


Abstract
The advent in high-throughput-sequencing (HTS) technologies has revolutionized conventional biodiversity research by enabling parallel capture of DNA sequences possessing species-level diagnosis. However, polymerase chain reaction (PCR)-based implementation is biased by the efficiency of primer binding across lineages of organisms. A PCR-free HTS approach will alleviate this artefact and significantly improve upon the multi-locus method utilizing full mitogenomes. Here we developed a novel multiplex sequencing and assembly pipeline allowing for simultaneous acquisition of full mitogenomes from pooled animals without DNA enrichment or amplification. By concatenating assemblies from three de novo assemblers, we obtained high-quality mitogenomes for all 49 pooled taxa, with 36 species >15 kb and the remaining >10 kb, including 20 complete mitogenomes and nearly all protein coding genes (99.6%). The assembly quality was carefully validated with Sanger sequences, reference genomes and conservativeness of protein coding genes across taxa. The new method was effective even for closely related taxa, e.g. three Drosophila spp., demonstrating its broad utility for biodiversity research and mito-phylogenomics. Finally, the in silico simulation showed that by recruiting multiple mito-loci, taxon detection was improved at a fixed sequencing depth. Combined, these results demonstrate the plausibility of a multi-locus mito-metagenomics approach as the next phase of the current single-locus metabarcoding method.
INTRODUCTION
Over the past few years, DNA metabarcoding—identifying mixed taxa using short DNA markers via high-throughput-sequencing (HTS)—has emerged as a fast and effective approach to characterizing bulk environmental samples (1). To date, most published works have relied on polymerase chain reaction (PCR) amplification of a single (typically standard) DNA marker, e.g. the CO1 ‘barcode’ fragment for animals (2). While enriching targeted gene fragments, PCR amplifications can introduce taxonomic biases (1,3,4) and chimeric sequences (5,6) due to varied primer binding efficiencies across taxa. When the target bulk sample contains organisms from a wide range of lineages, as is typical of many biodiversity surveys, such artefacts would cause systematic biases in the subsequent diversity analysis. For example, our recent work based on HTS of PCR amplicons of CO1 barcodes (7) showed significantly higher failure rate in hymenopterans (wasps and bees, 32%) relative to other mixed insects, even though the overall taxonomic recovery rate was improved from the previous method (8). Zhou et al. (9) demonstrated some success in identifying mixed species without PCR amplifications. However, in that study, a large proportion of potentially informative sequences (e.g. non-CO1 mitochondrial gene fragments) were ignored for species recovery because only CO1 barcodes were available as the reference.
A multi-locus identification approach has not only been promoted as a standard barcoding method for difficult groups, such as plants (10,11), but also improved barcoding efficiencies in insects (12) and fungi (13), where a single-locus approach has been predominantly applied. A multi-locus system has been argued to deliver better taxonomic resolution in general (14–17). In addition to improving taxonomic delineation, a multi-locus approach can also alleviate false negatives caused by random missing of a given target gene caused by insufficient sequencing or DNA degradation. Therefore, the acquisition of reference sequences for non-standard-barcode genes will greatly facilitate the expansion of the current single-gene barcoding approach by taking advantage of additional informative markers. Recent studies (15,17) have also discussed mechanisms in utilizing multiple markers in biodiversity analysis.
