

Abstract
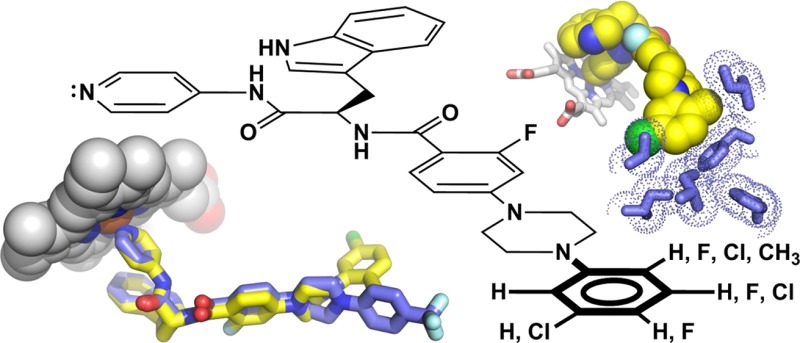
Chagas disease is a chronic infection in humans caused by Trypanosoma cruzi and manifested in progressive cardiomyopathy and/or gastrointestinal dysfunction. Limited therapeutic options to prevent and treat Chagas disease put 8 million people infected with T. cruzi worldwide at risk. CYP51, involved in the biosynthesis of the membrane sterol component in eukaryotes, is a promising drug target in T. cruzi. We report the structure–activity relationships (SAR) of an N-arylpiperazine series of N-indolyloxopyridinyl-4-aminopropanyl-based inhibitors designed to probe the impact of substituents in the terminal N-phenyl ring on binding mode, selectivity and potency. Depending on the substituents at C-4, two distinct ring binding modes, buried and solvent-exposed, have been observed by X-ray structure analysis (resolution of 1.95–2.48 Å). The 5-chloro-substituted analogs 9 and 10 with no substituent at C-4 demonstrated improved selectivity and potency, suppressing ≥99.8% parasitemia in mice when administered orally at 25 mg/kg, b.i.d., for 4 days.
Introduction
Chagas disease, prevalent in Latin America, is caused by chronic infection by Trypanosoma cruzi, a protozoon transmitted in nature by the bite of the Triatominae insect vector. Following an initial stage, which can be either acute or asymptomatic, T. cruzi invades the heart, gastrointestinal tract, or nervous system, where it may persist asymptomatically for years before manifesting in cardiomyopathy, megacolon, and megaesophagus syndromes.1 Nifurtimox and benznidazole are the only two medicines currently available for treatment of Chagas disease. Both drugs are notorious for adverse gastrointestinal and neurological side effects.2 Against the initial acute stage of infection benznidazole is about 80% effective,3 but in the much longer and epidemiologically prevalent chronic stage its efficacy is controversial. While the efficacy and safety of benznidazole in patients with chronic Chagas disease are being investigated in the ongoing clinical trials BENEFIT4,5 and TRAENA,6 significant efforts have been made to repurpose antifungal azole drugs for Chagas disease patients. These efforts led to recent clinical trials of posaconazole7,8 (Noxafil, Merck) and ravuconazole9 (Eisai, Tokyo). Both drugs target sterol 14-demethylase (CYP51), a clinically validated drug target for fungal infections and a promising therapeutic target in T. cruzi.10 However, both drugs’ disappointingly low efficacy against Chagas over long time frames demonstrates the need for further trials, at different doses or in combination with benznidazole.11 Discovery of new anti-Chagasic agents thus remains a high research priority.
While antifungal drugs like posaconazole and ravuconazole are effective against T. cruzi in vitro and in experimental animal models, they were not designed or optimized specifically for the T. cruzi CYP51 target. An alternative structure-based approach has emerged over the past decade that focuses on optimization of diverse new chemical scaffolds specifically targeting T. cruzi CYP51.12 This approach is augmented by monitoring the key pharmacokinetic parameters for anti-Chagas therapy: large volume of distribution and long terminal half-life.13−16 Starting points for these efforts have been identified via phenotypic or target-based screens or by repurposing drug candidates aimed at other diseases.12 Thus, promising lead compound series have emerged from the efforts of DNDi (fenarimol analogs),14−16 University of Washington (tipifarnib analogs),17−21 Vanderbilt (VNI/VNF),22−24 Northeastern (NEU321),25 and finally, the UCSF-Scripps Florida collaboration (N-indolyloxypyridinyl-4-aminopropanyl derivatives),13,26−28 among others (Figure 1). While sharing common structural features, the new compounds studied by these investigators belong to chemically distinct classes such as imidazole, triazole, pyridine, and pyrimidine derivatives.12 This classification is based on the structure of the heme Fe-coordinating aromatic heterocycle present in each inhibitor.
Figure 1.

CYP51 inhibitors aimed at kinetoplastid parasites. Distances between the Fe-coordinating nitrogen atom and ring 2 are expressed in the number of bonds (n) highlighted in blue. The PDB codes of corresponding structures are shown in parentheses. Ligands are labeled by their small-molecule codes. Images were generated using PYMOL.49 Primary citations for the cited structures are as following: 4C0C,134H6O,253GW9,503KSW,513ZG3,52 and 3TIK.53
The N-indolyloxopyridinyl-4-aminopropanyl-based series being developed de novo by our collaborative research team derives from the target-based high throughput screening hit LP10.29,30 Having carried out a detailed review of the features and binding mode of the S-enantiomers of first generation analogs of the initial hit,26 we switched to the R-enantiomer as the basis for design of next generation inhibitors and developed the R-enantiomer series of CYP51 inhibitors that are more than 1000 times more potent against T. cruzi infected cells than the first generation inhibitors.27 In the course of this work, further optimized inhibitors gained increased stability in liver microsome extracts, improved selectivity over human CYPs,27,28 and demonstrated oral bioavailability and potency in an animal model of T. cruzi infection.13 Binding modes of three different scaffold variants, the biaryl,26,27 sulfonamide,28 and N-arylpiperazine13 series, so named based on the structure of the longest substituent at the chiral carbon center, have been characterized by X-ray crystallography. Insertion of the piperazine moiety between the aryl groups of the biaryl scaffold enhanced drug–target interactions of the N-arylpiperazine inhibitors by increasing the number of target-specific contacts outside the heme pocket.13
To maximize target-specific interactions, modifications of the terminal N-aryl ring of the N-arylpiperazine scaffold were systematically explored in this work. As a result, >99.8% inhibition of parasitemia in mice was attained by oral administration of compounds 9 and 10 at 25 mg/kg, b.i.d., for 4 consecutive days. Significantly, for the first time with the N-indolyloxopyridinyl-4-aminopropanyl-based series, a substantial decline in off-target inhibition of the most persistent human CYP isoform, CYP2C9, was achieved (compound 9), while selectivity over other human drug-metabolizing CYPs notably improved. The binding mode of three new compounds was assessed by X-ray structure analysis to a resolution ranging between 1.95 and 2.48 Å, which demonstrated that substituents at C-4 of the terminal N-phenyl ring controlled a switch between the two distinct ring-binding modes, referred to here as buried and solvent-exposed. The most potent inhibitors in this series retained a 5-chloro substituent but, to gain access to the buried site, tolerated at most a small fluoro substituent at C-4. Correlation of inhibitor structure, binding mode, and potency emphasizes the indispensable role for cocrystal structures in the design of parasite-specific interactions into CYP51 inhibitors to advance the discovery of much-needed potent anti-Chagas antibiotics.
Results
Synthesis of New N-Arylpiperazine Compounds
Eleven new N-arylpiperazine analogs (3–13) with different substitution patterns in the terminal N-phenyl ring were synthesized and tested in this work (Table 1). Syntheses of compounds 3–13 are summarized in Scheme 1. Briefly, palladium-mediated coupling of methyl 4-bromo-2-fluorobenzoate (14) with various N-arylpiperazines served as the key step for the syntheses of intermediates 15a–25a. Intermediates 15a–25a were then hydrolyzed under basic conditions to provide carboxylic acid intermediates 15b–25b. These carboxylic acids were coupled with d-tryptophan derivative 26 to provide the final compounds 3–13. The detailed synthetic procedures and spectroscopic data for the final products and synthetic intermediates are available in Experimental Procedures or Supporting Information.
Table 1. Impact of the Terminal N-Phenyl Ring Structure on Biological Activity.
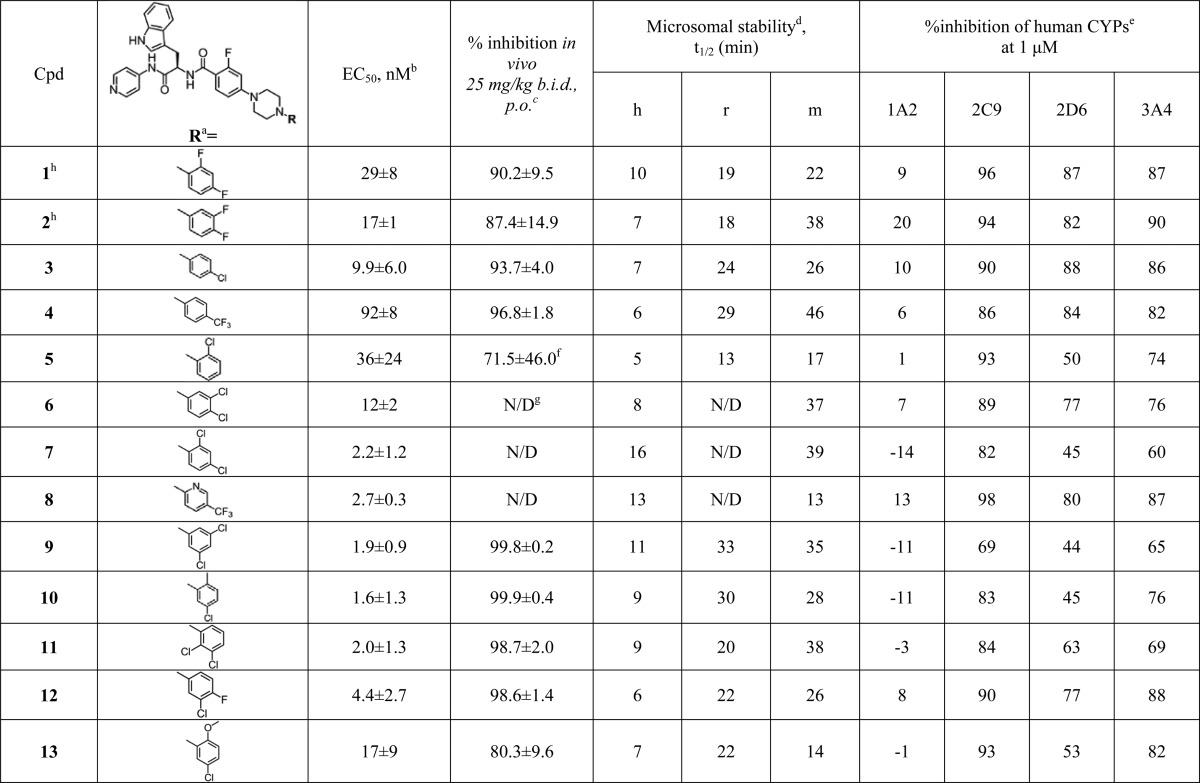
The terminal N-phenyl rings of the compounds are depicted in orientations deduced from the X-ray structure analysis and SAR.
