

Abstract
Fibroblast growth factor homologous factors (FHFs) form native intracellular complexes with the mitogen-activated protein kinase (MAPK) scaffold protein islet-brain 2 (IB2) in adult brain. FHF binding to IB2 facilitates recruitment of the MAPK p38δ (SAPK4), while failing to stimulate binding of JNK, the preferred kinase of the related scaffold IB1 (JIP-1). We now report further biochemical evidence supporting FHFs as regulators of IB2’s function as a scaffold for p38δ kinase activation. MLK3 and IB2 synergistically activate p38δ, but not the MAPKs JNK-1 and p38α. Binding of p38δ to IB2 is mediated by the carboxyterminal half of the scaffold (IB2Δ1–436). FHF2 also binds weakly to IB2Δ1–436, and can thereby increase p38δ interaction with IB2Δ1–436. FHF-induced recruitment of p38δ to IB2 is accompanied by increased levels of activated p38δ, and synergistic activation of p38δ by MLK3/IB2 is further enhanced by FHF2. Consistent with a role for FHFs as signaling molecules, FHF2 isolated from rat brain is serine/threonine phosphorylated, and FHF can serve as a substrate for p38δ in vitro. These results support the existence of a signaling module in which IB2 scaffolds a MLK3/MKK/p38δ kinase cascade. FHFs aid in recruitment of p38 to IB2 and may serve as kinase substrates.
INTRODUCTION
Cellular responses to extracellular signals include activation of mitogen activated protein kinase (MAPK) pathways that coordinate proliferation, differentiation, apoptosis and survival. In mammalian cells, the three known branches of MAPKs are ERKs, c-Jun amino-terminal kinases (JNKs) and p38 MAPKs, the latter two referred to collectively as stress-activated protein kinases (SAPK), as they mediate responses both to environmental stresses and to proinflammatory cytokines (1). Activated SAPKs phosphorylate a wide variety of proteins, including transcription factors and cytoskeletal proteins (reviewed in (2,3). MAPKs are typically activated through three-tiered kinase cascades: MAPK kinase kinases (MAPKKKs) activate MAPK kinases (MAPKKs), which in turn activate MAPKs (4,5). Several different MAPKKKs can activate SAPKs, including TAK1, ASK1 and members of the family of Mixed Lineage Kinases (MLKs). A set of MAPKKs mediates activation of SAPKs, with MKK4 and MKK7 generally activating JNKs, and MKK3 and MKK6 generally activating p38s.
JNKs are regulators of cell proliferation and apoptosis during development and in response to environmental stress (2). JNK-deficient mice display defects in developmental and excitotoxin-induced neuronal apoptosis (2) and in T-cell function and immune responses (6). Among the four p38 proteins, p38α and p38β play roles in cytokine- and stress-induced actin cytoskeletal reorganization (7–9), as well as in cell differentiation and apoptosis (3). Although no specific biological function has been uncovered for p38δ (also known as SAPK4) (10–12), several observations suggest p38δ to be somewhat different from the prototypical p38α and p38β. In vitro kinase assays have revealed a partially overlapping but distinct substrate specificity for p38δ, including the ability to phosphorylate certain microtubule associated proteins (13,14) and eukaryotic elongation factor 2 kinase (15). In addition, some studies suggest that p38δ may be activated by a broader spectrum of MAPKKs, including MKK4 and MKK7 (16).
Scaffold proteins contribute to signaling efficiency and/or specificity of MAP kinase signaling by sequestering MAPK and upstream activating kinases (17). JNK-Interacting Protein (JIP)-1, also known as islet-brain-1 (IB1), is a vertebrate scaffold protein which regulates JNK activity. IB1 was discovered as a JNK-binding protein that also binds MAPKKKs of the MLK family and the MAPKK MKK7 (18–20). IB1 facilitates JNK activation when overexpressed by transfection together with IB1-interacting upstream kinases (20,21). Forced overexpression of IB1 or its JNK-binding domain inhibits JNK-signaling in many types of cells (18,19,22). Endogenous IB1 appears to serve as both a positive and negative regulator of JNK activity. JNK activation in mechanically stressed urothelium is negatively regulated by IB1, as demonstrated by gain- and loss-of function experiments (23). By contrast, analysis of IB1-deficient mice showed IB1 to be required for JNK activation in hippocampal neurons following exposure to excitotoxins or to anoxic stress (20).
IB2 and its highly similar alterative splice isoform, JIP-2 (Figure 1A), were discovered through their homology with IB1. IB2 and IB1 share considerable amino acid sequence homology in an N-terminal region encompassing the JNK binding domain in IB1, and in the C-terminal half including, but not limited to, the SH3 and PTB domains (see Figure 1A). IB2 and JIP-2 interact with MLKs and MKK7, and initial studies had identified IB2 and JIP-2 as additional putative scaffolds regulating the JNK pathway (24,25). In contrast to IB1, however, IB2 interacts in vivo with fibroblast growth factor homologous factors (FHFs) (26). FHFs were intially identified by virtue of their sequence homology to fibroblast growth factors (FGFs) (27–29). However, FHFs are different from FGFs in several ways. FHF proteins are synthesized and localized within native or transfected expressing cells (26,27), as opposed to the function of FGFs as secreted growth factors acting on extracellular receptors (30). Consistent with FHF intracellular localization, we have shown that FHFs facilitate recruitment of a specific SAPK, p38δ, to IB2 in transfected 293T cells (26). These findings have revealed fundamental differences in the signaling complexes assembled by IB2 versus IB1.
Figure 1. Alignment of IB2 and IB1 sequences.

A) The predicted amino acid sequences of human IB2 and JIP-2 are aligned with murine IB1 (JIP-1b) (20). IB2 and JIP-2 (24) are transcribed from the same gene, and differ only in the presence of alternatively spliced exons in the aminoterminus such that JIP-2 bears a distinct 57 residue segment in place of the first 30 residues of IB2. Three regions of IB2/IB1 amino acid sequence homology are hatched. Regions I, II and III share 32%, 82% and 79% amino acid identity, respectively. A black box indicates a conserved RPTTL motif that is essential for JNK-binding to IB1 (18). Conserved phosphotyrosine binding (PTB) and Src Homology-3 (SH3) domains are indicated, as well as JNK-binding and FHF binding domains of IB1 and IB2, respectively. B) Schematic representation of mutant IB2 proteins and their relative magnitude of interaction with p38δ and FHF2. Previous data on FHF2 binding (26) are indicated in italics. The regions which span the major FHF binding domain in IB2 determined previously (26), a weak FHF binding region, as well as the region required for p38δ interaction are indicated. The MLK3 and MKK7 binding region is depicted as determined by Yasuda et al. (24).
Here, taking advantage of the identification of p38δ as a potential IB2-regulated kinase, we investigated the role of FHFs in recruitment of p38δ to IB2 and subsequent activation of p38δ in a tissue culture overexpression system. We show that FHF interaction with IB2 facilitates the association and subsequent activation of p38δ. We also show that p38δ interacts with the C-terminal half of IB2, which stands in contrast to JNK interaction with the aminoterminal region of IB1 (JIP-1b). Lastly, we show that FHF2 is serine/threonine phosphorylated in adult rat brain, consistent with an intracellular function for this IB2-interacting protein.
