


Abstract
Ets homologous factor (EHF) is an Ets family transcription factor expressed in many epithelial cell types including those lining the respiratory system. Disruption of the airway epithelium is central to many lung diseases, and a network of transcription factors coordinates its normal function. EHF can act as a transcriptional activator or a repressor, though its targets in lung epithelial cells are largely uncharacterized. Chromatin immunoprecipitation followed by deep sequencing (ChIP-seq), showed that the majority of EHF binding sites in lung epithelial cells are intergenic or intronic and coincide with putative enhancers, marked by specific histone modifications. EHF occupies many genomic sites that are close to genes involved in intercellular and cell–matrix adhesion. RNA-seq after EHF depletion or overexpression showed significant alterations in the expression of genes involved in response to wounding. EHF knockdown also targeted genes in pathways of epithelial development and differentiation and locomotory behavior. These changes in gene expression coincided with alterations in cellular phenotype including slowed wound closure and increased transepithelial resistance. Our data suggest that EHF regulates gene pathways critical for epithelial response to injury, including those involved in maintenance of barrier function, inflammation and efficient wound repair.
INTRODUCTION
The surface of the trachea and bronchial tree is covered with an epithelial layer that is critical for establishing and maintaining normal lung function. Not only does the epithelium provide a physical barrier between the outside environment and other tissues within the lung, it also makes a major contribution to the production and homeostasis of airway surface liquid (ASL), which is pivotal to a healthy respiratory environment (1). The structural integrity of the lung epithelium is maintained by intercellular tight junctions, and by additional mechanisms that adhere the epithelial cells to each other and to the underlying basement membrane (2). Under normal conditions, the epithelial cells play an important role in defense against external insults by driving the mucociliary escalator, which removes foreign particles and pathogens from the lung (3,4). Epithelial dysfunction underlies the pathology of several human respiratory diseases, including cystic fibrosis (CF), asthma and chronic obstructive pulmonary disease (COPD) (5–7). A significant component of dysfunction in these diseases is associated with impaired epithelial repair, inflammation and fibrosis (8). Epithelial cell function is regulated by networks of transcription factors that control gene expression (9–11) and show some common features across all epithelia in addition to organ-specific programs. The application of genome-wide approaches to study the critical transcription factors in lung epithelial differentiation is beginning to elucidate the molecular basis of these pathways.
Ets homologous factor (EHF) is a member of the epithelial-specific Ets transcription factor subfamily that is expressed in multiple epithelial cell types, including those in the lung (12–14). EHF has been shown to act at the promoter of genes to either activate or repress transcription (12,15,16). Moreover, predicted EHF binding sites are over-represented in intergenic open chromatin genome-wide in primary human tracheal and bronchial epithelial cells (9), suggesting that this factor plays an important role in the transcriptional program of these cells. EHF contributes to corneal epithelial cell fate (17), and in prostate cancer cells, loss of EHF promotes epithelial to mesenchymal transition (EMT) (18). During EMT, epithelial cells transition to a more mesenchymal phenotype, losing intercellular junctions and in some circumstances becoming more motile (19). Similar pathways are likely to be involved in lung epithelial repair after injury, and an exaggerated response may be associated with lung fibrosis, a prominent feature of multiple airway diseases (20,21).
Also relevant to inflammatory diseases of the airway is the regulation of EHF by cytokines in bronchial epithelial cells, where interleukin-1β (IL-1β) and/or tumor necrosis factor-α (TNF-α) increase EHF expression in an NF-κB dependent manner (22). Renewed interest in the potential importance of EHF in lung disease arose from a genome-wide association study (GWAS) to identify genetic markers of lung disease severity in the inherited disorder CF (23). Single nucleotide polymorphisms (SNPs) showing the strongest association with this trait mapped to an intergenic region of chromosome 11p13. The EHF gene maps immediately adjacent to this region on the 5' side and so became a candidate factor for an important role in lung epithelial function in health and disease. However, to date very little is known about the biological targets of EHF in airway epithelial cells and thus is the focus of this study. We hypothesize that through its direct interaction with cis-regulatory elements of critical genes, EHF controls essential cellular processes that coordinate the barrier function of the lung epithelium and participate in wound repair mechanisms.
We used Calu-3 lung adenocarcinoma cells for this analysis since they are highly differentiated and express critical markers of normal human airway epithelial function. Specifically, they form polarized monolayers in culture with tight junctions that lead to high transepithelial resistance, and they respond to cAMP agonists that drive chloride secretion across the epithelial layer (24). These properties of Calu-3 cells replicate key features of the normal lung epithelium.
Here, we show that EHF regulates genes that are important for normal epithelial function. Using chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) and RNA-sequencing (RNA-seq) after modulation of EHF levels, we identify a genome-wide binding signature for EHF and also determine its transcriptional targets. We also find that changes in gene expression correlate with phenotypic changes in airway epithelial cell behavior. In EHF-depleted lung adenocarcinoma cells, wound repair is delayed and transepithelial resistance (TER) is increased, suggesting that EHF is critical for maintenance of a differentiated phenotype in the airway epithelium.
MATERIALS AND METHODS
Cell culture
Calu-3 and A549 cells were obtained from ATCC and cultured by standard methods. A549 stable clones overexpressing EHF from pcDNA-EHF or control clones carrying pcDNA3.1 were generated by Lipofectin (Life Technologies (LT)) transfection, followed by G418 selection.
Western blots
Calu-3 cell lysates were analysed by standard methods. Antibodies used were specific for EHF (Clone 5A.5, Pierce), ITGA2 (ab133557, Abcam) and β-tubulin (T4026, Sigma-Aldrich).
ChIP-seq
The antibodies used for ChIP-seq were for EHF (Clone 5A.5), H3K4me1 (ab8895, Abcam) and H3K27ac (ab4729, Abcam). ChIP-seq was performed as described previously (25–27) with some modifications (see supplementary methods). Sequence data are summarized in Supplementary Table S1. Binding sites were identified using HOMER software (28). Two replicates of EHF ChIP-seq were performed. Peaks that were identified in both replicates were used for further analysis. To confirm results, ChIP was performed as described, followed by qPCR using primer pairs specific for each target (Supplementary Table S2) and SYBR green (LT). All data are deposited at GEO (http://www.ncbi.nlm.nih.gov/geo/).
DNAse-seq
DNase-seq was performed on Calu-3 cells as described previously (9,29). All data are deposited at GEO (http://www.ncbi.nlm.nih.gov/geo/).
RNA-seq
Calu-3 cells were treated with 30nM Silencer Select negative control siRNA #2 or EHF siRNA (s25399, both from Ambion) using RNAiMax transfection reagent (LT). 48 h post-transfection, RNA was isolated from five samples per treatment using Trizol (LT). RNA-seq libraries were prepared using the TruSeq RNA Sample Preparation Kit v2 per the manufacturer's Low-Throughput protocol (Illumina). The libraries were sequenced on Illumina HiSeq2500 machines. Data were analysed using TopHat and Cufflinks (30). qRT-PCR was performed with TaqMan Reverse Transcription reagents kit (LT), primer pairs specific for each target (Supplementary Table S2) and SYBR green. All data are deposited at GEO (http://www.ncbi.nlm.nih.gov/geo/).
