whether Epfn interacts with either Rb or Sp1 during interfollicular epidermal formation.
Lrig is a marker of epidermal stem cells and maintains stem cell quiescence by suppressing EGF–EGFR signaling in the basal epidermis (Jensen et al., 2009). Stem cells and transit amplifying cells express similar levels of EGFR and are equally exposed to EGF. Lrig1 or Lrig3 interacts with EGFR and leads to EGFR degradation through ubiquitylation (Gur et al., 2004; Laederich et al., 2004). The Epfn−/− keratinocytes and Epfn-knockdown HaCaT cells showed increased Lrig expression and reduced EGF-mediated proliferation (Fig. 3A; supplementary material Fig. S2; Fig. 4C–E). These results reveal the crucial roles of Epfn in controlling the proliferation of transit amplifying cells, which it does by acting as both a positive regulator through Rb phosphorylation and a negative regulator by suppressing Lrig expression.
Epfn is expressed in the basal layer keratinocytes and proliferating HaCaT cells, and its expression levels are increased during the differentiation of keratinocytes and HaCaT cell differentiation. These expression patterns suggest that Epfn is involved in keratinocyte differentiation processes. Indeed, we found that Epfn transfection into HaCaT cells induced the expression of keratinocyte differentiation markers such as Notch1 and K10, even when cultured in a low-Ca2+ medium. Therefore, in addition to proliferation, Epfn is required for the differentiation of epidermal keratinocytes. This result is in agreement with our previous report on tooth development, in which we show that Epfn is crucial for both proliferation of the inner dental epithelium – an ameloblast progenitor similar to the transit amplifying cells of the basal layer epidermis – and its differentiation into the enamel-secreting ameloblast during amelogenesis (Nakamura et al., 2008).
The Notch signaling pathway regulates cell fate determination during the development of many tissues (Artavanis-Tsakonas et al., 1999; Blanpain et al., 2006; Boulter et al., 2012; Fuchs, 2008; Moriyama et al., 2008; Xiong et al., 2013). In the epidermis, Notch1 signaling is involved in cell cycle exit and commitment to differentiation (Breunig et al., 2007; Georgia et al., 2006; Rangarajan et al., 2001). A conditional Notch1 deficiency in the basal epidermal layer of newborn mice results in the formation of multiple p63-expressing cell layers, delayed cell cycle exit and premature keratinocyte differentiation (Devgan et al., 2005; Rangarajan et al., 2001). In the Epfn−/− epidermis, Notch1 expression was significantly reduced and multiple p63-expressing cell layers were formed (Fig. 1Db,E), similar to the Notch1-deficient epidermis. In addition, expression of the cell cycle inhibitor p21, one of the targets of Notch1 in keratinocytes (Devgan et al., 2005; Dotto, 2008; Nicolas et al., 2003; Rangarajan et al., 2001), was reduced in the Epfn−/− epidermis and gave similar phenotypes to that of the Notch-deficient epidermis (Fig. 2E). Epfn induced Notch1 expression and promoted its transactivation activity as well as HaCaT cell differentiation (Fig. 6A,D). These results strongly suggest that Epfn is upstream of Notch1 and regulates Notch1 expression. Epfn binds to the Notch1 promoter, suggesting its involvement in the transcriptional activation of the Notch1 gene (Fig. 6E). The expression of p63 and Notch is mutually exclusive because of a negative regulatory loop (Nguyen et al., 2006). Therefore, Epfn-induced Notch1 expression probably also suppresses p63 expression. Our results reveal that Epfn promotes the commitment to keratinocyte differentiation through the induction of Notch1 transcription and promotion of the Notch1–p63 negative regulatory loop (Fig. 7).