MATERIALS AND METHODS
Nomenclature and sequences
Four murine and corresponding human FHF genes were described by Jeremy Nathans and coworkers (27). Our database searches identified three of these four FHF genes, which we had originally referred to as FGFs (FGF-11 = FHF3, FGF-12 = FHF1, FGF-13 = FHF2) (28,29,31), and FHF4 was given the alternative name FGF-14 (32). Since FHFs so far lack demonstrable FGF-like activity and since we have characterized an intracellular function for these factors, we now refer to these factors solely by the acronym FHF, so as to distinguish them from FGFs. Our previously described FGF-12A is now referred to as FHF1, while the alternatively spliced FGF-12B is herein referred to as FHF1B (28).
Alluding to its most prominent sites of expression, we (26) and others (25) have described islet-brain-2 (IB2), while an alternative splice form of this gene has been described as JIP-2 (24). In the absence of in vivo data implicating IB2 or JIP-2 in regulation of JNK, we have adopted the functionally neutral islet-brain nomenclature.
Mammalian expression vectors
Vectors to express wild type and mutant derivatives of human IB2, murine IB1(JIP-1B) (20), and human SHP-2 (33) as N-terminal Flag-tagged proteins have been described as well as vectors to express N-terminal 5X-Myc-tagged versions of human FGF1 (Genbank #E04557) and the long isoform of FHF2 (34).
pCDNA-T7-MLK3 was constructed by PCR modification of a human MLK3 cDNA template to add the N-terminal T7 tag (MASMTGGQQMG) prior to cloning into pCDNA3 (Invitrogen). The β1 isoform of human MKK7 (35) was RT-PCR amplified from human brain cDNA and cloned into pEBG (36) to attach an N-terminal GST-tag. All other kinase expression vectors, including pCDNA-HA-JNK1α1, pCEV29-HA-p38α, pCEFL-HA-p38δ and pCEFL-HA-MLK3, were gifts from R. Silvio Gutkind.
Antibodies
Antibodies to epitope tags: HA-tag, mouse monoclonal 12CA5, IgG2b, and Myc-tag, mouse monoclonal 9E10, IgG1 (Mt. Sinai School of Medicine Monoclonal Antibody Facility); Flag-tag, mouse monclonal M2, IgG1 (Sigma Chemical); Flag-tag, rabbit polyclonal (Zymed Corp.); T7-tag, mouse monoclonal, IgG2b (Novagen); GST-tag, mouse monoclonal, IgG1 (Santa Cruz Biotech.).
Rabbits were immunized against peptides corresponding to mouse FHF2 residues 234–244 (GGKSMSHNEST) synthesized with added N-terminal cysteine for sulfhydryl-mediated directional coupling to carrier protein (Research Genetics). Antibodies were affinity purified with corresponding peptides immobilized on activated agarose (Pierce Chemical). FHF2 antibodies recognized FHF2, but not FHF-1, in lysates from expression vector-transfected 293T cells. FHF2 antibodies were biotinylated using N-sulfo-biotin reagent (Pierce Chemical).
Western blotting employed horseradish peroxidase-conjugated secondary antibodies specific for mIgG1, mIgG2b, or rabbit IgG or (CALTAG), or horseradish peroxidase-conjugated streptavidin (CALTAG) to detect biotinylated FHF2 antibodies.
Protein complexes in transfected cells
293T cells seeded onto gelatinized or Primaria-coated 6-well cluster dishes (Falcon) were transiently transfected and subsequently harvested as described (34). Experiment to experiment small differences in cell density, growth rate, and response to transfection conditions substantially influenced the magnitude of responses reported. Therefore, each figure panel presents comparisons made among samples derived from the same transfection. Immunoprecipitation assays from transiently transfected 293T cell extracts as well as subsequent analysis by SDS PAGE and standard Western blot analysis have been described (34).
Assays for MAP kinase activity
Cells were stimulated before lysis with 0.5 mM H2O2 for 30 min or with 50 ng/ml anisomycin for 20 min. MAPK activities in transfected cells were detected by immune complex kinase assays performed as previously described (37). Cell extracts containing HA-tagged kinases were incubated overnight at 4°C with 2 μg 12CA5 anti-HA in lysis buffer and immunoprecipitated using GammaBindG-Sepharose. Immunoprecipitates were washed three times in PBS/ 0.1% NP40/ 2 mM sodium orthovanadate, once with 100 mM Tris-HCl pH 7.5 – 0.5M LiCl and once with kinase buffer (12.5 mM MOPS, pH 7.5 – 12.5 mM β–glycerophosphate - 7.5 mM MgCl2 - 0.5 mM sodium orthovanadate - 0.5 mM NaF - 0.5 mM EGTA). Pellets were incubated in 30 μl kinase buffer containing 1 μg substrate, 1 μCi [γ-32P] ATP, 3.3 mM dithiothreitol and 20 μM ATP for 30 minutes at 30°C on a shaking platform. Bacterially expressed GST-ATF-2 or GST-cJun (Santa Cruz Biotech) were used as substrate for p38s and JNK, respectively. Recombinant human FHF1B used as a substrate was purified from induced BL21(DE3)pLysS harboring pET-FHF1B vector. Assays were terminated by addition of Laemmli sample buffer, boiled for 5 minutes, and proteins resolved by SDS-PAGE and transfer to PVDF membrane. The incorporation of 32P in substrate was visualized by autoradiography, and quantitated after PhosphorImager exposure using ImageQuant software (Molecular Dynamics). Only extracts with comparable levels of exogenous HA-tagged kinases were compared, as determined by Western blotting of extracts and immunoprecipitated kinases.
Activity of HA-tagged p38δ or HA-tagged JNK1 was also assayed by electrophoresis, blotting, and probing with antibodies specific for threonine/tyrosine dually phosphorylated p38 and JNK proteins, respectively (New England Biolabs).
Assays to detect FHF phosphorylation
Protein extracts from 7 day old rat pup cerebella were prepared as described (34). Cerebellar extracts (1 mg protein) were incubated overnight at 4°C with 2 μg antibody in lysis buffer. Immune complexes were recovered the following day using ProteinG-Sepharose. Pellets were washed two times in lysis buffer, once in 20 mM Tris-HCl pH 7.4, 137 mM NaCl, 2 mM EDTA, 10% glycerol, 1% Triton X-100, 1 mM PMSF, 10 μg/ml aprotinin and 10 μg/ml leupeptin and once in 20 mM HEPES pH 7.5 – 0.1 M NaCl - 0.5 mM MnCl2 - 30 μM sodium orthovanadate – 5 mM benzamidine -1% NP40 - 2 mg/ml BSA (PP buffer). Pellets were incubated in 50 μl PP buffer in the presence of PP2a (generous gift from David Li) −/+ 50 nM okadaic acid for 20 min at 30°C on a shaking platform. Assays were terminated by addition of Laemmli sample buffer, boiled for 5 min, and proteins resolved by SDS-PAGE (12% gels) and transferred to PVDF membrane. Proteins species in immunoprecipitates or total lysates were identified by standard Western blot analysis, using primary biotinylated antibody, streptavidin-conjugated secondary antibodies (CALTAG), and chemiluminescence detection with ECL reagent (Amersham Pharmacia Biotech).
