

Abstract
DNA microarrays have been used for over a decade to profile gene expression on a genomic scale. While this technology has advanced our understanding of complex cellular function, the reliance of microarrays on hybridization kinetics results in several technical limitations. For example, knowledge of the sequences being probed is required, distinguishing similar sequences is difficult because of cross-hybridization, and the relatively narrow dynamic range of the signal limits sensitivity. Recently, new technologies have been introduced that are based on novel sequencing methodologies. These next-generation sequencing methods do not have the limitations inherent to microarrays. Next-generation sequencing is unique since it allows the detection of all known and novel RNAs present in biological samples without bias toward known transcripts. In addition, the expression of coding and noncoding RNAs, alternative splicing events, and expressed single nucleotide polymorphisms (SNPs) can be identified in a single experiment. Furthermore, this technology allows for remarkably higher throughput while lowering sequencing costs. This significant shift in throughput and pricing makes low-cost access to whole genomes possible and more importantly expands sequencing applications far beyond traditional uses (Morozova & Marra, 2008) to include sequencing the transcriptome (RNA-Seq), providing detail on gene structure, alternative splicing events, expressed SNPs, and transcript size (Mane et al., 2009; Tang et al., 2009; Walter et al., 2009), in a single experiment, while also quantifying the absolute abundance of genes, all with greater sensitivity and dynamic range than the competing cDNA microarray technology (Mortazavi, Williams, McCue, Schaeffer, & Wold, 2008).
1. OVERVIEW
RNA-Seq utilizes highly efficient sequencing techniques and subsequent mapping of short sequence reads to a reference genome, making it possible to identify exons and introns by mapping their boundaries of genes, which in turn allows investigation of the complexity of transcriptomes in unparalleled detail. Moreover, RNA-Seq enables identification of transcription initiation sites and new splicing variants and permits quantitative determination of exon and splicing isoform expression. This innovative technology facilitates detailed examination of individual expression differences in human brain and makes it possible to dissect the genetic complexities of alcoholism and a variety of physiological conditions (Wang, Gerstein, & Snyder, 2009).
This review addresses three critical barriers to progress in alcohol research: (1) Regulation of cell function often occurs at the level of alternative splicing of mRNAs (Hartmann & Valcárcel, 2009; Tazi et al., 2009), and emerging evidence indicates that this can be important for alcohol tolerance (Pietrzykowski et al., 2008), yet we have little information about splicing changes in human alcoholism. This can now be examined using next-generation sequencing of brain RNA from alcoholics and controls. (2) We do not know if our rodent and nonhuman primate models of alcohol consumption or dependence contain any of the molecular signatures found in human alcoholic brain. Because these animal models must serve as the basis for future medication development, it is essential to determine which, if any, display genomic convergence with human alcoholics. (3) Noncoding RNAs (ncRNAs) are emerging as “master regulators” of gene expression and may underlie many of the widespread genomic changes produced by chronic alcohol consumption, yet we have limited knowledge of changes in brain miRNA levels in human alcoholics or animal models and even less is understood regarding the behavioral significance of changes in ncRNAs.
2. RNA-SEQ OF POSTMORTEM BRAIN TISSUE
Transcriptome profiling of postmortem brain tissue from alcoholics and matched controls has revealed novel and detailed gene expression changes, generating new avenues for addiction research. Although there are certain difficulties inherent with using postmortem brain tissue, such as difficulty in obtaining samples and accounting for variable patterns of alcohol use and other human variables, postmortem brain tissue remains the gold standard against which all other model systems should be evaluated. Next-generation sequencing provides a more comprehensive and accurate tool for transcriptome analysis of this limited, valuable resource.
A first-pass examination of the transcriptome of alcoholics and matched controls identified a number of molecular constituents within a specific brain region (Fig. 11.1). The type of RNA molecules uncovered depends on the initial experimental design, but novel biological features may also be revealed. By design, RNA-Seq of the prefrontal cortex primarily identified protein-coding transcripts and also discerned an appreciable number of pseudogenes and small nucleolar RNAs (snoRNAs) (Fig. 11.1). In addition to having recognized roles in RNA processing and ribosomal RNA modification (Eddy, 2001; Kiss, 2002), snoRNAs are implicated in regulating CNS function (Cao, Yeo, Muotri, Kuwabara, & Gage, 2006; Rogelj, Hartmann, Yeo, Hunt, & Giese, 2003). The expression of snoRNAs and other ncRNAs may have important roles in alcoholism and other diseases.
Figure 11.1.

RNA-Seq detection of biological features for alcoholics and matched controls. Bar plot demonstrates the percentage of features detected in a representative control (blue (black in the print version)) and alcoholic (red (dark gray in the print version)) sample from a cohort of the prefrontal cortex. The left axis shows percentage of features for the top three biotypes with the right axis showing percentage of remaining biotypes (separated by dotted green (light gray in the print version) vertical line). Protein-coding transcripts were the predominant feature detected in both groups.
Comparing the overall expression of detected biological RNA categories within individual samples illustrates consistency among nonalcoholics and alcoholics (Fig. 11.2). Alternatively, any possible discrepancies that may need special consideration in downstream analyses may also be revealed through this type of comparison. The global expression level of transcriptome elements is stable among individuals from alcoholics and matched controls (Fig. 11.3A), and is also similar in terms of sensitivity, determined through the number of counts per million (CPM) mapped reads over varying levels of stringency (Fig. 11.3B). General agreement across the samples indicates the absence of a potential batch effect or outliers within the examined cohort. Sequencing depth, based on reliably and unambiguously mapped reads, is nearly uniform across controls and alcoholics for all of the biological units (Fig. 11.4A), as well as for only protein-coding transcripts (Fig. 11.4B). Although increased sequencing depth could improve expression estimates, continuity between specimens suggests reliable biotype measurements for comparing alcoholic and nonalcoholic subjects. Importantly, lack of overall expression differences in proportion to disease state does not exclude finding potential differences in discrete RNA molecules, which may be important players in the development of alcohol use disorder.
Figure 11.2.

Expression values of the detected biological features for alcoholics and matched controls. Box and whisker plots for expression of biological features in representative controls (A) and alcoholics (B) from the prefrontal cortex. Shown along the x-axis is the number of corresponding biotypes determined for all samples having greater than zero counts. The two groups have similar overall expression values for biological features.