In normal organogenesis, cell cycle exit is a prerequisite for differentiation. How does Epfn act as a positive regulator of both cell proliferation and differentiation? One possible explanation for this apparent contradiction is that Epfn expression levels might regulate the switch from proliferation to differentiation. Low levels of Epfn expression promoted proliferation of HaCaT cells, whereas high levels did not (Fig. 5B). Epfn interacted with E2F and reduced E2F transactivation activity in HaCaT cells (Fig. 6B,C), suggesting that Epfn inhibits the DNA binding of E2F by sequestration in a multiprotein complex. The inhibition of E2F activity reduced the expression of genes required for cell cycle progression and promoted cell cycle exit. We propose that low Epfn levels increase the amount of free active E2F through Rb phosphorylation and concomitant release of E2F from E2F–Rb complexes (which results in proliferation), whereas high Epfn expression increases the formation of inactive E2F–Epfn complexes, resulting in cell cycle exit (Fig. 7B). In addition, post-translational modifications of Epfn, such as phosphorylation and sumoylation, might also be involved in this functional switch in Epfn activity from promoting proliferation to promoting differentiation.
In summary, our data provide evidence for a novel mechanism that sequentially regulates the proliferation and differentiation of transit amplifying cells through multiple distinct functions of Epfn as a cell cycle regulator and a transcription factor (Fig. 7B). The switch from proliferation to differentiation is regulated in part by the level of Epfn expression. Our in vitro and in vivo studies suggest that Epfn promotes rapid proliferation of transit amplifying cells by enhancing Rb phosphorylation and suppressing expression of the EGFR antagonist Lrig1, and that Epfn promotes keratinocyte differentiation by inducing Notch1 transcription. Our findings suggest that Epfn regulates the p63–Notch axis in interfollicular epidermal formation. Thus, Epfn plays multiple roles in orchestrating keratinocyte proliferation and differentiation that are crucial to epidermal homeostasis and morphogenesis (Fig. 7).
MATERIALS AND METHODS
Mice and cells
Epfn−/− mice were generated as described previously (Nakamura et al., 2008). The animal protocol was approved by the National Institute of Dental and Craniofacial Research (NIDCR) Animal Care and Use Committee. All mice were housed in an animal facility that was approved by the American Association for the Accreditation of Laboratory Animal Care.
Primary keratinocytes were prepared from newborn Epfn+/+, Epfn+/− and Epfn−/− mice as described previously (Hennings et al., 1980), with minor modifications. Briefly, the mouse skin was floated on a layer of dispase (5 mg/ml) in Ca2+ and Mg2+-free PBS at 4°C overnight. The dermis and epidermis were separated and keratinocytes were collected by mincing the epidermis, placing it in 0.25% trypsin/0.5 mM EDTA and incubating for 15 min at 37°C. After resuspending in Ca2+ and Mg2+-free PBS, the keratinocytes were filtered through a 100-µm cell strainer (BD Dickinson, Franklin Lakes, NJ) and centrifuged at 100 g for 10 min. The keratinocytes were cultured in a serum-free and low-Ca2+ keratinocyte growth medium (KGM) (Invitrogen, Carlsbad, CA).
HaCaT cells, a spontaneously transformed human epithelial cell line from adult skin (Boukamp et al., 1988), were obtained from Silvio Gutkind (NIDCR, NIH). Primary human keratinocytes and HEK293T cells were obtained from Life Technologies (Invitrogen, Carlsbad, CA). Primary human keratinocytes were cultured in Epilife™ medium (Invitrogen, Carlsbad, CA) and HEK293T cells were cultured in DMEM (Invitrogen, Carlsbad, CA).
Transfection and proliferation assay
For transfection, exponentially growing HaCaT cells were electroporated with the Epfn expression vector (Epfn-pcDNA3.1/myc-His vector; Nakamura et al., 2004) or Halo-tagged Epfn vectors (Promega, Madison, WI) using Amaxa Nucleofector Solution V (Lonza, Walkersville, MD). For the gene knockdown studies, HaCaT cells or primary human keratinocytes were transfected with either the Epfn shRNA vector (ID: TRCN0000017806, Open Biosystems, Inc., Huntsville, AL) or control pLKO vector, and the resulting puromycin-resistant colonies were pooled and used for proliferation analysis. We also used another Epfn shRNA (ID: TRCN0000017807, Open Biosystems) and obtained similar results.
