

Abstract
Noonan syndrome is a genetic multisystem disorder characterised by distinctive facial features, developmental delay, learning difficulties, short stature, congenital heart disease, renal anomalies, lymphatic malformations, and bleeding difficulties. Mutations that cause Noonan syndrome alter genes encoding proteins with roles in the RAS–MAPK pathway, leading to pathway dysregulation. Management guidelines have been developed. Several clinically relevant genotype–phenotype correlations aid risk assessment and patient management. Increased understanding of the pathophysiology of the disease could help development of pharmacogenetic treatments.
Introduction
Noonan syndrome is an autosomal dominant, variably expressed, multisystem disorder with an estimated prevalence of 1 in 1000–2500.1 It was characterised by Jacqueline Noonan, who reported nine patients with pulmonary valve stenosis, small stature, hypertelorism, mild intellectual disability, ptosis, undescended testes, and skeletal malformations.2 Understanding of the molecular genetic causes of Noonan syndrome has increased greatly in the past decade, enabling study of the pathophysiological mechanisms underlying the varied medical and developmental features of the disorder.
The RAS–MAPK pathway is a well-studied, widely important signal transduction pathway through which extracellular ligands—such as some growth factors, cytokines, and hormones—stimulate cell proliferation, differentiation, survival, and metabolism (figure 1). Cell surface receptors are phosphorylated at sites within their cytoplasmic region after ligand binding. This binding leads to recruitment of adaptor proteins (eg, GRB2), which form a constitutive complex with guanine nucleotide exchange factors (eg, SOS) that convert inactive, GDP-bound RAS to its active GTP-bound form. Activated RAS proteins then activate the RAF–MEK–ERK cascade through a series of phosphorylation events, ending with activated ERK entering the nucleus to alter gene transcription and modulating the activity of cytoplasmic targets to cause the appropriate short-term and long-term cellular response to the stimulus. All the genes implicated in Noonan syndrome encode proteins integral to this pathway and disease-causing mutations usually enhance signal flow through this pathway.3,4
Figure 1. The RAS–MAPK signaling pathway.
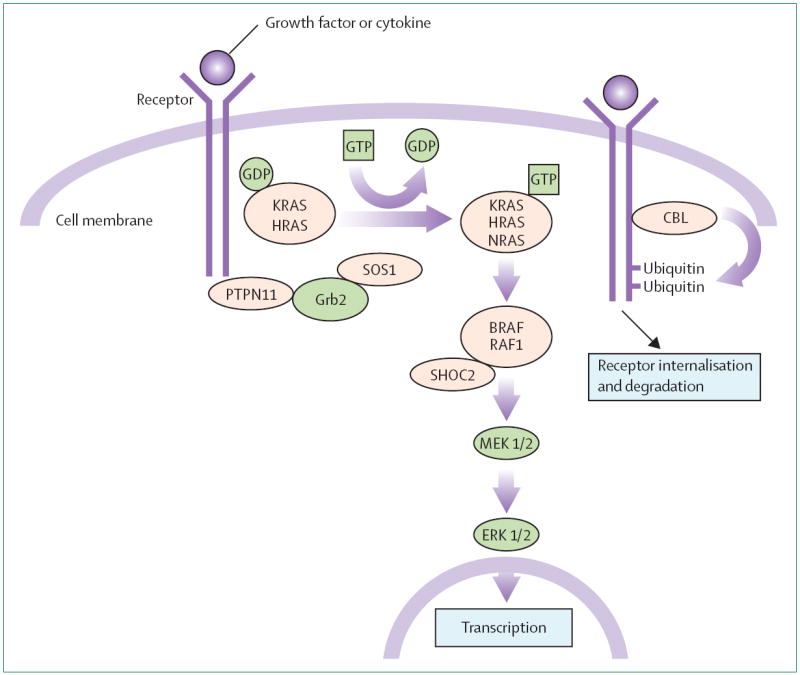
Growth signals are relayed from activated growth factor receptors to the nucleus. Mutations in PTPN11, KRAS, SOS, NRAS, and RAF1 are associated with Noonan syndrome and mutations in SHOC2 and CBL are associated with a Noonan syndrome-like phenotype.
Organ dysfunction
Roughly 10% of affected individuals have auditory deficits in the low frequency range caused by sensorineural hearing loss and 25% have deficits in the high frequency range.5 Inner ear structural abnormalities, including temporal bone abnormalities, have been reported.6,7
Noonan syndrome is the second most common syndromic cause of congenital heart disease, exceeded in prevalence only by trisomy 21.8 Several cardiovascular phenotypes occur in Noonan syndrome. The most common are pulmonary stenosis (often with dysplastic valves; 50–60%), hypertrophic cardiomyopathy (20%), and secundum atrial septal defect (6–10%), but ventricular septal defect, peripheral pulmonary stenosis, atrioventricular canal, aortic stenosis, mitral valve abnormalities, aortic coarctation, and coronary artery anomalies have also been noted.9-11 Hypertrophic cardiomyopathy can be mild or severe and can present from the prenatal period to late childhood. Almost 25% of patients die because of heart failure in the first year, although the rate of sudden death is lower than that for familial hypertrophic cardiomyopathy.9,10 Electrocardiograms often display wide QRS complexes with a predominantly negative pattern in the left precordial leads and left axis deviation with giant Q waves,11 sometimes even when the heart and chest wall are structurally normal. Cerebrovascular anomalies have been associated with Noonan syndrome, including arteriovenous malformations, aneurysms, hypoplasia of the posterior vessels, and moyamoya.12,13
Abnormal pigmentation can occur, including multiple pigmented nevi, café au lait spots, and lentigines. Keratosis pilaris of the upper arms and face is common and can impede hair and eyebrow growth.14 Hair is often thick and curly, although thin, sparse hair has also been reported.
Birthweight and body length are usually normal. Short stature is a common manifestation of Noonan syndrome although adult height is not always adversely affected. Because the pubertal growth spurt is often attenuated or delayed, the prevalence of short stature in Noonan syndrome is greatest during the age of normal puberty. Furthermore, bone ageing is delayed. Growth of many patients catches up in patient’s late teens. Growth hormone deficiency, neurosecretory dysfunction, and growth hormone resistance can occur in Noonan syndrome.15-17 Patients with PTPN11-associated Noonan syndrome often have normal or slightly increased concentrations of growth hormone and low concentrations of IGF1.17 A Ptpn11 Noonan syndrome mouse model had increased ERK activation in response to growth hormone; a potential cause of short stature and a promising treatment target. Growth improves significantly when ERK1/2 is inhibited in these mice.18
In 2007, the US Food and Drug Administration approved treatment of short stature caused by Noonan syndrome with recombinant human growth hormone in doses of up to 0·066 mg/kg per day. However, growth hormone treatment for Noonan syndrome is still controversial. Data are difficult to compare because of differing protocols and outcome criteria.19-21 In a report of final adult height of 73 patients with a clinical diagnosis of Noonan syndrome, 21 had an adult height in the normal range.22 Analysis of data from 370 children with Noonan syndrome in the National Cooperative Growth Study23—a post-marketing observational study started in 1985—showed a mean incremental height gain in near adult height for men of 10·9 cm higher than that projected in patients treated with recombinant human growth hormone and 9·2 cm in women.23 Because children treated with recombinant human growth hormone might also have or be at risk for hypertrophic cardiomyopathy or haematological malignancy, patients should be monitored for adverse complications. Increased biventricular hypertrophy has been reported in two patients and hypertrophic cardiomyopathy in another in the National Cooperative Growth Study cohort.
Mean age at onset of puberty is delayed in patients with Noonan syndrome compared with the general population; 35% of boys enter puberty after age 13·5 years and 44% of girls enter puberty after age 13 years.23 Case reports suggest that adults might be at risk of osteopenia associated with male hypo-oestrogenism and the resultant increased bone resorption.24
Most infants with Noonan syndrome have feeding difficulties that can lead to failure to thrive. Poor suck, prolonged feeding time, or recurrent vomiting have been reported and about 25% of infants need to be fed by tube for 2 weeks or longer.10,14 Gastroesophageal reflux is also common. These issues resolve in most affected children by age 15 months. Case reports exist of intestinal malrotation, immature gut motility, and delayed gastrointestinal motor development.