RESULTS
p38δ is an IB2 regulated kinase
IB2 can complex with p38δ, but not other MAPKs, including JNK1, in transfected 293T cells (26). These findings suggested that IB2 may regulate p38δ kinase activity. As a means to survey the putative involvement of IB2 in regulation of p38δ, we tested whether IB2 was capable of stimulating or inhibiting activation of p38δ in 293T cells. Basal activity of p38δ, detected by the presence of phospho-p38δ, was found to be weakly activated in some experiments by coexpression of IB2 (Figure 2A). Additionally, IB2 increased activation of p38δ induced by peroxide stress (Figure 2A). By contrast, activation of p38δ by anisomycin stress was inhibited by IB2 expression (Figure 2B), similar to findings reported for effects of IB1 (JIP-1) on JNK activation (20). These findings demonstrate that IB2 can positively or negatively modulate signaling pathways leading to p38δ activation.
Figure 2. p38δ is an IB2-regulated protein.
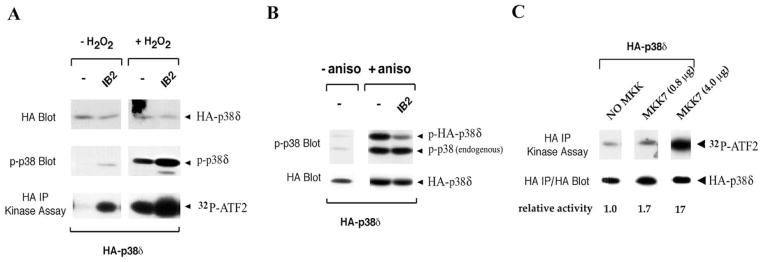
293T cells were co-transfected with epitope-tagged protein expression vectors, and aliquots of subsequent cell lysates were analyzed by Western blotting or in kinase assays. A) Cells were co-transfected with Flag-tagged IB2 and HA-tagged p38δ, stimulated with H2O2, and lysates were electrophoresed, blotted, and probed with anti-HA antibodies or with antibodies against phospho-p38 (p-p38). Anti-HA immunoprecipitates were added to kinase assay reactions containing GST-ATF-2 substrate and γ32P-ATP, which were subsequently electrophoresed and blotted. 32P incorporation into ATF-2 was visualized by autoradiography. B) Cells were cotransfected with Flag-tagged IB2 and HA-tagged p38δ, stimulated with anisomycin, and lysates were electrophoresed, blotted, and probed with anti-HA antibodies or with antibodies against p-p38. C) Lysates from cells co-transfected with HA-tagged p38δ together with either GST-tagged MKK7 or empty vector were immunoprecipitated with anti-HA antibodies and subjected to kinase assays as described above. Products were subsequently electrophoresed and blotted. 32P-ATF-2 levels were visualized by autoradiography and analyzed using PhosphorImager imaging and ImageQuant software, and recalculated as activity relative to the activity of p38δ in the absence of MKK7. The same blot was probed with anti-HA to detect HA-p38δ proteins.
IB2 binds to the MAPKKK MLK3 and related members of the family of mixed-lineage kinases, but not to other MAPKKKs (24), and to the MAPKK MKK7, but not to other MKKs [(24,25), and JS and MG, unpublished data]. Hence, IB2 is envisaged to scaffold a MAPK pathway, enlisting MLKs and MKK7 as upstream kinases of p38δ. However, the published data regarding the ability of MKK7 to activate p38δ is conflicting (10,16). To address this issue we tested directly MKK7’s ability to activate p38δ. Upon coexpression of both kinases in 293T cells, protein kinase activity of p38δ (measured on immunoprecipitates using ATF-2 as in vitro substrate) was easily demonstrated (Figure 2C).
In order to test the potential of IB2 to facilitate activation of p38δ by upstream kinases, we coexpressed IB2 and p38δ proteins in 293T cells together with increasing amounts of an upstream activator kinase, MLK3. Parallel transfections were performed using JNK1 in place of p38δ as a negative control. MAPK activities were measured with antibodies specific for either phospho-p38 or phospho-JNK. In the absence of IB2, MLK3 activated JNK-1 or p38δ in a concentration-dependent manner (Figure 3A,B). Activation of JNK by increasing amounts of MLK3 was not enhanced by the inclusion of IB2 (Figure 3B), consistent with the absence of detectable IB2/JNK binding in 293T cells (26). By contrast, IB2 significantly enhanced the weak activation of p38δ by MLK3 (Figure 3A). Together, these findings provide evidence that IB2 assembles a signaling cascade in which MLK3 activates p38δ, presumably through the MAPKK MKK7.
Figure 3. Synergistic activation of p38δ by MLK-3 and IB2.

Activation of p38δ by MLK-3 and IB2. 293T cells were transfected with plasmids expressing HA-p38δ or HA-p38δ, Flag-tagged IB2 and T7-tagged MLK-3 in different combinations as indicated and serum starved for 5 hours before lysis. HA-p38δ or HA-JNK activity in extracts was assayed by probing Western blots of electrophoresed extracts with antibodies specific for dually phosphorylated p38 or JNK, respectively. The same blots were probed with tag-specific antibodies to detect IB2 and JNK1 or p38δ proteins.
FHF-dependence for IB2/p38δ interaction is overcome by elevated IB2 concentration
We have previously shown that FHFs promote IB2/p38δ interaction, identifying FHFs as cofactors in the recruitment of p38δ into a heterotrimeric IB2/FHF/p38δ complex (26). The FHF-independent activation of p38δ by MLK3 and IB2 seen above (Figure 2A) might have been the consequence of high IB2 expression in these experiments together with inherently weak IB2 affinity for p38δ. We tested this possibility by analyzing IB2/ p38δ complex formation in transfected 293T cells, varying the amounts of IB2 and FHF2 proteins. At lower IB2 protein levels, weak p38δ interaction was potentiated by FHF2 (Figure 4A, lanes 1 and 3), as reported previously (26). By contrast, IB2/p38δ interaction could be rendered FHF-independent when IB2 expression levels were increased (Figure 4A). At high scaffold concentration, IB2 still could not complex significantly with other p38s nor with JNK1, ERK2, or BMK/ERK5 (data not shown).
Figure 4. Carboxyl-terminal half of IB2 mediates p38δ binding.

A) IB2/p38δ complex formation is FHF-independent at high IB2 concentration. 293T cells were transfected with plasmids expressing HA-p38δ (0.05 μg), Flag-IB2 (0.5 or 1.5 μg), and Myc-FHF2 (0 or 2.5 μg), and lysates were immunoprecipitated with anti-Flag and blot probed with anti-HA to detect IB2/p38δ complexes, and analyzed for expression of all proteins by appropriate blots, as indicated. B) The C-terminal half of IB2 is essential for p38δ interaction. 293T cells were transfected with plasmids expressing HA-p38δ and Flag-tagged IB2 (wild-type or mutants; Figure 1B) at high IB2 plasmid concentrations (1.5 μg). Anti-flag immunoprecipitation and Western blot probing with anti-HA assayed for IB2/p38δ complexes, and Western blots of total cell lysates were probed to detect p38δ and IB2 proteins. (C) Heterotrimeric FHF/IB2/p38δ complex detection. 293T cells were transfected with expression plasmids for HA-p38δ, Myc-tagged FHF2 or FGF1, and Flag-tagged IB2 proteins (wild-type or mutant, Figure 1B). Lysates were analyzed for expression of all proteins with appropriate antibodies, as indicated, and also used in anti-Myc immunoprecipitation for Western blot probing with anti-HA to detect FHF/IB2/p38δ heterotrimeric complexes. D) Potentiation of IB2Δ1–436/p38δ complex formation by FHF2. Lysates from 293T cells cotransfected with plasmids for HA-p38δ and Flag-IB2Δ1–436 in the presence or absence of Myc-FHF2 were immunoprecipitated with anti-Flag and Western blot probed with anti-HA to detect IB2Δ1–436/p38δ complexes, and samples of total lysates were used to detect expression of all proteins.