Wound repair assay
Wound closure assay was performed as previously described (31) with modifications. Cells were treated with siRNA as above. 72 h post-transfection, the confluent lawn was damaged with a comb and then monitored by microscopy. Scratch area was measured using ImageJ software.
Transepithelial resistance measurements
Calu-3 cells were grown on Transwell cell culture inserts coated with collagen I (Sigma, C3867–1VL) and Collagen IV (Sigma, C6745-1ML). Triplicate cultures were reverse transfected using siRNAs described above. An epithelial voltohmmeter (EVOM2, World Precision Instruments) was used to take four resistivity readings (Ohms) per Transwell every 24 h for 96 h and TER values were calculated by multiplying resistivity by the insert's surface area (0.31 cm2).
RESULTS
ChIP-seq identifies sites of EHF occupancy genome-wide
EHF targets in the lung epithelium are poorly characterized. To investigate the genome-wide binding profile of EHF in airway epithelia, ChIP-seq was performed in Calu-3 lung adenocarcinoma cells (24). The antibody used in the ChIP experiments was first tested for specificity for EHF using western blots of protein lysates from cells in which EHF was depleted by a specific siRNA (Supplementary Figure S1). Two independent ChIP-seq experiments were performed. To identify EHF binding sites, the Homer software package (28) was used to call peaks for the two samples (1537 and 5216, respectively) and ChIP input DNA was used as a background control. Overlapping segments of peaks called in both samples were considered high-confidence EHF binding sites and used for further analysis. 768 binding sites were identified in both samples at a false discovery rate (FDR) of 1% (Supplementary Table S3). Moreover, there was significant correlation between tag counts at peaks found in both samples (Figure 1A).
Figure 1.
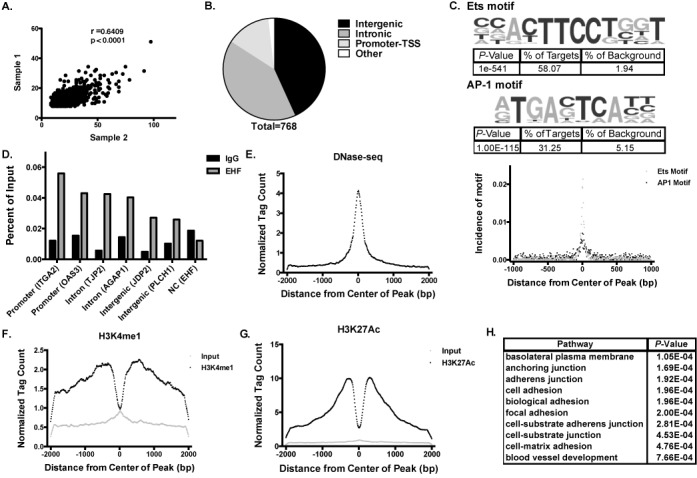
ChIP-seq in airway epithelial cells identifies genome-wide binding sites of EHF. (A) Comparison of tag counts between peaks identified in two biological replicates of EHF ChIP-seq experiments. (B) Number of EHF peaks found at intergenic sites, in introns, promoters and other sites (including exons, 5'UTRs and 3'UTRs). A total of 768 peaks were identified. Most peaks were found in either intergenic or intronic regions. (C) (Top) De novo motif analysis shows that EHF ChIP-seq peaks are enriched for an Ets motif similar to the consensus sequence for epithelial-specific Ets transcription factor subfamily proteins (58.07% of peaks within a 50 bp window, 82.29% within a 200 bp window) and the AP-1 binding motif (31.25% of peaks within a 200 bp window). (Bottom) Incidence of motifs measured as distance from the center of EHF ChIP-seq peaks in base pairs (bp). (D) ChIP-qPCR validated EHF binding to 6 peaks identified by ChIP-seq (n = 2). Peaks are labeled based on their location with the nearest gene in parentheses. An intergenic negative control site showed no enrichment. (E) Calu-3 DNase-seq tag counts measured from the center of EHF peaks. (F) As (E) for H3K4me1 ChIP-seq. (G) As (E) for H3K27ac ChIP-seq. (H) Gene ontology analysis by DAVID on the nearest genes to each EHF peak.
Peaks were annotated based on their genomic location. The majority of EHF peaks were found at either intergenic or intronic sites, as predicted for cell-type specific regulatory elements (9,32), although many binding sites at promoters (defined as −1 kb to +100 bp relative to the transcription start site) were also identified (Figure 1B). De novo motif analysis of the significant peaks seen in both ChIP-seq samples found that an Ets motif similar to the consensus binding sites for the epithelial-specific Ets transcription factor subfamily members (EHF, Elf3 and Elf5) was significantly enriched within these regions (P = 1e−541, Figure 1C). This consensus sequence coincided with an EHF binding motif characterized previously (33) and was found in 58.07% of peaks within 50 bp of the center in the current data set. Also enriched within EHF ChIP-seq peaks was the binding matrix for the activator protein 1 (AP-1) transcription factor complex (P = 1e−115, 1C), found in 31.25% of sites within 200 bp of the center of the peak. Sixteen percent of the peaks contain both an EHF and AP-1 motif. This is consistent with earlier findings that EHF binds near AP-1 motifs in repressive contexts (12).
To validate the ChIP-seq findings, ChIP followed by quantitative PCR (qPCR) was performed on two independent Calu-3 chromatin samples. The sites tested included 2 intronic (in integrin, α2 (ITGA2) and 2'-5'-oligoadenylate synthetase 3 (OAS3)), 2 intergenic (near tight junction protein 2 (TJP2) and ArfGAP with GTPase domain, ankyrin repeat and PH domain 1 (AGAP1)), and 2 promoter sites (in Jun dimerization protein 2 (JDP2) and phospholipase C, eta 1 (PLCH1)). These sites were chosen due to their location near genes with a significant change in expression following EHF depletion (ITGA2, OAS3, JDP2, TJP2) and/or because they contained an EHF motif (AGAP1, JDP2). For comparison, one negative control (NC) region (near EHF) was selected, which lacked an EHF ChIP-seq peak, a predicted EHF binding motif and H3K4me1/H3K27ac enrichment. Peaks identified in ChIP-seq were enriched over an IgG control in both replicates, at all six sites tested, while no enrichment was seen in the negative control (Figure 1D).
Next, the EHF ChIP-seq peaks in Calu-3 cells were intersected with open chromatin data for this cell line, generated by DNase I digestion followed by deep sequencing (DNase-seq). EHF occupancy sites showed increased DNase hypersensitivity, suggesting that this factor binds to open chromatin (Figure 1E). To determine whether the EHF ChIP-seq peaks contained enhancer elements, as might be predicted since many of them were intergenic or intronic, we next performed ChIP-seq for the enhancer-specific histone modifications H3K4me1 and H3K27ac in Calu-3 cells. Histone modification tag counts per base pair per peak were plotted as distance from the center of EHF peaks. The results showed a bimodal distribution of enrichment for the H3K4me1 and H3K27ac modifications surrounding the center of EHF ChIP-seq peaks (Figure 1F and G), which is characteristic of enhancers and was not seen in the input control. Among the 768 EHF binding sites identified by ChIP-seq, 591 coincided with peaks of H3K27ac enrichment. Sixty-four overlapped regions marked by H3K4me1, of which 53 also showed H3K27ac enrichment. Thus, though the majority of EHF binding sites coincide with active histone marks, a substantial minority (166 peaks) lack them, and these sites may contribute to EHF-mediated repression of gene expression.