Up to 80% of boys diagnosed with Noonan syndrome have unilateral or bilateral cryptorchidism. Male gonadal dysfunction has been reported and is suggested to be caused by primary Sertoli cell dysfunction rather than cryptorchidism.25 Fertility is not impaired in women with Noonan syndrome. 10% of patients with Noonan syndrome have renal abnormalities but most do not need treatment.14
Although infants with Noonan syndrome are predisposed to several haematological abnormalities, including transient monocytosis, thrombocytopenia, and myeloproliferative disorder,26 the most common haematologic disorders are abnormalities of bleeding caused by coagulation defects. In a study of 72 individuals with Noonan syndrome, 65% had abnormal bruising or bleeding, 40% had prolonged activated partial thromboplastin time, and 50% had abnormalities of the intrinsic pathway.27 Factor deficiencies and platelet defects affect about a third of patients.27,28 Patients should consult a haematologist before having invasive procedures to ensure that complications from bleeding are minimised.29
Lymphatic abnormalities, most commonly peripheral lymphoedema, are present in less than 20% of individuals but can cause substantial morbidity. Peripheral lymphoedema can occur in infants and resolves in the first few years of life or it can develop in adolescence or adulthood.30 Less commonly reported are hydrops fetalis; pulmonary, testicular, or intestinal lymphangiectasia; chylous effusions of the pleural space and peritoneum; hypoplastic leg lymphatic vessels; anomalous thoracic cage lymphatic vessels; aplasia or absence of the thoracic duct; hypoplastic inguinal and iliac lymphatic vessels; and lymphoedema of the scrotum or vulva.31,32
Multiple giant cell lesions are associated with Noonan syndrome, caused by mutations in PTPN11 or SOS1.33,34 Pigmented villonodular synovitis—a proliferative synovial lesion involving joints, tendons, and bursae—has been reported in patients with Noonan syndrome and, unlike in isolated cases, is often polyarticular.33,35 An estimated 30% of children have a spinal deformity with surgical correction recommended in two thirds of cases.36 Chest deformity (superior pectus carinatum and inferior pectus excavatum), widely spaced nipples, cubitus valgus, and genu valgum have also been reported.14,37
In most affected individuals, intelligence is within the normal range, with intelligence quotient generally varying between 70 and 120.38,39 In one study,40 six of ten individuals had a significant difference between verbal and nonverbal IQ but without a consistent pattern. In these patients, social cognition was moderately impaired, with particular deficits in recognition of emotions, leading to alexithymia (an inability to express emotions verbally). In small group analyses, mood disturbances, social difficulties, communication difficulties, issues with executive function, attention deficit/hyperactivity disorder, and difficulties with social interaction have been reported.27,35-41 Studies of cognitive ability in individuals with clinically diagnosed Noonan syndrome suggest that the prevalence of intellectual impairment (intelligence quotient <70) is about 20%.38,42,43 Other studies have noted difficulties with social competence, especially emotional perception of self and others.37,39,40,42
A study of the language phenotype of children and adults with Noonan syndrome showed that language impairments are more common in people with the disease than in the general population and, when present, are associated with a high risk of reading and spelling difficulties.44 Results of a survey45 show that many patients with Noonan syndrome have impaired or below average performance in a delayed verbal free recall memory task, and that delayed visual recognition memory is recurrently compromised.
Several haematological cancers have been reported in patients with Noonan syndrome, particularly during childhood, at a proportion slightly above that in the general population, including juvenile myelomonocytic leukaemia, acute myelogenous leukaemia, and B-cell acute lymphoblastic leukaemia.26,46 Myeloproliferative disorder in patients with Noonan syndrome and juvenile myelomonocytic leukaemia is usually benign compared with patients with juvenile myelomonocytic leukaemia only.47-50
Cases of embryonal rhabdomyosarcoma (of the duodenum, bladder, urachus, orbit, vagina, and abdomen) have been reported, three in association with an SOS1 germline mutation.51-55 Granular cell tumour in an affected mother and child, pilocytic astrocytoma, and Sertoli tumour in a cryptorchid testis have also been reported.56 Three cases of neuroblastoma have been reported in.57,58 SOS1 germline mutation has also been reported in patients with Noonan syndrome with Sertoli cell testis tumour and granular cell tumours of the skin.51 Three cases of glial tumours in Noonan syndrome have been recorded, two in association with a germline PTPN11 mutation.59,60
A large study of a cohort of 297 Dutch patients with Noonan syndrome and a pathogenic PTPN11 mutation (mean age 18 years) calculated a 3·5 times increased risk of developing a cancer compared with the general population.61 Cases of a malignant mastocytosis and malignant epithelioid angiosarcoma have also been reported.
Up to 95% of affected individuals will have at least one characteristic eye finding including strabismus, refractive errors, amblyopia, or nystagmus. Two thirds of patients develop anterior chamber abnormalities including cataracts. Fundal changes, including optic head drusen, optic disk hypoplasia, colobomas, and myelinated nerves, occur in 20% of patients.14,62
Natural history
Long-term follow-up data are scarce. Shaw and colleagues10 studied 112 British individuals with a clinical or molecular genetic diagnosis of Noonan syndrome (mean age at assessment 25·3 years, mean follow-up 12 years) in the largest study of its kind. The mean final adult height was 1·70 m for men and 1·53 m for women. Feeding difficulties in infancy were an early marker of delayed language development and long-term educational achievement. Most patients gained a diploma: 43% gained a General Certificate of Secondary Education, 8% an A Level, and 16% had a higher educational qualification. Roughly a third of adults had attended a school for children with learning difficulties and another 20% attended a mainstream school but needed extra help. Of those adults who were no longer in school, 60% had a full-time job.
About a third of patients with pulmonary stenosis needed surgery or a repeat procedure. Those with hypertrophic cardiomyopathy had much the same yearly mortality rate as non-syndromic patients but with no sudden death and arrhythmia. Adults with pulmonary stenosis had disorders including severe pulmonary insufficiency, exercise intolerance, and right ventricular dysfunction.63 Adults need long-term cardiac follow-up because a third have ongoing cardiac disorders requiring drugs for heart failure or arrhythmias, a defibrillator, or a pacemaker.62 Positive prognostic features included absence of symptoms, normal resting cardiac output, peak right ventricular pressure of less than 100 mm Hg, and normal pulmonary artery pressure.10,38 Other cardiac complications reported in adults with Noonan syndrome include mild aortic insufficiency, substantial right ventricular outflow tract obstruction caused by subpulmonary or pulmonary valve stenosis, aortic root dilation, aortic dissection, giant aneurysm of the sinuses of Valsalva, dilated cardiomyopathy evolving from hypertrophic cardiomyopathy, restrictive cardiomyopathy, constrictive pericarditis, and idiopathic pulmonary hypertension.63
The mortality rate in the study by Shaw and colleagues was 9%, with age of death from a few months to 61 years. Four deaths were attributed to cardiac complications (severe hypertrophic cardiomyopathy in three, ischaemic heart disease in one).
Diagnosis
Many of the recognisable features of Noonan syndrome could be the consequence of lymphatic obstruction or dysfunction during development, including webbing of the neck and prominence of the trapezius, cryptorchidism, widely spaced nipples, low-set and posteriorly rotated ears, hypertelorism, and ptosis.38 Other key features of Noonan syndrome include congenital heart defects, superior pectus carinatum with inferior pectus excavatum, developmental delay, short stature, and lymphatic dysplasias. The facial features associated with Noonan syndrome change with age (table 1, figure 2).14,64
Table 1. Characteristic facial features of Noonan syndrome by age.
| Forehead, face, hair | Eyes | Ears | Nose | Mouth | Neck | |
|---|---|---|---|---|---|---|
| Newborn baby* | Tall forehead, low posterior hairline | Hypertelorism, downslanting palpebral fissures, epicanthal folds | ·· | Short and broad, depressed root, upturned tip | Deeply grooved philtrum, high wide peaks of the vermilion, micrognathia | Excessive nuchal skin |
| Infancy (2–12 months) | Large head, tall and prominent forehead | Hypertelorism, ptosis, or thick-hooded eyelids | ·· | Short, wide, depressed nasal root | ·· | ·· |
| Childhood (1–12 years) | Features might appear coarse, elongated face | ·· | ·· | ·· | ·· | ·· |
| Adolescence (12–18 years) | Myopathic facies | ·· | ·· | Bridge is high and thin | ·· | Webbing obvious |
| Adulthood (>18 years) | Distinguishing facial features are subtle, skin appears thin and transparent | ·· | ·· | Prominent nasolabial fold | ·· | ·· |
| All ages | ·· | Blue green irises, arched and diamond-shaped eyebrows | Low-set, posteriorly rotated, thick helices | ·· | ·· | ·· |
Features can be subtle or absent.