The main impediment for a wide application of a multi-locus identification system does not lie in the lack of scientific motivation but rather in challenges in practical logistics (cost, technical difficulties, etc.). While significant progress has been made toward constructing DNA reference libraries useful for taxonomic identification, e.g. the International Barcode of Life (iBOL, http://ibol.org) project, such endeavors are primarily focused on carefully selected markers, e.g. CO1 barcode for the animals, leaving other taxonomically informative genes aside. Although other mitochondrial genes (e.g. CYTB, ND1) have been demonstrated effectively in both species delineation (18,19) and phylogenetic reconstruction (20,21), Sanger-sequencing-based reference construction for each of the additional MT genes will require similar global investment as the DNA barcoding initiative. Alternatively, whole mitochondrial genome sequencing can produce a full set of references in one shot, including protein coding genes, ribosomal DNA (16 and 12S), tRNA genes and the hyper-variable control region. Conventional methods in obtaining whole mitogenome sequences include primer-walking and long-range PCR coupled with Sanger or Next generation sequencing (NGS) (22,23). These time-consuming pipelines typically also require high-quality mitochondrial DNA to ensure the success of targeted PCR amplifications, which rules out the utility of many preserved specimens in less optimal conditions. Furthermore, for taxa containing high variability in gene sequences and gene orders, primer optimization is difficult (24). A whole-genome shotgun approach employing second generation sequencing technologies has been successfully applied in assembling full mitogenomes. However, most previous work has only dealt with a single taxon at a time (24–26) or a limited number of pooled taxa (27,28), resulting in high analytical cost for each genome. An in silico test containing 100 species (29) showed the pooling strategy might enable simultaneous construction of many distantly related taxa but this has not been demonstrated with real data.