Each measurement performed in triplicate (see Supporting Information).
Each measurement is an average of five mice treated with 25 mg/kg (20% Kolliphor), po, b.i.d., for 4 days.
Stability of compounds in human (h), rat (r), and mouse (m) liver microsomes as evaluated compared to the sunitinib reference.
Inhibition of CYPs as evaluated in human liver microsomes using selective marker substrates for each CYP.
There was one outlier in this group with no effect of the compound (signal comparable to untreated controls, see Figure S3).
N/D: not determined.
Compounds 1 and 2 have been previously characterized.13
Scheme 1. Synthesis of the N-Arylpiperazine Compounds 3–13.

Reagents and conditions: (a) 1-(aryl)piperazine, Pd(OAc)2, P(o-tolyl)3, Cs2CO3, toluene, 50 °C, 48 h, ∼60%; (b) 10% NaOH (aq), MeOH/THF (1/1), 60 °C, 3 h, >98%; (c) 15b, 16b, 17b, 18b, 19b, 20b, 21b, 22b, 23b, 24b, or 25b (as appropriate), PyBOP, HOBt, Et3N, CH2Cl2, 23 °C, 1 h, ∼60%.
Binding to the CYP51 Target
As previously discussed,13,31 because of low sensitivity of the UV–vis assay, true KD values cannot be deduced from UV–vis binding curves of the tight-binding CYP51 inhibitors. Accordingly, the binding curves obtained by the UV–vis titration of the CYP51 target at 1 μM (Figure S1A) reached a plateau at equimolar concentrations (with the exception of compounds 5, 6, 7, and 9), suggesting low nanomolar binding affinity. The binding curves for 5, 6, 7, and 9 did not reach saturation at equimolar concentrations, suggesting a drop in binding affinity or possibly compound solubility issues. The binding behavior correlated well with the EC50 values in the single digit nanomolar range for the majority of compounds (Table 1). Slightly higher EC50 values were recorded for compounds 4 (92 nM) and 5 (36 nM), compared to other compounds presented in this study.
Anti T. cruzi Activity in Cell-Based Assay
All 11 newly synthesized N-arylpiperazine analogs were tested in our standard cell-based T. cruzi assay; the EC50 curve for each compound is provided in Figure S1B. Compounds 3 and 7–12 demonstrated EC50 in the single digit nanomolar range (Table 1), an order of magnitude improvement compared to the previously reported analogs 1 (29 nM) and 2 (17 nM).13 Compounds 6 (12 nM) and 13 (17 nM) retained potency at compound 2 level, while compounds 4 (92 nM) and 5 (36 nM) were notably less potent among those featured in Table 1. Potency in the cell-based assay is consistent with tight binding affinity.
Metabolic Stability and CYP Selectivity
Attenuated metabolic stability and the selectivity of human drug-metabolizing CYPs are two major drawbacks of the previously studied N-arylpiperazine scaffold members that were specifically addressed in this work. In the newly synthesized and tested compound series, compound 9 showed improvement in both microsome stability and CYP selectivity compared to the previously characterized compounds 1 and 2.13 Inhibition of CYP2C9, the most persistent human isoform, dropped to 69% at 1 μM, which favorably compares to the ≥90% inhibition of CYP2C9 previously observed for the majority of N-indolyloxopyridinyl-4-aminopropanyl inhibitors.13,26−28 Half-life in liver microsome preparations also improved, reaching >30 min in both mouse and rat liver microsomes. Stability of these compounds in human liver microsomes remained comparable to that of compound 1.
Pharmacokinetic Properties
Pharmacokinetic (PK) parameters in mice were calculated from plasma concentration versus time curves obtained for two compounds, 9 and 10, orally single-dosed at 25 mg/kg as 5 mg/mL suspensions in 20% Kolliphor (Table 2). Both compounds had good oral exposure and were absorbed over an extended period, resulting in low micromolar drug levels in plasma between 2 and 8 h (Figure S2). Relatively stable submicromolar drug concentrations were maintained up to 12 h. After 24 h, plasma concentrations were still an order of magnitude above EC50. Compound 9 was metabolically more stable than 10, had higher serum concentration 24 h postexposure, and had higher volume of distribution (VD) of 3.97 versus 3.44, respectively (Table 2). The determined values suggest extensive extravascular distribution and penetration into tissues, with the qualification that they may be affected by an error associated with oral absorption; VD calculation assumes 100% absorption and is commonly determined upon iv injection. Consistent with large volume of distribution, high tissue tropism was previously observed in mice for the closely related analogs.13 For comparison, posaconazole VD in humans varies from approximately 5 to 25 L/kg depending on dosage regimen and food.32 Posaconazole absorption is enhanced by food, with 400% increase in bioavailability when administered with a high-fat meal.33
Table 2. PK Parameters for Compounds 9 and 10.
| single
dose PK parametersa |
||||||
|---|---|---|---|---|---|---|
| compd | T1/2 (h) | Tmax (h) | Cmax (μM) | AUClast (μM·h) | VD (L/kg) | Clobs (mL min–1 kg–1) |
| 9 | 3.86 | 4.67 | 1.85 | 22.48 | 3.97 | 30.50 |
| 10 | 2.99 | 3.33 | 1.8 | 19.01 | 3.44 | 35.73 |
Each measurement is an average of three mice that received a single 25 mg/kg dose of test compound at 5 mg/mL suspension in 20% Kolliphor. See Figure S2 for details.
Efficacy in the Animal Model
Selected compounds 3–5 and 9–13 were tested at 25 mg/kg, po, b.i.d., in a 4-day animal model of infection by a transgenic T. cruzi strain expressing firefly luciferase. (Figure 2 and Figure S3). The parental T. cruzi strain was originally designated as T. cruzi Y (TcII),34 but strain identity was later confirmed to be Brazil (TcI).35 This identity clarification is consistent with attenuated infectivity and self-clearance of parasites documented in our earlier work,13 which contrasts with typical behavior of T. cruzi Y strain. In this work we continued to use the same strain, now called T. cruzi Brazil luc, for short-term assessment of bioavailability and potency of test compounds. A 25 mg/kg dose, proven more informative for rank ordering of the N-indolyloxopyridinyl-4-aminopropanyl inhibitors than a 50 mg/kg dose,13 was used in these studies. The ability of most of the newly synthesized compounds to suppress parasite growth in the 4-day animal model was improved relative to previously tested analogs 1 (90.2%) and 2 (87.4%), with the exception of compounds 5 and 13 which showed 71.5% and 80.3% inhibition, respectively (Table 1). In particular, compounds 9–12, inhibiting >98.5% parasitemia, were more potent than compounds 3 (93.7%) and 4 (96.8%). The most efficacious compounds 9 and 10 attained ≥99.8% inhibition and were superior to benznidazole administered at 25 mg/kg (Figure 2 and Figure S3). On the basis of these SAR data, we conclude that the 5-chloro substituent on the terminal phenyl ring, combined with unmodified C-4 or C-4 substituted with a small fluorine atom, was a prerequisite for the highest anti-T. cruzi activity.
Figure 2.

Anti T. cruzi efficacy of compounds in 4-day mouse model of infection. In two independent experiments (A, B), compounds were administered at 25 mg/kg, po, b.i.d. Each data point is an average of five mice (see Figure S3 for original animal images). Benznidazole (BNZ) at 25 and 50 mg/kg served as a positive control. Percent inhibition for each compound is calculated relative to the vehicle-treated control on day 7 postinfection. Percent values are significantly different than vehicle-treated control (p ≤ 0.05) except for compound 5 where one mice in the group failed treatment (Figure S3).