Figure 2. Five generations of a family with Noonan syndrome caused by an SOS1 mutation.

Distinguishing facial features become milder with age.
Prenatal features are nonspecific but include polyhydramnios, hydronephrosis, pleural effusion, oedema, cardiac defects, distended jugular lymphatic sacs, cystic hygroma, and increased nuchal translucency.65,66 For fetuses with normal chromosomes, Noonan syndrome will be diagnosed in roughly 1–3% of cases with first trimester nuchal oedema and in up to 10% of second trimester fetuses with a cystic hygroma.65,67-70
Eight genes in the RAS–MAPK signalling pathway cause Noonan syndrome or closely related conditions (PTPN11, SOS1, KRAS, NRAS, RAF1, BRAF, SHOC2, and CBL). Noonan syndrome has been linked to the chromosomal band 12q24.1 and PTPN11—which encodes the protein SHP2—is within this region. Because SHP2 has essential roles in signal transduction pathways that control several developmental processes, including cardiac semilunar valvulogenesis, PTPN11 was deemed an excellent candidate gene. Studies suggest that 50% of cases of Noonan syndrome are caused by missense, gain-of-function mutations in PTPN11.71-73 These missense mutations cluster in and around the interacting portions of the N terminus of SH2 and the phosphotyrosine phosphatase domains of SHP2, which are involved in switching the protein between its inactive and active conformations. Mutations typically perturb equilibrium, causing constitutive or prolonged activation of the protein.72,73 Somatically acquired PTPN11 mutations occur in haematological cancers.74 However, some aminoacid substitutions are specifically or preferentially associated with Noonan syndrome or cancer, and most germline mutations that cause Noonan syndrome are less able to cause SHP2 gain of function than are somatic mutations associated with leukaemia.50
Noonan syndrome in patients with PTPN11 mutations is more likely than Noonan syndrome without PTPN11 mutations to be familial, to be associated with pulmonary stenosis or atrial septal defect (ostium secundum type), bleeding diathesis, and juvenile myelomonocytic leukaemia, and is negatively associated with hypertrophic cardiomyopathy and coarctation of the aorta (table 2).73,75-77 The most common features include cardiac defects (74%), low set ears (80%), low posterior hairline (68%), down-slanting palpebral fissures (68%), cryptorchidism (94% of boys), and short stature (93%).75,76,78
Table 2. Correlations between genotype and phenotype in Noonan syndrome.
| Cardiovascular | Growth | Developmental | Skin and hair | Other | |
|---|---|---|---|---|---|
| PTPN11 (roughly 50%) | More pulmonary stenosis; less hypertrophic cardiomyopathy, and atrial septal defect (ostium secundum type) | More short stature; lower IGF1 concentrations | Patients with N308D and N308S have little or no intellectual disability | ·· | More bleeding diathesis and juvenile myelomonocytic leukaemia |
| SOS1 (roughly 10%) | Less atrial septal defect | Less short stature | Less intellectual disability, language delays | Similar to cardiofaciocutaneous syndrome | ·· |
| RAF1 (roughly 10%) | More hypertrophic cardiomyopathy | ·· | ·· | More naevi, lentigines, café au lait spots | ·· |
| KRAS (<2%) | ·· | ·· | More severe cognitive delay | Similar to cardiofaciocutaneous syndrome | ·· |
| NRAS (<1%) | ·· | ·· | ·· | ·· | ·· |
Percentages in parentheses are the proportion of patients with Noonan syndrome who have the mutation.
RAS proteins act as molecular switches, cycling between inactive GDP-bound and active, GTP-bound states.79 In its active form, RAS can interact with more than 20 effectors, which activates the MEK–ERK cascade and promotes several cellular processes. Germline mutations in KRAS and NRAS have been detected in patients with Noonan syndrome, each accounting for less than 2% of cases.80-82 Detailed biochemical and structural analyses have shown that germline KRAS mutations generally confer milder gain-of-function effects than do somatically acquired cancer-associated mutations.83
19 cases of Noonan syndrome have been caused by a KRAS mutation.80,82,84-86 Early studies suggested patients with KRAS mutations are more severely medically and cognitively affected than most patients with Noonan syndrome (table 2).80 In 2009, Kratz and colleagues87 reported the unusual feature of craniosynostosis and a missense KRAS mutation in two unrelated patients with Noonan syndrome.87 85% of patients with Noonan syndrome who have KRAS mutations have cardiac involvement,84 and all have mild-to-moderate intellectual disability.86
In an analysis of 917 individuals with Noonan syndrome or a related disorder, four unrelated individuals (three de novo and one familial segregating with the mutation) had one of two mutations in NRAS (Cys149Thr or Gly179Ala).81 Too few cases have been reported to establish genotype–phenotype correlations.
The protein product of SOS1 is a guanine nucleotide exchange factor that activates RAS proteins by displacing GDP, enabling GTP to bind through mass action.88,89 SOS1 is a complex protein with several domains that participate in autoinhibition. Missense mutations in SOS1 account for roughly 10% of cases of Noonan syndrome.90,91 The altered aminoacid residues might relieve autoinhibition, leading to constitutive activation.92-94
SOS1-associated Noonan syndrome is characterised by a higher prevalence of ectodermal abnormalities but less intellectual disability, short stature, and atrial septal defect than does PTPN11-associated Noonan syndrome (table 2).92,93,95 In a cognitive assessment of 65 people with Noonan syndrome (aged 4–18 years), those with SOS1 mutations (n=6) scored within the average range or higher and did not differ from people without Noonan syndrome. The SOS1 group scored significantly higher than the PTPN11 group on both verbal and nonverbal cognitive scales.
The RAF–MEK–ERK cascade is the best characterised RAS effector pathway. Three RAF serine-threonine kinases (ARAF, BRAF, and RAF1) activate the MEK–ERK cascade.96-98 Mutations in RAF1 cause Noonan syndrome in 5–15% of people.99,100 Because phosphorylation of Ser259 contributes to RAF1’s autoinhibition, mutations that prevent phosphorylation at that site cause gain of function of the kinase. Mutations affecting RAF1’s activation loop reduce activity of the kinase.90,99
Hypertrophic cardiomyopathy is over-represented in RAF1-associated Noonan (75% vs 20% overall) and seems to be allele specific. It is associated with mutations in the Ser259 and Ser621 hotspots.91,99 Multiple naevi, lentigines, or café au lait spots are reported in a third of patients with RAF1-associated Noonan syndrome (table 2).
Although most commonly associated with the phenotypically related cardiofaciocutaneous syndrome, mutations in BRAF have also been reported in individuals meeting clinical diagnostic criteria for Noonan syndrome, though rarely.100-102 BRAF mutation-associated Noonan syndrome is characterised by neonatal growth failure, feeding difficulties, short stature, dolichocephaly, distinctive facial features, multiple naevi and dark-coloured lentigines, mild-to-moderate cognitive deficits, skeletal anomalies, reduced muscle tone, and absence of cardiofaciocutaneous syndrome-related skin anomalies, polyhydramnios, and hypertrophic cardiomyopathy.102
Genetic testing is widely available and chip-based sequencing, which enables the simultaneous assessment of all genes related to diseases caused by changes in RAS, has reduced the cost compared with individual sequencing of each gene. Genetic testing can be useful in several scenarios. Because the presentation of cardiofaciocutaneous and Costello syndromes overlaps substantially in the first year of life, genotyping can aid diagnosis. If a patient has a mild or atypical presentation, genotyping could establish the diagnosis. For an adult with suspected Noonan syndrome, establishing the molecular genetic cause will enable preimplantation, prenatal, or postnatal testing if desired. The specific genotype of a child with Noonan syndrome is useful to know in order to provide specific guidance—for example, to address the increased prevalence of hypertrophic cardiomyopathy in RAF1-associated Noonan syndrome or short stature and growth hormone abnormalities in PTPN11-associated Noonan syndrome. Rarely, a variant of unknown importance might be detected by gene sequencing. Testing family members to see if the variant segregates only with those affected can help to establish pathogenicity.