The main motivation for multiplex sequencing and reconstructing mitogenomes from pooled taxa is to reduce analytical cost on individual library construction required for HTS. In principle, the more taxa that can be pool-sequenced, the less the average cost for each species, to the point where the main cost per taxon is mainly determined by its sequencing volume and the associated computational cost. In practice, a number of factors must be balanced: total number of pooled taxa, phylogenetic distance among taxa, DNA quality and quantity, and total sequencing volume. Empirical analysis will also need to consider specific features associated to the employed sequencing technology and assembly programs. In this study, we seek to answer these questions and develop a new pipeline for rapid and accurate reconstruction of multiplex mitogenomes from pooled taxa without relying on any DNA enrichment or amplification. In addition, we explore the plausibility of a multi-locus identification approach that integrates full mitogenome sequences or ‘mito-metagenomics’.
MATERIALS AND METHODS
Raw data (SRA174290), Sanger sequences (KM207019–KM207147) and assembled mitogenomes (KM244654–KM244713) are available on GenBank.
Taxon selection
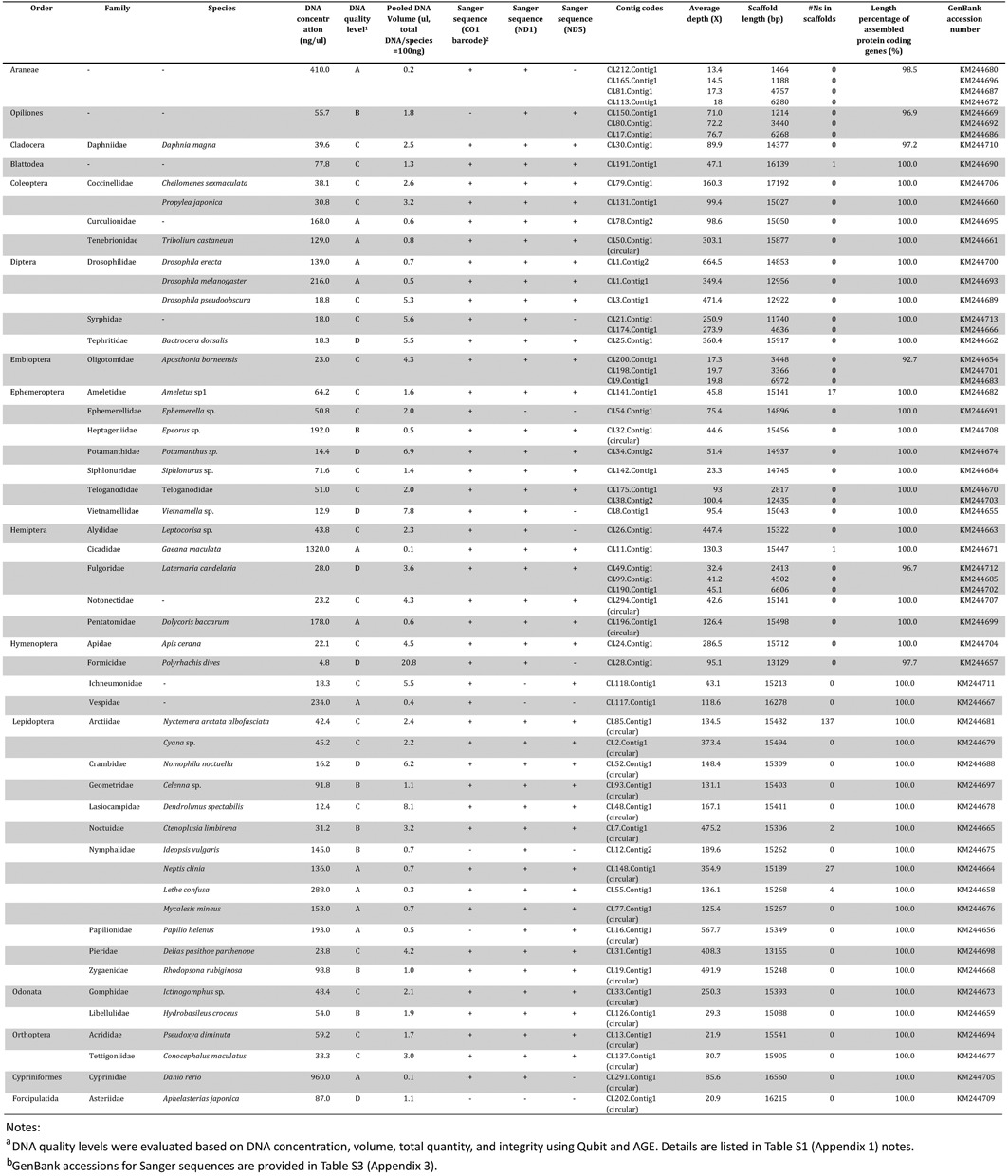
The schematic analytical pipeline is illustrated in Figure 1. The level of phylogenetic distance among pooled taxa can potentially impact both shotgun-read assembly and subsequent taxonomic assignments for assembled scaffolds. A total of 49 animal species (primarily insects, Table 1 and Supplementary Table S1 in Appendix 1) were selected from 47 genera and 42 families, with most taxa representing a single family while a number of them were chosen from the same family, subfamily (e.g. Cheilomenes sexmaculata and Propylea japonica, Lethe confusa and Mycalesis mineus) or genus (e.g. three Drosophila spp.)Such sampling strategy enables us to understand the influence of pooling closely related species on mitogenome assembly. Samples used in this work include recently collected specimens and preserved tissues (collected in 2009 and 2010, see Supplementary Table S1 in Appendix 1 for details).
Figure 1.
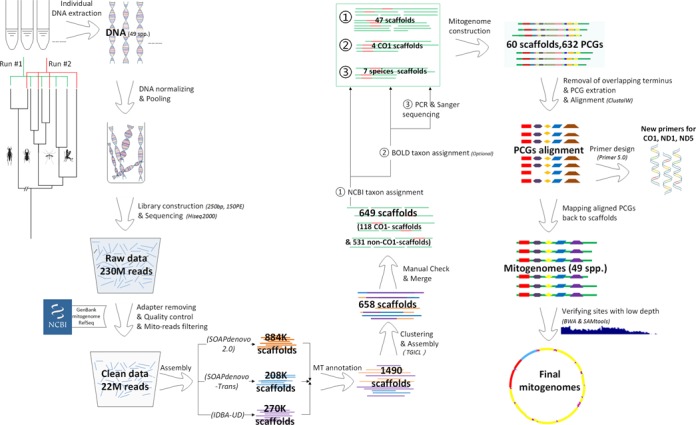
Schematic illustration of the pipeline.
Table 1. List of taxa analyzed and corresponding assembly results.