Figure 11.3.

Comparison of individual samples for overall expression and sensitivity. (A) Box and whisker plots for overall expression within individual samples and (B) stacked bar plot of binned expression based on counts per million (CPM) mapped reads across individuals to determine intersample consistency and the percentage of low expression values that may interfere with downstream analyses. Horizontal lines depict the percentage of CPM expressed in at least one specimen to help determine an appropriate range of sensitivity.
Figure 11.4.

Saturation plots for assessing quality control across individual samples and disease groups. Number of detected features compared with sequencing depth in million mapped reads for controls (left), alcoholics (middle), and all samples (right) for all biological features detected (A) and “protein-coding” transcripts only (B). Figures demonstrate all samples have comparable saturation slopes and can be included in downstream analysis.
3. DETECTION OF TECHNICAL BIASES IN RNA-SEQ DATA
Obtaining an accurate assessment of RNA molecules that correspond with disease is not a trivial undertaking and should include a comprehensive evaluation of expression estimates for potential areas of artificial biases (Ozsolak & Milos, 2011). Transcript length and guanine–cytosine content (GC content) are two particular characteristics that may influence the quantification of RNA-Seq data (Oshlack & Wakefield, 2009; Pickrell et al., 2010). Nonnormalized expression counts follow a similar trend for alcoholics and matched controls with respect to the length (Fig. 11.5A) and percentage of GC content of identified transcripts (Fig. 11.6A). The length and GC content for mapped features, without normalization, are significantly associated with expression for both groups (Figs. 11.5B and 11.6B). Correcting expression estimates based on the number of collected reads per kilobase per million (RPKM) mapped reads, one method accounting for molar concentration and transcript length (Mortazavi, Williams, McCue, Schaeffer, & Wold, 2008), effectively alleviated the significant bias introduced by transcript length within controls and alcoholics (Fig. 11.5D). Utilizing RPKM values also blunted the relationship between GC content and computed expression values (Fig. 11.6D), although not to the same degree as the length of expressed biotypes. The effect of GC content on expression may be minimized with additional processing/normalization strategies (Hansen, Irizarry, & Wu, 2012; Risso, Schwartz, Sherlock, & Dudoit, 2011). At first glance, expression may appear nearly indistinguishable between alcoholics and controls (Figs. 11.5 and 11.6); however, detailed examination of RNA-Seq can expose biases that may affect expression estimates, and an appropriate normalization strategy is therefore crucial.
Figure 11.5.

Assessment of gene expression for bias in sequencing length. The size of detected biotypes is binned along the x-axis and compared to mean expression values of raw counts (top) and normalized expression by reads per kilobase per million (RPKM) mapped reads (bottom). Alcoholic and control samples follow similar trends in raw mean expression values (A) and RPKM values (C). Mean raw count values in controls and alcoholics are highly associated with feature length (B); however, feature length is not strongly correlated with mean RPKM (D).
Figure 11.6.

Assessment of gene expression for bias in GC content. The GC% of detected biotypes is binned along the x-axis and compared to mean expression values of raw counts (top) and normalized expression by reads per kilobase per million (RPKM) mapped reads (bottom). Alcoholic and control samples follow similar trends in raw mean expression values (A) and RPKM values (C) in relation to GC%. Mean raw count values in controls and alcoholics are persistently correlated with % of GC content (B); however, GC% shows lower correlation with normalized expression (D).
4. NORMALIZATION OF RNA-SEQ DATA
No single procedure has yet emerged as a gold standard for RNA-Seq analyses. Differing methodologies for profiling expression can reveal discrepant findings in the identification of differentially expressed genes from the same experimental dataset (Rapaport et al., 2013; Soneson & Delorenzi, 2013; Tarazona, García-Alcalde, Dopazo, Ferrer, & Conesa, 2011). In order to adequately manage bioinformatics pipelines, multiple in silico experimental designs, rather than a one size fits all approach, may initially need to be explored before selecting a suitable model of normalization. RNA-Seq expression data from the prefrontal cortex are illustrated using different representative methods of normalization. The intersample correlations among controls and alcoholics fluctuate according to the normalization strategy and impact the extent of within group variation (Fig. 11.7). RPKM values have the highest proportion of variability, which may impede the identification of differentially expressed features between alcoholics and controls. A comprehensive evaluation of normalization techniques for RNA-Seq data has previously suggested the RPKM approach is ineffective and should cease to be used for evaluating differential expression (Dillies et al., 2013). Additionally, RPKM data may fail to adequately account for RNA composition bias (Robinson & Oshlack, 2010) or gene length (Bullard, Purdom, Hansen, & Dudoit, 2010) in the detection of differentially expressed features. Practical recommendations are available for generating fairly robust datasets (Dillies et al., 2013) and will continue to evolve as RNA-Seq is adopted in a larger number of laboratories. Selecting the appropriate statistical method for minimizing the effects of technical error will also depend upon additional known sources of systematic variation.
Figure 11.7.

Comparison of normalization strategies for assessing gene expression in controls and alcoholics. Box and whisker plots for intersample Pearson correlation coefficients of control and alcoholic prefrontal cortex gene expression across multiple strategies for normalizing RNA-Seq count data. Differing methods of normalization exhibit differing median intersample consistency and within group variation, which may affect experimental outcomes. The appropriate method should be based on the quality control measures and hypothesis in question.
5. ALTERNATIVE SPLICING AND DIFFERENTIAL EXPRESSION
Once an acceptable normalization method is determined, summarized read counts can be evaluated for divergent expression profiles between two or more conditions. RNA-Seq is a powerful tool for the detection of differentially expressed features, capable of capturing weakly expressed genes and alternatively splices transcripts within a single experiment (Bottomly et al., 2011; Marioni, Mason, Mane, Stephens, & Gilad, 2008). Although it is challenging to use short-read sequencers to quantify splice variants having identical exons, several algorithms exist for computing the expression of full-length isoforms (Garber, Grabherr, Guttman, & Trapnell, 2011; Trapnell et al., 2012; Xing et al., 2006). Recognition of alternatively spliced transcripts, and their individual exons, is an important aspect for interpreting the neurobiology of disease. The human transcriptome is able to generate a tremendous degree of biodiversity, with ~95% of all multiexon genes undergoing alternative splicing (Pan, Kaiguo, Razak, Westwood, & Gerlai, 2011). Humans, and closely related primates, exhibit the greatest degree of complexity in splicing, with the human brain being the most diverse among several tissue types (Barbosa-Morais et al., 2012). The higher rate of alternative splicing in human brain may underscore evolutionary remodeling for higher cognitive function while generating greater susceptibility to neuropsychiatric diseases.