The carboxyl-terminal half of IB2 mediates binding to p38δ
In order to map the region of IB2 required for p38δ interaction we employed several terminal and internal deletion derivatives of human IB2 (Figure 1B) in cotransfection with p38δ. The analysis was guided by the presence of a major FHF-binding domain within the region spanning IB2 residues 212–471 (26) and three regions of IB2/IB1 homology (see Figure 1A). Region I spans IB2 residues 55–141 and includes residues homologous to the JNK-binding domain on IB1 (18), region II spans IB2 residues 477–504, and region III spans the C-terminal 263 residues of IB2 (including both an SH3 and a PTB domain). IB2/p38δ interactions were assayed after cotransfection of flag-tagged IB2 mutants, HA-tagged p38δ, and in some cases myc-tagged FHF2. We manipulated IB2 expression to higher or lower levels, which allowed comparison of FHF-independent (Figure 4B) to FHF-induced (Figure 4C, D) interaction with p38δ. IB2/p38δ complexes were assayed by anti-HA Western blot after anti-flag immunoprecipitation (Figure 4B, D), while heterotrimeric FHF/IB2/p38δ complexes were assayed by anti-HA Western blot after anti-myc (FHF) immunoprecipitation (Figure 4C). These data are also summarized in Figure 1B.
An IB2 mutant (IB2Δ90–174) harboring a deletion in region I was as efficient as wild-type IB2 for p38δ binding (Figure 4B). This mutant also effectively mediated formation of a FHF2/IB2Δ90–174/p38δ heterotrimeric complex (Figure 4C). Hence, despite sequence homology between IB1 and IB2 in the region encompassing the JNK-binding domain of IB1, the corresponding domain of IB2 is dispensable for binding of p38δ.
Further mutational analysis demonstrated that p38δ binding requires more C-terminal IB2 sequence elements. IB2Δ212–471 complexed with p38δ as efficiently as did full-length IB2 (Figure 4B). However, IB2Δ459–529 and IB2Δ514–797, bearing deletions spanning region II or III, respectively, displayed reduced abilities to bind p38δ, while IB2Δ459–797 could not interact with p38δ at all (Figure 4B). Moreover, formation of a FHF2/ IB2Δ459–529/p38δ heterotrimeric complex was very inefficient as compared to wildtype IB2 (Figure 4C). Consistent with the above findings, a IB2 fragment spanning regions II + III (IB2Δ1–436) retained the ability to interact with p38δ (Figure 4B). These data define the IB2 C-terminal homology regions II and III as bearing the determinants for p38δ interaction.
A weak FHF-binding site on IB2 involved in formation of FHF/IB2/p38δ complexes
FHF2, IB2 and p38δ assemble into a heterotrimeric complex when overexpressed in 293T cells, and deletion of the major FHF binding domain (residues 212–471) nearly abolishes IB2’s ability to assemble such complex (26)(and Figure 4C). IB2 deletion of part of the p38δ binding domain (IB2Δ459–529) severely impaired heterotrimeric complex formation as anticipated (Figure 4C). Surprisingly, IB2Δ1–436 was able to assemble into heterotrimeric complexes with FHF and p38δ (Figure 4C). This finding suggested that an additional binding site for FHF might be present within IB2Δ1–436. We screened for additional FHF binding regions in IB2, overexpressing tagged proteins in cotransfected 293T cells. As reported before, wildtype IB2 binds FHF2 strongly, while IB2Δ212–471 (missing the major FHF binding domain) did not bind at all (Figure 5). The C-terminal segment of IB2 resulting from further truncation of the aminoterminus (IB2Δ1–436) bound FHF weakly (Figure 5). This finding together with previous mapping of FHF/IB2 interaction domains [(26); summarized in Figure 1B], suggested that the C-terminal half of IB2 harbors a weak binding site for FHF as well as p38δ, and that the weak FHF-binding site is masked by a more aminoterminal region of IB2. Consistent with the presence of both a weak FHF binding site and a p38δ binding site in the C-terminal half of IB2, interaction between IB-2Δ1–436 and p38δ was modestly potentiated by cotransfection of FHF2 (Figure 4D). These results suggest that the weak FHF-binding domain in the C-terminal half of IB2 contributes to FHF-induced recruitment of p38δ to IB2.
Figure 5. Carboxyl-terminal half of IB2 contains a low-affinity FHF binding site.

Lysates from 293T cells cotransfected with plasmids encoding Myc-tagged FHF2 and Flag-tagged proteins (wild-type IB2, IB2 mutants, or SHP-2) were electrophoresed directly for Western blot detection of tagged proteins or immunopreciptated with anti-Flag and Western blot probed with anti-Myc to detect protein complexes (IB2/FHF2).
FHF promotes p38δ activation through recruitment of p38δ to IB2
In order to determine whether FHF-induced recruitment of p38δ to IB2 favors p38δ activation, we expressed tagged versions of these signaling proteins in 293T cells at levels sensitive to FHF induction of scaffold/kinase complexes. Subsequently, p38δ kinase activity was assayed by Western blot using antibodies against phospho-p38, or in kinase assays using ATF-2 as in vitro substrate. When cells were transfected with p38δ and IB2, a low basal level of p38δ activity was observed. In the presence of increasing concentrations of FHF2, p38δ phosphorylation and kinase activity increased commensurately (Figure 6A).
Figure 6. IB2-mediated p38δ activation.