DNA extraction and sequencing
Genomic DNA of each individual specimen was extracted separately following Ivanova et al. (30). All genomic DNA extracts were quantified using Qubit 2.0 (Invitrogen, Life technologies). DNA quality was categorized as levels A, B, C and D based on quantity and level of degradation (see notes in Supplementary Table S1 in Appendix 1). A total of 100 ng of each DNA was then pooled and used for Hiseq DNA library construction with an insert size of 250 bp following manufacturer's instruction. The library was sequenced on an Illumina HiSeq 2000 with the strategy of 150 paired-end (PE) at BGI-Shenzhen, China.
De novo assembly and taxonomic assignments of mitochondrial scaffolds
Scripts and Shell command lines are provided in ‘Appendix 2_Supplementary notes.pdf’. All relevant script files and sequence alignments are available at: http://sourceforge.net/projects/mt10k/files/?sourceg304=navbar.
Data filtering and parallel assembling using multiple assemblers
Pre-analysis data filtering includes: (i) Reads with adapter contamination and ploy-Ns (≥5) and PE reads with >10 bases of low quality scores (<20) were removed from raw data following Zhou et al. (9); (ii) Clean data were then compared with reference mitogenomes downloaded from GenBank (716 RefSeq genomes, including 699 arthropods, seven starfish and 10 cyprinid fish accessed on 10 March 2014) to screen out candidate mitochondrial reads using a relaxed criteria: blast identity >30% and E-value ≤ 10−5; (iii) 51-mer set was then generated from these candidate mito-reads and used as references for a second round of data filtering for the discarded reads from step 2; (iv) The combined clean reads from steps 2 and 3 were used for de novo assembling.
High quality mito-reads were assembled by SOAPdenovo 2.0 (31,32) (-K 61, -k 45), SOAPdenovo-Trans (33) (-K 71, -L 100, -t 1) and IDBA-UD (34) (kMaxShortSequence = 256, –num_threads 12), respectively. Three sets of assemblies produced by these programs were annotated separately using a Perl script described by Zhou et al. (9) and a mitogenome reference database containing full mitogenomes (RefSeq) from 604 arthropod species, two asteriid starfish and the zebrafish downloaded from GenBank on 13 June 2013. Only scaffolds of mitochondrial origin were kept for subsequent taxonomic assignments.
Scaffold concatenation
All scaffolds containing mitochondrial proteins (mito-scaffolds) were clustered and re-assembled into concatenated scaffolds using TGICL (35) (-l 100 -c 10 -v 10 000 -p 99 -O ‘-repeat_stringency 0.95 -minmatch 35 -minscore 35’). An additional manual examination was performed afterward to combine overlapping scaffolds missed by TGICL. Concatenated scaffolds were annotated again to identify regions of protein coding genes.
Taxonomic assignments for protein coding genes and scaffolds
The taxonomic assignment pipeline is summarized in Supplementary Figure S1 (Appendix 2). Briefly, all protein coding genes were aligned by ‘megablast’ to a mitochondrial protein coding gene reference database containing 886 010 sequences downloaded from GenBank on Feb. 25th, 2014, including all arthropods, starfish and the zebrafish. For a given protein coding gene, the best blast match (top hit) was selected for subsequent taxonomic assignment: if the best-matched species was listed in our input taxa table, a species-level assignment was made for the protein coding gene; otherwise the associated higher taxonomic hierarchy of the best-matched species (i.e. Genus, Subfamily, Family, Order) were used to compare against the input taxon list until a match was achieved. Unassigned CO1 sequences were also compared with the Barcode of Life Data Systems (BOLD, http://boldsystems.org) for further taxonomic assignments. Taxonomic assignment of scaffolds was made primarily based on CO1 (when available) and confirmed by other protein coding genes assembled on the same scaffold on a majority consensus basis (section S2 in Appendix 2).