Differential expression between alcoholics and controls is able to distinguish ~1000 genes and ~1200 significant alternatively spliced transcripts (Fig. 11.8). Genes and corresponding spliced isoforms tend to follow similar patterns of differential expression in relation to alcohol dependence. The majority of genes and splice variants with a p-value ≤0.05 have only a modest, less than twofold change in expression. Statistically significant genes, or alternatively spliced transcripts, with larger fold changes in expression are usually weaker in overall expression. In some circumstances, it may be advantageous to remove missing or low-level counts; however, some methods of statistical inference may account for extreme or missing variables (Anders et al., 2013). Individual exons are more abundant in RNA-Seq counts, making no assumption of its interconnection with other units to form a functional RNA molecule. Approximately, 11,000 individual exons are differentially expressed in the prefrontal cortex of alcoholics (Fig. 11.8). Additionally, a greater number of exon features have fold-change values >2, which may suggest some gene or isoform reconstructions underestimate some of the differences occurring with alcohol dependence. Although individual exons cannot function solely on their own accord, these molecular units might serve as surrogate markers for differences in the functional transcript or gene product. Altered expression of individual exons may be of substantial interest, especially if these changes coincide with the site of activation, a site of intramolecule docking, or an alcohol-binding site for the fully formed protein substrate.
Figure 11.8.
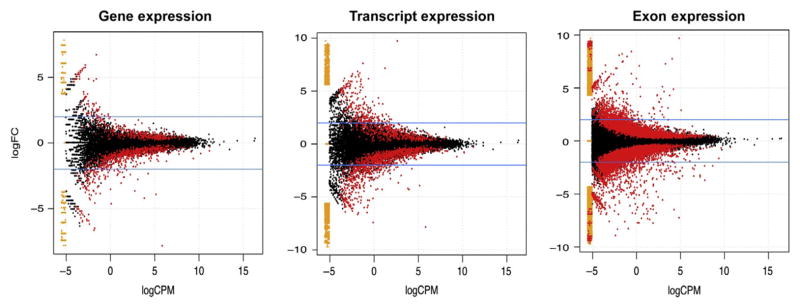
Differential expression for gene, transcript, and exon models of RNA-Seq data. The mean log CPM is plotted against the log fold change in gene (left), alternatively spliced transcript (middle), and exon (right) expression for alcoholics versus controls in the prefrontal cortex. Horizontal blue (light gray in the print version) lines depict a twofold change in expression (increased or decreased) between disease groups and red (dark gray in the print version) dots indicate features with a p-value ≤ 0.05.
6. LONG NONCODING RNA
In the absence of forming a functional protein, intracellular molecules can still function as ncRNAs (Mattick & Makunin, 2006). ncRNAs make up a sizeable share of the transcriptional landscape (Carninci et al., 2005; ENCODE Project Consortium et al., 2012, 2007), but the precise function of many noncoding elements remains largely unknown. Defining the diverse biological roles carried out by multiple classes of ncRNAs is a burgeoning aspect of transcriptomics that will likely match or rival the large number and diversity represented by the proteome. Long noncoding RNAs (lncRNAs) represent one of the most abundant classes of nonprotein-coding RNAs in the brain (Jia et al., 2010; Ravasi et al., 2006). Similar to protein-coding transcripts, lncRNAs can be found within specific neuroanatomical regions (Belgard et al., 2011; Mercer, Dinger, Sunkin, Mehler, & Mattick, 2008). A study of human alcoholic brain tissue showed an increase in the expression of the lncRNA MALAT1 within multiple brain regions (Kryger, Fan, Wilce, & Jaquet, 2012). Overall expression of lncRNAs may be lower than protein-coding transcripts, but can be dynamically regulated in alcoholic brain tissue (Fig. 11.9). Although the role of lncRNAs on alcohol dependence and drug addiction is still unclear, lncRNAs are known to (1) mediate control of epigenetic factors for regulating gene expression (Khalil et al., 2009; Lee, 2012; Wang et al., 2011), (2) act as endogenous competitors (Cesana et al., 2011), (3) regulate alternative splicing events (Barry et al., 2013; Massone et al., 2011; Tripathi et al., 2010), (4) control neuronal development (Pollard et al., 2006), and (5) guide synaptic plasticity (Bond et al., 2009). These diverse roles make it likely that even low-to-moderate changes in lncRNA expression could significantly impact alcohol use disorders and other psychiatric diseases.
Figure 11.9.

Fold change of long noncoding RNAs within the prefrontal cortex. Bar plot of fold change in expression of the top 15 long noncoding RNAs (lncRNAs) between alcoholics and controls within prefrontal cortex. Eleven lncRNAs show increased expression in the prefrontal cortex of alcoholics compared to controls, while four lncRNAs show decreased expression.
7. NOVEL THREE PRIME UNTRANSLATED REGIONS
RNA-Seq can generate rich expression maps for annotated and unannotated regions of the transcriptional landscape (Nagalakshmi et al., 2008). The expression of transcribed RNA features can be extensively regulated across human tissues and cell types (Djebali et al., 2012; Wang et al., 2008), which may involve alternative splicing of exons or pervasive variation within the 3′-UTR of transcripts. Differences in the 3′-UTR of transcripts are known to contribute to expression instability, translation, and act as sites of posttranscriptional regulation (Jackson, 1993). Unbiased transcriptome sequencing of the human brain can identify novel 3′-UTRs for candidate genes (Fig. 11.10). Further characterization of the transcriptome across assorted brain structures, experimental circumstances, and individuals may reveal unique 3′-UTRs or other features for transcribed elements. Probing the neurobiology of novel 3′-UTRs, though time consuming, could eventually expose distinct mechanisms of neuronal function. For example, a short 3′-UTR of Bdnf RNA is restricted to neuronal somata, while a long form of the 3′-UTR is trafficked to the dendrites where it can act upon spine morphology and synaptic transmission (An et al., 2008). Local translation of BDNF, delivered via a long form of the 3′-UTR, can modulate GABAergic transmission (Waterhouse et al., 2012), a well-characterized neurotransmitter system targeted by alcohol and other drugs of abuse (Davies et al., 2003; Harris, Trudell, & Mihic, 2008; Kauer & Malenka, 2007).