Annotation of EHF binding sites
Next, the nearest gene annotation method was used to predict which genes were likely regulated by occupancy of EHF at cis-regulatory elements. This method has inherent limitations since many regulatory elements lie very far from the genes they control and can be located within introns of irrelevant genes. The nearest gene to each EHF binding peak was identified and a gene ontology process enrichment analysis was performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID) (34) to determine the pathways that were enriched among these annotated genes (Figure 1H, Supplementary Table S4). Genes located nearest to EHF binding sites were enriched for pathways involved in maintenance of the epithelial monolayer and intercellular, cell–matrix and cell–substrate junctions.
An increase or decrease in EHF alters expression of genes important for epithelial function
EHF depletion
To identify target genes regulated by EHF, either directly or indirectly, a specific siRNA was used to deplete EHF in Calu-3 cells (Figure 2A). Total RNA was isolated from five negative control (NC) siRNA and five EHF targeted siRNA samples (>90% depletion), and RNA quality was confirmed by Nanodrop measurement of OD 260/280 and 260/230 ratios. RNA-seq was performed on libraries generated from the 10 samples yielding ∼2.6–3.5 x107 reads per library. Gene expression values in the format of Fragments Per Kilobase per Million mapped fragments (FPKM) obtained from TopHat and Cufflinks (30) were compiled into an expression value matrix. Principle component analysis (PCA) on the FPKM values showed that the five samples within each treatment group clustered together and the different treatment groups segregated along PC #2 (Supplementary Figure S2). CuffDiff (30) was used to determine differentially regulated genes following EHF depletion. A total of 256 genes showed a greater than 1.5-fold difference in expression following EHF knockdown (Figure 2B, Supplementary Table S5). One hundred and twenty one and 135 genes showed decreased or increased expression, respectively, suggesting that EHF can positively or negatively regulate gene expression. The average distance between EHF bound sites and EHF-activated or -repressed genes (2930 kb and 2524 kb, respectively) was not significantly different (P = 0.435, Supplementary Figure S3). This observation is consistent with a role for EHF in both gene activation and repression.
Figure 2.
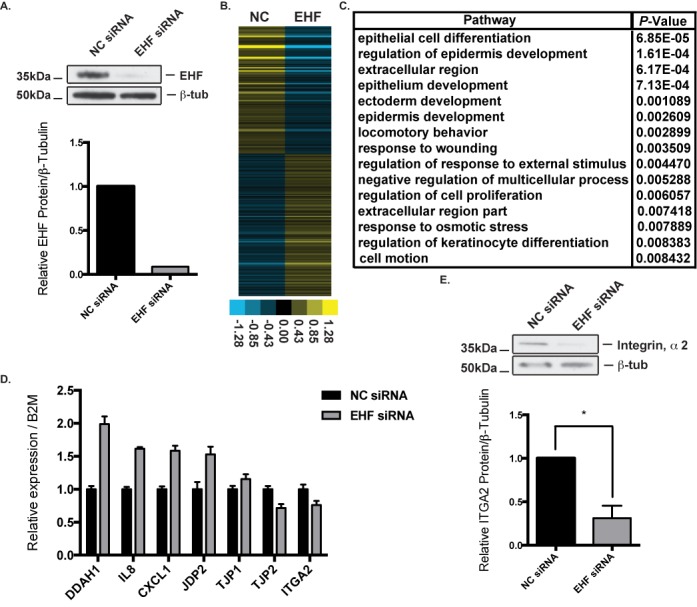
EHF depletion followed by RNA-sequencing identifies EHF as a regulator of pathways important for lung epithelial function. (A) siRNA depletion of EHF in Calu-3 cells measured by western blot compared to β-tubulin (top) and quantified using gel densitometry (bottom). (B) Relative expression of 256 genes that showed greater than 1.5-fold change in expression following siRNA depletion of EHF. Each row represents a different gene. (C) Gene ontology analysis by DAVID of differentially expressed genes (DEGs) with greater than 1.5-fold change in expression following EHF knockdown. (D) EHF siRNA knockdown followed by qPCR confirmed changes in gene expression identified by RNA-seq (n = 2). All 7 genes assayed showed changes in expression consistent with RNA-seq results. (E). Integrin, α2 protein expression significantly decreased following EHF knockdown (n = 3), corresponding to the observed decrease in RNA abundance for this gene. * P<0.05 by an unpaired two-tailed Student's t test, also used in Figures 4 and 5.
A gene ontology process enrichment analysis was performed on the differentially expressed genes (DEGs) following EHF knockdown using DAVID (Figure 2C, Supplementary Table S6). The DEGs were enriched for gene ontology pathways crucial for lung development and repair. Genes involved in epithelial cell differentiation and development included frizzled class receptor 1 (FZD1), and keratins 4, 5 and 14 (KRT4, KRT5, KRT14). Pathways involved in locomotory behavior and response to wounding were also enriched, with DEGs including S100 calcium binding protein A8 and A9 (S100A8, S100A9), chemokine (C-X-C motif) ligands 1 and 6 (CXCL1, CXCL6) and chemokine (C-C Motif) ligand 5 (CCL5).
Gene expression changes following EHF depletion were validated using siRNA knockdown (KD) followed by RT-qPCR. DEGs involved in cell–cell junctions, immune function and intracellular signaling were tested. Expression changes seen in the RNA-seq data were replicated in the qPCR validation (Figure 2D). ITGA2, JDP2, TJP2, CXCL1, interleukin-8 (IL-8), dimethylarginine dimethylaminohydrolase 1 (DDAH1) and tight junction protein 1 (TJP1) showed changes in expression that corresponded to RNA-seq results. The RT-qPCR and RNA-seq signals are significantly correlated (r = 0.81, P = 0.027). ITGA2 showed decreased expression following EHF KD, and contains an EHF binding site at its promoter. To confirm that transcriptional changes identified in the RNA-seq data were also seen at the protein level, whole cell lysates from negative control and EHF siRNA-treated samples were analysed by western blot to look for changes in integrin, α2 protein levels. A specific antibody showed that integrin, α2 protein expression significantly decreased (P<0.05) in comparison to the β-tubulin control following EHF depletion, consistent with the mRNA data (Figure 2E).
EHF overexpression
A549 cell clones were generated that stably overexpress EHF from a transfected plasmid (pcDNA 3.1). This alveolar-like lung adenocarcinoma cell line (35) was chosen due to its low levels of endogenous EHF, in contrast to the high levels seen in Calu-3 cells. Three A549 clones carrying vector only and 3 overexpressing EHF were analysed further, and western blots showed ∼ 11-fold more EHF protein in the latter clones (Figure 3A). Total RNA from 3–4 replicates of each clone was isolated and used for RNA-seq. At the transcript level (measured by FPKM), there was an 8.6-fold increase in EHF in the overexpressing clones. A total of 1153 genes showed a significant change in expression of >1.5-fold in the EHF overexpressing clones as compared to the vector clones (Figure 3B, Supplementary Table S7), with 761 increasing and 392 decreasing. These data confirm that EHF can positively or negatively regulate gene expression.