Inhibition of Sterol Biosynthesis in T. cruzi Amastigotes
To confirm the mechanism of action, the two most potent compounds, 9 and 10, have been tested for disruption of the sterol biosynthesis in intracellular T. cruzi amastigotes. The assay was conducted as described elsewhere.13,31,36 Lipids extracted from intracellular amastigotes were analyzed by gas chromatography and mass spectrometry (GC–MS) and identified as described previously.37 DMSO (vehicle), K777, and benznidazole were used as negative controls; posaconazole served as positive control. The major sterol observed in untreated amastigotes was episterol (Figure 3, peak d), followed by approximately equal amounts of fecosterol (e) and cholesta-7,24-dien-3β-ol (peak a). As a result of treatment, two 14-methylated precursors, lanosterol (f) and eburicol (h), dominated the GC–MS traces of the CYP51 inhibitors posaconazole and compounds 9 and 10, with concomitant decline in episterol (d) and virtually disappearance of fecosterol (e) and other 14-demethylated intermediates. Cholesterol was the only peak originating from the host cells. No changes in lipid composition have been observed in K777- and benznidazole-treated samples. Both control drugs have different mechanism of action. K777 (also known as K11777) is a potent T. cruzi inhibitor targeting protease cruzain,38 while benznidazole acts nonspecifically via production of toxic metabolites.39 On the basis of this experiment, we conclude that in the course of hit-to-lead optimization compounds of the N-indolyloxopyridinyl-4-aminopropanyl-based SAR series retained the mechanism of action of the parental hit LP1037 and specifically target T. cruzi CYP51.
Figure 3.

Sterol profile of T. cruzi amastigotes infecting mouse myoblasts analyzed by GC–MS. C2C12 uninfected mouse myoblasts display a large peak corresponding to cholesterol (Chol). This is the only peak originating from the host cells. T. cruzi infected cultures also show parasite-specific lipids corresponding to chromatographic peaks labeled as the following: (a) cholesta-7,24-dien-3β-ol, [M]•+ = m/z 454, tR = 12.51–12.74; (b) cholesta-8,24-diene-3β-ol (zymosterol), m/z = 470, tR = 12.81–12.86 min; (c) 24-methyl-7-en-cholesta-en-3β-ol, m/z = 472, tR = 12.91–12.93 min; (d) ergosta-7,24-diene-3-β-ol (episterol), m/z = 470, tR = 13.2–13.5 min; (e) ergosta-8,24-diene-3-β-ol (fecosterol), m/z = 470, tR = 13.5–13.7 min; (f) lanosterol, m/z 498, tR = 13.95–14.06 min; (g) 4-methylepisterol, m/z = 484, tR = 14.55–14.75 min; (h) eburicol, m/z = 512, tR = 14.8–14.86 min; (i) 24-ethyl-7,24(24′)-en-cholestadiene-3β-ol, m/z = 484, tR = 15.14–15.30 min. Treatment of cultures was performed as indicated in each panel. DMSO (vehicle), benznidazole, and K777 were used as negative controls. Posaconazole, a potent CYP51 inhibitor, was used as positive control. Treatment of cultures with compounds 9 and 10 resulted in accumulation of lanosterol (f) and eburicol (h) and decline of episterol (d), fecosterol (e), and other 14-demethylated intermediates (a, b), indicating the inhibition of CYP51.
Buried and Exposed Binding Modes Determined by X-ray Crystallography
Three new drug–target complexes for compounds 3, 4, and 10 have been structurally characterized to resolution of 2.04, 1.95, and 2.48 Å, respectively (Table 3). Together with the cocrystal structures for compounds 1 and 2 reported previously,13 the compound 10 cocrystal structure reported here details at the atomic level the interactions of the substituted terminal N-phenyl ring with the amino acid landscape of the buried cavity at the end of the hydrophobic tunnel spanning the β-domain of CYP51 (Figure 4A). Remarkably, substituents at C-4 larger than fluorine could not be accommodated in this buried cavity. Thus, the 4-chloro- and 4-trifluoromethyl-substituted terminal rings of 3 and 4 respectively were expelled from the protein interior into the solvent-exposed entrance of the tunnel separating the α- and β-domains (Figure 4B). The binding mode switch was enabled by the flexibility of the piperazine ring, which in 3 and 4 restored the coplanar, sp2-hybridized arrangement with the nitrogen-linked terminal N-phenyl ring, thereby alleviating the conformational distortion of this unit imposed in the bound poses of compounds 1, 2, and now 10.13 The exposure to the bulk solvent in 3 and 4 is reminiscent of that in posaconazole40 (Figure 5), with the qualification that the N-arylpiperazine moiety of 3 and 4 is less accessible to the bulk solvent and makes fewer contacts with the binding tunnel mouth than the posaconazole terminal unit.40
Table 3. Data Collection and Refinement Statisticsa.
| protein |
|||
|---|---|---|---|
| CYP51 | CYP51 | CYP51 | |
| PDB code | 4C28 | 4C27 | 4UVR |
| compd | 3 | 4 | 10 |
| small molecule indentification | TW5 | 26N | J5Y |
| Data Collection | |||
| space group | P212121 | P212121 | P6322 |
| cell dimensions | |||
| a, b, c (Å) | 72.95, 79.00, 176.84 | 73.1, 79.16, 176.95 | 128.12, 128.12, 117.78 |
| α, β, γ (deg) | 90, 90, 90 | 90, 90, 90 | 90, 90, 120 |
| molecules in au | 2 | 2 | 1 |
| wavelength (Å) | 1.115 87 | 1.115 87 | 1.115 87 |
| resolution (Å) | 2.04 | 1.95 | 2.48 |
| Rsym or Rmerge (%) | 6.9 (53.5) | 16.9 (146) | 21.6 (179.5) |
| I/σI | 9.9 (1.6) | 8.3 (1.5) | 11.5 (1.8) |
| completeness (%) | 86.4 (47.8) | 99.5 (96.4) | 100 (100) |
| redundancy | 3.5 (2.0) | 7.4 (6.7) | 15.2 (15.6) |
| Crystallization Conditions | |||
| 0.4 M ammonium acetate | 0.4 M ammonium acetate | 0.25 M ammonium sulfate | |
| 0.1 M sodium acetate, pH 4.5 | 0.1 M sodium acetate, pH 4.5 | 0.1 M sodium acetate, pH 5.6 | |
| 28% PEG 3350 | 28% PEG 3350 | 25% PEG 3350 | |
| 5% Jeffamine M-600, pH 7.0 | 2% 2,5-hexanediol | ||
| Refinement | |||
| no. reflections | 54 178 | 71 040 | 19 783 |
| Rwork/Rfree (%) | 18.6/23.7 | 18.3/23.3 | 19.2/25.3 |
| no. atoms | |||
| protein | 7063 | 7161 | 3397 |
| heme | 86 | 86 | 43 |
| inhibitor | 172 | 138 | 44 |
| solvent | 410 | 427 | 33 |
| mean B value | 25.02 | 28.178 | 41.1 |
| B-factors | |||
| protein | 25.2 | 28.3 | 42.4 |
| heme | 18.1 | 21.6 | 29.0 |
| inhibitor | 18.3 | 21.5 | 36.5 |
| solvent | 26.3 | 31.6 | 32.1 |
| rms deviation | |||
| bond length (Å) | 0.017 | 0.019 | 0.014 |
| bond angle (deg) | 1.911 | 1.979 | 1.687 |
Values in parentheses are for highest-resolution.
Figure 4.
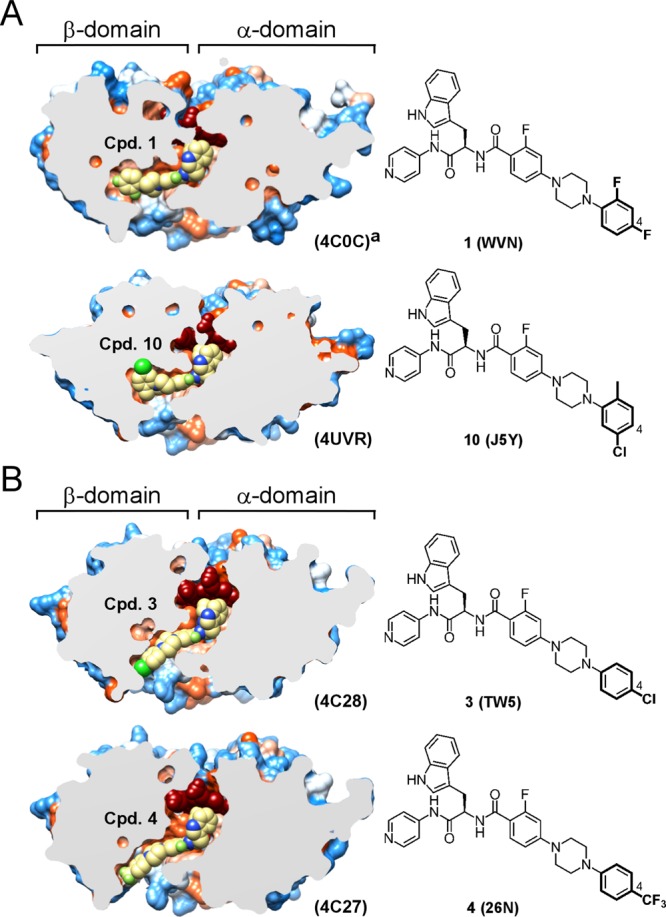
Terminal phenyl ring structures and binding modes: (A) buried binding mode; (B) solvent-exposed binding mode. Slices through the binding site show bound inhibitors (yellow spheres) and the protein surface colored by hydrophobicity; hydrophobic areas are in orange, and hydrophilic areas are in blue. Heme is in dark-red spheres. C-4 position in the terminal phenyl ring is labeled by a number. PDB codes for protein structures and small-molecule ligands are shown in parentheses. Images were generated using CHIMERA.54 Cocrystal structure with compound 1 was previously published.13
Figure 5.
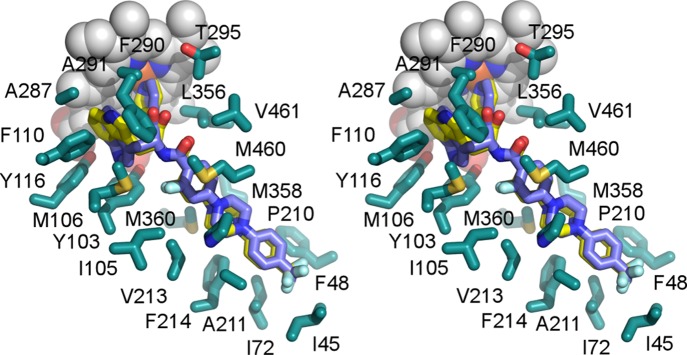
Stereogram of the compound 4 binding site. Two alternative binding poses of 4 (PDB code 4C27) are depicted in yellow and blue, emphasizing flipping of the indole moiety in the vicinity of the heme macrocycle. The top face of the 4-trifluoromethyl substituted terminal unit facing the viewer is solvent-exposed. Amino acid side chains (cyan) are shown within 5 Å. Heme is in gray spheres.