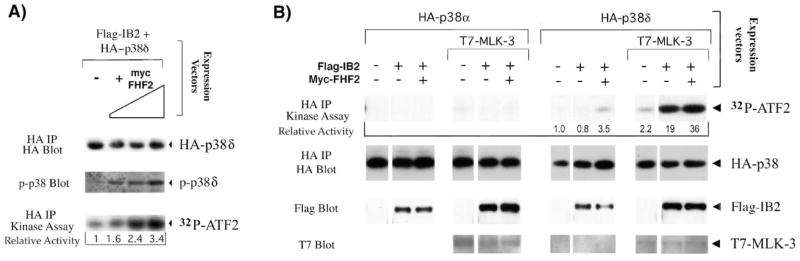
A) FHF2 induces activation of p38δ. 293T cells were transfected with plasmids expressing HA-p38δ, Flag-tagged IB2 and increasing amounts of Myc-tagged FHF2 and serum starved for 5 hours before cell lysis. HA-p38δ activity in extracts was assayed by probing Western blots of electrophoresed extracts with antibodies specific for dually phosphorylated p38. Alternatively, HA-p38δ activity was assayed by its ability to phosphorylate GST-ATF-2 in kinase assays as described in Figure 2A. 32P-ATF-2 levels were visualized by autoradiography and analyzed using PhosphorImager imaging and ImageQuant software, and recalculated as activity relative to the activity of each kinase in the absence of FHF2. Western blot probing with anti-HA after anti-HA immunoprecipitation was used to detect the amount of HA-p38δ proteins assayed in the kinase assay. Extracts analyzed in this panel have also been used to demonstrate FHF-2-mediated recruitment of p38δ to IB2 (26). B) Synergistic activation of p38δ by MLK-3 and IB2 is stimulated by FHF. 293T cells were transfected with plasmids expressing HA-p38δ or HA-p38δ, Flag-tagged IB2, T7-tagged MLK-3 and Myc-tagged FHF2 in different combinations as indicated and serum starved for 5 hours. HA-p38δ activity in extracts was assayed in kinase assays as described in the legend to Figure 2A. 32P incorporation into ATF-2 was visualized and analyzed as described above and recalculated as activity relative to the activity of each kinase in the absence of other transfected proteins to determine the relative p38 activities (kinase activity in absence of cotransfected proteins = 1.0). The same blot was probed with anti-HA to detect HA-p38 proteins. Western blots of total cell lysates were probed with anti-Flag or anti-T7 to detect IB2 and MLK-3 proteins, respectively.
We next tested the separate and combined effects of FHF2, MLK3 and IB2 on activation of p38δ. As shown in panel B of Figure 6, expression of IB2 by itself did not stimulate p38δ activity and expression of MLK3 alone stimulated p38δ activity only 2.2-fold. By contrast, coexpression of MLK3 with IB2 stimulated p38δ activity 19-fold. In the absence of IB2, FHF was unable to stimulate p38δ activity, nor could it potentiate the weak activation of p38δ by MLK3. By contrast, FHF2 augmented the IB2-mediated activation of p38δ by MLK3 (Figure 6B). Reflecting the binding specificity of IB2 for p38δ, IB2 could not potentiate MLK3-mediated activation of p38α (Figure 6B). These results demonstrate that FHF-mediated recruitment of p38δ to IB2 enhances the signaling efficiency of the IB2 module.
FHF phosphorylation
For each FHF gene, mRNAs generated by alternative splicing of transcripts encode variant isoforms differing in N-terminal sequence (28,32,38); for FHF2, the predicted isoforms are 21.5 and 27.5 kD (Genbank Accessions NM_033642 and NM_010200, respectively). We investigated the expression and sizes of FHF2 isoforms in extract of rat cerebral cortex by Western blot using antibodies raised against an epitope in the common C-terminus of FHF2 variants.
FHF2 species corresponding to both predicted isoforms were detected (Figure 7A, lane 1). However, when the extracts were subjected to immunoprecipitation with the same antibody prior to Western blot, the approximately 28 kD species was significantly underrepresented (Figure 7A, lane 2). Treatment of immunoprecipitates with phosphatase PP2a restored detection of the 28 kD species (Figure 7A, lane 3), and this effect was blocked when the phosphatase inhibitor okadaic acid was included in the treatment (Figure 7A, lane 4). These findings demonstrate that the longer FHF2 isoform is phosphorylated on serine or threonine residues, rendering its immunoprecipitation and detection by antibodies inefficient. FHF2 is most likely phosphorylated within the C-terminal epitope which served as immunogen, thereby masking antibody recognition.
Figure 7. FHF2 is a phosphoprotein.

A) Dephosphorylation of FHF2 by PP2a enhances immunodetection. Rat cerebellum extract was assayed for FHF2 by Western blot analysis using biotinylated anti-FHF2 antibodies. Lane 1, total extract, lanes 2–4, anti-FHF2 immunoprecipitates. Washed immunoprecipitates were left untreated (lane2), treated with PP2a (lane 3) or with PP2a in the presence of phosphatase inhibitor okadaic acid (lane 4). A prominent band at 20 kD is indicated as well as the lower and upper bands of a 28–34 kD ladder. The 28 kD band is also indicated in lane 3. B) FHF1 is a substrate for p38δ in vitro. 293T cells were transfected with plasmids expressing HA-p38δ and left untreated or induced with H2O2. HA-p38δ activity in extracts was assayed in kinase assays containing bacterially expressed FHF1 as a substrate and γ32P-ATP, which were subsequently electrophoresed and blotted. 32P incorporation into FHF1 was visualized by autoradiography. The same blot was probed with anti-HA to detect HA-p38δ proteins.
Additional FHF2 species ranging from 30,000 to 34,000 apparent molecular weight were also detected in the brain extracts, indicating additional FHF post-translational modifications (Figure 7, lanes 1,2). Phosphatase pretreatment of immunoprecipitates reduces detection of the 32,000 apparent molecular weight species (Figure 7, lane 3). This finding suggests that additional serine/threonine phosphorylation results in retarded FHF electrophoretic mobility.
As FHF associates with the IB2 scaffold and is phosphorylated in vivo, we considered the possibility that FHFs are substrates of the scaffolded kinase cascade. To preliminarily address this question, HA-tagged p38δ from untreated or peroxide-treated cells was immunoprecipitated and incubated in vitro with recombinant, bacterially expressed FHF1. Phosphorylation of FHF1 was detected after incubation with peroxide-activated p38δ, but not with inactive p38δ (Figure 7B), demonstrating that FHF-1 can serve as a p38δ substrate in vitro. Phosphorylation of FHFs in vivo and in vitro provides further evidence for intracellular FHF function and suggests that this function may be regulated by the kinase complex associated with IB2.
DISCUSSION
A p38δ kinase cascade coordinated by IB2
Islet-brain proteins are kinase scaffolds and mediate MAPK activation through MLKs and MKK7 in transfected cells. JNKs have been identified as the exclusive targets of IB1 regulation (24). We identified IB2 as an FHF binding protein and showed it to bind p38δ in contrast to JNKs or other MAPKs (26). We have now shown that FHF-induced binding of IB2 to p38δ is associated with activation of p38δ, and that IB2 can facilitate activation of p38δ by MLK3. We thereby suggest p38δ to be an IB2-regulated MAPK, making IB2 the first vertebrate scaffold protein found to bear specificity for a member of the p38 subfamily of MAPKs. This model is also consistent with the ability of MKK7 to activate p38δ [Figure 1, and (16)]. The coupling of MLK3 and MKK7 to JNK and p38δ by IB1 and IB2, respectively, extends to vertebrates a principle of kinase target specificity first appreciated in yeast, wherein the MAPKKK STE11 is channelled towards FUS3 or HOG1 MAPK in response to different cell stimuli acting through different kinase scaffolds (39).
Previous studies from Yasuda et al. (24) reported weak binding of the N-terminal IB2 variant JIP-2 to JNKs in transfected COS cells, and IB2 potentiated MLK3-induced JNK activation in these cells. By contrast, we find that IB2 fails to bind JNK1 in 293T cells (26). Moreover, in many experiments in 293T cells with varying levels of IB2, JNK and upstream kinase, JNK activity was not significantly enhanced in the presence of IB2 (Figure 2, and J.S., unpublished data). IB2, instead, could bind to p38δ and facilitate p38δ activation [(26), and this paper]. Furthermore, the amino-terminal variant JIP-2 binds to p38δ as effectively as does IB2 in 293T cells (J.S., unpublished data). We suggest that the differences between our findings and those of Yasuda et al. (24) may result from different scaffold/kinase interactions in different cellular contexts.