Figure 3.
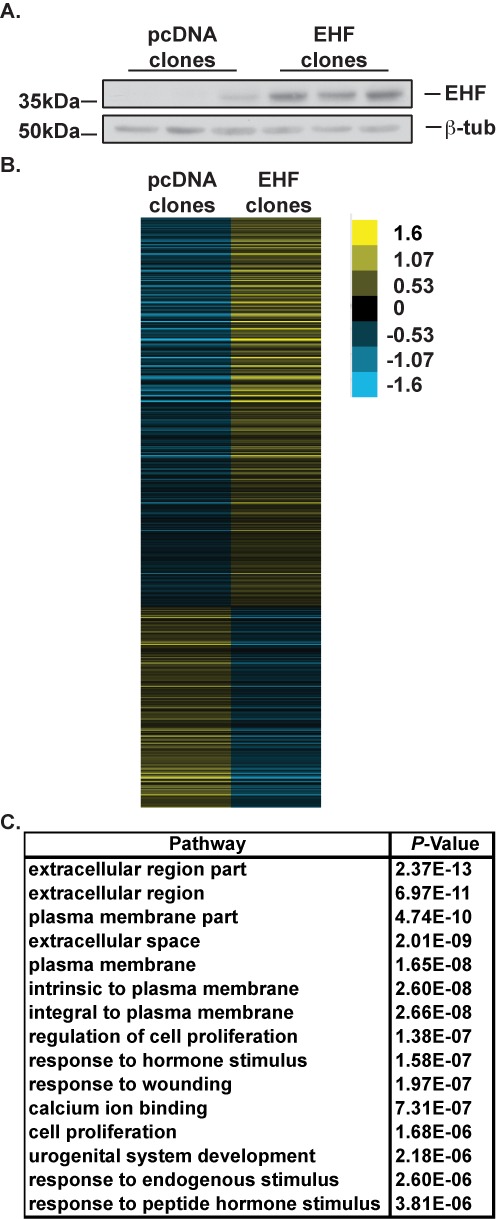
EHF overexpression followed by RNA-sequencing confirms that EHF controls genes integral to airway epithelial function. (A). Western blot probed with an antibody to EHF shows low levels of EHF in A549 cell clones containing the pcDNA 3.1 vector alone (pcDNA clones) and higher levels in clones expressing pcDNA-EHF (EHF clones). β-tubulin provided the loading control. (B) Relative expression of 1153 genes that showed greater than 1.5-fold change in expression in EHF overexpressing clones versus control clones. Each row represents a different gene. (C) Gene ontology analysis by DAVID of DEGs with greater than 1.5-fold change in expression in EHF overexpressing clones.
A gene ontology process enrichment analysis was performed on the DEGs using DAVID (Figure 3C, Supplementary Table S8). These genes were enriched for pathways involved in extracellular matrix properties, plasma membrane structure and function, cell proliferation and response to wounding. Genes involved in the extracellular region include multiple collagens (COL4A3, COL5A1, COL5A2, COL7A1 and COL12A1), mucins (MUC1, MUC3A, MUC5AC and MUC13) interleukins (IL6, IL11 and IL15) and members of the tumor necrosis factor (ligand) superfamily (TNFSF9, TNFSF15 and TNFSF13). Transcripts related to the plasma membrane include matrix metallopeptidase 14 (MMP14), CD44 and multiple solute carrier family members relevant to lung epithelial function (SLC6A14, SLC7A2, SLC7A7, SLC12A2, SLC16A7 and SLC23A2). Moreover, SLC6A14 is a pleiotropic gene modifier of lung disease severity and age of first Pseudomonas aeruginosa infection in CF patients (36). These genes and pathways are critical for response to the environment, maintenance of the epithelial barrier and extracellular matrix production.
Common targets of EHF modulation across cell types
Fifty-eight genes were positively regulated in both Calu-3 and A549 cells (that is, expression decreased in EHF depleted Calu-3 cells and increased in EHF-overexpressing A549 clones, Supplementary Table S9). A gene ontology enrichment analysis showed that these 58 genes were associated with processes of epithelial differentiation, anchoring junction and regulation of cell proliferation (Supplementary Table S10). Furthermore, 28 genes were negatively regulated in both cell types, many of which encode cell surface proteins (Supplementary Table S11).
EHF depletion slows the rate of wound repair in Calu-3 cells
Our RNA-seq data suggest that EHF is important in epithelial barrier integrity and regulates genes important for cell motility, which are essential components of wound repair. Shortly after epithelial injury, nearby epithelial cells migrate to repopulate the damaged area (37), a process that can be monitored in vitro using a wound scratch assay (31). To determine the effect of EHF depletion on wound repair in airway epithelial cells, a wound scratch assay was performed in Calu-3 cells. Cells were transfected with NC or EHF siRNA and grown to confluence. 72 h after transfection, the cell monolayer was wounded in a uniform manner using a comb. Scratches were then imaged at 3, 6, 9 and 12 h after transfection (Figure 4A). At 3 h, the average diameter for negative controls was 0.60 mm2 and the average for EHF siRNA is 0.53 mm2. These values are not significantly different (P = 0.25). Cells were grown for 27 h post-scratch to ensure wound closure and then lysed to measure EHF depletion. A comparison of the migration rate between NC and EHF siRNA-treated cells shows that the latter migrated at 67% the rate of NC treated cells at 6 h (P<0.01) and 71% at 9 h (P<0.001) (Figure 4B). This difference was no longer apparent at 12 hours. Western blots of cell lysates probed with an antibody specific for EHF and a β-tubulin control antibody showed that EHF depletion was maintained throughout the wound repair assay; 27 h after wounding, EHF protein levels were 15% of those in negative control treated cells (Figure 4C).
Figure 4.