Collectively, the 1, 2, and 10 cocrystal structures demonstrated that in the buried mode fluorine and methyl at C-2, fluorine at C-3 and C-4, and chlorine at C-5 positions of the terminal N-phenyl ring are tolerated (Figure 6A). The bulky 5-chloro substituent established multiple van der Waals contacts with V77, M358, M360, I379, and A381 (Figure 6B), enhancing target-specific interactions, consistent with gain of biological activity for compounds 9–12 carrying the 5-chloro substituent (Table 1). The less extensive range of van der Waals interactions of the 2-methyl substituent in compound 10 was limited to I45 and F48 and was established at the cost of repositioning the F48 side chain from its arrangement in analogs 1 and 2 (Figure 6A). No other major differences have been observed between the cocrystal structures of 1, 2, and 10. The increased size of the 2-oxymethyl substituent in the terminal aryl ring of compound 13 is consistent with a notable drop of the in vivo activity (Table 1), likely due to interference with I45.
Figure 6.
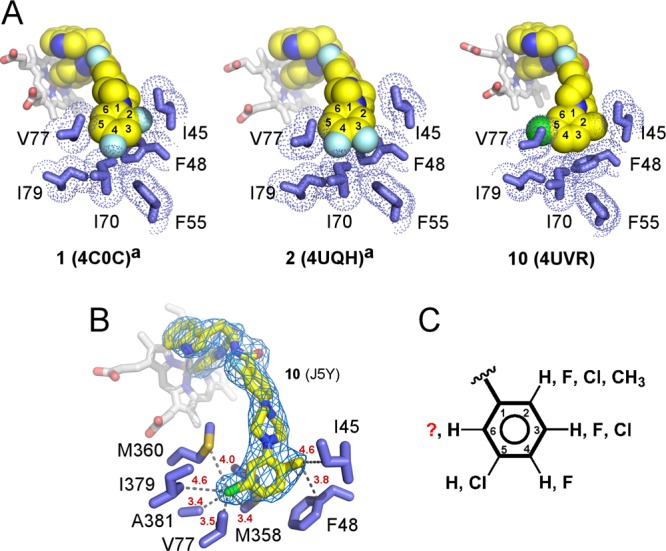
Buried binding mode. (A) van der Waals interactions between the terminal N-phenyl rings of 1, 2, and 10 (yellow spheres) and amino acid side chains (blue sticks). PDB codes of the corresponding structures are shown in parentheses. (B) The fragment of the 2fo – fc electron density map (blue mesh) contoured at 1σ demonstrates unambiguous orientation of the terminal ring placing the 5-chloro substituent at van der Waals distances of V77, M358, M360, I379, and A381. Heteroatoms are colored by types: oxygen in red, nitrogen in blue, fluorine in cyan, chlorine in green. Heme is shown in gray sticks. Distances in red are in angstroms. (C) Collective substitution pattern of the terminal N-phenyl ring deduced from the X-ray structure analysis and SAR. Superscript “a” indicates that cocrystal structures with compounds 1 and 2 were previously published.13
Terminal N-Phenyl Ring Substitution Pattern
With no ambiguity in terminal ring orientation in any of the reported X-ray structures, we assume that compounds 6–8, all carrying bulky substituents at C-4, bind in solvent-exposed mode, while compounds 9, 11, and 12 are likely to bind in buried mode. On the basis of these assumptions, we speculate that large substituents, such as Cl or CH3, can be tolerated at C-2, C-3, and C-5 but not at C-4 (Figure 6C). Hypothetically, if the 5-chloro substituent of compound 11 makes the same contacts as its counterpart in 10, a bulky chloro substituent should be tolerated at C-6. Otherwise, the terminal N-phenyl ring of 11 may be flipped in the buried cavity compared to the ring orientation observed in 10, placing its two chloro-substituents at C-2 and C-3 positions. We have not obtained a cocrystal structure of compound 11 to resolve this potential ambiguity and will rely on molecular docking-driven hit-to-lead optimization to find out if further ring chlorination will increase potency of the N-arylpiperazine series, as may be assumed based on the current studies.
Induced Fit Binding Mechanism
Differences in the protein conformation between buried and solvent-exposed binding modes were observed, which are likely due to the induced fit mechanism associated with inhibitor binding. Collectively, data accumulated by our research team on the N-indolyloxopyridinyl-4-aminopropanyl-based inhibitors13,28 challenge speculations in the literature about CYP51’s “rigidity”.41,42 As suggested by X-ray structure analysis, the conformational dynamics of the CYP51 backbone are rather typical for the P450 protein family; particularly affected are the A′-helix in the β-domain and the F- and G-helices and the spanning loop accommodating the short F′- and F″-helices in the α-domain (Figure 7). The F″-helix experienced ∼35° rotation angle difference between the buried and surface-exposed terminal N-phenyl ring binding modes, resulting in restructuring of the FG-loop and ∼10 Å repositioning of the P216-R228 fragment.
Figure 7.
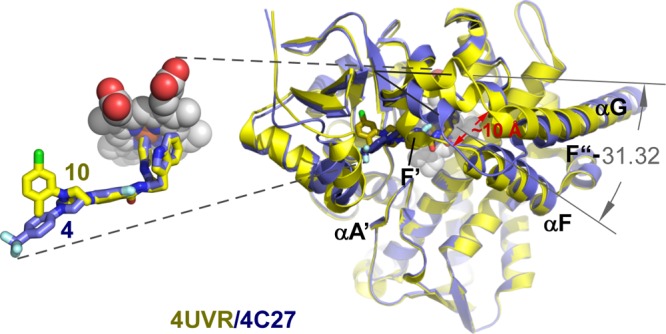
Solvent-exposed binding mode. (A) Superimposition of the buried (yellow) and solvent-exposed (blue) binding mode structures highlights protein conformational changes induced by the compound 10, compared to the compound 4 binding. Protein is represented by the ribbon. Heme is in gray spheres. Distance in red is in angstrom. The angle between F″ helices in superimposed structures is in degrees. An insert on the left zooms in on the superimposed ligands (stick mode) emphasizing differences between buried and solvent-exposed conformations of the terminal N-phenyl ring.
Furthermore, conformational changes triggered by binding of the terminal ring in the buried site affected the binding mode of the invariant N-indolyloxopyridinyl-4-aminopropanyl portion of the skeleton. Thus, in the 1, 2, and now 10 complexes, the indole ring adopted a single well-defined conformation, tightly enclosed by the electron-rich residues Y103, M106, F110, and Y116.13 In the 3 and 4 complexes, binding ambiguity was manifested in flipping of the indole ring (Figure 5). Repositioning of the bulky aromatic side chains of Y103, F110, and Y116, in response to the long substituent binding, enlarged the size of the chamber adjacent to heme, allowing the indole ring to flip. The same phenomenon has been reported previously for the sulfonamide analog.28
Discussion
Development of the N-indolyloxopyridinyl-4-aminopropanyl-based series of CYP51 inhibitors was previously reported, emphasizing the medicinal chemistry, drug metabolism, PK, and CYP-binding properties of these compounds.13,26−28 Rounds of molecular modeling and inhibitor synthesis to improve stability, selectivity, and potency ultimately led to enhanced binding to the T. cruzi CYP51 target, enabling high-resolution crystal structures to be obtained for the drug–target complexes. One of the three newly reported X-ray structures pushed the resolution limit for this protein drug target beyond 2.0 Å. Collectively, the cocrystal structures determined for the N-arylpiperazine SAR series revealed a correlation between the modification pattern of the terminal N-phenyl ring, binding mode, and anti-T. cruzi activity of the compounds. The following discussion is informed by the drug–target interactions observed in our studies and by others.