Cell proliferation was assessed using the Cell Counting Kit-8 containing WST-8, according to the manufacturer's manual (Dojindo Laboratories, Kumamoto, Japan). EGF (Sigma, St Louis, MO) was added to the cell cultures as a mitogenic cytokine (10 ng/ml). The cells were cultured in the absence or presence of EGF for three days and then the proliferation activities were analyzed. To test HaCaT cell proliferation associated with various Epfn expression levels, we prepared Halo-tag Epfn expression vectors driven by full-length CMV promoter or CMV promoters with various deletions (CMV, CMV-d1, -d2 and -d3) (Promega, Madison, WI). In these experiments, we used the reverse transfection method with Lipofectamine LTX reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. The transfection mixtures for individual vectors and HaCaT cells (10,000 cells/well) were incubated with KGM. The cell proliferation assay was performed on days 0, 1 and 2 after the medium change to KGM. The protein expression of Halo-tagged Epfn in HaCaT cells was examined on day 1 by western blot analysis with anti-Epfn and anti-β-actin antibodies. For transfection of primary human keratinocytes, ViaFect™ (Promega, Madison, WI) was employed with the reverse-transfection method.
Tissue sections, immunohistochemistry and antibodies
Tissues were fixed with 4% paraformaldehyde in PBS at 4°C overnight. For histological analysis, sections were stained with Harris hematoxylin and eosin Y. For immunohistochemistry, sections were boiled with a targeted retrieval solution (Dako, Carpinteria, CA) and incubated in 1% bovine serum albumin (BSA) in PBS (blocking reagent) for 1 h before incubation with the primary antibody. We used antibodies against cytokeratin 10 (Sigma, St Louis, MO), cytokeratin 5, involucrin, filaggrin and loricrin (all from Covance, Berkeley, CA), p63 (BD Pharmingen, San Jose, CA), Notch1 (Cell Signaling Technology, Danvers, MA; BD Pharmingen, San Jose, CA), Hes1 (Millipore, Billerica, MA), PCNA (Zymed, Carlsbad, CA), Ki67 (Abcam, Cambridge, MA) and BrdU (Roche, Indianapolis, IN). The affinity-purified rabbit polyclonal antibody against Epfn was as described previously (Nakamura et al., 2008). Primary antibodies were detected with Cy-3-conjugated and Cy-5-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA). Nuclear staining was performed with Hoechst 33258 or DAPI (Sigma, St Louis, MO). All the immunopositive cells within the epidermis grid (600 µm×600 µm) were counted and the positive cell ratio was calculated by dividing by the total number of cells in the grid. To calculate the ratio, at least four independent regions were counted.
Western blotting, immunoprecipitation assay and kinase assay
Total cellular proteins were prepared from exponentially growing HaCaT cells cultured in KGM with either 0 mM, 0.7 mM or 1.2 mM Ca2+ using NP-40 lysis buffer with a protease inhibitor cocktail (Roche, Indianapolis, IN). Protein extracts were separated on 4–12% NuPAGE gels (Invitrogen, Carlsbad, CA), followed by western blotting. Kinase assays were performed using subconfluent 293T cells transfected with both HA-tagged CDK4 and Epfn expression vectors. Cells were washed in cold PBS and incubated on ice for 10 min with 500 µl of immunoprecipitation buffer (20 mM HEPES pH 7.5, containing 10 mM EGTA, 40 mM β-glycerophosphate, 1 mM DTT and 1% NP-40), supplemented with a protease inhibitor cocktail (Roche, Indianapolis, IN) and 2 mM orthovanadate. Protein extracts were incubated with an anti-HA-antibody (Thermo Scientific, Rockford, IL) at 4°C for an additional 1 h. Immune complexes were pulled down with Protein-G–Sepharose beads and washed twice with immunoprecipitation buffer. Immune complexes on the beads were equilibrated with Rb kinase buffer (50 mM HEPES pH 7.5, containing 2.5 mM EGTA, 10 mM β-glycerophosphate, 10 mM MgCl2, 5 mM MnCl2, 1 mM DTT, 0.1 mM sodium orthovanadate, 1 mM NaF and 2 µCi of [γ-32P]ATP). The beads were resuspended in Rb kinase buffer containing 2 µg of GST–Rb (Millipore, Billerica, MA) and were subsequently incubated at 30°C for 30 min. To reduce the background signal, 5 mM cold ATP was added to each reaction mixture. The reaction was stopped by adding SDS sample buffer. The samples were boiled for 5 min and resolved by SDS-10% PAGE. The gels were dried and visualized by using autoradiography on BioMAX MR film. The co-immunoprecipitation assay was performed with an HA-Tag Co-IP kit (Thermo Scientific, Rockford, IL) using subconfluent HaCaT cells transfected with both the HA-tagged E2F1 expression vector, which was kindly gifted from Kristian Helin (University of Copenhagen, Denmark), and the VSV-tagged Epfn expression vector. The immune complexes on the beads were resuspended in lysis buffer and the samples were boiled for 5 min and resolved using NuPage 4–12% gradient gels (Invitrogen, Carlsbad, CA). Immunoblotting was performed with antibodies against phosphorylated Rb, Rb, CDK4 (Cell Signaling Technology, Danvers, MA), VSV (Sigma-Aldrich, St Louis, MO), E2F1 (Santa Cruz Biotechnology, Dallas, TX), GAPDH (Santa Cruz Biotechnology, Dallas, TX) and β-actin (Cell Signaling Technology, Danvers, MA).