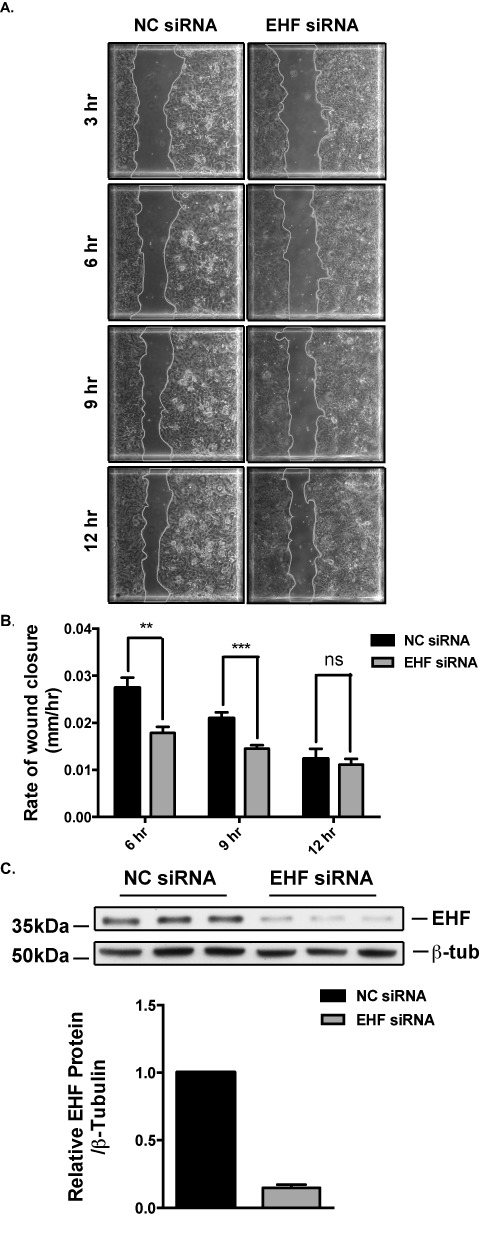
Effect of EHF depletion on wound repair in Calu-3 cells. (A) Images of negative control (NC) and EHF siRNA treated cells at 3, 6, 9 and 12 h after wounding. The outline of the wound is traced with a gray line. (B) Depletion of EHF significantly reduced the rate of wound closure at 6 and 9 h after wounding (n = 3). This difference was no longer evident 12 h after wounding. **P<0.01, ***P<0.001. (C) siRNA knockdown of EHF was measured by western blot in lysates taken 27 h after wounding and the signal was quantified using densitometry. EHF depletion was maintained throughout the experiment.
Transepithelial resistance is increased following depletion of EHF in Calu-3 cells
Experimental modulation of EHF shows that this factor regulates genes encoding proteins that establish and maintain intercellular junctions. Tight junctions are critical for preserving a selectively permeable barrier between the luminal and basolateral surfaces of the intact epithelium. These barrier functions are monitored by transepithelial resistance (TER) measurements of cells grown in vitro on permeable supports. TER measurements were performed on post-confluent Calu-3 cells to investigate the effect of EHF depletion on epithelial cell monolayer and tight-junction formation. Calu-3 cells were seeded onto collagen coated Transwell inserts and reverse transfected with either NC or EHF siRNA in triplicate. Resistivity measurements were taken from each insert in quadruplicate at 24, 48, 72 and 96 h post-transfection and TER values were calculated. EHF depleted Calu-3 cells showed higher TER compared to the negative control cells at multiple timepoints (Figure 5A). Normalization across 3 independent experiments revealed that TER values were significantly higher in EHF-depleted Calu-3 than controls at 48 h (P<0.05), 72 h (P<0.001) and 96 h (P<0.0001) (Figure 5B). Cells were lysed at 96 h and EHF/β-tubulin western blots confirmed EHF siRNA knockdown (Figure 5C).
Figure 5.
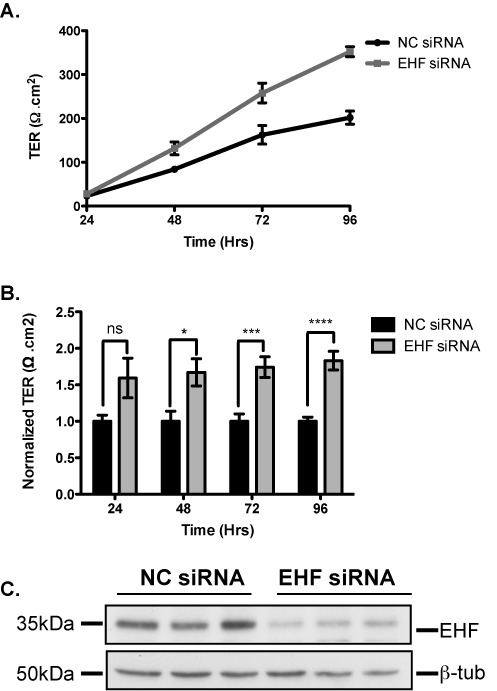
Effect of EHF depletion on transepithelial resistance in Calu-3 cells. (A) TER measurements in a single experiment show that EHF depleted Calu-3 cells (EHF siRNA, gray) had higher TER values compared to negative control cells (NC siRNA, black) at 48 to 96 h post-transfection. (B) TER measurements EHF siRNA normalized to NC; EHF depletion resulted in a significantly higher TER than NC-treated cells at 48 (*P< 0.05), 72 (***P<0.001) and 96 h (****P<0.0001) (n = 3). (C.) Western blot shows NC and EHF siRNA transfected Calu-3 cells from a single TER experiment (in triplicate) lysed at 96 h and depletion of EHF protein relative to β-tubulin was confirmed.
DISCUSSION
Understanding regulation of gene expression in lung epithelial cells is critical to determining how these cell types develop, differentiate and respond to the environment. Integration of these individual processes is required for normal airway function and their perturbation results in airway pathology. Several transcription factors are known to have an important role in lung epithelial development and function, including FOXA1/A2 (38), SOX family members (39) and Grainyhead (40), among others. Here, we characterize the role of EHF in coordinating gene expression profiles in lung epithelial cells. Our studies show that this transcription factor regulates genes and pathways involved in maintaining epithelial cell identity.
Previous studies on the function of EHF focused on its role at gene promoters, where it was shown to act as either a gene activator or repressor depending on the cellular context (12,15,16,18). However, the majority of EHF binding sites identified here by ChIP-seq were either intronic or intergenic. This is consistent with our data showing that cell-type selective open chromatin in primary airway epithelial cells is predominantly located outside gene promoters (9). The observation that EHF binding sites were enriched for H3K4me1 and H3K27ac histone modifications further reinforces the prediction that this transcription factor binds to distal elements to regulate gene expression. H3K4me1 is a general marker for enhancers, whereas H3K27ac tends to mark active regulatory elements (41,42). Moreover, EHF ChIP-seq peaks in Calu-3 cells also showed increased DNase I hypersensitivity when compared to background, suggesting that the factor binds to nucleosome-depleted regions in these cells. Thus, EHF binds to active enhancers, promoters and potentially other cis-regulatory elements in open chromatin sites to orchestrate its transcriptional program.
EHF ChIP-seq peaks are enriched for AP-1 binding sites, suggesting that members of this complex (which includes heterodimers of the c-Fos, c-Jun, ATF and JDP families) may be interacting partners of EHF in regulating gene expression. This prediction is consistent with a previous study showing that EHF represses promoters containing AP-1 binding motifs (12). In general, promoters containing AP-1 sites are responsive to mitogen-activated protein (MAP)-kinase signaling, which plays an important role in a diverse array of cell processes including differentiation, proliferation and migration. AP-1 and Ets family members have been shown to interact directly in vitro and in activated T-cells (43) and to co-localize in genome-wide ChIP-seq studies in prostate cancer cell lines (44). The extrapolation from our data that AP-1 and EHF may interact directly genome-wide to regulate gene expression in the lung epithelium warrants further study. Moreover, the mechanism whereby these factors could co-regulate gene expression through cis-regulatory elements and promoters is intriguing.