The major structural elements defining inhibitor interactions with the CYP51 drug target include (i) coordination to the heme iron by the aromatic nitrogen atom of ring 1, (ii) both nonspecific (with respect to the heme macrocycle) and target-specific interactions of ring 2, and (iii) target-specific contacts mediated by a third substituent at the molecular branching point, often a carbon chiral center or tertiary amine, as in fenarimol analogs (Figure 1). Both five-membered and six-membered heterocycles are employed in CYP51 inhibitors as iron-coordinating units.12 They provide nonspecific drug–metal interactions which are modulated by repulsive or attractive forces contributed by second aromatic functionality, ring 2, or its fused equivalent in NEE and VNF. The geometry of ring 2 interactions with the heme macrocycle varies significantly depending on the degree of freedom allowed by the inhibitor structure which is roughly proportional to the distance between the Fe-ligating aromatic nitrogen in ring 1 and ring 2 functionality, expressed as number of bonds (n) separating them: the shorter is the distance, the more coplanar is the interaction between ring 2 and the heme macrocycle. Thus, an energetically unfavorable sandwich geometry between two aromatic systems is observed in UDD (Figure 1E) and JKF (Figure 1F), n = 4, while in VNI, ring 2, having one extra bond separating two functionalities, tilts with respect to the heme tending to adopt a more attractive edge-on geometry (Figure 1C). Disposition of ring 2 and the heme macrocycle is also affected by the rigidity of the connector. Thus, in the more rigid structures of NEE (Figure 1B) and VNF (Figure 1D) (n = 8 and n = 7, respectively), the ring 2 fused equivalent tends to offset the heme macrocycle to establish energetically more favorable contacts. Finally, WVN (Figure 1A), n = 8, has the most favorable T-shape stacking configuration, which is consistent with about 2 orders of magnitude gain in binding affinity, achieved in the parental scaffold by replacing the ethyl moiety with tryptophan at the early stage of scaffold development.29,37
The N-indoyloxypyridinyl-4-aminopropanyl-based scaffold is one of two classes of CYP51 inhibitors that utilize six-membered heterocycles to coordinate to the heme iron, the other being the fenarimol-based scaffold.14−16 These two compound series have significant differences in heme binding, which have been previously reviewed.12,13 The energetically more favorable edge-to-face π–π stacking interaction between the heme macrocycle and the indole ring of the N-indolyloxopyridinyl-4-aminopropanyl-based inhibitors (Figure 1A) contrasts with the virtually coplanar aromatic systems in fenarimol analogs (Figure 1E). These differences may explain, at least in part, differences in selectivity profiles of these inhibitor classes over a panel of human drug-metabolizing CYPs. Strong but nonspecific, the drug–heme interactions may contribute to the attenuated selectivity of the N-indoyloxypyridinyl-4-aminopropanyl-based compounds over a panel of human CYPs.13
In general, all three major aspects of drug–target interactions in CYP51 inhibitors are subject to optimization. In this work, we focused on the drug–target contacts mediated by a third substituent at the molecular branching point of the N-indoyloxypyridinyl-4-aminopropanyl-based scaffold while retaining favorable T-shape geometry of the indole moiety contact with heme macrocycle. This third and longest substituent at the chiral carbon center confers the majority of target-specific interactions and significantly contributes to binding affinity.13 To gain binding specificity, a series of new analogs focusing on the substitution pattern of the terminal N-phenyl ring was synthesized and tested. The terminal N-phenyl ring was shown previously to bind distal to the heme buried hydrophobic cavity, the least conserved portion of the substrate binding site across the CYP51 family.13 Depending on the ring substitution pattern, an alternative ring binding mode was observed in these studies. In compounds 3 and 4, carrying a large chloro- or trifluoromethyl substituent at C-4, respectively, the terminal ring was expelled from a buried into a solvent-exposed environment, while in the analogs 1, 2, and 10, with no substituent or a small substituent at C-4, the ring was accommodated in a buried site (Figure 4).
On the basis of the X-ray structure analysis and accumulated SAR data, an assumption was made that compounds 9 and 11–12 may also bind fully buried in the protein interior, which would be consistent with their high potency both in cell-based assay and in the animal model (Table 1). Two of the most optimized compounds belonging to this group, 9 and 10, suppressed ≥99.8% parasitemia when administered at 25 mg/kg, b.i.d., orally for 4 consecutive days. The observed trend of potency and selectivity expanding upon increasing the size of substituents in certain ring positions now allows fine-tuning drug–target interactions to maximize selectivity and potency of this class of T. cruzi inhibitors (Figure 5). For instance, the accumulated data did not resolve a potential ambiguity which may be caused by the compound 11 terminal N-phenyl ring flipping within the buried site, suggesting that further ring chlorination may be beneficial for the drug–target fit and hence potency and specificity of this compound series. The atomic details of drug–target interactions attained in this work demonstrate the utility of cocrystal structures of the multiple closely related representatives of the SAR series for refining hit-to-lead optimization strategy.
Finally, binding in the buried mode of the terminal N-phenyl unit in compounds 1, 2, and 10 allosterically stabilized in a single conformation the invariable portion of the inhibitor skeleton, the indolyl moiety, bound in vicinity of the heme macrocycle. In contrast, binding of the terminal N-phenyl ring of compounds 3 and 4 in the solvent-exposed mode was accompanied by concomitant flipping of the indolyl moiety, creating ambiguity illustrated by two alternative conformations of 4 in Figure 5. On the basis of these observations, we speculate that the allosteric component may be a key to induced fit mechanism characterizing binding of the most potent CYP51 inhibitors. This represents an additional challenge to CYP51-targeted drug design, as modification introduced at one end of a molecule may modulate the binding mode at the other end.
Conclusions
The N-indoyloxypyridinyl-4-aminopropanyl-based inhibitors represent a promising anti T. cruzi lead series, where potency, selectivity, and metabolic stability are accreted through medicinal chemistry efforts guided by the cocrystal structures of the drug–target complexes. The SAR studies reported here focused on the terminal N-phenyl ring of the N-arylpiperazine series. A key role for the C-4 ring position in switching between two alternative ring binding modes, buried and solvent-exposed, has been recognized. Buried binding mode is characterized by the terminal N-phenyl ring buried in the protein interior. Binding of the terminal unit in the buried mode induces concomitant stabilization of the binding of the inhibitor indolyl moiety in the heme pocket. Inhibitors with the bulky 5-chloro substituent bound in the buried mode were more potent in the T. cruzi cell-based assay, exhibited higher metabolic stability, and had better selectivity over human CYPs. Particularly notable was the decline in inhibition of the most persistent human isoform, CYP2C9, achieved by compound 9. Compounds 9 and 10 also had excellent oral bioavailability and exposure and higher efficacy in the animal model compared to the previously reported closely related analogs 1 and 2. Further, an attractive feature of the buried binding mode is its reduced propensity for drug resistance, as the inhibitor binds within a portion of the substrate envelope that is less prone to mutagenesis as a result of prolonged drug exposure. The knowledge gained in these studies opens the door to SAR fine-tuning and rigorous assessment of compounds of the N-indoyloxypyridinyl-4-aminopropanyl-based scaffold for parasitological cure in long-term animal models harboring more virulent T. cruzi strains.
Experimental Procedures
Chemistry. General Procedure for the Synthesis of Inhibitors 1–13
To a solution of the appropriate benzoic acid (∼1.2 equiv), PyBOP (∼1.4 equiv), and HOBt (∼10 mol %) in dry CH2Cl2 (5 mL) was slowly added triethylamine (∼4 equiv) at ambient temperature. The reaction mixture was stirred for 15 min until it became homogeneous. d-Tryptophan derivative 26 was added, and the reaction mixture was stirred at room temperature for 1 h. After confirmation that the reaction was complete by using TLC analysis, the solvent was removed under reduced pressure. Ethyl acetate (10 mL) was added to the crude product mixture, and this solution was then washed with saturated aqueous NaHCO3 (2 mL × 2) and brine (2 mL × 2). The organic layer was concentrated in vacuo, and the crude product was directly subjected to flash chromatographic purification to provide the title products.
(R)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-4-(4-(3,5-dichlorophenyl)piperazin-1-yl)-2-fluorobenzamide TFA (9)
The general procedure was followed using carboxylic acid 21b as the acylating agent. The crude product was further purified by HPLC to afford 9 as a white solid (54%): 1H NMR (400 MHz, DMSO-d6) δ 11.47 (s, 1H), 10.93 (d, J = 2.5 Hz, 1H), 8.75–8.62 (m, 2H), 8.10–7.90 (m, 3H), 7.60 (dd, J = 8.5, 6.7 Hz, 2H), 7.33 (d, J = 8.1 Hz, 1H), 7.26 (d, J = 2.4 Hz, 1H), 7.05 (ddd, J = 8.1, 7.0, 1.2 Hz, 1H), 7.00 (d, J = 1.8 Hz, 2H), 6.94 (ddd, J = 8.0, 6.9, 1.0 Hz, 1H), 6.89 (t, J = 1.7 Hz, 1H), 6.87–6.76 (m, 2H), 4.86 (q, J = 6.7 Hz, 1H), 3.57–3.18 (m, 10H); 13C NMR (176 MHz, DMSO-d6) δ 172.98, 163.26, 162.19, 160.79, 158.06, 157.88, 154.06, 153.99, 152.28, 151.53, 143.88, 136.11, 134.67, 131.67, 131.65, 127.06, 124.10, 121.06, 118.33, 117.79, 117.21, 116.09, 114.36, 113.09, 111.42, 109.97, 109.90, 109.74, 108.89, 100.74, 100.58, 79.16, 78.97, 78.78, 55.47, 46.60, 46.18, 27.06; MS (ESI) m/z 631.2 [M + H]+; HRMS (ESI) m/z for C33H31N6O2FCl2 [M + H]+ calcd 631.1791, found 631.1782.
(R)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-4-(4-(5-chloro-2-methylphenyl)piperazin-1-yl)-2-fluorobenzamide TFA (10)
The general procedure was followed using carboxylic acid 22b as the acylating agent. The crude product was further purified by HPLC to afford 10 as a white solid (60%): 1H NMR (400 MHz, DMSO-d6) δ 11.55 (s, 1H), 10.94 (d, J = 2.5 Hz, 1H), 8.77–8.59 (m, 2H), 8.06–8.02 (m, 2H), 7.99 (t, J = 6.8 Hz, 1H), 7.61 (dt, J = 9.1, 5.1 Hz, 2H), 7.33 (d, J = 8.1 Hz, 1H), 7.27 (d, J = 2.4 Hz, 1H), 7.25–7.17 (m, 1H), 7.09–7.01 (m, 3H), 6.98–6.90 (m, 1H), 6.90–6.78 (m, 2H), 4.87 (q, J = 6.7 Hz, 1H), 3.43 (dd, J = 6.5, 3.5 Hz, 4H), 3.39–3.18 (m, 2H), 2.97 (t, J = 4.9 Hz, 4H), 2.26 (s, 3H); 13C NMR (176 MHz, DMSO-d6) δ 173.04, 163.30, 162.17, 160.76, 158.09, 157.90, 154.47, 154.40, 152.26, 151.80, 143.61, 136.11, 132.29, 131.64, 131.61, 130.76, 130.66, 127.06, 124.12, 122.77, 121.07, 119.07, 118.33, 114.41, 111.43, 110.07, 109.99, 109.85, 108.89, 100.86, 100.70, 55.50, 50.68, 47.17, 27.03, 17.20; MS (ESI) m/z 611.2 [M + H]+; HRMS (ESI) m/z for C34H34N6O2FCl [M + H]+ calcd 611.2338, found 611.2336.