Management and differential diagnosis
Management guidelines for Noonan syndrome have been developed (table 3).38,103,104 Although the facial features and many of the medical and developmental complications associated with Noonan syndrome-like disorders are the same for Noonan syndrome, mutations in two genes—SHOC2 and CBL—are associated with additional features not seen in Noonan syndrome.
Table 3.
Management guidelines by system
| At diagnosis | After diagnosis | If symptomatic | |
|---|---|---|---|
| General | Complete physical and neurological examination; medical genetics consultation to confirm diagnosis, consider molecular genetic testing and genetic counselling | Yearly complete physical and neurological examination; return to geneticist if genotype negative or for multisystem assessment; genetic counselling at adolescence or when a young adult | Orchiopexy by age 1 year for cryptorchidism; if lymphoedema, refer to specialty clinic; brain and cervical spine MRI if intracranial pressure increases; electroencephalogram and referral to neurologist if seizures suspected |
| Developmental | Multidisciplinary developmental assessment | Developmental screening yearly for children aged 5–18 years | Neuropsychologist testing if screening abnormal; referral to early intervention if delays detected before age 3 years; individual education plan for children aged 5–18 years with delays |
| Dental | First dental assessment between age 1 year and 2 years | Yearly dental assessment | ·· |
| Growth and feeding | Plot growth on curves for Noonan syndrome | Plot growth on curves for Noonan syndrome three times per year until age 3 years, then yearly | Refer to gastroenterologist for feeding problems or recurrent vomiting, or if evidence of growth failure without comorbid cause exists; thyroid function tests if signs or symptoms of hypothyroidism |
| Cardiovascular | Cardiac examination, electrocardiogram, echocardiogram | Follow up on the basis of initial findings. If initial assessment normal, repeat every 5 years | ·· |
| Ophthalmological | Baseline eye examination Baseline audiology examination | Repeat every 2 years, sooner if indicated | ·· |
| Audiological | Baseline audiology examination | Repeat if recurrent otitis media or speech delay | Refer to ear, nose, and throat specialist for recurrent otitis media or serous otitis; hearing aids or classroom interventions for hearing loss |
| Haematological | Complete blood cell count with differential, and prothrombin time or activated partial thromboplastin time | Repeat complete blood cell count with differential and prothrombin time or activated partial thromboplastin time if aged 6–12 months at initial screen; pre- operatively: complete blood cell count with differential and prothrombin time or activated partial thromboplastin time, second tier (in consultation with haematologist) factor IX, XI, and XII concentrations, von Willebrand factor, platelet aggregation | Prothrombin time or activated partial thromboplastin time if bleeding abnormal or persistent, refer to haematologist; complete blood cell count with differential for splenomegaly; complete blood cell count with differential and liver function studies for hepatosplenomegaly |
| Renal | Kidney ultrasound | ·· | ·· |
| Skeletal | Clinical assessment of spine with radiology if indicated by examination | Repeat spinal examination yearly through adolescence; radiology and referral to orthopaedic specialist if abnormal | ·· |
SHOC2 is a widely expressed protein and positive modulator of the RAS–MAPK signalling cascade. The recurrent missense SHOC2 mutation, 4A→G (Ser2Gly), occurs in a subgroup of patients with features of Noonan syndrome but also growth hormone deficiency, distinctive hyperactive behaviour, loose anagen hair, darkly pigmented skin with eczema or ichthyosis, hypernasal voice, and more mitral valve dysplasia and cardiac septal defects than is associated with classic Noonan syndrome.105 The Ser2Gly substitution introduces an N-myristoylation site and results in aberrant targeting of SHOC2 to the plasma membrane and impaired translocation to the nucleus after stimulation by growth factor.
CBL is a ubiquitously expressed E3 ubiquitin ligase that negatively regulates intracellular signalling downstream of receptor tyrosine kinases.106,107 Missense CBL mutations that change evolutionarily conserved residues cause impaired growth, developmental delay, and cryptorchidism resembling Noonan syndrome.107-109 In addition to distinctive facial features, those affected also have an enlarged left atrium, transient chaotic ventricular dysrhythmia, delayed brain myelination, cerebellar vermis hypoplasia, bicuspid aortic valve with stenosis, average adult height, mitral valve insufficiency, and predisposition to juvenile myelomonocytic leukaemia.107-109
LEOPARD syndrome is an autosomal dominant disorder resembling Noonan syndrome but with additional unique phenotypic features. The acronym stands for lentigines, electrocardiogram abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal genitalia, retardation of growth, and deafness. Many affected individuals have hypertrophic cardiomyopathy. Because lentigines are associated with age, an individual with LEOPARD syndrome might be thought to have Noonan syndrome in early life. LEOPARD syndrome is mainly caused by PTPN11 missense mutations and, less often, by RAF1 mutations.99,110,111
The syndromes with the most overlap with Nonnan syndrome include cardiofaciocutaneous syndrome, Costello syndrome, Turner syndrome, and Aarskog syndrome. Features of cardiofaciocutaneous syndrome (Online Mendelian Inheritance in Man reference 115150) include multiple congenital anomalies and intellectual disability, failure to thrive and short stature, congenital heart defects, and a characteristic facial appearance. The phenotypic presentation overlaps substantially with Noonan syndrome, particularly in young children. Patients with cardiofaciocutaneous syndrome have a rounder, more bulbous nasal tip, wider nasal base, fuller lips, and coarser facial features.112 They also often have follicular hyperkeratosis, sparse eyebrows and lashes, and ichthyosis. Most affected infants have severe and often long-lasting feeding difficulties and go on to develop moderate intellectual disability. Heterozygous missense mutations in BRAF, KRAS, MEK1, and MEK2 cause cardiofaciocutaneous syndrome (figure 1).113,114
Patients with Costello syndrome (Online Mendelian Inheritance in Man reference 218040) have high birthweight and delayed growth, developmental delay, coarse facial features, wide nasal bridge, loose and soft skin, increased pigmentation over time, deep palmar and plantar creases, facial or perianal papillomata, premature ageing and hair loss, moderate intellectual disability, flexion or ulnar deviation of the wrist and fingers, and cardiac abnormalities (most commonly pulmonic stenosis and hypertrophic cardiomyopathy).103,115,116 The most common arrhythmia reported in Costello syndrome is supraventricular or paroxysmal tachycardia and the most distinctive is chaotic atrial rhythm or multifocal atrial tachycardia, or ectopic atrial tachycardia.
The dysmorphic facial features, skin abnormalities, short stature, and cardiac abnormalities associated with Costello syndrome have prompted comparisons with Noonan syndrome and cardiofaciocutaneous syndrome.117 Mutations in the HRAS gene cause of roughly 85% of cases of Costello syndrome (figure 1).118 Children with Costello syndrome are prone to some cancers, including rhabdomyosarcoma, neuroblastoma, and bladder carcinoma.119
Girls with Turner syndrome have substantial phenotypic overlap with girls with Noonan syndrome, including widely spaced, down-slanting eyes, short webbed neck, widely spaced nipples, and shield-like chest. Those with Turner syndrome typically have left-sided heart lesions (compared with right-sided lesions in Noonan syndrome), have no or arrested pubertal development, and have gonadal dysgenesis.120 Turner syndrome is caused by loss of a sex chromosome.
The facial features and short stature of Noonan syndrome and Aarskog syndrome (also known as faciodigitogenital syndrome, Online Mendelian Inheritance in Man reference 305400; an X-linked disorder caused by FGD1 mutations121) are similar but those with Aarskog syndrome do not have congenital heart disease and affected boys have a shawl scrotum.