Figure 11.10.

Example of a novel three prime untranslated region (3′-UTR) from RNA-Seq data. Snapshot of RNA-Seq counts for three representative samples of a novel long form of a 3′-UTR. Blue (black in the print version) boxes indicate the presence of the last exon and currently annotated 3′-UTR; however, RNA-Seq may detect previously unknown details regarding transcript expression that may impact function.
8. GENETIC VARIATION AND ALCOHOL DEPENDENCE
Single nucleotide polymorphisms (SNPs) within GABA receptors, and several other candidate genes, are likely contributors in susceptibility to the development of alcohol dependence (Dick & Foroud, 2003). Similar to other psychiatric diseases, alcoholism is influenced by multiple genes with low-to-moderate effect (Sullivan, Daly, & O’Donovan, 2012). Polygenic factors can account for 40–60% of the risk for developing alcohol dependence (Schuckit, 2009); however, SNPs associated with disease usually reside within noncoding regions. Surveying the alcoholic transcriptome for genetic variants further corroborates this assertion (Fig. 11.11). The largest percentage of detected variants is located with sequencing reads mapped to intronic regions, followed by areas located up- or downstream of coding elements and intergenic regions. Introns are typically removed through RNA splicing events, but may be retained within individual isoforms harboring cis-acting SNPs controlling their expression (Dieter & Estus, 2010). High-throughput sequencing of human populations, as well as other model systems, is beginning to pinpoint numerous sites of nucleotide variation, that regardless of genomic loci, are capable of gene, alternative splicing, and downstream expression (Gerstein et al., 2010; Graveley et al., 2011; Lappalainen et al., 2013). However, linking any causal points of genetic inference with disease remains a significant challenge in the modern era for quantitative biology. Isolated studies often lack statistical power to definitively link any single SNP, let alone the interaction among multiple SNPs, with disease progression. Strategies are emerging to overcome these types of hurdles and identify unanticipated points of genetic interaction (Pan, 2008; Pandey et al., 2010). Similar to other complex traits, alcoholism is driven by the interaction of countless SNPs and competing environmental influences, which shape the transcriptome and regulate neurobiological functions. Although a number of differences may exist between human DNA and RNA sequences from the same individual (Li et al., 2011), sequencing the transcriptome in alcoholic brain tissue will continue to provide a valuable resource that represents the multidimensional factors operating in alcohol use disorders.
Figure 11.11.

Classifying genetic variants within alcoholic prefrontal cortex. Genetic variants detected within alcoholic prefrontal cortex classified by currently annotated gene regions: intergenic, upstream, 5′-UTR, exon, splice site donor, intron, splice site acceptor, 3′-UTR, and downstream. Genetic variants are primarily located in unannotated genomic areas or noncoding/intronic regions.
9. BIOLOGICAL COEXPRESSION NETWORKS
The number of genes implicated in alcohol dependence and other psychiatric illnesses continues to grow, with no single factor being uniquely responsible for the genotype–phenotype relationship. This is not a surprising notion, given that genes and their ensuing proteins do not exist in isolation, but work through coordinated pathways to govern cellular actions. Current canonical pathways, although useful to some extent, are becoming increasingly inadequate to account for the multitude of factors driving cellular behavior and manifesting phenotypes (Califano, Butte, Friend, Ideker, & Schadt, 2012). Using a variety of high-throughput approaches, both expected and unexpected connections can be simultaneously established among multiple cellular substrates to define biological networks for nearly any condition (Barabási & Oltvai, 2004; Vidal, Cusick, & Barabási, 2011). Not all genetic perturbations may be of equal value but may spread their effects across a web of neighboring genes to propagate disease symptomology. Disease-associated genes form an extended network that surrounds highly connected hub genes, which are essential to influence multiple biochemical pathways for development and survival (Goh et al., 2007). Understanding phenotypes across a spectrum of human disorders will require understanding the corresponding network architecture of related diseases.
The human brain transcriptome represents highly organized gene coexpression networks that are consistent across individuals (Hawrylycz et al., 2012; Oldham et al., 2008). Defining gene coexpression patterns for human diseases has revealed convergent molecular profiles (Voineagu et al., 2011), predicted causal systems in neuropathology (Zhang et al., 2013), and unveiled distinct network structures for similar phenotypes (Parikshak et al., 2013). Gene coexpression networks of alcoholic brain tissue, determined with microarray profiling, generated a systemic view of gene expression alterations spanning multiple cell types and brain regions (Ponomarev, Wang, Zhang, Harris, & Mayfield, 2012). A significant portion of transcripts coregulated by chronic alcohol abuse may be conserved within animal models of alcohol consumption (Nunez et al., 2013), permitting an experimentally tractable mode of elucidating gene networks in complex behaviors. Mouse models of alcohol-responsive gene networks can genetically dissect the interrelationship among endophenotypes (Wolen et al., 2012) and clarify networks of candidate genes in alcohol-responsive behaviors (Farris & Miles, 2013). Most network models of acute and chronic alcohol exposure have relied primarily upon microarrays, excluding many available ncRNA substrates. RNA-Seq-derived coexpression networks for alcohol-related phenotypes are still emerging, but should offer greater insight into the whole transcriptome (Giorgi, Del Fabbro, & Licausi, 2013; Iancu et al., 2012). Leveraging the network structure, in addition to differential expression, of the complete transcriptome using RNA-Seq will facilitate a more comprehensive assessment of transcribed features involved in alcoholism and drug conditions.