BrdU and TUNEL assays
BrdU (100 mg/kg body weight) was intraperitoneally injected into P13 mice. The mice were euthanized 4 h later, and the skin was dissected and fixed with 4% paraformaldehyde. BrdU incorporation was visualized with diaminobenzidine (DAB) using the BrdU detection kit (Roche, Indianapolis, IN). TUNEL assays (Roche, Indianapolis, IN) were performed on paraffin-embedded skin sections from P3 and P7 mice. The sections were counterstained with hematoxylin. The ratios of the BrdU- and TUNEL-positive cells within the grid in the epidermis (600 µm×600 µm) were calculated by dividing by the total number of cells in the grid.
Flow cytometry
Primary keratinocytes (2×106 cells) from either Epfn+/− or Epfn−/− mice were cultured for 3 days in type I collagen-coated dishes (BD Bioscience, Franklin Lakes, NJ) and washed three times in PBS, followed by fixation with ice-cold 70% ethanol for 30 min. After fixative removal, cells were centrifuged (100 g for 10 min), and the pellet was resuspended in 500 µl of propidium iodide solution (50 µg/ml in PBS) together with RNase A (10 mg/ml; Invitrogen, Carlsbad, CA) and incubated on ice for 30 min before cell cycle analysis using the FACSCalibur (BD Biosciences, Franklin Lakes, NJ).
Real-time quantitative PCR and microarray analysis
Primary keratinocytes or HaCaT cells were transiently transfected with Epfn vectors. After 48 h, total RNA was extracted using the TRIzol reagent (Invitrogen, Carlsbad, CA). After DNase I (Sigma) treatment, 2 µg of total RNA was used for reverse transcription to generate cDNA, which was subsequently used as a template for PCR reactions with gene-specific primers (supplementary material Table S1). For expression analysis, real-time quantitative (q)PCR was performed using the 2XSYBR green supermixture (BioRad, Hercules, CA) and the Chromo4 thermocycler (MJ Research, Waltham, MA). For microarray analysis, total RNA was extracted from the primary keratinocytes of newborn Epfn+/+ and Epfn−/− mice. Affymetrix mouse 430 2.0 array chips were used to analyze gene expression profiles. Data analysis was performed using Ingenuity Pathway Analysis software.
Luciferase assay
E2F and RBP-Jk activities were analyzed using the Cignal E2F and RBP-Jk Reporter (luc) Kit (Qiagen, Valencia, CA). The E2F and RBP-Jk reporter plasmids containing the control Renilla luciferase construct were transfected into HaCaT cells in a 48-well plate using Lipofectamine LTX with PLUS reagent. The activities of the firefly and Renilla luciferases were determined 48 h after transfection by using the dual luciferase reporter assay system (Promega, Madison, WI) using a luminometer (Promega, Madison, WI). The luciferase activities were normalized for the Renilla luciferase activity of the internal control.
Notch1 promoter analysis and ChIP assay
A 4-kb human Notch1 promoter sequence was analyzed with Genomatix software (Genomatix, Germany) to predict potential factor binding sites. Multiple Sp- or Klf-binding elements were predicted 3.2 kb upstream of the transcription start site and at the proximal promoter of the human Notch1 gene. Chromatin DNA and nuclear protein complexes were extracted from the HaCaT cells transfected with His-tagged Epfn or empty expression vectors, and ChIP assays were conducted with an anti-His antibody (Novagen, Madison, WI) and EZ ChIP kit (Millipore, Billerica, MA). ChIP primer sequences are shown in supplementary material Table S2 (Lefort et al., 2007).