Sequencing the genes implicated in cardiofaciocutaneous, Costello, and Aarskog syndromes and completing a chromosome analysis to rule out Turner syndrome can help diagnosis of these four disorders, each of which have unique medical and developmental complications requiring periodic monitoring and anticipatory guidance.
Conclusion
With a prevalence of one in 1000–2500, Noonan syndrome is a disorder that most doctors will encounter during their careers. Because presentation can be mild and the typical facial features recede with age, the diagnosis might be overlooked. Regular detailed follow-up with a multidisciplinary approach is often needed to address the medical and developmental complications of Noonan syndrome. Much progress has been made in our understanding of the molecular genetic causes of Noonan syndrome in the past decade. We hope that with continued research, targeted pharmacogenomic approaches will be developed on the basis of a detailed understanding of the different disease-causing changes to Ras–MAPK.
Acknowledgments
BDG was partly funded by a grant from the NIH (HL71207). MT was partly funded by grants from Telethon-Italy (GGP10020), European commission sixth framework programme, ERA-net for research programmes on rare diseases (NSEuroNet), and Associazone Italiana Sindrome di Costello. AER was partly funded by a Children’s Hospital Boston Clinical Research Program Grant. We thank The Noonan Syndrome Support Group and the many individuals with Noonan syndrome and their families who participated in the research studies cited in this report.
Footnotes
Contributors
AER searched the published work and wrote the first draft. The other authors interpreted retrieved publications and revised and edited the report.
Conflicts of interest
BDG and MT have received royalties for genetic testing for Noonan syndrome from the following: GeneDx, Prevention Genetics, Correlagen, Harvard/Partners, and Baylor College of Medicine. AER and JEA declare that they have no conflicts of interest.
Contributor Information
Amy E Roberts, Department of Cardiology and Division of Genetics, Children’s Hospital Boston, Boston, MA, USA.
Prof Judith E Allanson, Department of Genetics, Children’s Hospital of Eastern Ontario, Ottawa, ON, Canada.
Marco Tartaglia, Dipartimento di Ematologia, Oncologia e Medicina Molecolare, Istituto Superiore di Sanità, Rome, Italy.
Prof Bruce D Gelb, Child Health and Development Institute, Departments of Pediatrics and Genetics & Genomic Sciences, Mount Sinai School of Medicine, New York, NY, USA.
References
- 1.Mendez HM, Opitz JM. Noonan syndrome: a review. Am J Med Genet. 1985;21:493–506. doi: 10.1002/ajmg.1320210312. [DOI] [PubMed] [Google Scholar]
- 2.Noonan JA, Ehmke DA. Associated noncardiac malformations in children with congenital heart disease. Midwest Soc Pediatr Res. 1963;63:468–70. [Google Scholar]
- 3.Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- 4.Matozaki T, Murata Y, Saito Y, Okazawa H, Ohnishi H. Protein tyrosine phosphatase SHP-2: a proto-oncogene product that promotes Ras activation. Cancer Sci. 2009;100:1786–93. doi: 10.1111/j.1349-7006.2009.01257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qiu WW, Yin SS, Stucker FJ. Audiologic manifestations of Noonan syndrome. Otolaryngol Head Neck Surg. 1998;118:319–23. doi: 10.1016/S0194-59989870308-0. [DOI] [PubMed] [Google Scholar]
- 6.Cremers CW, van der Burgt CJ. Hearing loss in Noonan syndrome. Int J Pediatr Otorhinolaryngol. 1992;23:81–84. doi: 10.1016/0165-5876(92)90082-z. [DOI] [PubMed] [Google Scholar]
- 7.Naficy S, Shepard NT, Telian SA. Multiple temporal bone anomalies associated with Noonan syndrome. Otolaryngol Head Neck Surg. 1997;116:265–67. doi: 10.1016/S0194-59989770339-5. [DOI] [PubMed] [Google Scholar]
- 8.Marino B, Digilio MC, Toscano A, Giannotti A, Dallapiccola B. Congenital heart diseases in children with Noonan syndrome: an expanded cardiac spectrum with high prevalence of atrioventricular canal. J Pediatr. 1999;135:703–06. doi: 10.1016/s0022-3476(99)70088-0. [DOI] [PubMed] [Google Scholar]
- 9.Patton MA. Noonan syndrome: a review. Growth Genet Horm. 1994;33:1–3. [Google Scholar]
- 10.Shaw AC, Kalidas K, Crosby AH, Jeffery S, Patton MA. The natural history of Noonan syndrome: a long-term follow-up study. Arch Dis Child. 2007;92:128–32. doi: 10.1136/adc.2006.104547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanchez-Cascos A. The Noonan syndrome. Eur Heart J. 1983;4:223–29. doi: 10.1093/oxfordjournals.eurheartj.a061452. [DOI] [PubMed] [Google Scholar]
- 12.Schon F, Bowler J, Baraitser M. Cerebral arteriovenous malformation in Noonan’s syndrome. Postgrad Med J. 1992;68:37–40. doi: 10.1136/pgmj.68.795.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ganesan V, Kirkham FJ. Noonan syndrome and moyamoya. Pediatr Neurol. 1997;16:256–58. doi: 10.1016/s0887-8994(97)89980-8. [DOI] [PubMed] [Google Scholar]
- 14.Sharland M, Burch M, McKenna WM, Paton MA. A clinical study of Noonan syndrome. Arch Dis Child. 1992;67:178–83. doi: 10.1136/adc.67.2.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Romano AA, Blethen SL, Dana K, Noto RA. Growth hormone treatment in Noonan syndrome: the National Cooperative Growth Study experience. J Pediatr. 1996;128:S18–21. doi: 10.1016/s0022-3476(96)70005-7. [DOI] [PubMed] [Google Scholar]
- 16.Noordam C, van der Burgt I, Sweep CG, Delemarre-van de Waal HA, Sengers RC, Otten BJ. Growth hormone (GH) secretion in children with Noonan syndrome: frequently abnormal without consequences for growth or response to GH treatment. Clin Endocrinol (Oxf) 2001;54:53–59. doi: 10.1046/j.1365-2265.2001.01188.x. [DOI] [PubMed] [Google Scholar]
- 17.Binder G, Neuer K, Ranke MB, Wittekindt NE. PTPN11 mutations are associated with mild growth hormone resistance in individuals with Noonan syndrome. J Clin Endocrinol Metab. 2005;90:5377–81. doi: 10.1210/jc.2005-0995. [DOI] [PubMed] [Google Scholar]
- 18.De Rocca Serra-Nedelec A, Edouard T, Treguer K, et al. Noonan syndrome-causing SHP2 mutants inhibit insulin-like growth factor 1 release via growth hormone-induced ERK hyperactivation, which contributes to short stature. Proc Natl Acad Sci USA. 2012;109:4257–62. doi: 10.1073/pnas.1119803109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kirk JM, Betts PR, Butler GE, et al. Short stature in Noonan syndrome: response to growth hormone therapy. Arch Dis Child. 2001;84:440–43. doi: 10.1136/adc.84.5.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noordam C, Peer PG, Francois I, De Schepper J, van den Burgt I, Otten BJ. Long-term GH treatment improves adult height in children with Noonan syndrome with and without mutations in protein tyrosine phosphatase, non-receptor-type 11. Eur J Endocrinol. 2008;159:203–08. doi: 10.1530/EJE-08-0413. [DOI] [PubMed] [Google Scholar]
- 21.Raaijmakers R, Noordam C, Karagiannis G, et al. Response to growth hormone treatment and final height in Noonan syndrome in a large cohort of patients in the KIGS database. J Pediatr Endocrinol Metab. 2008;21:267–73. doi: 10.1515/jpem.2008.21.3.267. [DOI] [PubMed] [Google Scholar]