10. FUTURE DIRECTIONS
Realizing the full potential of RNA-Seq will eventually involve incorporating multiple levels of discrete data types and model systems (Fig. 11.12). The entire complement of RNA molecules, including miRNAs and lncRNAs, exists as a highly orchestrated network, regulated in part by genetic variants or epigenetic phenomena spread throughout the genome. Alternative splicing of mature RNA enables considerable bio-diversity of protein products and protein–protein interaction networks. The human proteome (Rual et al., 2005) is far from complete and will likely evolve in parallel with information gleaned from the transcriptome. In the long term, such information will further inform the interpretation of neurophysiological and neuroanatomical studies, including large-scale initiatives like the Human Connectome Project (Van Essen et al., 2013, 2012), in human health. A major challenge will be distilling the vast amount of biological data that bridge multiple scales and also are linked to discrete phenotypes. Focusing on intermediate phenotypes of complex traits may be useful for discovering large, consistent effects exerted by gene networks.
Figure 11.12.

Utilizing RNA-Seq in the context of a multiscale systems approach for understanding the neurobiology of alcohol dependence. Differing brain regions of alcoholics and controls can be evaluated using high-throughput sequencing for regulation of DNA and RNA expression. Information from sequencing data can then be layered with current protein data, brain imaging, physiological function, a variety of phenotypic traits (i.e., drinking behavior, withdrawal, craving), and additional influences (non-CNS tissues, human microbiota, and environmental pressures). Pooling resources from both clinical and preclinical sources can clarify points of convergent validity to determine individualized treatment plans incorporating behavioral therapy, current FDA approved compounds, or designer compounds that best target the underlying structure of an individual’s disease.
Although it should be emphasized that many of the biological effects seen in neuropsychiatric diseases may be specific to humans, model systems will continue to serve a fundamental role in the post transcriptomic era of modern biology. For example, systematically combining multiple biological networks within a yeast reference population clearly demonstrated that integrating several datasets can improve prediction of causal regulators of complex system behavior (Zhu et al., 2008). Combining information from human and animal models can ascertain core networks affecting disease (Emilsson et al., 2008). Alternatively, animal models explicitly created for a desired attribute may be sequenced to find novel causal contributors. Studying alcohol preferring and nonpreferring rats identified a stop codon within the metabotropic glutamate receptor 2 (Grm2) that controls protein expression and alcohol-drinking behavior (Zhou et al., 2013). This is just one example of the presumably large collection of variants that will be identified in alcohol consumption, which may eventually intersect with those recognized in human populations. Identifying networks with convergent validity across model organisms and humans (Fig. 11.12) has the potential to isolate systems for therapeutic intervention tailored to the specific needs of the individual.
Whole-transcriptome sequencing has far reaching effects in both clinical and preclinical applications. As a foundation for basic sciences, RNA-Seq continues to impart a deeper appreciation for the vast transcriptional structure of genes and quantification of transcript expression throughout differing cells, tissues, and species. Although still in the early stages of use, RNA-Seq can monitor spatial organization of the transcriptome (Lee et al., 2014) and extract some of the subtle expression differences induced by individual neurons and their microenvironments (Lovatt et al., 2014). With psychiatric illnesses representing one of the most challenging areas of medicine, sophisticated tools such as those furnished by deep sequencing technologies are essential for deciphering all of the converging elements that orchestrate these diseases.
References
- An JJ, Gharami K, Liao GY, Woo NH, Lau AG, Vanevski F, et al. Distinct role of long 3′ UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell. 2008;134(1):175–187. doi: 10.1016/j.cell.2008.05.045. http://dx.doi.org/10.1016/j.cell.2008.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, et al. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nature Protocols. 2013;8(9):1765–1786. doi: 10.1038/nprot.2013.099. http://dx.doi.org/10.1038/nprot.2013.099. [DOI] [PubMed] [Google Scholar]
- Barabási AL, Oltvai ZN. Network biology: Understanding the cell’s functional organization. Nature Reviews. Genetics. 2004;5(2):101–113. doi: 10.1038/nrg1272. http://dx.doi.org/10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, et al. The evolutionary landscape of alternative splicing in vertebrate species. Science (New York, NY) 2012;338(6114):1587–1593. doi: 10.1126/science.1230612. http://dx.doi.org/10.1126/science.1230612. [DOI] [PubMed] [Google Scholar]
- Barry G, Briggs JA, Vanichkina DP, Poth EM, Beveridge NJ, Ratnu VS, et al. The long non-coding RNA Gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Molecular Psychiatry. 2013;19:486–494. doi: 10.1038/mp.2013.45. http://dx.doi.org/10.1038/mp.2013.45. [DOI] [PubMed] [Google Scholar]
- Belgard TG, Marques AC, Oliver PL, Abaan HO, Sirey TM, Hoerder-Suabedissen A, et al. A transcriptomic atlas of mouse neocortical layers. Neuron. 2011;71(4):605–616. doi: 10.1016/j.neuron.2011.06.039. http://dx.doi.org/10.1016/j.neuron.2011.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond AM, Vangompel MJW, Sametsky EA, Clark MF, Savage JC, Disterhoft JF, et al. Balanced gene regulation by an embryonic brain ncRNA is critical for adult hippocampal GABA circuitry. Nature Neuroscience. 2009;12(8):1020–1027. doi: 10.1038/nn.2371. http://dx.doi.org/10.1038/nn.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottomly D, Walter NAR, Hunter JE, Darakjian P, Kawane S, Buck KJ, et al. Evaluating gene expression in C57BL/6J and DBA/2J mouse striatum using RNA-Seq and microarrays. PLoS One. 2011;6(3):e17820. doi: 10.1371/journal.pone.0017820. http://dx.doi.org/10.1371/journal.pone.0017820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics. 2010;11:94. doi: 10.1186/1471-2105-11-94. http://dx.doi.org/10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Califano A, Butte AJ, Friend S, Ideker T, Schadt E. Leveraging models of cell regulation and GWAS data in integrative network-based association studies. Nature Genetics. 2012;44(8):841–847. doi: 10.1038/ng.2355. http://dx.doi.org/10.1038/ng.2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Yeo G, Muotri AR, Kuwabara T, Gage FH. Noncoding RNAs in the mammalian central nervous system. Annual Review of Neuroscience. 2006;29:77–103. doi: 10.1146/annurev.neuro.29.051605.112839. http://dx.doi.org/10.1146/annurev.neuro.29.051605.112839. [DOI] [PubMed] [Google Scholar]