Statistical analysis
The data were analyzed with Prism 4 software. Differences between groups were analyzed for statistical significance using Student's t-tests.
Supplementary Material
Acknowledgments
We wish to thank Kenneth Yamada, Thomas Bugge, Karin List, Roman Szabo (National Institute of Dental and Craniofacial Research, Bethesda, MD) and Maurizio Pacifici (Children's Hospital of Philadelphia Research Institute, PA) for their valuable suggestions.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
T.N., S.F., and Y.Yamada conceived of the study. T.N. performed the majority of the experiments and analyzed the data, with some contributions from Y.Yoshitomi. and V.P. T.N., Y.Yoshitomi, V.P., K.S. and Y.Yamada interpreted the data. T.N. and Y.Yamada wrote the manuscript.
Funding
This work was supported in part by the Intramural Research Program of the National Institutes of Health, National Institute of Dental and Craniofacial Research [grant numbers DE000483-26 and DE000720-08 to Y.Yamada]; and grants-in-aid [grant numbers 24390441 and 24659908 to T.N.] from the Ministry of Education, Science and Culture of Japan; and the NEXT program [grant number LS010 to S.F.]; as well as a fellowship from the Japan Society for the Promotion of Science (to T.N. and Y.Yoshitomi). Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.156778/-/DC1
References
- Artavanis-Tsakonas S., Rand M. D., Lake R. J. (1999). Notch signaling: cell fate control and signal integration in development. Science 284, 770–776 10.1126/science.284.5415.770 [DOI] [PubMed] [Google Scholar]
- Bergh G., Ehinger M., Olsson I., Jacobsen S. E., Gullberg U. (1999). Involvement of the retinoblastoma protein in monocytic and neutrophilic lineage commitment of human bone marrow progenitor cells. Blood 94, 1971–1978. [PubMed] [Google Scholar]
- Blanpain C., Lowry W. E., Pasolli H. A., Fuchs E. (2006). Canonical notch signaling functions as a commitment switch in the epidermal lineage. Genes Dev. 20, 3022–3035 10.1101/gad.1477606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukamp P., Petrussevska R. T., Breitkreutz D., Hornung J., Markham A., Fusenig N. E. (1988). Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 106, 761–771 10.1083/jcb.106.3.761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulter L., Govaere O., Bird T. G., Radulescu S., Ramachandran P., Pellicoro A., Ridgway R. A., Seo S. S., Spee B., Van Rooijen N. et al. (2012). Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat. Med. 18, 572–579 10.1038/nm.2667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breunig J. J., Silbereis J., Vaccarino F. M., Sestan N., Rakic P. (2007). Notch regulates cell fate and dendrite morphology of newborn neurons in the postnatal dentate gyrus. Proc. Natl. Acad. Sci. USA 104, 20558–20563 10.1073/pnas.0710156104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgeson R. E., Christiano A. M. (1997). The dermal-epidermal junction. Curr. Opin. Cell Biol. 9, 651–658 10.1016/S0955-0674(97)80118-4 [DOI] [PubMed] [Google Scholar]
- Cai M., Han L., Chen R., Ye F., Wang B., Han F., Lei T., Guo D. (2009). Inhibition of LRIG3 gene expression via RNA interference modulates the proliferation, cell cycle, cell apoptosis, adhesion and invasion of glioblastoma cell (GL15). Cancer Lett. 278, 104–112 10.1016/j.canlet.2009.01.001 [DOI] [PubMed] [Google Scholar]
- Classon M., Kennedy B. K., Mulloy R., Harlow E. (2000). Opposing roles of pRB and p107 in adipocyte differentiation. Proc. Natl. Acad. Sci. USA 97, 10826–10831 10.1073/pnas.190343597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Pompa J. L., Wakeham A., Correia K. M., Samper E., Brown S., Aguilera R. J., Nakano T., Honjo T., Mak T. W., Rossant J. et al. (1997). Conservation of the Notch signalling pathway in mammalian neurogenesis. Development 124, 1139–1148. [DOI] [PubMed] [Google Scholar]