Hepatic Microsomal Stability
Hepatic microsomal stability was addressed as previously described.26
CYP Inhibition
Cytochrome P450 inhibition was evaluated in human liver microsomes using four selective marker substrates for CYP1A2, CYP2C9, CYP2D6, and CYP3A4, as previously described.26 Compounds were tested at 1 μM.
Binding Affinity by UV–Visible Spectroscopy
Binding affinity of compounds to CYP51 was approximated from the spectrophotometry titration curves as previously described.28,31 Spectra were recorded using a Cary scanning spectrophotometer (Varian) in a 1 cm path length quartz cuvette at 23 °C. Protein at 1 μM concentration in 1 mL of 100 mM potassium phosphate buffer (pH 7.5) containing 10% glycerol was titrated with 1 μL aliquots of test compound (100 μM in DMSO) ranging from 0.1 to 4 μM. The organic solvent effect was compensated by adding the same volume of DMSO in the reference cuvette containing protein.
Anti T. cruzi Activity in Cell-Based Assay
EC50 values of compounds were determined in a cell-based assay performed in triplicate as described elsewhere.31 Briefly, mouse C2C12 myoblasts (ATCC no. CRL-1772) used to harbor parasites were cultivated in Dulbecco’s modified Eagle medium H-21 containing 4.5 g/L glucose (DMEM H-21), supplemented with 5% fetal bovine serum (FBS), 25 mM HEPES, 2 mM l-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. T. cruzi CA-I/72 trypomastigotes were obtained from infected-culture supernatants after 4–7 days of infection. Cultures were maintained at 37 °C with 5% CO2. The concentrations of trypomastigotes and C2C12 cells were determined using a Neubauer hemocytometer. Sterile, black 384-well plates with clear-bottom wells (Greiner Bio-One) were seeded with 500 cells/well and then were infected with 2500 parasites/well in a final volume of 50 μL/well. Culture plates were incubated at 37 °C with 5% CO2 for 24 h. After that, culture medium was removed and test compounds were added in fresh medium. For this, an intermediate plate (384-well plate) was prepared by serial dilution (10 mM, 2 mM, 400 μM, 80 μM, 16 μM, 3 μM, 128 nM, 25.6 nM, 5.1 nM) for all the compounds in 100% DMSO. Then an amount of 50 nL of each sample was diluted in 50 μL of medium (DMEM H-21) and added to the experimental plate followed by incubation at 37 °C with 5% CO2 for 72 h. Wells containing noninfected cells were used as a positive control (100% cell survival), while T. cruzi infected but untreated cells (0% cell survival) were used as a negative control. Cells were then fixed for 2 h with 4% paraformaldehyde and rinsed with a solution of 150 mM NaCl, 100 mM NH4Cl, 0.1% Triton X-100, and 0.1% NaN3. After that, they were treated for 4 h with 0.2 μg/mL the DNA fluorescent dye, DAPI (4,6-diamidino-2-phenylindole), diluted in the same solution. Plates were kept at ambient temperature until image acquisition was performed. Images were acquired by an IN Cell Analyzer 2000 (GE Healthcare), and the procedure and analyses were performed according to previously described.31
Inhibition of Sterol Biosynthesis in Amastigotes
Sterol profiling was performed on T. cruzi whole-cell lipid extracts, prepared as described previously.51 Posaconazole (100 nM) was used as a positive control, while benznidazole (5 μM) and K777 (1.6 μM) served as negative controls. Compounds 9 and 10 were tested at 100 nM. Briefly, C2C12 mouse myoblasts were infected with T. cruzi (CAI/72 strain) and treated with compounds after 72 h of infection. After 24 h of treatment, the myoblast cells were detached, and the lipids from cell pellet were extracted with chloroform/methanol, chloroform, and acetonitrile, each step followed by several rounds of washes with water to extract polar molecules. The organic layer was then dried under nitrogen gas and subsequently treated with 75 μL of N,N-bis(trimethylsilyl)-2,2,2-trifluoroacetamide (BSTFA) for 2 h at 37 °C to facilitate chemical derivatization with trimethylsilyl (TMS) groups (BSTFA, Sigma-Aldrich). The TMS-derivatized lipid mixture was analyzed by injecting 3 μL directly into an Agilent HP5790 gas chromatography system outfitted with a DB5-MS analytical column (30 m, 0.25 mm i.d., 0.33 μm film thickness, Agilent) coupled to a mass selective detector. The lipids were separated on the analytical column using a temperature profile that begins at 200 °C for 1 min, increases by 15 °C/min to 300 °C, and then holds at 300 °C for 20 min. The inlet temperatures of the GC instrument and the MSD were held at 250 and 300 °C, respectively. The mass spectrometer scanned from m/z 50–750 during the course of analysis.
Animal Model
To assess in vivo efficacy of test compounds, a 4-day mouse model of infection by transgenic T.cruzi Brazil luc strain, former T. cruzi Y luc,34,35 expressing firefly luciferase was used as previously described.13 Eight-week-old female Swiss Webster albino mice (average weight 20 g) were obtained from Simonsen Labs (Gilroy, CA). All animal protocols were approved and carried out in accordance with the guidelines established by the Institutional Animal Care and Use Committee from UCSF (approval no. AN087605-01). Mice were housed at a maximum of 5 per cage and kept in a specific-pathogen free (SPF) room at 20–24 °C under a 12 h light/12 h dark cycle and provided with sterilized water and chow ad libitum. To infect the mice, trypomastigote strains were harvested from culture supernatant and injected intraperitonealy, 105 trypomastigotes per mouse. Three control groups included untreated mice, which received a vehicle, 20% Kolliphor HS 15 (also known as Solutol), and the positive control groups, which received 25 or 50 mg/kg benznidazole, all via oral gavage (po), twice a day (b.i.d). Starting on day 4 the infected mice were treated with test compounds at 25 mg/kg administered in 20% Kolliphor, po, b.i.d., for 4 consecutive days. On day 7 postinfection, the luminescent signal in the mice was read upon injection of d-luciferin. The absolute numbers of (measured photons/s)/cm2 were averaged between all five mice in each group and compared directly between compound-treated mice and the control groups. Two-tailed paired Student t test was used to assess statistical significance between luminescence values from vehicle-treated and compound-treated groups on day 7 postinfection; values are statistically significant when p ≤ 0.05.
Single Dose PK
Compounds were dose at 25 mg/kg in 8-week-old female Swiss Webster albino mice over oral gavage. All compounds were formulated to a concentration of 5 mg/mL in 20% Kolliphor/80% water. Three mice were dosed with each compound, and n = 3 plasma samples were collected at approximately 0.25, 0.5, 1, 2, 4, 6, 8, 12, and 24 h after exposure. Plasma samples were treated with 5× v/v acetonitrile to precipitate protein and filtered through a 0.2 μm filter prior to analysis by LC–MS/MS using ABSciex 5500.
CYP51 Expression and Purification
Heterologous expression and purification of recombinant T. cruzi CYP51 modified by replacing the first 31 residues upstream of Pro32 with the fragment MAKKTSSKGKL43 and by inserting a His6-tag at the C-terminus were carried out according to the protocol described elsewhere.31,40 Briefly, 6 L of Terrific Broth medium supplemented with 100 μg/mL ampicillin, 1 mM thiamine, and trace elements was inoculated with 60 mL of the overnight culture and was incubated at 28 °C, 230 rpm until OD600nm reached 1.0. CYP51 expression was induced by adding 0.25 mM isopropyl-β-thiogalactopyranoside (IPTG), and 1 mM δ-aminolevulinic acid, a precursor in heme biosynthesis, was added at that time. After induction, the growth was continued at 18 °C at 180 rpm for 48 h. Cells were harvested, resuspended in 50 mM Tris, pH 8.5, 1 mM EDTA, 100 mM NaCl, 0.5 mM PMSF, 1 mM DTT and lysed using a microfluidizer. After centrifugation, the soluble fraction was purified by conventional Ni-NTA agarose chromatography using a linear gradient of imidazole (0–0.5 M) in 50 mM potassium phosphate, pH 8.0, 10% glycerol, 1 mM DTT, 0.5 mM EDTA, 500 mM NaCl. After dialysis overnight against 20 mM potassium phosphate, pH 7.5, 10% glycerol, 1 mM DTT, 0.5 mM EDTA, the sample was applied on MonoQ column. The flow-through fractions were applied on Mono S column, and the protein was eluted in the same buffer using linear NaCl gradient (0–0.5 M). Fractions containing CYP51 were combined and concentrated using Centriprep-50K concentrating device (Millipore). These samples were stored at −80 °C and used as needed for cocrystallization and binding assays.
X-ray Crystallography
To analyze the inhibitor binding mode, recombinant T. cruzi CYP51 was used to obtain the cocrystal structures with inhibitors. Concentrated purified protein samples were diluted prior to crystallization to 0.1 mM by mixing with 20 mM potassium phosphate, pH 7.5, 10% glycerol, 1 mM DTT, 0.5 mM EDTA, 300 mM NaCl supplemented with equimolar inhibitor. Crystallization conditions were determined using commercial high-throughput screening kits available in deep-well format (Hampton Research), a nanoliter drop-setting Mosquito robot (TTP LabTech) operating with 96-well plates, and a hanging drop crystallization protocol. Crystals were further optimized in 96-well plates for diffraction data collection and harvested directly from the 200 nL drops. Prior to data collection, crystals were cryoprotected by plunging them into a drop of reservoir solution supplemented with 20% ethylene glycol, then flash-frozen in liquid nitrogen.