- 22.Noonan JA, Raaijmakers R, Hall BD. Adult height in Noonan syndrome. Am J Med Genet A. 2003;123:68–71. doi: 10.1002/ajmg.a.20502. [DOI] [PubMed] [Google Scholar]
- 23.Romano AA, Dana K, Bakker B, et al. Growth response, near-adult height, and patterns of growth and puberty in patients with noonan syndrome treated with growth hormone. J Clin Endocrinol Metab. 2009;94:2338–44. doi: 10.1210/jc.2008-2094. [DOI] [PubMed] [Google Scholar]
- 24.Takagi M, Miyashita Y, Koga M, Ebara S, Arita N, Kasayama S. Estrogen deficiency is a potential cause for osteopenia in adult male patients with Noonan’s syndrome. Calcif Tissue Int. 2000;66:200–03. doi: 10.1007/s002230010040. [DOI] [PubMed] [Google Scholar]
- 25.Marcus KA, Sweep CG, van der Burgt I, Noordam C. Impaired Sertoli cell function in males diagnosed with Noonan syndrome. J Pediatr Endocrinol Metab. 2008;21:1079–84. doi: 10.1515/jpem.2008.21.11.1079. [DOI] [PubMed] [Google Scholar]
- 26.Tartaglia M, Gelb BD. Germ-line and somatic PTPN11 mutations in human disease. Eur J Med Genet. 2005;48:81–96. doi: 10.1016/j.ejmg.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 27.Sharland M, Patton MA, Talbot S, Chitolie A, Bevan DH. Coagulation-factor deficiencies and abnormal bleeding in Noonan’s syndrome. Lancet. 1992;339:19–21. doi: 10.1016/0140-6736(92)90141-o. [DOI] [PubMed] [Google Scholar]
- 28.Witt DR, McGillivray BC, Allanson JE, et al. Bleeding diathesis in Noonan syndrome: a common association. Am J Med Genet. 1988;31:305–17. doi: 10.1002/ajmg.1320310208. [DOI] [PubMed] [Google Scholar]
- 29.Tofil NM, Winkler MK, Watts RG, Noonan J. The use of recombinant factor VIIa in a patient with Noonan syndrome and life-threatening bleeding. Pediatr Crit Care Med. 2005;6:352–54. doi: 10.1097/01.PCC.0000160656.71424.D1. [DOI] [PubMed] [Google Scholar]
- 30.Miller M, Motulsky AC. Noonan syndrome in an adult family presenting with chronic lymphedema. Am J Med. 1978;65:379–83. doi: 10.1016/0002-9343(78)90836-7. [DOI] [PubMed] [Google Scholar]
- 31.White SW. Lymphedema in Noonan’s syndrome. Int J Dermatol. 1984;23:656–57. doi: 10.1111/j.1365-4362.1984.tb01226.x. [DOI] [PubMed] [Google Scholar]
- 32.Pardo JM. Spontaneous chylothorax in a male newborn with Noonan syndrome. Int Pediatr. 1994;9:55–58. [Google Scholar]
- 33.Cohen MM, Jr, Gorlin RJ. Noonan-like/multiple giant cell lesion syndrome. Am J Med Genet. 1991;40:159–66. doi: 10.1002/ajmg.1320400208. [DOI] [PubMed] [Google Scholar]
- 34.Hanna N, Parfait B, Talaat IM, Vidaud M, Elsedfy HH. SOS1: a new player in the Noonan-like/multiple giant cell lesion syndrome. Clin Genet. 2009;75:568–71. doi: 10.1111/j.1399-0004.2009.01149.x. [DOI] [PubMed] [Google Scholar]
- 35.Bertola DR, Kim CA, Pereira AC, et al. Are Noonan syndrome and Noonan-like/multiple giant cell lesion syndrome distinct entities? Am J Med Genet. 2001;98:230–34. doi: 10.1002/1096-8628(20010122)98:3<230::aid-ajmg1080>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 36.Lee CK, Chang BS, Hong YM, Yang SW, Lee CS, Seo JB. Spinal deformities in Noonan syndrome: a clinical review of sixty cases. J Bone Joint Surg Am. 2001;83:1495–502. [PubMed] [Google Scholar]
- 37.Allanson JE. Noonan syndrome. J Med Genet. 1987;24:9–13. doi: 10.1136/jmg.24.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allanson JE. Noonan syndrome. Am J Med Genet C Semin Med Genet. 2007;145:274–79. doi: 10.1002/ajmg.c.30138. [DOI] [PubMed] [Google Scholar]
- 39.Cesarini L, Alfieri P, Pantaleoni F, et al. Cognitive profile of disorders associated with dysregulation of the RAS/MAPK signaling cascade. Am J Med Genet A. 2009;149:140–46. doi: 10.1002/ajmg.a.32488. [DOI] [PubMed] [Google Scholar]
- 40.Verhoeven W, Wingbermuhle E, Egger J, Van der Burgt I, Tuinier S. Noonan syndrome: psychological and psychiatric aspects. Am J Med Genet A. 2008;146:191–96. doi: 10.1002/ajmg.a.32115. [DOI] [PubMed] [Google Scholar]
- 41.Wood A, Massarano A, Super M, Harrington R. Behavioural aspects and psychiatric findings in Noonan’s syndrome. Arch Dis Child. 1995;72:153–55. doi: 10.1136/adc.72.2.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee DA, Portnoy S, Hill P, Gillberg C, Patton MA. Psychological profile of children with Noonan syndrome. Dev Med Child Neurol. 2005;47:35–38. doi: 10.1017/s001216220500006x. [DOI] [PubMed] [Google Scholar]
- 43.Pierpont EI, Pierpont ME, Mendelsohn NJ, Roberts AE, Tworog-Dube E, Seidenberg MS. Genotype differences in cognitive functioning in Noonan syndrome. Genes Brain Behav. 2009;8:275–82. doi: 10.1111/j.1601-183X.2008.00469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pierpont EI, Weismer SE, Roberts AE, et al. The language phenotype of children and adolescents with Noonan syndrome. J Speech Lang Hear Res. 2010;53:917–32. doi: 10.1044/1092-4388(2009/09-0046). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alfieri P, Cesarini L, Mallardi M, et al. Long term memory profile of disorders associated with dysregulation of the RAS–MAPK signaling cascade. Behav Genet. 2011;41:423–29. doi: 10.1007/s10519-011-9446-5. [DOI] [PubMed] [Google Scholar]
- 46.Kratz CP. Myeloproliferative Disease and Cancer in Persons with Noonan Syndrome and Related Disorders. In: Zenker M, editor. Noonan Syndrome and Related Disorders. Basel: Karger; 2009. pp. 119–27. [Google Scholar]
- 47.Bader-Meunier B, Tchernia G, Mielot F, et al. Occurrence of myeloproliferative disorder in patients with Noonan syndrome. J Pediatr. 1997;130:885–89. doi: 10.1016/s0022-3476(97)70273-7. [DOI] [PubMed] [Google Scholar]
- 48.Fukuda M, Horibe K, Miyajima Y, Matsumoto K, Nagashima M. Spontaneous remission of juvenile chronic myelomonocytic leukemia in an infant with Noonan syndrome. J Pediatr Hematol Oncol. 1997;19:177–79. doi: 10.1097/00043426-199703000-00019. [DOI] [PubMed] [Google Scholar]
- 49.Choong K, Freedman MH, Chitayat D, Kelly EN, Taylor G, Zipursky A. Juvenile myelomonocytic leukemia and Noonan syndrome. J Pediatr Hematol Oncol. 1999;21:523–27. [PubMed] [Google Scholar]
- 50.Tartaglia M, Martinelli S, Stella L, et al. Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease. Am J Hum Genet. 2006;78:279–90. doi: 10.1086/499925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Denayer E, Devriendt K, de Ravel T, et al. Tumor spectrum in children with Noonan syndrome and SOS1 or RAF1 mutations. Genes Chromosomes Cancer. 2010;49:242–52. doi: 10.1002/gcc.20735. [DOI] [PubMed] [Google Scholar]
- 52.Jongmans MC, Hoogerbrugge PM, Hilkens L, et al. Noonan syndrome, the SOS1 gene and embryonal rhabdomyosarcoma. Genes Chromosomes Cancer. 2010;49:635–41. doi: 10.1002/gcc.20773. [DOI] [PubMed] [Google Scholar]
- 53.Jung A, Bechthold S, Pfluger T, Renner C, Ehrt O. Orbital rhabdomyosarcoma in Noonan syndrome. J Pediatr Hematol Oncol. 2003;25:330–32. doi: 10.1097/00043426-200304000-00014. [DOI] [PubMed] [Google Scholar]