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, et al. The transcriptional landscape of the mammalian genome. Science (New York, NY) 2005;309(5740):1559–1563. doi: 10.1126/science.1112014. http://dx.doi.org/10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147(2):358–369. doi: 10.1016/j.cell.2011.09.028. http://dx.doi.org/10.1016/j.cell.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies AG, Pierce-Shimomura JT, Kim H, VanHoven MK, Thiele TR, Bonci A, et al. A central role of the BK potassium channel in behavioral responses to ethanol in C. elegans. Cell. 2003;115(6):655–666. doi: 10.1016/s0092-8674(03)00979-6. [DOI] [PubMed] [Google Scholar]
- Dick DM, Foroud T. Candidate genes for alcohol dependence: A review of genetic evidence from human studies. Alcoholism, Clinical and Experimental Research. 2003;27(5):868–879. doi: 10.1097/01.ALC.0000065436.24221.63. http://dx.doi.org/10.1097/01.ALC.0000065436.24221.63. [DOI] [PubMed] [Google Scholar]
- Dieter LS, Estus S. Isoform of APOE with retained intron 3; quantitation and identification of an associated single nucleotide polymorphism. Molecular Neurodegeneration. 2010;5:34. doi: 10.1186/1750-1326-5-34. http://dx.doi.org/10.1186/1750-1326-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillies MA, Rau A, Aubert J, Hennequet-Antier C, Jeanmougin M, Servant N, et al. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Briefings in Bioinformatics. 2013;14(6):671–683. doi: 10.1093/bib/bbs046. http://dx.doi.org/10.1093/bib/bbs046. [DOI] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, et al. Landscape of transcription in human cells. Nature. 2012;489(7414):101–108. doi: 10.1038/nature11233. http://dx.doi.org/10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. Non-coding RNA genes and the modern RNA world. Nature Reviews. Genetics. 2001;2(12):919–929. doi: 10.1038/35103511. http://dx.doi.org/10.1038/35103511. [DOI] [PubMed] [Google Scholar]
- Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J, et al. Genetics of gene expression and its effect on disease. Nature. 2008;452(7186):423–428. doi: 10.1038/nature06758. http://dx.doi.org/10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium. Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. http://dx.doi.org/10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium. Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816. doi: 10.1038/nature05874. http://dx.doi.org/10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris SP, Miles MF. Fyn-dependent gene networks in acute ethanol sensitivity. PLoS One. 2013;8(11):e82435. doi: 10.1371/journal.pone.0082435. http://dx.doi.org/10.1371/journal.pone.0082435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber M, Grabherr MG, Guttman M, Trapnell C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nature Methods. 2011;8(6):469–477. doi: 10.1038/nmeth.1613. http://dx.doi.org/10.1038/nmeth.1613. [DOI] [PubMed] [Google Scholar]
- Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science (New York, NY) 2010;330(6012):1775–1787. doi: 10.1126/science.1196914. http://dx.doi.org/10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi FM, Del Fabbro C, Licausi F. Comparative study of RNA-seq- and microarray-derived coexpression networks in Arabidopsis thaliana. Bioinformatics (Oxford, England) 2013;29(6):717–724. doi: 10.1093/bioinformatics/btt053. http://dx.doi.org/10.1093/bioinformatics/btt053. [DOI] [PubMed] [Google Scholar]
- Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabási AL. The human disease network. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(21):8685–8690. doi: 10.1073/pnas.0701361104. http://dx.doi.org/10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, et al. The developmental transcriptome of Drosophila melanogaster. Nature. 2011;471(7339):473–479. doi: 10.1038/nature09715. http://dx.doi.org/10.1038/nature09715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Irizarry RA, Wu Z. Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics (Oxford, England) 2012;13(2):204–216. doi: 10.1093/biostatistics/kxr054. http://dx.doi.org/10.1093/biostatistics/kxr054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RA, Trudell JR, Mihic SJ. Ethanol’s molecular targets. Science Signaling. 2008;1(28):re7. doi: 10.1126/scisignal.128re7. http://dx.doi.org/10.1126/scisignal.128re7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann B, Valcárcel J. Decrypting the genome’s alternative messages. Current Opinion in Cell Biology. 2009;21(3):377–386. doi: 10.1016/j.ceb.2009.02.006. http://dx.doi.org/10.1016/j.ceb.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489(7416):391–399. doi: 10.1038/nature11405. http://dx.doi.org/10.1038/nature11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iancu OD, Kawane S, Bottomly D, Searles R, Hitzemann R, McWeeney S. Utilizing RNA-Seq data for de novo coexpression network inference. Bioinformatics (Oxford, England) 2012;28(12):1592–1597. doi: 10.1093/bioinformatics/bts245. http://dx.doi.org/10.1093/bioinformatics/bts245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ. Cytoplasmic regulation of mRNA function: The importance of the 3′ untranslated region. Cell. 1993;74(1):9–14. doi: 10.1016/0092-8674(93)90290-7. [DOI] [PubMed] [Google Scholar]
- Jia H, Osak M, Bogu GK, Stanton LW, Johnson R, Lipovich L. Genome-wide computational identification and manual annotation of human long non-coding RNA genes. RNA (New York, NY) 2010;16(8):1478–1487. doi: 10.1261/rna.1951310. http://dx.doi.org/10.1261/rna.1951310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nature Reviews. Neuroscience. 2007;8(11):844–858. doi: 10.1038/nrn2234. http://dx.doi.org/10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(28):11667–11672. doi: 10.1073/pnas.0904715106. http://dx.doi.org/10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss T. Small nucleolar RNAs: An abundant group of noncoding RNAs with diverse cellular functions. Cell. 2002;109(2):145–148. doi: 10.1016/s0092-8674(02)00718-3. [DOI] [PubMed] [Google Scholar]