- Devgan V., Mammucari C., Millar S. E., Brisken C., Dotto G. P. (2005). p21WAF1/Cip1 is a negative transcriptional regulator of Wnt4 expression downstream of Notch1 activation. Genes Dev. 19, 1485–1495 10.1101/gad.341405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotto G. P. (2008). Notch tumor suppressor function. Oncogene 27, 5115–5123 10.1038/onc.2008.225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs E. (2008). Skin stem cells: rising to the surface. J. Cell Biol. 180, 273–284 10.1083/jcb.200708185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgia S., Soliz R., Li M., Zhang P., Bhushan A. (2006). p57 and Hes1 coordinate cell cycle exit with self-renewal of pancreatic progenitors. Dev. Biol. 298, 22–31 10.1016/j.ydbio.2006.05.036 [DOI] [PubMed] [Google Scholar]
- Guo C. S., Degnin C., Fiddler T. A., Stauffer D., Thayer M. J. (2003). Regulation of MyoD activity and muscle cell differentiation by MDM2, pRb, and Sp1. J. Biol. Chem. 278, 22615–22622 10.1074/jbc.M301943200 [DOI] [PubMed] [Google Scholar]
- Gur G., Rubin C., Katz M., Amit I., Citri A., Nilsson J., Amariglio N., Henriksson R., Rechavi G., Hedman H. et al. (2004). LRIG1 restricts growth factor signaling by enhancing receptor ubiquitylation and degradation. EMBO J. 23, 3270–3281 10.1038/sj.emboj.7600342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D. (2000). Benefits of bad telomeres. Nature 406, 573–574 10.1038/35020662 [DOI] [PubMed] [Google Scholar]
- Harbour J. W., Luo R. X., Dei Santi A., Postigo A. A., Dean D. C. (1999). Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 98, 859–869 10.1016/S0092-8674(00)81519-6 [DOI] [PubMed] [Google Scholar]
- Hennings H., Michael D., Cheng C., Steinert P., Holbrook K., Yuspa S. H. (1980). Calcium regulation of growth and differentiation of mouse epidermal cells in culture. Cell 19, 245–254 10.1016/0092-8674(80)90406-7 [DOI] [PubMed] [Google Scholar]
- Hertveldt V., Louryan S., van Reeth T., Drèze P., van Vooren P., Szpirer J., Szpirer C. (2008). The development of several organs and appendages is impaired in mice lacking Sp6. Dev. Dyn. 237, 883–892 10.1002/dvdy.21355 [DOI] [PubMed] [Google Scholar]
- Ibarretxe G., Aurrekoetxea M., Crende O., Badiola I., Jimenez-Rojo L., Nakamura T., Yamada Y., Unda F. (2012). Epiprofin/Sp6 regulates Wnt-BMP signaling and the establishment of cellular junctions during the bell stage of tooth development. Cell Tissue Res. 350, 95–107 10.1007/s00441-012-1459-8 [DOI] [PubMed] [Google Scholar]
- Jensen K. B., Watt F. M. (2006). Single-cell expression profiling of human epidermal stem and transit-amplifying cells: Lrig1 is a regulator of stem cell quiescence. Proc. Natl. Acad. Sci. USA 103, 11958–11963 10.1073/pnas.0601886103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K. B., Collins C. A., Nascimento E., Tan D. W., Frye M., Itami S., Watt F. M. (2009). Lrig1 expression defines a distinct multipotent stem cell population in mammalian epidermis. Cell Stem Cell 4, 427–439 10.1016/j.stem.2009.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P. H., Watt F. M. (1993). Separation of human epidermal stem cells from transit amplifying cells on the basis of differences in integrin function and expression. Cell 73, 713–724 10.1016/0092-8674(93)90251-K [DOI] [PubMed] [Google Scholar]
- Koster M. I., Kim S., Mills A. A., DeMayo F. J., Roop D. R. (2004). p63 is the molecular switch for initiation of an epithelial stratification program. Genes Dev. 18, 126–131 10.1101/gad.1165104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurata S., Okuyama T., Osada M., Watanabe T., Tomimori Y., Sato S., Iwai A., Tsuji T., Ikawa Y., Katoh I. (2004). p51/p63 Controls subunit alpha3 of the major epidermis integrin anchoring the stem cells to the niche. J. Biol. Chem. 279, 50069–50077 10.1074/jbc.M406322200 [DOI] [PubMed] [Google Scholar]