The IB2 scaffold and FHFs are principally expressed in the nervous system (25,28) along with several known or putative scaffold-associated kinases, i.e. DLK, MLK3, JNK and p38δ (2,12,40). The assignment of specificity of IB2 scaffold towards the JNK or p38δ kinases is based so far on evidence obtained in overexpression assays in non-neuronal tissue culture cells. While interaction of endogenous IB2 with FHFs in the brain has been validated (26), identification of the actual MAPKs assembled by IB2 in vivo still await confirmation.
Kinase interactions with IB scaffolds
A short aminoterminal segment of IB1 (JIP-1B, residues 152–162) has been described as its JNK-binding domain (18,20). Although this segment bears 55% sequence identity to a corresponding segment of IB2 (residues 106–116), we failed to detect direct binding of p38δ to this homology region in IB2. The p38δ-binding domain of IB2 includes residues C-terminal to the major FHF-binding domain, which are related to IB1 (homology regions II, III). In other words, despite well-aligned regions of sequence homology, the MAPK-binding segments of IB2 and IB1 appear unrelated in position and sequence. Despite these unanticipated findings, it is still possible that IB1 and IB2 fold to create similar MAPK binding surfaces consisting of N-terminal region I and more C-terminal elements; within this common fold, IB2 and IB1 could differ in the residues which primarily contribute to stabilizing interactions with their respective kinases. We also note that the ability of IB2 to homooligomerize or heterooligomerize with IB1 (JIP-1) [(24); and J. S., unpublished data] raises the possilibity that these scaffolds may coordinately activate both JNK and p38δ.
IB kinase scaffolds were initially modelled as bearing multiple domains each able to bind all kinases in a cascade simultaneously (39). However, IB scaffolds may bind kinases sequentially, as JNK binding to IB1 decreases the affinity of DLK or MLK3 to the scaffold by an unknown mechanism, resulting in their activation (21). Preliminary studies have identified the C-terminal half of IB2(JIP-2) containing homology domains II and III, as the binding region for MLK3, MKK7 (24) and p38δ (this study). Our preliminary analysis in 293T cells, using IB2 mutants lacking either domain II or III, has failed to assign independent separable binding sites for each of these kinases; each of these mutants (IB2Δ459–529 and IB2Δ514–797) displayed reduced binding to both p38δ (Figure 4B, C) and MLK3 (J.S., unpublished data). In other words, homology domains II and III may contribute to a binding surface for multiple kinases. Our findings leave open the possibility that IB2 scaffold binding sites for different kinases overlap and preclude simultaneous occupancy. If so, a signaling unit containing multiple kinases may be assembled through oligomerization of IB2 chains, each harboring different kinases.
FHFs function through interactions with intracellular proteins
Although FHFs bear substantial sequence homology with the core region of FGFs, FHFs complex with IB2 to recruit p38δ, pointing towards an intracellular role in MAPK signaling (26). The relationship of FHFs to protein phosphorylation is now further supported by additional findings: 1) FHF2 positively regulates IB2 function, facilitating IB2’s ability to mediate the activation of p38δ, and 2) FHF is a phosphoprotein in vivo.
Analysis of truncated IB2 proteins has defined the C-terminal 360 residues of IB2 as possessing the binding site for p38δ (Figure 4B, C), along with a minor binding site for FHF (Figure 4D). Hence, IB2Δ1–436 can form heterotrimeric complexes with FHF and p38δ, and FHF can potentiate IB2Δ1–436/p38δ interaction. More upstream IB2 sequences, within a region bearing no sequence homology to IB1, are required for strong interaction with FHF. Potentially, FHFs may interact with both major and minor FHF binding domains on IB2 to promote a stronger affinity of the complex towards p38δ.
Whereas IB2 is expressed in apparently all neurons of the central and peripheral nervous systems (M.G., unpublished data), FHF expression is restricted to subsets of neurons (27,28,32). Regulation of FHF expression in neurons may be a direct stimulus for MAPK activation on IB2. Alternatively, FHF may serve as cofactor for MAPK recruitment to IB2, allowing for kinase activation by other stimuli in FHF-positive neurons. Furthermore, FHF phosphorylation in brain and by p38δ in vitro opens to speculation the possibility that FHF is a target of IB2-scaffolded kinases.
Potential function of the IB2 signaling module
The JNK scaffold IB1 is essential for stress-induced apoptosis in hippocampal neurons and has been suggested to connect kinesin motors for microtubular transport to vesicular cargos. By contrast, the biological functions of IB2 are currently unknown. Here we speculate regarding IB2 function, based upon current knowledge of IB1 function together with our findings.
Kainate stress results in JNK- and c-Jun-mediated neuronal apoptosis in brain that is dependent upon IB1. IB1 is also required for activation of JNK and subsequent apoptosis induced by anoxia combined with glucose deprivation in brain and hippocampal neurons, as may happen during ischemia (2,41). By contrast, IB1 is not required for UV light- or anisomycin-induced JNK activation in hippocampal neurons (41), indicating that specific SAPK pathways are engaged by different stress stimuli. We speculate that IB2 may play a role in neuronal pathophysiological stress responses in reaction to specific stimuli that might not engage IB1. If so, IB2 and FHFs may direct signaling towards a different output as compared to IB1 through regulation of p38δ.
Trafficking of IB1 to growth cones in differentiating neurons depends upon IB1 interaction with the cargo-binding domain of the light-chain subunit of kinesin I, a major motor protein for axonal transport (42). As IB1 also interacts with the intracellular domains of transmembrane proteins in vitro (43,44), it has been inferred that the IB1 scaffold might connect kinesin motors to vesicular cargos (42,45,46). This might illustrate a more generalized function for kinase scaffolds, as a role of the structurally unrelated JNK-scaffold JIP-3 in axonal transport has been firmly established (47,48). Directed transport of IB1 may serve to deliver preformed signaling complexes to defined intracellular locations. Alternatively, IB1-associated kinases may regulate the transport process itself or association of the scaffold to vesicular cargo (42).
Consistent with kinesin-dependent trafficking of IB2 to a peripheral location in neuronal cells (42) and conservation of the amino acids required for kinesin binding in IB scaffolds, IB2 can be assumed to be transported along microtubules by mechanisms similar to IB1. However, IB2 may not function identically to IB1. The localization of IB2 is overlapping with but distinct from IB1 in pancreatic β cells (24) and in the axons of cerebral cortical neurons (M.G., unpublished data). Moreover, the affinity of IB2 for at least some transmembrane proteins in putative vesicular cargos is different from IB1 (49). Finally, IB2 possesses the unique property of regulating p38δ in conjunction with FHFs, potentially allowing regulation of distinct targets such as microtubule-associated proteins involved in transport (13,14). Altogether IB2 might add to regulation of intracellular traffic and, in conjunction with FHFs, to localized SAPK signaling as part of the pleiotropic cellular responses to pathophysiological stress.