- 54.Khan S, McDowell H, Upadhyaya M, Fryer A. Vaginal rhabdomyosarcoma in a patient with Noonan syndrome. J Med Genet. 1995;32:743–45. doi: 10.1136/jmg.32.9.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moschovi M, Touliatou V, Papadopoulou A, Mayakou MA, Nikolaidou-Karpathiou P, Kitsiou-Tzeli S. Rhabdomyosarcoma in a patient with Noonan syndrome phenotype and review of the literature. J Pediatr Hematol Oncol. 2007;29:341–44. doi: 10.1097/MPH.0b013e31805d8f57. [DOI] [PubMed] [Google Scholar]
- 56.Fryssira H, Leventopoulos G, Psoni S, Kitsiou-Tzeli S, Stavrianeas N, Kanavakis E. Tumor development in three patients with Noonan syndrome. Eur J Pediatr. 2008;167:1025–31. doi: 10.1007/s00431-007-0636-3. [DOI] [PubMed] [Google Scholar]
- 57.Cotton JL, Williams RG. Noonan syndrome and neuroblastoma. Arch Pediatr Adolesc Med. 1995;149:1280–81. doi: 10.1001/archpedi.1995.02170240098019. [DOI] [PubMed] [Google Scholar]
- 58.Ijiri R, Tanaka Y, Keisuke K, Masuno M, Imaizumi K. A case of Noonan’s syndrome with possible associated neuroblastoma. Pediatr Radiol. 2000;30:432–33. doi: 10.1007/s002470050781. [DOI] [PubMed] [Google Scholar]
- 59.Sanford RA, Bowman R, Tomita T, De Leon G, Palka P. A 16-year-old male with Noonan’s syndrome develops progressive scoliosis and deteriorating gait. Pediatr Neurosurg. 1999;30:47–52. doi: 10.1159/000028761. [DOI] [PubMed] [Google Scholar]
- 60.Sherman CB, Ali-Nazir A, Gonzales-Gomez I, Finlay JL, Dhall G. Primary mixed glioneuronal tumor of the central nervous system in a patient with noonan syndrome: a case report and review of the literature. J Pediatr Hematol Oncol. 2009;31:61–64. doi: 10.1097/MPH.0b013e31818ab2cf. [DOI] [PubMed] [Google Scholar]
- 61.Jongmans MC, van der Burgt I, Hoogerbrugge PM, et al. Cancer risk in patients with Noonan syndrome carrying a PTPN11 mutation. Eur J Hum Genet. 2011;19:870–74. doi: 10.1038/ejhg.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Noonan JA. Noonan Syndrome. In: Goldstein SR, Reynolds CR, editors. Handbook of Neurodevelopmental and Genetic Disorders in Adults. New York: The Guilford Press; 2005. pp. 308–19. [Google Scholar]
- 63.Lin AE, Basson CT, Goldmuntz E, et al. Adults with genetic syndromes and cardiovascular abnormalities: clinical history and management. Genet Med. 2008;10:469–94. doi: 10.1097/GIM.0b013e3181772111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Allanson JE, Hall JG, Hughes HE, Preus M, Witt RD. Noonan syndrome: the changing phenotype. Am J Med Genet. 1985;21:507–14. doi: 10.1002/ajmg.1320210313. [DOI] [PubMed] [Google Scholar]
- 65.Nisbet DL, Griffin DR, Chitty LS. Prenatal features of Noonan syndrome. Prenat Diagn. 1999;19:642–47. doi: 10.1002/(sici)1097-0223(199907)19:7<642::aid-pd610>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 66.Houweling AC, de Mooij YM, van der Burgt I, Yntema HG, Lachmeijer AM, Go AT. Prenatal detection of Noonan syndrome by mutation analysis of the PTPN11 and the KRAS genes. Prenat Diagn. 2010;30:284–86. doi: 10.1002/pd.2458. [DOI] [PubMed] [Google Scholar]
- 67.Adekunle O, Gopee A, el-Sayed M, Thilaganathan B. Increased first trimester nuchal translucency: pregnancy and infant outcomes after routine screening for Down’s syndrome in an unselected antenatal population. Br J Radiol. 1999;72:457–60. doi: 10.1259/bjr.72.857.10505009. [DOI] [PubMed] [Google Scholar]
- 68.Hiippala A, Eronen M, Taipale P, Salonen R, Hiilesmaa V. Fetal nuchal translucency and normal chromosomes: a long-term follow-up study. Ultrasound Obstet Gynecol. 2001;18:18–22. doi: 10.1046/j.1469-0705.2001.00481.x. [DOI] [PubMed] [Google Scholar]
- 69.Reynders CS, Pauker SP, Benacerraf BR. First trimester isolated fetal nuchal lucency: significance and outcome. J Ultrasound Med. 1997;16:101–05. doi: 10.7863/jum.1997.16.2.101. [DOI] [PubMed] [Google Scholar]
- 70.Lee KA, Williams B, Roza K, et al. PTPN11 analysis for the prenatal diagnosis of Noonan syndrome in fetuses with abnormal ultrasound findings. Clin Genet. 2009;75:190–94. doi: 10.1111/j.1399-0004.2008.01085.x. [DOI] [PubMed] [Google Scholar]
- 71.Maheshwari M, Belmont J, Fernbach S, et al. PTPN11 mutations in Noonan syndrome type I: detection of recurrent mutations in exons 3 and 13. Hum Mutat. 2002;20:298–304. doi: 10.1002/humu.10129. [DOI] [PubMed] [Google Scholar]
- 72.Tartaglia M, Mehler EL, Goldberg R, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29:465–68. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 73.Tartaglia M, Kalidas K, Shaw A, et al. PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am J Hum Genet. 2002;70:1555–63. doi: 10.1086/340847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tartaglia M, Niemeyer CM, Fragale A, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34:148–50. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- 75.Sznajer Y, Keren B, Baumann C, et al. The spectrum of cardiac anomalies in Noonan syndrome as a result of mutations in the PTPN11 gene. Pediatrics. 2007;119:e1325–31. doi: 10.1542/peds.2006-0211. [DOI] [PubMed] [Google Scholar]
- 76.Yoshida R, Hasegawa T, Hasegawa Y, et al. Protein-tyrosine phosphatase, nonreceptor type 11 mutation analysis and clinical assessment in 45 patients with Noonan syndrome. J Clin Endocrinol Metab. 2004;89:3359–64. doi: 10.1210/jc.2003-032091. [DOI] [PubMed] [Google Scholar]
- 77.Zenker M, Buheitel G, Rauch R, et al. Genotype-phenotype correlations in Noonan syndrome. J Pediatr. 2004;144:368–74. doi: 10.1016/j.jpeds.2003.11.032. [DOI] [PubMed] [Google Scholar]
- 78.Zandrino F, Curone P, Benzi L, Musante F. Value of an early arteriographic acquisition for evaluating the splanchnic vessels as an adjunct to biphasic CT using a multislice scanner. Eur Radiol. 2003;13:1072–79. doi: 10.1007/s00330-002-1566-0. [DOI] [PubMed] [Google Scholar]
- 79.Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294:1299–304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- 80.Carta C, Pantaleoni F, Bocchinfuso G, et al. Germline missense mutations affecting KRAS isoform B are associated with a severe Noonan syndrome phenotype. Am J Hum Genet. 2006;79:129–35. doi: 10.1086/504394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cirstea IC, Kutsche K, Dvorsky R, et al. A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat Genet. 2010;42:27–29. doi: 10.1038/ng.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schubbert S, Zenker M, Rowe SL, et al. Germline KRAS mutations cause Noonan syndrome. Nat Genet. 2006;38:331–36. doi: 10.1038/ng1748. [DOI] [PubMed] [Google Scholar]
- 83.Gremer L, Merbitz-Zahradnik T, Dvorsky R, et al. Germline KRAS mutations cause aberrant biochemical and physical properties leading to developmental disorders. Hum Mutat. 2011;32:33–43. doi: 10.1002/humu.21377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lo FS, Lin JL, Kuo MT, et al. Noonan syndrome caused by germline KRAS mutation in Taiwan: report of two patients and a review of the literature. Eur J Pediatr. 2009;168:919–23. doi: 10.1007/s00431-008-0858-z. [DOI] [PubMed] [Google Scholar]