- Laederich M. B., Funes-Duran M., Yen L., Ingalla E., Wu X., Carraway K. L., 3rd, Sweeney C. (2004). The leucine-rich repeat protein LRIG1 is a negative regulator of ErbB family receptor tyrosine kinases. J. Biol. Chem. 279, 47050–47056 10.1074/jbc.M409703200 [DOI] [PubMed] [Google Scholar]
- Lefort K., Mandinova A., Ostano P., Kolev V., Calpini V., Kolfschoten I., Devgan V., Lieb J., Raffoul W., Hohl D. et al. (2007). Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev. 21, 562–577 10.1101/gad.1484707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F. Q., Coonrod A., Horwitz M. (2000). Selection of a dominant negative retinoblastoma protein (RB) inhibiting satellite myoblast differentiation implies an indirect interaction between MyoD and RB. Mol. Cell. Biol. 20, 5129–5139 10.1128/MCB.20.14.5129-5139.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S. Y., Black A. R., Kostic D., Pajovic S., Hoover C. N., Azizkhan J. C. (1996). Cell cycle-regulated association of E2F1 and Sp1 is related to their functional interaction. Mol. Cell. Biol. 16, 1668–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowell S., Jones P., Le Roux I., Dunne J., Watt F. M. (2000). Stimulation of human epidermal differentiation by delta-notch signalling at the boundaries of stem-cell clusters. Curr. Biol. 10, 491–500 10.1016/S0960-9822(00)00451-6 [DOI] [PubMed] [Google Scholar]
- Mills A. A., Zheng B., Wang X. J., Vogel H., Roop D. R., Bradley A. (1999). p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 398, 708–713 10.1038/19531 [DOI] [PubMed] [Google Scholar]
- Moriyama M., Durham A. D., Moriyama H., Hasegawa K., Nishikawa S., Radtke F., Osawa M. (2008). Multiple roles of Notch signaling in the regulation of epidermal development. Dev. Cell 14, 594–604 10.1016/j.devcel.2008.01.017 [DOI] [PubMed] [Google Scholar]
- Muller S., Lindquist D., Kanter L., Flores-Staino C., Henriksson R., Hedman H., Andersson S. (2013). Expression of LRIG1 and LRIG3 correlates with human papillomavirus status and patient survival in cervical adenocarcinoma. Int. J. Oncol. 42, 247–252 10.3892/ijo.2012.1702 [DOI] [PubMed] [Google Scholar]
- Nakamura T., Unda F., de-Vega S., Vilaxa A., Fukumoto S., Yamada K. M., Yamada Y. (2004). The Krüppel-like factor epiprofin is expressed by epithelium of developing teeth, hair follicles, and limb buds and promotes cell proliferation. J. Biol. Chem. 279, 626–634 10.1074/jbc.M307502200 [DOI] [PubMed] [Google Scholar]
- Nakamura T., de Vega S., Fukumoto S., Jimenez L., Unda F., Yamada Y. (2008). Transcription factor epiprofin is essential for tooth morphogenesis by regulating epithelial cell fate and tooth number. J. Biol. Chem. 283, 4825–4833 10.1074/jbc.M708388200 [DOI] [PubMed] [Google Scholar]
- Nguyen B. C., Lefort K., Mandinova A., Antonini D., Devgan V., Della Gatta G., Koster M. I., Zhang Z., Wang J., Tommasi di Vignano A. et al. (2006). Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev. 20, 1028–1042 10.1101/gad.1406006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas M., Wolfer A., Raj K., Kummer J. A., Mill P., van Noort M., Hui C. C., Clevers H., Dotto G. P., Radtke F. (2003). Notch1 functions as a tumor suppressor in mouse skin. Nat. Genet. 33, 416–421 10.1038/ng1099 [DOI] [PubMed] [Google Scholar]