References
- 1.Tibbles LA, Woodgett JR. Cell Mol Life Sci. 1999;55:1230–1254. doi: 10.1007/s000180050369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davis RJ. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. http://www.cell.com/cgi/content/full/103/2/239. [DOI] [PubMed] [Google Scholar]
- 3.Nebreda AR, Porras A. Trends Biochem Sci. 2000;25:257–260. doi: 10.1016/s0968-0004(00)01595-4. http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6TCV-40GH8K7-1&_user=30742&_coverDate=06%2F01%2F2000&_alid=46147934&_rdoc=1&_fmt=full&_orig=search&_qd=1&_cdi=5180&_sort=d&_acct=C000000333&_version=1&_urlVersion=0&_userid=30742&md5=771bae0e13f160ed1568894363123ddd. [DOI] [PubMed] [Google Scholar]
- 4.Treisman R. Curr Opin Cell Biol. 1996;8:205–215. doi: 10.1016/s0955-0674(96)80067-6. [DOI] [PubMed] [Google Scholar]
- 5.Widmann C, Gibson S, Jarpe MB, Johnson GL. Physiol Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. http://physrev.physiology.org/cgi/content/full/79/1/143. [DOI] [PubMed] [Google Scholar]
- 6.Dong C, Yang DD, Wysk M, Whitmarsh AJ, Davis RJ, Flavell RA. Science. 1998;282:2092–2095. doi: 10.1126/science.282.5396.2092. http://www.sciencemag.org/cgi/content/full/282/5396/2092. [DOI] [PubMed] [Google Scholar]
- 7.Huot J, Houle F, Rousseau S, Deschesnes RG, Shah GM, Landry J. J Cell Biol. 1998;143:1361–1373. doi: 10.1083/jcb.143.5.1361. http://www.jcb.org/cgi/content/full/143/5/1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rousseau S, Houle F, Landry J, Huot J. Oncogene. 1997;15:2169–2177. doi: 10.1038/sj.onc.1201380. [DOI] [PubMed] [Google Scholar]
- 9.Zhu T, Lobie PE. J Biol Chem. 2000;275:2103–2114. doi: 10.1074/jbc.275.3.2103. http://www.jbc.org/cgi/content/full/275/3/2103. [DOI] [PubMed] [Google Scholar]
- 10.Goedert M, Cuenda A, Craxton M, Jakes R, Cohen P. Embo J. 1997;16:3563–3571. doi: 10.1093/emboj/16.12.3563. http://emboj.oupjournals.org/cgi/content/full/16/12/3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang Y, Gram H, Zhao M, New L, Gu J, Feng L, di Padova F, Ulevitch RJ, Han J. J Biol Chem. 1997;272:30122–30128. doi: 10.1074/jbc.272.48.30122. http://www.jbc.org/cgi/content/full/272/48/30122. [DOI] [PubMed] [Google Scholar]
- 12.Wang XS, Diener K, Manthey CL, Wang S, Rosenzweig B, Bray J, Delaney J, Cole CN, Chan-Hui PY, Mantlo N, Lichenstein HS, Zukowski M, Yao Z. J Biol Chem. 1997;272:23668–23674. doi: 10.1074/jbc.272.38.23668. http://www.jbc.org/cgi/content/full/272/38/23668. [DOI] [PubMed] [Google Scholar]
- 13.Parker CG, Hunt J, Diener K, McGinley M, Soriano B, Keesler GA, Bray J, Yao Z, Wang XS, Kohno T, Lichenstein HS. Biochem Biophys Res Commun. 1998;249:791–796. doi: 10.1006/bbrc.1998.9250. http://www.idealibrary.com/links/citation/0006-291X/249/791. [DOI] [PubMed] [Google Scholar]
- 14.Goedert M, Hasagawa M, Jakes R, Lawler S, Cuenda A, Cohen P. FEBS Lett. 1997;409:57–62. doi: 10.1016/s0014-5793(97)00483-3. http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6T36-3TGVYT1-F&_coverDate=06%2F02%2F1997&_alid=46149447&_rdoc=1&_fmt=summar. [DOI] [PubMed] [Google Scholar]
- 15.Knebel A, Morrice N, Cohen P. EMBO J. 2001;20:4360–4369. doi: 10.1093/emboj/20.16.4360. http://emboj.oupjournals.org/cgi/content/full/20/16/4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu MC, Wang YP, Mikhail A, Qiu WR, Tan TH. J Biol Chem. 1999;274:7095–7102. doi: 10.1074/jbc.274.11.7095. http://www.jbc.org/cgi/content/full/274/11/7095. [DOI] [PubMed] [Google Scholar]
- 17.Garrington TP, Johnson GL. Curr Opin Cell Biol. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6VRW-3WWCKH7-8&_coverDate=04%2F30%2F1999&_alid=46149838&_rdoc=1&_fmt=summary&_orig=search&_qd=1&_cdi=6245&_sort=d&_acct=C000000333&_version=1&_urlVersion=0&_userid=30742&md5=8befd8844b0062de58a7fae0debab17a. [DOI] [PubMed] [Google Scholar]
- 18.Dickens M, Rogers JS, Cavanagh J, Raitano A, Xia Z, Halpern JR, Greenberg ME, Sawyers CL, Davis RJ. Science. 1997;277:696–696. doi: 10.1126/science.277.5326.693. http://www.sciencemag.org/cgi/content/full/277/5326/693. [DOI] [PubMed] [Google Scholar]
- 19.Bonny C, Nicod P, Waeber G. J Biol Chem. 1998;273:1843–1846. doi: 10.1074/jbc.273.4.1843. http://www.jbc.org/cgi/content/full/273/4/1843. [DOI] [PubMed] [Google Scholar]
- 20.Whitmarsh AJ, Cavanagh J, Tournier C, Yasuda J, Davis RJ. Science. 1998;281:1671–1674. doi: 10.1126/science.281.5383.1671. http://www.sciencemag.org/cgi/content/full/281/5383/1671. [DOI] [PubMed] [Google Scholar]
- 21.Nihalani D, Meyer D, Pajni S, Holzman LB. Embo J. 2001;20:3447–3458. doi: 10.1093/emboj/20.13.3447. http://www.emboj.org/cgi/content/full/20/13/3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonny C, Oberson A, Steinmann M, Schorderet DF, Nicod P, Waeber G. J Biol Chem. 2000;275:16466–16472. doi: 10.1074/jbc.M908297199. http://www.jbc.org/cgi/content/full/275/22/16466. [DOI] [PubMed] [Google Scholar]
- 23.Tawadros T, Formenton A, Dudler J, Thompson N, Nicod P, Leisinger HJ, Waeber G, Haefliger JA. J Cell Sci. 2002;115:385–393. doi: 10.1242/jcs.115.2.385. http://jcs.biologists.org/cgi/content/full/115/2/385. [DOI] [PubMed] [Google Scholar]
- 24.Yasuda J, Whitmarsh AJ, Cavanagh J, Sharma M, Davis RJ. Mol Cell Biol. 1999;19:7245–7254. doi: 10.1128/mcb.19.10.7245. http://mcb.asm.org/cgi/content/full/19/10/7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Negri S, Oberson A, Steinmann M, Sauser C, Nicod P, Waeber G, Schorderet DF, Bonny C. Genomics. 2000;64:324–330. doi: 10.1006/geno.2000.6129. http://www.idealibrary.com/links/citation/0888-7543/64/324. [DOI] [PubMed] [Google Scholar]