We found that EHF bound to 768 high-confidence sites in human lung epithelial cells, which was less than the number seen in EHF ChIP-seq performed in the mouse cornea epithelium (17). However, the previous study used a higher FDR (5%) to call peaks in comparison to the 1% used in our analysis. Moreover, we reduced the number of sites significantly by only considering peaks that overlapped in two independent experiments (1537 and 5216 peaks, respectively), while the former analysis did not require peaks to be present in two biological replicates.
We showed that depletion of EHF in Calu-3 cells altered the expression of genes in pathways that are important for epithelial and ectoderm development, and response to wounding. EHF overexpression in A549 cells differentially regulated genes involved in response to wounding and cell proliferation. The EHF depletion and overexpression experiments were performed in different cell types (with high or low endogenous EHF levels, respectively) to facilitate the greatest expression changes. Thus, under baseline conditions many EHF-regulated genes may differ between them. Despite this, many genes were similarly regulated in both cell types. EHF positively regulated genes involved in epithelial differentiation and cell junctions in both cell types.
Our data correlate well with the reported effects of EHF knockdown in corneal epithelium (17). Although the specialized biological properties of the airway and eye epithelium are different, they share common features of all epithelial layers in the body. EHF depletion in immortalized prostate epithelial cells caused up-regulation of genes involved in epithelial-mesenchymal transition (18), suggesting that one important property of this transcription factor is the maintenance of epithelial phenotype. The mechanism whereby EHF coordinates its transcriptional targets to achieve this is of interest, since the factor is known to be either an activator or repressor of gene expression. Our data show that though EHF occupies the promoter of some DEGs identified by RNA-seq after EHF-depletion, many lacked an EHF binding site at their promoter. However, the predominance of EHF ChIP-seq peaks in intronic and intergenic regions rather than promoters suggests that the majority of DEGs may be controlled through distal EHF-regulated cis-elements. It is also likely that some genes are indirect targets of EHF.
Of the genes tested to validate the RNA-seq results, DDAH1 showed the greatest change in expression following EHF depletion. DDAH1 metabolizes asymmetric dimethylarginine (ADMA) a competitive inhibitor of nitric oxide synthase (NOS). ADMA potentiates lung inflammation in a mouse model of allergic asthma (45). Furthermore, DDAH1 overexpression attenuates airway inflammation in a different mouse model of asthma by decreasing IgE expression and eosinophil migration in the lung. It also causes a decrease in expression of genes encoding cytokines, chemokines and matrix metalloproteinases (46). Since EHF apparently represses DDAH1 in our studies, this suggests a role for the transcription factor in potentiating allergic lung inflammation.
In Calu-3 cells, EHF regulated several genes critical for normal cell motility and wound repair, including ITGA2, S100A8 and S100A9. Integrin, α2, which decreases following EHF depletion, contributes to alveolar epithelial cell migration on collagens I and IV (47,48). We found that wound closure was impaired upon EHF knockdown, consistent with down-regulation of genes that promote cell motility. This phenotype may be due to changes in ITGA2 or other genes involved in cell motility or proliferation.
Among genes comprising the GO:0009611 response to wounding pathway, 14 are differentially expressed after EHF depletion, including a number of chemokines (Supplementary Table S6). Furthermore, 60 were differentially expressed in response to EHF overexpression (Supplementary Table S8). This suggests that EHF regulates wound healing across multiple lung epithelial cell types. Three genes showing altered expression after EHF depletion in Calu-3 cells, CXCL1, CXCL6 and CCL5, encode chemokines expressed in bronchial epithelium. Their release is increased in response to inflammatory mediators (49,50), and while CXCL1 and CXCL6 are neutrophil chemoattractants, CCL5 mobilizes monocytes, T cells, basophils and eosinophils. Inflammation is an integral part of the response to wounding and determines, in part, whether a damaged epithelium undergoes proper repair or fibrosis (51). CXCL1 is over-expressed in fibrotic versus non-fibrotic lungs (52). CXCL6 expression is increased in patients with idiopathic fibrosis, and its inactivation can attenuate inflammation and fibrosis in the bleomycin mouse lung fibrosis model (53). IL-6 and IL-11 showed increased expression in EHF-overexpressing clones as compared with control clones. Overexpression of these cytokines in the mouse lung leads to structural changes and altered responsiveness to methacholine in the airway (54). Our results show that EHF regulates all of these inflammatory mediators, which could directly contribute to inflammation and fibrosis during wound repair. The impact of EHF in the wound scratch assay supports a general role in controlling response to epithelial damage. Of particular relevance to the role of genetic elements at 11p13 in modifying CF lung disease severity, aberrant wound repair is integral to the pathology of lung disease, including CF, in which cell migration is impaired (55). Therefore, this might be one of the mechanisms by which EHF contributes to dysfunction in this tissue.
Our ChIP-seq results show that EHF binds near genes involved in cell–cell and cell–substrate/matrix junctions, suggesting that this factor has an important role in maintaining the epithelial cell barrier. This was confirmed by measurements of transepithelial resistance in post-confluent Calu-3 monolayers, which showed that EHF depletion increased TER. Maintenance of the epithelial barrier is critical for normal lung function and abnormal intercellular adhesion is linked to lung disease and infection (2). Moreover, a reduction in TER is associated with the F508del mutation in cell culture models of CF airway epithelia (56). Coordination of expression of genes that encode components of junctional complexes may be a mechanism whereby EHF regulates barrier function of the lung epithelium. This capability of EHF is equally relevant to the potential mechanisms whereby genes at 11p13 contribute to disease severity in the CF lung. Since EHF regulates pathways of inflammation, motility and barrier function, it may be pivotal in coordinating processes involved in lung epithelial repair.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Dr G. E. Crawford and L. C. Jones for generating the DNase-seq data and RNA-seq libraries, respectively; also Drs G. Barish and M. Schipma and T. Skimina for helpful discussions.
FUNDING
National Institute of Health (NIH) [R01HL117843, R01HL094585, R01HD068901 to A.H.]; [NIDDK P30DK065988 to M.R.K.]; Cystic Fibrosis Foundation Harris [11G0 to A.H, CFF-RDP R026 to M.R.K.]. Funding for open access charge: NIH, USA.
Conflict of interest statement. None declared.