- Papadimou E., Ménard C., Grey C., Pucéat M. (2005). Interplay between the retinoblastoma protein and LEK1 specifies stem cells toward the cardiac lineage. EMBO J. 24, 1750–1761 10.1038/sj.emboj.7600652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini G., Dellambra E., Golisano O., Martinelli E., Fantozzi I., Bondanza S., Ponzin D., McKeon F., De Luca M. (2001). p63 identifies keratinocyte stem cells. Proc. Natl. Acad. Sci. USA 98, 3156–3161 10.1073/pnas.061032098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell A. E., Wang Y., Li Y., Poulin E. J., Means A. L., Washington M. K., Higginbotham J. N., Juchheim A., Prasad N., Levy S. E. et al. (2012). The pan-ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell 149, 146–158 10.1016/j.cell.2012.02.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangarajan A., Talora C., Okuyama R., Nicolas M., Mammucari C., Oh H., Aster J. C., Krishna S., Metzger D., Chambon P. et al. (2001). Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J. 20, 3427–3436 10.1093/emboj/20.13.3427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano R. A., Smalley K., Magraw C., Serna V. A., Kurita T., Raghavan S., Sinha S. (2012). ΔNp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development 139, 772–782 10.1242/dev.071191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondahl V., Holmlund C., Karlsson T., Wang B., Faraz M., Henriksson R., Hedman H. (2013). Lrig2-deficient mice are protected against PDGFB-induced glioma. PLoS ONE 8, e73635 10.1371/journal.pone.0073635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotheneder H., Geymayer S., Haidweger E. (1999). Transcription factors of the Sp1 family: interaction with E2F and regulation of the murine thymidine kinase promoter. J. Mol. Biol. 293, 1005–1015 10.1006/jmbi.1999.3213 [DOI] [PubMed] [Google Scholar]
- Ruiz S., Santos M., Segrelles C., Leis H., Jorcano J. L., Berns A., Paramio J. M., Vooijs M. (2004). Unique and overlapping functions of pRb and p107 in the control of proliferation and differentiation in epidermis. Development 131, 2737–2748 10.1242/dev.01148 [DOI] [PubMed] [Google Scholar]
- Suske G., Bruford E., Philipsen S. (2005). Mammalian SP/KLF transcription factors: bring in the family. Genomics 85, 551–556 10.1016/j.ygeno.2005.01.005 [DOI] [PubMed] [Google Scholar]
- Suzuki Y., Miura H., Tanemura A., Kobayashi K., Kondoh G., Sano S., Ozawa K., Inui S., Nakata A., Takagi T. et al. (2002). Targeted disruption of LIG-1 gene results in psoriasiform epidermal hyperplasia. FEBS Lett. 521, 67–71 10.1016/S0014-5793(02)02824-7 [DOI] [PubMed] [Google Scholar]
- Talamillo A., Delgado I., Nakamura T., de-Vega S., Yoshitomi Y., Unda F., Birchmeier W., Yamada Y., Ros M. A. (2010). Role of Epiprofin, a zinc-finger transcription factor, in limb development. Dev. Biol. 337, 363–374 10.1016/j.ydbio.2009.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong A. B., Kretz M., Ridky T. W., Kimmel R., Khavari P. A. (2006). p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes Dev. 20, 3185–3197 10.1101/gad.1463206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt F. M., Jensen K. B. (2009). Epidermal stem cell diversity and quiescence. EMBO Mol. Med. 1, 260–267 10.1002/emmm.200900033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong V. W., Stange D. E., Page M. E., Buczacki S., Wabik A., Itami S., van de Wetering M., Poulsom R., Wright N. A., Trotter M. W. et al. (2012). Lrig1 controls intestinal stem-cell homeostasis by negative regulation of ErbB signalling. Nat. Cell Biol. 14, 401–408 10.1038/ncb2464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W., Morillo S. A., Rebay I. (2013). The Abelson tyrosine kinase regulates Notch endocytosis and signaling to maintain neuronal cell fate in Drosophila photoreceptors. Development 140, 176–184 10.1242/dev.088799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A., Schweitzer R., Sun D., Kaghad M., Walker N., Bronson R. T., Tabin C., Sharpe A., Caput D., Crum C. et al. (1999). p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398, 714–718 10.1038/19539 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.