- Kryger R, Fan L, Wilce PA, Jaquet V. MALAT-1, a non protein-coding RNA is upregulated in the cerebellum, hippocampus and brain stem of human alcoholics. Alcohol. 2012;46(7):629–634. doi: 10.1016/j.alcohol.2012.04.002. http://dx.doi.org/10.1016/j.alcohol.2012.04.002. [DOI] [PubMed] [Google Scholar]
- Lappalainen T, Sammeth M, Friedländer MR, ’t Hoen PAC, Monlong J, Rivas MA, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501(7468):506–511. doi: 10.1038/nature12531. http://dx.doi.org/10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT. Epigenetic regulation by long noncoding RNAs. Science (New York, NY) 2012;338(6113):1435–1439. doi: 10.1126/science.1231776. http://dx.doi.org/10.1126/science.1231776. [DOI] [PubMed] [Google Scholar]
- Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, et al. Highly multiplexed subcellular RNA sequencing in situ. Science (New York, NY) 2014;343:1360–1363. doi: 10.1126/science.1250212. http://dx.doi.org/10.1126/science.1250212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Wang IX, Li Y, Bruzel A, Richards AL, Toung JM, et al. Widespread RNA and DNA sequence differences in the human transcriptome. Science (New York, NY) 2011;333(6038):53–58. doi: 10.1126/science.1207018. http://dx.doi.org/10.1126/science.1207018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovatt D, Ruble BK, Lee J, Dueck H, Kim TK, Fisher S, et al. Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nature Methods. 2014;11(2):190–196. doi: 10.1038/nmeth.2804. http://dx.doi.org/10.1038/nmeth.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mane SP, Evans C, Cooper KL, Crasta OR, Folkerts O, Hutchison SK, et al. Transcriptome sequencing of the microarray quality control (MAQC) RNA reference samples using next generation sequencing. BMC Genomics. 2009;10(1):264. doi: 10.1186/1471-2164-10-264. http://dx.doi.org/10.1186/1471-2164-10-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Research. 2008;18(9):1509–1517. doi: 10.1101/gr.079558.108. http://dx.doi.org/10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massone S, Vassallo I, Fiorino G, Castelnuovo M, Barbieri F, Borghi R, et al. 17A, a novel non-coding RNA, regulates GABA B alternative splicing and signaling in response to inflammatory stimuli and in Alzheimer disease. Neurobiology of Disease. 2011;41(2):308–317. doi: 10.1016/j.nbd.2010.09.019. http://dx.doi.org/10.1016/j.nbd.2010.09.019. [DOI] [PubMed] [Google Scholar]
- Mattick JS, Makunin IV. Non-coding RNA. Human Molecular Genetics. 2006;15(Spec No 1):R17–R29. doi: 10.1093/hmg/ddl046. http://dx.doi.org/10.1093/hmg/ddl046. [DOI] [PubMed] [Google Scholar]
- Mercer TR, Dinger ME, Sunkin SM, Mehler MF, Mattick JS. Specific expression of long noncoding RNAs in the mouse brain. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(2):716–721. doi: 10.1073/pnas.0706729105. http://dx.doi.org/10.1073/pnas.0706729105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozova O, Marra MA. Applications of next-generation sequencing technologies in functional genomics. Genomics. 2008;92(5):255–264. doi: 10.1016/j.ygeno.2008.07.001. http://dx.doi.org/10.1016/j.ygeno.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. http://dx.doi.org/10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, et al. he transcriptional landscape of the yeast genome defined by RNA sequencing. Science (New York, NY) 2008;320(5881):1344–1349. doi: 10.1126/science.1158441. http://dx.doi.org/10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez YO, Truitt JM, Gorini G, Ponomareva ON, Blednov YA, Harris RA, et al. Positively correlated miRNA-mRNA regulatory networks in mouse frontal cortex during early stages of alcohol dependence. BMC Genomics. 2013;14:725. doi: 10.1186/1471-2164-14-725. http://dx.doi.org/10.1186/1471-2164-14-725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, et al. Functional organization of the transcriptome in human brain. Nature Neuroscience. 2008;11(11):1271–1282. doi: 10.1038/nn.2207. http://dx.doi.org/10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshlack A, Wakefield MJ. Transcript length bias in RNA-seq data confounds systems biology. Biology Direct. 2009;4:14. doi: 10.1186/1745-6150-4-14. http://dx.doi.org/10.1186/1745-6150-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F, Milos PM. RNA sequencing: Advances, challenges and opportunities. Nature Reviews. Genetics. 2011;12(2):87–98. doi: 10.1038/nrg2934. http://dx.doi.org/10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan W. Network-based model weighting to detect multiple loci influencing complex diseases. Human Genetics. 2008;124(3):225–234. doi: 10.1007/s00439-008-0545-1. http://dx.doi.org/10.1007/s00439-008-0545-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Kaiguo M, Razak Z, Westwood JT, Gerlai R. Chronic alcohol exposure induced gene expression changes in the zebrafish brain. Behavioural Brain Research. 2011;216(1):66–76. doi: 10.1016/j.bbr.2010.07.017. http://dx.doi.org/10.1016/j.bbr.2010.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey G, Zhang B, Chang AN, Myers CL, Zhu J, Kumar V, et al. An integrative multi-network and multi-classifier approach to predict genetic interactions. PLoS Computational Biology. 2010;6(9) doi: 10.1371/journal.pcbi.1000928. http://dx.doi.org/10.1371/journal.pcbi.1000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155(5):1008–1021. doi: 10.1016/j.cell.2013.10.031. http://dx.doi.org/10.1016/j.cell.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, et al. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464(7289):768–772. doi: 10.1038/nature08872. http://dx.doi.org/10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrzykowski AZ, Friesen RM, Martin GE, Puig SI, Nowak CL, Wynne PM, et al. Posttranscriptional regulation of BK channel splice variant stability by miR-9 underlies neuroadaptation to alcohol. Neuron. 2008;59(2):274–287. doi: 10.1016/j.neuron.2008.05.032. http://dx.doi.org/10.1016/j.neuron.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Salama SR, Lambert N, Lambot MA, Coppens S, Pedersen JS, et al. An RNA gene expressed during cortical development evolved rapidly in humans. Nature. 2006;443(7108):167–172. doi: 10.1038/nature05113. http://dx.doi.org/10.1038/nature05113. [DOI] [PubMed] [Google Scholar]