Diffraction data were collected at 100–110 K at beamline 8.3.1, Advanced Light Source, Lawrence Berkeley National Laboratory, USA. Data indexing, integration, and scaling were conducted using MOSFLM44 and the programs implemented in the ELVES software suite.45 The crystal structures were determined by molecular replacement using diffraction data processed in the corresponding space groups and atomic coordinates of T. cruzi CYP51 (PDB code 4C0C) as a search model. The final model was built using COOT,46 and refinement was performed by using REFMAC5 software.47,48 Data collection and refinement statistics are shown in Table 3.
Acknowledgments
We thank Potter Wickware for proofreading of the manuscript and the staff members of beamline 8.3.1, James Holton, George Meigs, and Jane Tanamachi, at the Advanced Light Source at Lawrence Berkeley National Laboratory for assistance with data collection. The authors also thank the Ortiz de Montellano laboratory at University of California—San Francisco for use of the GC–MS instrument, and Li Lin, Susan Khan, and Claudia Ruiz, Department of Molecular Therapeutics, Scripps Florida, Scripps Research Institute, for assistance with the in vivo pharmacokinetics and in vitro microsomal stability and CYP inhibition tests. This work was supported by NIH RO1 Grant AI095437 (to L.M.P and W.R.R.). C.M.C. was supported by Conselho Nacional de Desenvolvimento Cientifico e Tecnologico (CNPq) and FIOCRUZ. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract DE-AC02-05CH11231. Molecular graphics images were produced in part using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California—San Francisco (supported by NIH P41 Grant RR001081).
Glossary
Abbreviations Used
- T. cruzi
Trypanosoma cruzi
- TcI and TcII
T. cruzi discrete typing units I and II
- CYP
cytochrome P450 family
- CYP51
cytochrome P450 family 51
- CYP1A2, CYP2C9, CYP2D6, and CYP3A4
cytochrome P450 isoforms 1A2, 2C9, 2D6, and 3A4
- SAR
structure–activity relationship
- EC50
half maximal effective concentration
- VD
volume of distribution
Supporting Information Available
Synthetic procedures and spectroscopic data for final products and synthetic intermediates, UV–vis titration curves, EC50 curves, pharmacokinetic (PK) curves, animal model images, and a csv file containing molecular formula strings (SMILES). This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The atomic coordinates and structure factors (PDB codes 4C27, 4C28, and 4UVR) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
Author Present Address
∞ J.L.S.-N., J.H.M., and L.M.P.: Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California—San Diego, La Jolla, CA 92093, U.S.
Author Present Address
× D.K.: Five Prime Therapeutics, San Francisco, CA 94080, U.S.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Rassi A. Jr.; Rassi A.; Marin-Neto J. A. Chagas disease. Lancet 2010, 37597231388–1402. [DOI] [PubMed] [Google Scholar]
- Castro J. A.; de Mecca M. M.; Bartel L. C. Toxic side effects of drugs used to treat Chagas’ disease (American trypanosomiasis). Hum. Exp. Toxicol. 2006, 258471–479. [DOI] [PubMed] [Google Scholar]
- Cancado J. R. Long term evaluation of etiological treatment of chagas disease with benznidazole. Rev. Inst. Med. Trop. Sao Paulo 2002, 44129–37. [PubMed] [Google Scholar]
- Marin-Neto J. A.; Rassi A. Jr.; Avezum A. Jr.; Mattos A. C.; Rassi A.; Morillo C. A.; Sosa-Estani S.; Yusuf S. The BENEFIT trial: testing the hypothesis that trypanocidal therapy is beneficial for patients with chronic Chagas heart disease. Mem. Inst. Oswaldo Cruz 2009, 104Suppl. 1319–324. [DOI] [PubMed] [Google Scholar]
- BENEFIT trial: Evaluation of the use of antiparasital drug (benznidazole) in the treatment of chronic Chagas’ disease. http://clinicaltrials.gov/ (Identifier: NCT00123916. Sponser: Population Health Research Institute).
- TRAENA trial: Riarte A.TRAENA: Placebo-controlled evaluation of impact of benznidazole treatment on long-term disease progression in adults with chronic Chagas disease. Presented at the 62nd Annual Meeting of the American Society of Tropical Medicine and Hygiene, November 13–17, 2013.
- CHAGASAZOL trial (Posaconazole, Spain): Molina I.First clinical trial with posaconazole and benznidazole for the treatment of chronic Chagas disease. Presented at the XVIII International Congress of Tropical Medicine and Malaria, Rio de Janeiro, Brazil, September 23–27, 2012; http://ictmm2012.ioc.fiocruz.br/program_25_sept.html.; Clinical trial for the treatment of chronic Chagas disease with posaconazole and benznidazole (CHAGASAZOL). http://clinicaltrials.gov/ (Identifier: NCT01162967. Sponser: Hospital Universitari Vall d’Hebron Research Institute).
- Stop Chagas trial (Merck, Latin America and Spain): A study of the use of oral posaconazole (POS) in the treatment of asymptomatic chronic Chagas disease (P05267) (STOP CHAGAS). http://clinicaltrials.gov/ (Identifier: NCT01377480. Sponsor: Merck Sharp & Dohme Corp.).
- E1224/benznidazole trial (DNDi-Eisai, Bolivia): Torrico F.E1224—Results of proof of concept clinical trial in patients with chronic indeterminate Chagas disease. Presented at the 62nd Annual Meeting of the American Society of Tropical Medicine and Hygiene, Washington, DC, November 13–17, 2013.; Proof-of-concept study of E1224 to treat adult patients with Chagas Disease. http://clinicaltrials.gov/ (Identifier: NCT01489228. Sponsor: Drugs for Neglected Diseases).
- Buckner F. S.; Urbina J. A. Recent developments in sterol 14-demethylase inhibitors for Chagas Disease. Int. J. Parasitol.: Drugs Drug Resist. 2012, 2, 236–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drug trial for leading parasitic killer of the Americas shows mixed results but provides new evidence for improved therapy. DNDi: http://www.dndi.org/media-centre/press-releases/1700-e1224.html.
- Choi J. Y.; Podust L. M.; Roush W. R., Drug strategies targeting CYP51 in neglected tropical diseases. Chem. Rev. 2014, DOI: 10.1021/cr5003134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvet C. M.; Vieira D. F.; Choi J. Y.; Kellar D.; Cameron M. D.; Siqueira-Neto J. L.; Gut J.; Johnston J. B.; Lin L.; Khan S.; McKerrow J. H.; Roush W. R.; Podust L. M. 4-Aminopyridyl-based CYP51 inhibitors as anti-Trypanosoma cruzi drug leads with improved pharmacokinetic profile and in vivo potency. J. Med. Chem. 2014, 57166989–7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan M.; Abbott M. J.; Alexander P. W.; Armstrong T.; Best W. M.; Berven B.; Botero A.; Chaplin J. H.; Charman S. A.; Chatelain E.; von Geldern T. W.; Kerfoot M.; Khong A.; Nguyen T.; McManus J. D.; Morizzi J.; Ryan E.; Scandale I.; Thompson R. A.; Wang S. Z.; White K. L. Analogues of fenarimol are potent inhibitors of Trypanosoma cruzi and are efficacious in a murine model of Chagas disease. J. Med. Chem. 2012, 5594189–4204. [DOI] [PubMed] [Google Scholar]
- Keenan M.; Chaplin J. H.; Alexander P. W.; Abbott M. J.; Best W. M.; Khong A.; Botero A.; Perez C.; Cornwall S.; Thompson R. A.; White K. L.; Shackleford D. M.; Koltun M.; Chiu F. C.; Morizzi J.; Ryan E.; Campbell M.; von Geldern T. W.; Scandale I.; Chatelain E.; Charman S. A. Two analogues of fenarimol show curative activity in an experimental model of Chagas disease. J. Med. Chem. 2013, 562410158–10170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan M.; Alexander P. W.; Diao H.; Best W. M.; Khong A.; Kerfoot M.; Thompson R. C.; White K. L.; Shackleford D. M.; Ryan E.; Gregg A. D.; Charman S. A.; von Geldern T. W.; Scandale I.; Chatelain E. Design, structure–activity relationship and in vivo efficacy of piperazine analogues of fenarimol as inhibitors of Trypanosoma cruzi. Bioorg. Med. Chem. 2013, 2171756–1763. [DOI] [PubMed] [Google Scholar]