- 26.Schoorlemmer J, Goldfarb M. Curr Biol. 2001;11:793–797. doi: 10.1016/s0960-9822(01)00232-9. http://www.current-biology.com/cgi/content/full/11/10/793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smallwood PM, Munoz-Sanjuan I, Tong P, Macke JP, Hendry SH, Gilbert DJ, Copeland NG, Jenkins NA, Nathans J. Proc Natl Acad Sci U S A. 1996;93:9850–9857. doi: 10.1073/pnas.93.18.9850. http://www.pnas.org/cgi/reprint/93/18/9850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hartung H, Feldman B, Lovec H, Coulier F, Birnbaum D, Goldfarb M. Mech Dev. 1997;64:31–39. doi: 10.1016/s0925-4773(97)00042-7. http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6T9H-3RJXPWM-4&_user=30742&_coverDate=06%2F30%2F1997&_rdoc=4&_fmt=summary&_orig=browse&_srch=%23toc%235115%231997%23999359998%23497!&_cdi=5115&_sort=d&_docanchor=&_acct=C000000333&_version=1&_urlVersion=0&_userid=30742&md5=f9273f46de9c7a13907f24ffa36c46af. [DOI] [PubMed] [Google Scholar]
- 29.Coulier F, Pontarotti P, Roubin R, Hartung H, Goldfarb M, Birnbaum D. J Mol Evolution. 1997;44:43–56. doi: 10.1007/pl00006120. http://link.springer-ny.com/link/service/journals/00239/papers97/44n1p43.html. [DOI] [PubMed] [Google Scholar]
- 30.Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, Gao G, Goldfarb M. J Biol Chem. 1996;271:15292–15297. doi: 10.1074/jbc.271.25.15292. http://www.jbc.org/cgi/content/full/271/25/15292. [DOI] [PubMed] [Google Scholar]
- 31.Verdier AS, Mattei MG, Lovec H, Hartung H, Goldfarb M, Birnbaum D, Coulier F. Genomics. 1997;40:151–154. doi: 10.1006/geno.1996.4492. [DOI] [PubMed] [Google Scholar]
- 32.Wang Q, McEwen DG, Ornitz DM. Mech Dev. 2000;90:283–287. doi: 10.1016/s0925-4773(99)00241-5. http://www.elsevier.com:80/cgi-bin/cas/tree/store/mod/cas_sub/browse/browse.cgi?year=2000&volume=90&issue=2&aid=1305. [DOI] [PubMed] [Google Scholar]
- 33.Lechleider RJ, Freeman RMJ, Neel BG. J Biol Chem. 1993;268:13434–13438. [PubMed] [Google Scholar]
- 34.Schoorlemmer J, Goldfarb M. Supplement Materials: Curr Biol. 2001 doi: 10.1016/s0960-9822(01)00232-9. http://images.cellpress.com/supmat/cub/11jSchoorlemmer_793.pdf. [DOI] [PMC free article] [PubMed]
- 35.Tournier C, Whitmarsh AJ, Cavanagh J, Barrett T, Davis RJ. Mol Cell Biol. 1999;19:1569–1581. doi: 10.1128/mcb.19.2.1569. http://mcb.asm.org/cgi/content/full/19/2/1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanchez MP, Tapley P, Saini SS, He B, Pulido D, Barbacid M. Proc Natl Acad Sci USA. 1994;91:1819–1823. doi: 10.1073/pnas.91.5.1819. http://www.pnas.org/cgi/reprint/91/5/1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coso OA, Chiariello M, Yu JC, Teramoto H, Crespo P, Xu N, Miki T, Gutkind JS. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- 38.Munoz-Sanjuan I, Smallwood PM, Nathans J. J Biol Chem. 2000;275:2589–2597. doi: 10.1074/jbc.275.4.2589. http://www.jbc.org/cgi/content/full/275/4/2589. [DOI] [PubMed] [Google Scholar]
- 39.Elion EA. Science. 1998;281:1625–1626. doi: 10.1126/science.281.5383.1625. [DOI] [PubMed] [Google Scholar]
- 40.Xu Z, Maroney AC, Dobrzanski P, Kukekov NV, Greene LA. Mol Cell Biol. 2001;21:4713–4724. doi: 10.1128/MCB.21.14.4713-4724.2001. http://mcb.asm.org/cgi/content/full/21/14/4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Whitmarsh AJ, Kuan CY, Kennedy NJ, Kelkar N, Haydar TF, Mordes JP, Appel M, Rossini AA, Jones SN, Flavell RA, Rakic P, Davis RJ. Genes Dev. 2001;15:2421–2432. doi: 10.1101/gad.922801. http://www.genesdev.org/cgi/content/full/15/18/2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verhey KJ, Meyer D, Deehan R, Blenis J, Schnapp BJ, Rapoport TA, Margolis B. J Cell Biol. 2001;152:959–970. doi: 10.1083/jcb.152.5.959. http://www.jcb.org/cgi/content/full/152/5/959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gotthardt M, Trommsdorff M, Nevitt MF, Shelton J, Richardson JA, Stockinger W, Nimpf J, Herz J. J Biol Chem. 2000;275:25616–25624. doi: 10.1074/jbc.M000955200. http://www.jbc.org/cgi/content/full/275/33/25616. [DOI] [PubMed] [Google Scholar]
- 44.Matsuda S, Yasukawa T, Homma Y, Ito Y, Niikura T, Hiraki T, Hirai S, Ohno S, Kita Y, Kawasumi M, Kouyama K, Yamamoto T, Kyriakis JM, Nishimoto I. J Neurosci. 2001;21:6597–6607. doi: 10.1523/JNEUROSCI.21-17-06597.2001. http://www.jneurosci.org/cgi/content/full/21/17/6597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goldstein LS. Science. 2001;291:2102–2103. doi: 10.1126/science.1059766. http://www.sciencemag.org/cgi/content/full/291/5511/2102. [DOI] [PubMed] [Google Scholar]
- 46.Hollenbeck PJ. J Cell Biol. 2001;152:F25–28. doi: 10.1083/jcb.152.5.f25. http://www.jcb.org/cgi/content/full/152/5/F25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bowman AB, Kamal A, Ritchings BW, Philp AV, McGrail M, Gindhart JG, Goldstein LS. Cell. 2000;103:583–594. doi: 10.1016/s0092-8674(00)00162-8. http://www.cell.com/cgi/content/full/103/4/583. [DOI] [PubMed] [Google Scholar]
- 48.Byrd DT, Kawasaki M, Walcoff M, Hisamoto N, Matsumoto K, Jin Y. Neuron. 2001;32:787–800. doi: 10.1016/s0896-6273(01)00532-3. http://www.neuron.org/cgi/content/full/32/5/787. [DOI] [PubMed] [Google Scholar]
- 49.Taru H, Iijima KI, Hase M, Kirino Y, Yagi Y, Suzuki T. J Biol Chem. 2002;277:20070–20078. doi: 10.1074/jbc.M108372200. http://www.jbc.org/cgi/content/full/277/22/20070. [DOI] [PubMed] [Google Scholar]