REFERENCES
- 1.Widdicombe J.H. Regulation of the depth and composition of airway surface liquid. J. Anat. 2002;201:313–318. doi: 10.1046/j.1469-7580.2002.00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rezaee F., Georas S.N. Breaking barriers. New insights into airway epithelial barrier function in health and disease. Am. J. Respir. Cell Mol. Biol. 2014;50:857–869. doi: 10.1165/rcmb.2013-0541RT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knowles M.R., Boucher R.C. Mucus clearance as a primary innate defense mechanism for mammalian airways. J. Clin. Invest. 2002;109:571–577. doi: 10.1172/JCI15217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fahy J.V., Dickey B.F. Airway mucus funciton and dysfunction. N. Engl. J. Med. 2010;363:2233–2247. doi: 10.1056/NEJMra0910061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim V., Rogers T.J., Criner G.J. New concepts in the pathobiology of chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2008;5:478–485. doi: 10.1513/pats.200802-014ET. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holgate S.T. The sentinel role of the airway epithelium in asthma pathogenesis. Immunol. Rev. 2011;242:205–219. doi: 10.1111/j.1600-065X.2011.01030.x. [DOI] [PubMed] [Google Scholar]
- 7.Regamey N., Jeffery P.K., Alton E.W., Bush A., Davies J.C. Airway remodelling and its relationship to inflammation in cystic fibrosis. Thorax. 2011;66:624–629. doi: 10.1136/thx.2009.134106. [DOI] [PubMed] [Google Scholar]
- 8.Beers M.F., Morrisey E.E. The three R's of lung health and disease: repair, remodeling, and regeneration. J. Clin. Invest. 2011;121:2065–2073. doi: 10.1172/JCI45961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bischof J.M., Ott C.J., Leir S.H., Gosalia N., Song L., London D., Furey T.S., Cotton C.U., Crawford G.E., Harris A. A genome-wide analysis of open chromatin in human tracheal epithelial cells reveals novel candidate regulatory elements for lung function. Thorax. 2012;67:385–391. doi: 10.1136/thoraxjnl-2011-200880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ren X., Shah T.A., Ustiyan V., Zhang Y., Shinn J., Chen G., Whitsett J.A., Kalin T.V., Kalinichenko V.V. FOXM1 promotes allergen-induced goblet cell metaplasia and pulmonary inflammation. Mol. Cell. Biol. 2013;33:371–386. doi: 10.1128/MCB.00934-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korfhagen T.R., Kitzmiller J., Chen G., Sridharan A., Haitchi H.-M., Hegde R.S., Divanovic S., Karp C.L., Whitsett J.A. SAM-pointed domain ETS factor mediates epithelial cell-intrinsic innate immune signaling during airway mucous metaplasia. Proc. Natl Acad. Sci. U.S.A. 2012;109:16630–16635. doi: 10.1073/pnas.1208092109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tugores A., Le J., Sorokina I., Snijders A.J., Duyao M., Reddy P.S., Carlee L., Ronshaugen M., Mushegian A., Watanaskul T., et al. The epithelium-specific ETS protein EHF/ESE-3 is a context-dependent transcriptional repressor downstream of MAPK signaling cascades. J. Biol. Chem. 2001;276:20397–20406. doi: 10.1074/jbc.M010930200. [DOI] [PubMed] [Google Scholar]
- 13.Kas K., Finger E., Grall F., Gu X., Akbarali Y., Boltax J., Weiss A., Oettgen P., Kapeller R., Libermann T.A. ESE-3, a novel member of an epithelium-specific ets transcription factor subfamily, demonstrates different target gene specificity from ESE-1. J. Biol. Chem. 2000;275:2986–2998. doi: 10.1074/jbc.275.4.2986. [DOI] [PubMed] [Google Scholar]
- 14.Silverman E.S., Baron R.M., Palmer L.J., Le L., Hallock A., Subramaniam V., Riese R.J., McKenna M.D., Gu X., Libermann T.A., et al. Constitutive and cytokine-induced expression of the ETS transcription factor ESE-3 in the lung. Am. J. Respir. Cell Mol. Biol. 2002;27:697–704. doi: 10.1165/rcmb.2002-0011OC. [DOI] [PubMed] [Google Scholar]
- 15.Taniue K., Oda T., Hayashi T., Okuno M., Akiyama T. A member of the ETS family, EHF, and the ATPase RUVBL1 inhibit p53-mediated apoptosis. EMBO Rep. 2011;12:682–689. doi: 10.1038/embor.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kunderfranco P., Mello-Grand M., Cangemi R., Pellini S., Mensah A., Albertini V., Malek A., Chiorino G., Catapano C.V., Carbone G.M. ETS transcription factors control transcription of EZH2 and epigenetic silencing of the tumor suppressor gene Nkx3.1 in prostate cancer. PloS One. 2010;5:e10547. doi: 10.1371/journal.pone.0010547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stephens D.N., Klein R.H., Salmans M.L., Gordon W., Ho H., Andersen B. The Ets transcription factor EHF as a regulator of cornea epithelial cell identity. J. Biol. Chem. 2013;288:34304–34324. doi: 10.1074/jbc.M113.504399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Albino D., Longoni N., Curti L., Mello-Grand M., Pinton S., Civenni G., Thalmann G., D'Ambrosio G., Sarti M., Sessa F., et al. ESE3/EHF controls epithelial cell differentiation and its loss leads to prostate tumors with mesenchymal and stem-like features. Cancer Res. 2012;72:2889–2900. doi: 10.1158/0008-5472.CAN-12-0212. [DOI] [PubMed] [Google Scholar]
- 19.Kalluri R., Neilson E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim K.K., Kugler M.C., Wolters P.J., Robillard L., Galvez M.G., Brumwell A.N., Sheppard D., Chapman H.A. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc. Natl Acad. Sci. U.S.A. 2006;103:13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willis B.C., Borok Z. TGF-β-induced EMT: mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007;293:L525–L534. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- 22.Wu J., Duan R., Cao H., Field D., Newnham C.M., Koehler D.R., Zamel N., Pritchard M.A., Hertzog P., Post M., et al. Regulation of epithelium-specific Ets-like factors ESE-1 and ESE-3 in airway epithelial cells: potential roles in airway inflammation. Cell Res. 2008;18:649–663. doi: 10.1038/cr.2008.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wright F.A., Strug L.J., Doshi V.K., Commander C.W., Blackman S.M., Sun L., Berthiaume Y., Cutler D., Cojocaru A., Collaco J.M., et al. Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat. Genet. 2011;43:539–546. doi: 10.1038/ng.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen B.-Q., Finkbeiner W.E., Wine J.J., Mrsny R.J., Widdicombe J.H. Calu-3: a human airway epithelial cell line that shows cAMP-dependent Cl− secretion. Am. J. Physiol. Lung Cell. Mol. Physiol. 1994;10:L493–L501. doi: 10.1152/ajplung.1994.266.5.L493. [DOI] [PubMed] [Google Scholar]
- 25.Mortazavi A., Leeper Thompson E.C., Garcia S.T., Myers R.M., Wold B. Comparative genomics modeling of the NRSF/REST repressor network: from single conserved sites to genome-wide repertoire. Genome Res. 2006;16:1208–1221. doi: 10.1101/gr.4997306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson D.S., Mortazavi A., Myers R.M., Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 27.Ford E., Nikopoulou C., Kokkalis A., Thanos D. A method for generating highly multiplexed ChIP-seq libraries. BMC Res. Notes. 2014;7:312. doi: 10.1186/1756-0500-7-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boyle A.P., Davis S., Shulha H.P., Meltzer P., Margulies E.H., Weng Z., Furey T.S., Crawford G.E. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132:311–322. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leir S.H., Holgate S.T., Lackie P.M. Inflammatory cytokines can enhance CD44-mediated airway epithelial cell adhesion independently of CD44 expression. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003;285:L1305–L1311. doi: 10.1152/ajplung.00255.2002. [DOI] [PubMed] [Google Scholar]