- Buckner F.; Yokoyama K.; Lockman J.; Aikenhead K.; Ohkanda J.; Sadilek M.; Sebti S.; Van Voorhis W.; Hamilton A.; Gelb M. H. A class of sterol 14-demethylase inhibitors as anti-Trypanosoma cruzi agents. Proc. Natl. Acad. Sci. U.S.A. 2003, 1002515149–15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chennamaneni N. K.; Arif J.; Buckner F. S.; Gelb M. H. Isoquinoline-based analogs of the cancer drug clinical candidate tipifarnib as anti-Trypanosoma cruzi agents. Bioorg. Med. Chem. Lett. 2009, 19, 6582–6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucke O.; Gelb M. H.; Verlinde C. L.; Buckner F. S. The protein farnesyltransferase inhibitor tipifarnib as a new lead for the development of drugs against Chagas disease. J. Med. Chem. 2005, 48175415–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus J. M.; Tatipaka H. B.; McGuffin S. A.; Chennamaneni N. K.; Karimi M.; Arif J.; Verlinde C. L.; Buckner F. S.; Gelb M. H. Second generation analogues of the cancer drug clinical candidate tipifarnib for anti-Chagas disease drug discovery. J. Med. Chem. 2010, 53103887–3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus J. M.; Verlinde C. L.; Karimi M.; Lepesheva G. I.; Gelb M. H.; Buckner F. S. Rational modification of a candidate cancer drug for use against Chagas disease. J. Med. Chem. 2009, 5261639–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepesheva G. I.; Ott R. D.; Hargrove T. Y.; Kleshchenko Y. Y.; Schuster I.; Nes W. D.; Hill G. C.; Villalta F.; Waterman M. R. Sterol 14alpha-demethylase as a potential target for antitrypanosomal therapy: enzyme inhibition and parasite cell growth. Chem. Biol. 2007, 14111283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalta F.; Dobish M. C.; Nde P. N.; Kleshchenko Y. Y.; Hargrove T. Y.; Johnson C. A.; Waterman M. R.; Johnston J. N.; Lepesheva G. I. VNI cures acute and chronic experimental Chagas disease. J. Infect. Dis. 2013, 2083504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeiro Mde N.; de Souza E. M.; da Silva C. F.; Batista Dda G.; Batista M. M.; Pavao B. P.; Araujo J. S.; Aiub C. A.; da Silva P. B.; Lionel J.; Britto C.; Kim K.; Sulikowski G.; Hargrove T. Y.; Waterman M. R.; Lepesheva G. I. In vitro and in vivo studies of the antiparasitic activity of sterol 14alpha-demethylase (CYP51) inhibitor VNI against drug-resistant strains of Trypanosoma cruzi. Antimicrob. Agents Chemother. 2013, 5794151–4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andriani G.; Amata E.; Beatty J.; Clements Z.; Coffey B. J.; Courtemanche G.; Devine W.; Erath J.; Juda C. E.; Wawrzak Z.; Wood J. T.; Lepesheva G. I.; Rodriguez A.; Pollastri M. P. Antitrypanosomal lead discovery: identification of a ligand-efficient inhibitor of Trypanosoma cruzi CYP51 and parasite growth. J. Med. Chem. 2013, 5662556–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J. Y.; Calvet C. M.; Gunatilleke S. S.; Ruiz C.; Cameron M. D.; McKerrow J. H.; Podust L. M.; Roush W. R. Rational development of 4-aminopyridyl-based inhibitors targeting Trypanosoma cruzi CYP51 as anti-Chagas agents. J. Med. Chem. 2013, 56197651–7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J. Y.; Calvet C. M.; Vieira D. F.; Gunatilleke S. S.; Cameron M. D.; McKerrow J. H.; Podust L. M.; Roush W. R. R-Configuration of 4-aminopyridyl-based inhibitors of CYP51 confers superior efficacy against Trypanosoma cruzi. ACS Med. Chem. Lett. 2014, 54434–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira D. F.; Choi J. Y.; Roush W. R.; Podust L. M. Expanding the binding envelope of CYP51 inhibitors targeting Trypanosoma cruzi with 4-aminopyridyl-based sulfonamide derivatives. ChemBioChem 2014, 1581111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.-K.; Doyle P. S.; Yermalitskaya L. V.; Mackey Z. B.; Ang K. K. H.; McKerrow J. H.; Podust L. M. Trypanosoma cruzi CYP51 inhibitor derived from a Mycobacterium tuberculosis screen hit. PLoS Neglected Trop. Dis. 2009, 32e372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podust L. M.; von Kries J. P.; Nasser Eddine A.; Kim Y.; Yermalitskaya L. V.; Kuehne R.; Ouellet H.; Warrier T.; Altekoster M.; Lee J.-S.; Rademann J.; Oschkinat H.; Kaufmann S. H. E.; Waterman M. R. Small molecule scaffolds for CYP51 inhibitors identified by high-throughput screening and defined by X-ray crystallography. Antimicrob. Agents Chemother. 2007, 51113915–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunatilleke S. S.; Calvet C. M.; Johnston J. B.; Chen C. K.; Erenburg G.; Gut J.; Engel J. C.; Ang K. K.; Mulvaney J.; Chen S.; Arkin M. R.; McKerrow J. H.; Podust L. M. Diverse inhibitor chemotypes targeting Trypanosoma cruzi CYP51. PLoS Neglected Trop. Dis. 2012, 67e1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Theuretzbacher U.; Clancy C. J.; Nguyen M. H.; Derendorf H. Pharmacokinetic/pharmacodynamic profile of posaconazole. Clin. Pharmacokinet. 2010, 496379–396. [DOI] [PubMed] [Google Scholar]
- Courtney R.; Radwanski E.; Lim J.; Laughlin M. Pharmacokinetics of posaconazole coadministered with antacid in fasting or nonfasting healthy men. Antimicrob. Agents Chemother. 2004, 483804–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andriani G.; Chessler A. D.; Courtemanche G.; Burleigh B. A.; Rodriguez A. Activity in vivo of anti-Trypanosoma cruzi compounds selected from a high throughput screening. PLoS Neglected Trop. Dis. 2011, 58e1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andriani G.; Chessler A. D.; Courtemanche G.; Burleigh B. A.; Rodriguez A. Correction: Activity in vivo of anti-Trypanosoma cruzi compounds selected from a high throughput screening. PLoS Neglected Trop. Dis. 2014, 810e3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy J. W.; Bryant C.; Calvet C. M.; Doyle P. S.; Gunatilleke S. S.; Leung S. S.; Ang K. K.; Chen S.; Gut J.; Oses-Prieto J. A.; Johnston J. B.; Arkin M. R.; Burlingame A. L.; Taunton J.; Jacobson M. P.; McKerrow J. M.; Podust L. M.; Renslo A. R. Chemical–biological characterization of a cruzain inhibitor reveals a second target and a mammalian off-target. Beilstein J. Org. Chem. 2013, 9, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle P. S.; Chen C.-K.; Johnston J. B.; Hopkins S. D.; Leung S. S. F.; Jacobson M. P.; Engel J. C.; McKerrow J. H.; Podust L. M. A nonazole CYP51 inhibitor cures Chagas’ disease in a mouse model of acute infection. Antimicrob. Agents Chemother. 2010, 5462480–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKerrow J. H.; Doyle P. S.; Engel J. C.; Podust L. M.; Robertson S. A.; Ferreira R.; Saxton T.; Arkin M.; Kerr I. D.; Brinen L. S.; Craik C. S. Two approaches to discovering and developing new drugs for Chagas disease. Mem. Inst. Oswaldo Cruz 2009, 1044263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson S. R.; Bot C.; Kelly J. M.; Hall B. S. Trypanocidal activity of nitroaromatic prodrugs: current treatments and future perspectives. Curr. Top. Med. Chem. 2011, 11162072–2084. [DOI] [PubMed] [Google Scholar]
- Chen C.-K.; Leung S. S. F.; Guilbert C.; Jacobson M. P.; McKerrow J. H.; Podust L. M. Structural characterization of CYP51 from Trypanosoma cruzi and Trypanosoma brucei bound to the antifungal drugs posaconazole and fluconazole. PLoS Neglected Trop. Dis. 2010, 4, e651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargrove T. Y.; Kim K.; de Nazare Correia Soeiro M.; da Silva C. F.; Batista D. D.; Batista M. M.; Yazlovitskaya E. M.; Waterman M. R.; Sulikowski G. A.; Lepesheva G. I. CYP51 structures and structure-based development of novel, pathogen-specific inhibitory scaffolds. Int. J. Parasitol.: Drugs Drug Resist. 2012, 2, 178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepesheva G. I.; Waterman M. R. Sterol 14alpha-demethylase (CYP51) as a therapeutic target for human trypanosomiasis and leishmaniasis. Curr. Top. Med. Chem. 2011, 11162060–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Wachenfeldt C.; Richardson T. H.; Cosme J.; Johnson E. F. Microsomal P450 2C3 is expressed as a soluble dimer in Escherichia coli following modification of its N-terminus. Arch. Biochem. Biophys. 1997, 3391107–114. [DOI] [PubMed] [Google Scholar]
- Leslie A. G. W., Recent changes to the MOSFLM package for processing film and image plate data. Jt. CCP4 ESF-EAMCB Newsl. Protein Crystallogr. 1992, No. 26. [Google Scholar]
- Holton J.; Alber T. Automated protein crystal structure determination using ELVES. Proc. Natl. Acad. Sci. U.S.A. 2004, 10161537–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60Part 12, Part 12126–2132. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53Part 3240–255. [DOI] [PubMed] [Google Scholar]
- The CCP4 suite: programs for protein crystallography. Acta Crysallogr., Sect. D: Biol. Crystallogr. 1994, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- DeLano W. L.The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, U.S., 2002.
- Lepesheva G. I.; Park H. W.; Hargrove T. Y.; Vanhollebeke B.; Wawrzak Z.; Harp J. M.; Sundaramoorthy M.; Nes W. D.; Pays E.; Chaudhuri M.; Villalta F.; Waterman M. R. Crystal structures of Trypanosoma brucei sterol 14alpha-demethylase and implications for selective treatment of human infections. J. Biol. Chem. 2010, 28531773–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepesheva G. I.; Hargrove T. Y.; Anderson S.; Kleshchenko Y.; Furtak V.; Wawrzak Z.; Villalta F.; Waterman M. R. Structural insights into inhibition of sterol 14alpha-demethylase in the human pathogen Trypanosoma cruzi. J. Biol. Chem. 2010, 2853325582–25590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargrove T. Y.; Wawrzak Z.; Alexander P. W.; Chaplin J. H.; Keenan M.; Charman S. A.; Perez C. J.; Waterman M. R.; Chatelain E.; Lepesheva G. I. Complexes of Trypanosoma cruzi sterol 14alpha-demethylase (CYP51) with two pyridine-based drug candidates for Chagas disease: structural basis for pathogen selectivity. J. Biol. Chem. 2013, 2884431602–31615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner F. S.; Bahia M. T.; Suryadevara P. K.; White K. L.; Shackleford D. M.; Chennamaneni N. K.; Hulverson M. A.; Laydbak J. U.; Chatelain E.; Scandale I.; Verlinde C. L.; Charman S. A.; Lepesheva G. I.; Gelb M. H. Pharmacological characterization, structural studies, and in vivo activities of anti-chagas disease lead compounds derived from tipifarnib. Antimicrob. Agents Chemother. 2012, 5694914–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25131605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.