- 85.Nava C, Hanna N, Michot C, et al. Cardio-facio-cutaneous and Noonan syndromes due to mutations in the RAS/MAPK signalling pathway: genotype-phenotype relationships and overlap with Costello syndrome. J Med Genet. 2007;44:763–71. doi: 10.1136/jmg.2007.050450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zenker M, Lehmann K, Schulz AL, et al. Expansion of the genotypic and phenotypic spectrum in patients with KRAS germline mutations. J Med Genet. 2007;44:131–35. doi: 10.1136/jmg.2006.046300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kratz CP, Zampino G, Kriek M, et al. Craniosynostosis in patients with Noonan syndrome caused by germline KRAS mutations. Am J Med Genet A. 2009;149:1036–40. doi: 10.1002/ajmg.a.32786. [DOI] [PubMed] [Google Scholar]
- 88.Egan SE, Weinberg RA. The pathway to signal achievement. Nature. 1993;365:781–83. doi: 10.1038/365781a0. [DOI] [PubMed] [Google Scholar]
- 89.Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev. 2001;81:153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- 90.Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev. 2009;19:230–36. doi: 10.1016/j.gde.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tartaglia M, Zampino G, Gelb BD. Noonan syndrome: clinical aspects and molecular pathogenesis. Mol Syndromol. 2010;1:2–26. doi: 10.1159/000276766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Roberts AE, Araki T, Swanson KD, et al. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet. 2007;39:70–74. doi: 10.1038/ng1926. [DOI] [PubMed] [Google Scholar]
- 93.Tartaglia M, Pennacchio LA, Zhao C, et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet. 2007;39:75–79. doi: 10.1038/ng1939. [DOI] [PubMed] [Google Scholar]
- 94.Lepri F, De Luca A, Stella L, et al. SOS1 mutations in Noonan syndrome: molecular spectrum, structural insights on pathogenic effects, and genotype-phenotype correlations. Hum Mutat. 2011;32:760–72. doi: 10.1002/humu.21492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zenker M, Horn D, Wieczorek D, et al. SOS1 is the second most common Noonan gene but plays no major role in cardio-faciocutaneous syndrome. J Med Genet. 2007;44:651–56. doi: 10.1136/jmg.2007.051276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875–85. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- 97.Schreck R, Rapp UR. Raf kinases: oncogenesis and drug discovery. Int J Cancer. 2006;119:2261–71. doi: 10.1002/ijc.22144. [DOI] [PubMed] [Google Scholar]
- 98.Leicht DT, Balan V, Kaplun A, et al. Raf kinases: function, regulation and role in human cancer. Biochim Biophys Acta. 2007;1773:1196–212. doi: 10.1016/j.bbamcr.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pandit B, Sarkozy A, Pennacchio LA, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007;39:1007–12. doi: 10.1038/ng2073. [DOI] [PubMed] [Google Scholar]
- 100.Razzaque MA, Nishizawa T, Komoike Y, et al. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet. 2007;39:1013–17. doi: 10.1038/ng2078. [DOI] [PubMed] [Google Scholar]
- 101.Nystrom AM, Ekvall S, Berglund E, et al. Noonan and cardio-faciocutaneous syndromes: two clinically and genetically overlapping disorders. J Med Genet. 2008;45:500–06. doi: 10.1136/jmg.2008.057653. [DOI] [PubMed] [Google Scholar]
- 102.Sarkozy A, Carta C, Moretti S, et al. Germline BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: molecular diversity and associated phenotypic spectrum. Hum Mutat. 2009;30:695–702. doi: 10.1002/humu.20955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Romano AA, Allanson JE, Dahlgren J, et al. Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics. 2010;126:746–59. doi: 10.1542/peds.2009-3207. [DOI] [PubMed] [Google Scholar]
- 104.van der Burgt I. Noonan syndrome. Orphanet J Rare Dis. 2007;2:4. doi: 10.1186/1750-1172-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cordeddu V, Di Schiavi E, Pennacchio LA, et al. Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet. 2009;41:1022–26. doi: 10.1038/ng.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dikic I, Schmidt MH. Malfunctions within the Cbl interactome uncouple receptor tyrosine kinases from destructive transport. Eur J Cell Biol. 2007;86:505–12. doi: 10.1016/j.ejcb.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 107.Martinelli S, De Luca A, Stellacci E, et al. Heterozygous germline mutations in the CBL tumor-suppressor gene cause a Noonan syndrome-like phenotype. Am J Hum Genet. 2010;87:250–57. doi: 10.1016/j.ajhg.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Niemeyer CM, Kang MW, Shin DH, et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet. 2010;42:794–800. doi: 10.1038/ng.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Perez B, Mechinaud F, Galambrun C, et al. Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J Med Genet. 2010;47:686–91. doi: 10.1136/jmg.2010.076836. [DOI] [PubMed] [Google Scholar]
- 110.Digilio MC, Conti E, Sarkozy A, et al. Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am J Hum Genet. 2002;71:389–94. doi: 10.1086/341528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Legius E, Schrander-Stumpel C, Schollen E, Pulles-Heintzberger C, Gewillig M, Fryns JP. PTPN11 mutations in LEOPARD syndrome. J Med Genet. 2002;39:571–74. doi: 10.1136/jmg.39.8.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Allanson JE, Bohring A, Dorr HG, et al. The face of Noonan syndrome: does phenotype predict genotype. Am J Med Genet A. 2010;152:1960–66. doi: 10.1002/ajmg.a.33518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Niihori T, Aoki Y, Narumi Y, et al. Germline KRAS and BRAF mutations in cardio-faciocutaneous syndrome. Nat Genet. 2006;38:294–96. doi: 10.1038/ng1749. [DOI] [PubMed] [Google Scholar]
- 114.Rodriguez-Viciana P, Tetsu O, Tidyman WE, et al. Germline mutations in genes within the MAPK pathway cause cardio-faciocutaneous syndrome. Science. 2006;311:1287–90. doi: 10.1126/science.1124642. [DOI] [PubMed] [Google Scholar]
- 115.Lin AE, Grossfeld PD, Hamilton RM, et al. Further delineation of cardiac abnormalities in Costello syndrome. Am J Med Genet. 2002;111:115–29. doi: 10.1002/ajmg.10558. [DOI] [PubMed] [Google Scholar]
- 116.Hennekam RC. Costello syndrome: an overview. Am J Med Genet C Semin Med Genet. 2003;117:42–48. doi: 10.1002/ajmg.c.10019. [DOI] [PubMed] [Google Scholar]
- 117.Narumi Y, Aoki Y, Niihori T, et al. Molecular and clinical characterization of cardio-facio-cutaneous (CFC) syndrome: overlapping clinical manifestations with Costello syndrome. Am J Med Genet A. 2007;143:799–807. doi: 10.1002/ajmg.a.31658. [DOI] [PubMed] [Google Scholar]
- 118.Aoki Y, Niihori T, Kawame H, et al. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet. 2005;37:1038–40. doi: 10.1038/ng1641. [DOI] [PubMed] [Google Scholar]
- 119.Gripp KW. Tumor predisposition in Costello syndrome. Am J Med Genet C Semin Med Genet. 2005;137:72–77. doi: 10.1002/ajmg.c.30065. [DOI] [PubMed] [Google Scholar]
- 120.Davenport ML. Approach to the patient with Turner syndrome. J Clin Endocrinol Metab. 2010;95:1487–95. doi: 10.1210/jc.2009-0926. [DOI] [PubMed] [Google Scholar]
- 121.Orrico A, Galli L, Falciani M, et al. A mutation in the pleckstrin homology (PH) domain of the FGD1 gene in an Italian family with faciogenital dysplasia (Aarskog-Scott syndrome) FEBS Lett. 2000;478:216–20. doi: 10.1016/s0014-5793(00)01857-3. [DOI] [PubMed] [Google Scholar]