- 32.Gaulton K.J., Nammo T., Pasquali L., Simon J.M., Giresi P.G., Fogarty M.P., Panhuis T.M., Mieczkowski P., Secchi A., Bosco D., et al. A map of open chromatin in human pancreatic islets. Nat. Genet. 2010;42:255–259. doi: 10.1038/ng.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei G.H., Badis G., Berger M.F., Kivioja T., Palin K., Enge M., Bonke M., Jolma A., Varjosalo M., Gehrke A.R., et al. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J. 2010;29:2147–2160. doi: 10.1038/emboj.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dennis G., Jr, Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C., Lempicki R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 35.Smith B.T. Cell line A549: a model system for the study of alveolar type II cell function. Am. Rev. Respir. Dis. 1977;115:285–293. doi: 10.1164/arrd.1977.115.2.285. [DOI] [PubMed] [Google Scholar]
- 36.Li W., Soave D., Miller M.R., Keenan K., Lin F., Gong J., Chiang T., Stephenson A.L., Durie P., Rommens J., et al. Unraveling the complex genetic model for cystic fibrosis: pleiotropic effects of modifier genes on early cystic fibrosis-related morbidities. Hum. Genet. 2014;133:151–161. doi: 10.1007/s00439-013-1363-7. [DOI] [PubMed] [Google Scholar]
- 37.Zahm J.-M., Chevillard M., Edith Puchelle E. Wound repair of human surface respiratory epithelium. Am. J. Respir. Cell. Mol. Biol. 1991;5:242–248. doi: 10.1165/ajrcmb/5.3.242. [DOI] [PubMed] [Google Scholar]
- 38.Wan H., Dingle S., Xu Y., Besnard V., Kaestner K.H., Ang S.L., Wert S., Stahlman M.T., Whitsett J.A. Compensatory roles of Foxa1 and Foxa2 during lung morphogenesis. J. Biol. Chem. 2005;280:13809–13816. doi: 10.1074/jbc.M414122200. [DOI] [PubMed] [Google Scholar]
- 39.Park K.S., Wells J.M., Zorn A.M., Wert S.E., Laubach V.E., Fernandez L.G., Whitsett J.A. Transdifferentiation of ciliated cells during repair of the respiratory epithelium. Am. J. Respir. Cell. Mol. Biol. 2006;34:151–157. doi: 10.1165/rcmb.2005-0332OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao X.G., Vockley C.M., Pauli F., Newberry K.M., Xue Y., Randell S.H., Reddy T.E., Hogan B.L. Evidence for multiple roles for grainyheadlike 2 in the establishment and maintenance of human mucociliary airway epithelium. Proc. Natl Acad. Sci. U.S.A. 2013;110:9356–9361. doi: 10.1073/pnas.1307589110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heintzman N.D., Stuart R.K., Hon G., Fu Y., Ching C.W., Hawkins R.D., Barrera L.O., Van Calcar S., Qu C., Ching K.A., et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 42.Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A., et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl Acad. Sci. U.S.A. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bassuk A.G., Leiden J.M. A direct physical association between ETS and AP-1 transcription factors in normal human T cells. Immunity. 1995;3:223–237. doi: 10.1016/1074-7613(95)90092-6. [DOI] [PubMed] [Google Scholar]
- 44.Hollenhorst P.C., Ferris M.W., Hull M.A., Chae H., Kim S., Graves B.J. Oncogenic ETS proteins mimic activated RAS/MAPK signaling in prostate cells. Genes Dev. 2011;25:2147–2157. doi: 10.1101/gad.17546311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klein E., Weigel J., Buford M.C., Holian A., Wells S.M. Asymmetric dimethylarginine potentiates lung inflammation in a mouse model of allergic asthma. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010;299:L816–L825. doi: 10.1152/ajplung.00188.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kinker K.G., Gibson A.M., Bass S.A., Day B.P., Deng J., Medvedovic M., Figueroa J.A.L., Hershey G.K.K., Chen W. Overexpression of dimethylarginine dimethylaminohydrolase 1 attenuates airway inflammation in a mouse model of asthma. PloS One. 2014;9:e85148. doi: 10.1371/journal.pone.0085148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim H.J., Henke C.A., Savik S.K., Ingbar D.H. Integrin mediation of alveolar epithelial cell migration on fibronectin and type I collagen. Am. J. Physiol. 1997;273:L134–L141. doi: 10.1152/ajplung.1997.273.1.L134. [DOI] [PubMed] [Google Scholar]
- 48.White S.R., Dorscheid D.R., Rabe K.F., Wojcik K.R., Hamann K.J. Role of very late adhesion integrins in mediating repair of human airway epithelial cell monolayers after mechanical injury. Am. J. Respir. Cell Mol. Biol. 1999;20:787–796. doi: 10.1165/ajrcmb.20.4.3318. [DOI] [PubMed] [Google Scholar]
- 49.Wang J.H., Devalia J.L., Xia C., Sapsford R.J., Davies R.J. Expression of RANTES by human bronchial epithelial cells in vitro and in vivo and the effect of corticosteroids. Am. J. Respir. Cell. Mol. Biol. 1996;14:27–35. doi: 10.1165/ajrcmb.14.1.8534483. [DOI] [PubMed] [Google Scholar]
- 50.Prause O., Laan M., Lötvall J., Lindén A. Pharmacological modulation of interleukin-17-induced GCP-2-, GRO-α- and interleukin-8 release in human bronchial epithelial cells. Eur. J. Pharmacol. 2003;462:193–198. doi: 10.1016/s0014-2999(03)01341-4. [DOI] [PubMed] [Google Scholar]
- 51.Crosby L., Waters C.M. Epithelial repair mechanisms in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010;298:L715–L731. doi: 10.1152/ajplung.00361.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zuo F., Kaminski N., Eugui E., Allard J., Yakhini Z., Ben-Dor A., Lollini L., Morris D., Kim Y., DeLustro B., et al. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc. Natl Acad. Sci. U.S.A. 2002;99:6292–6297. doi: 10.1073/pnas.092134099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Besnard A.G., Struyf S., Guabiraba R., Fauconnier L., Rouxel N., Proost P., Uyttenhove C., Van Snick J., Couillin I., Ryffel B. CXCL6 antibody neutralization prevents lung inflammation and fibrosis in mice in the bleomycin model. J. Leukocyte Biol. 2013;94:1317–1323. doi: 10.1189/jlb.0313140. [DOI] [PubMed] [Google Scholar]
- 54.Kuhn C., III, Homer R.J., Zhu Z., Ward N., Flavell R.A., Geba G.P., Elias J.A. Airway hyperresponsiveness and airway obstruction in transgenic mice: morphologic correlates in mice overexpressing interleukin (IL)-11 and IL-6 in the lung. Am. J. Respir. Cell. Mol. Biol. 2000;22:289–295. doi: 10.1165/ajrcmb.22.3.3690. [DOI] [PubMed] [Google Scholar]
- 55.Schiller K.R., Maniak P.J., O'Grady S.M. Cystic fibrosis transmembrane conductance regulator is involved in airway epithelial wound repair. Am. J. Physiol. Cell. Physiol. 2010;299:C912–C921. doi: 10.1152/ajpcell.00215.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.LeSimple P., Liao J., Robert R., Gruenert D.C., Hanrahan J.W. Cystic fibrosis transmembrane conductance regulator trafficking modulates the barrier function of airway epithelial cell monolayers. J. Physiol. 2010;588:1195–1209. doi: 10.1113/jphysiol.2009.182246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.