- Ponomarev I, Wang S, Zhang L, Harris RA, Mayfield RD. Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2012;32(5):1884–1897. doi: 10.1523/JNEUROSCI.3136-11.2012. http://dx.doi.org/10.1523/JNEUROSCI.3136-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapaport F, Khanin R, Liang Y, Pirun M, Krek A, Zumbo P, et al. Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biology. 2013;14(9):R95. doi: 10.1186/gb-2013-14-9-r95. http://dx.doi.org/10.1186/gb-2013-14-9-r95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravasi T, Suzuki H, Pang KC, Katayama S, Furuno M, Okunishi R, et al. Experimental validation of the regulated expression of large numbers of non-coding RNAs from the mouse genome. Genome Research. 2006;16(1):11–19. doi: 10.1101/gr.4200206. http://dx.doi.org/10.1101/gr.4200206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso D, Schwartz K, Sherlock G, Dudoit S. GC-content normalization for RNA-Seq data. BMC Bioinformatics. 2011;12:480. doi: 10.1186/1471-2105-12-480. http://dx.doi.org/10.1186/1471-2105-12-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biology. 2010;11(3):R25. doi: 10.1186/gb-2010-11-3-r25. http://dx.doi.org/10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogelj B, Hartmann CEA, Yeo CH, Hunt SP, Giese KP. Contextual fear conditioning regulates the expression of brain-specific small nucleolar RNAs in hippocampus. The European Journal of Neuroscience. 2003;18(11):3089–3096. doi: 10.1111/j.1460-9568.2003.03026.x. [DOI] [PubMed] [Google Scholar]
- Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437(7062):1173–1178. doi: 10.1038/nature04209. http://dx.doi.org/10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- Schuckit MA. Alcohol-use disorders. Lancet. 2009;373(9662):492–501. doi: 10.1016/S0140-6736(09)60009-X. http://dx.doi.org/10.1016/S0140-6736(09)60009-X. [DOI] [PubMed] [Google Scholar]
- Soneson C, Delorenzi M. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinformatics. 2013;14:91. doi: 10.1186/1471-2105-14-91. http://dx.doi.org/10.1186/1471-2105-14-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF, Daly MJ, O’Donovan M. Genetic architectures of psychiatric disorders: The emerging picture and its implications. Nature Reviews. Genetics. 2012;13(8):537–551. doi: 10.1038/nrg3240. http://dx.doi.org/10.1038/nrg3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nature Methods. 2009;6(5):377–382. doi: 10.1038/nmeth.1315. http://dx.doi.org/10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- Tarazona S, García-Alcalde F, Dopazo J, Ferrer A, Conesa A. Differential expression in RNA-seq: A matter of depth. Genome Research. 2011;21(12):2213–2223. doi: 10.1101/gr.124321.111. http://dx.doi.org/10.1101/gr.124321.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tazi J, Tazi J, Bakkour N, Bakkour N, Stamm S, Stamm S. Alternative splicing and disease. Biochimica et Biophysica Acta. 2009;1792(1):14–26. doi: 10.1016/j.bbadis.2008.09.017. http://dx.doi.org/10.1016/j.bbadis.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols. 2012;7(3):562–578. doi: 10.1038/nprot.2012.016. http://dx.doi.org/10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Molecular Cell. 2010;39(6):925–938. doi: 10.1016/j.molcel.2010.08.011. http://dx.doi.org/10.1016/j.molcel.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Essen DC, Smith SM, Barch DM, Behrens TEJ, Yacoub E, Ugurbil K, et al. The WU-Minn Human Connectome project: An overview. NeuroImage. 2013;80:62–79. doi: 10.1016/j.neuroimage.2013.05.041. http://dx.doi.org/10.1016/j.neuroimage.2013.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Essen DC, Ugurbil K, Auerbach E, Barch D, Behrens TEJ, Bucholz R, et al. The Human Connectome Project: A data acquisition perspective. NeuroImage. 2012;62(4):2222–2231. doi: 10.1016/j.neuroimage.2012.02.018. http://dx.doi.org/10.1016/j.neuroimage.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M, Cusick ME, Barabási AL. Interactome networks and human disease. Cell. 2011;144(6):986–998. doi: 10.1016/j.cell.2011.02.016. http://dx.doi.org/10.1016/j.cell.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474(7351):380–384. doi: 10.1038/nature10110. http://dx.doi.org/10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter NAR, Bottomly D, Laderas T, Mooney MA, Darakjian P, Searles RP, et al. High throughput sequencing in mice: A platform comparison identifies a preponderance of cryptic SNPs. BMC Genomics. 2009;10:379. doi: 10.1186/1471-2164-10-379. http://dx.doi.org/10.1186/1471-2164-10-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: A revolutionary tool for trans-criptomics. Nature Reviews. Genetics. 2009;10(1):57–63. doi: 10.1038/nrg2484. http://dx.doi.org/10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–476. doi: 10.1038/nature07509. http://dx.doi.org/10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472(7341):120–124. doi: 10.1038/nature09819. http://dx.doi.org/10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse EG, An JJ, Orefice LL, Baydyuk M, Liao GY, Zheng K, et al. BDNF promotes differentiation and maturation of adult-born neurons through GABAergic transmission. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience. 2012;32(41):14318–14330. doi: 10.1523/JNEUROSCI.0709-12.2012. http://dx.doi.org/10.1523/JNEUROSCI.0709-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolen AR, Phillips CA, Langston MA, Putman AH, Vorster PJ, Bruce NA, et al. Genetic dissection of acute ethanol responsive gene networks in prefrontal cortex: Functional and mechanistic implications. PLoS One. 2012;7(4):e33575. doi: 10.1371/journal.pone.0033575. http://dx.doi.org/10.1371/journal.pone.0033575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y, Yu T, Wu YN, Roy M, Kim J, Lee C. An expectation-maximization algorithm for probabilistic reconstructions of full-length isoforms from splice graphs. Nucleic Acids Research. 2006;34(10):3150–3160. doi: 10.1093/nar/gkl396. http://dx.doi.org/10.1093/nar/gkl396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013;153(3):707–720. doi: 10.1016/j.cell.2013.03.030. http://dx.doi.org/10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Karlsson C, Liang T, Xiong W, Kimura M, Tapocik JD, et al. Loss of metabotropic glutamate receptor 2 escalates alcohol consumption. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(42):16963–16968. doi: 10.1073/pnas.1309839110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Zhang B, Smith EN, Drees B, Brem RB, Kruglyak L, et al. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nature Genetics. 2008;40(7):854–861. doi: 10.1038/ng.167. http://dx.doi.org/10.1038/ng.167. [DOI] [PMC free article] [PubMed] [Google Scholar]