Finally, the remaining unassigned scaffolds were made subject to Sanger sequence verification. Consulting results from missing taxa (i.e. species without any associated mito-scaffolds after the above protein coding gene and scaffold taxon assignments) and missing protein coding genes, we amplified and Sanger sequenced three sets of markers: CO1, ND1 and ND5. These optional genes were selected to obtain an even coverage for the mitochondrial genomes revealed in general arthropod mitochondrial structure. Primers used in this study are listed in Supplementary Table S2 (Appendix 3). These Sanger sequences were then used to identify mito-scaffolds missed from previous taxonomic assignment procedures. Finally, all mito-scaffolds that were assigned to the input taxa were used to construct the super-scaffold for each of the pooled species.
Alignment and validation of mitogenome sequences
Because none of the current de novo genome assemblers was designed for handling circular genomes, complete linear mitogenomes usually contained repetitive overlaps on the terminuses. If the terminuses of a linear super-scaffold contained overlapping sequences of >25 bp, the corresponding assembly was considered a complete circular genome. Sometimes TGICL produces longer-than-usual (e.g. 20 kb) scaffolds due to its incapability in recognizing real ends of the genome. Thus repetitive sequences need to be removed from the final genome assemblies. Automated Perl scripts (section S1 in Appendix 2) were developed to identify unusually long scaffolds containing identical overlaps. A manual inspection using ‘Geneious’ (36) was followed for remaining scaffolds of >15 kb after the automated step to identify overlapping terminal regions with Ns or mismatched nucleotides (artefacts produced in IDBA-UD assembling or concatenation of different assemblies by TGICL). Mito-scaffolds with redundant sequences removed from the overlapping terminuses were used for subsequent analysis.
Each of the 13 MT protein coding genes extracted from the mito-scaffolds was aligned individually across all assembled taxa by ‘ClustalW’ (37) using reference protein coding gene sequences from six model organisms (Drosophila melanogaster, Drosophila simulans, Drosophila pseudoobscura, Aedes aegypti, Danaus plexippus and Tribolium castaneum) and by MEGA (38) to ensure correct translation frames for amino acids. Indels created by assembly programs based on Hiseq reads’ paired-end information were validated by the global alignment results: redundant Ns were removed and alignment gaps were inserted. Stop codons were also examined as a hint for erroneous assemblies. Aligned protein coding genes were then placed back to the mitogenomes. Finally, a manual procedure was taken to assure assembly quality: the three assembly versions were compared to the final corresponding mitogenome and filtered mito-reads were mapped to the genome to examine uneven sequence depth (section S1 in Appendix 2). When a particular region was assembled only by one of the three assemblers with low read coverage (e.g. close to 0), we considered it as a false assembly and corrected it according to the other two programs. As a final step, reads were mapped to the mitogenomes using BWA (39) to identify regions with exceptionally low coverage relative to adjacent regions. These problematic regions were examined using SAMtools (40) to investigate potential conflicting allelic variations (including both natural polymorphic alleles and artefacts). Nucleotide suggested by SAMtools was subsequently chosen as the consensus base for the final assembly.
Reference mito-genomes of six species (D. melanogaster, Drosophila erecta, D. pseudoobscura, T. castaneum, Bactrocera dorsalis and Danio rerio) were downloaded from GenBank and compared to our final assemblies. Both nucleotide and amino acid sequences were examined with the recognized possibility of intraspecific variation in the mitogenomes. Finally, Sanger sequences from fragments of genes CO1, ND1 and ND5 were obtained for validating the final assembly quality. Primers were designed using Primer 5.0 (Supplementary Table S2 in Appendix S3). With the combined evidence, we examined all mitogenome scaffolds for hints of local assembly errors and chimeras. The assemblies of the all input taxa were also annotated for protein coding genes, tRNA, rRNA genes and the control regions using ‘Geneious’.
In silico simulation for multi-locus mitochondrial metagenomics
To evaluate whether and how multiple mitochondrial loci could improve biodiversity recovery for mixed animal samples, we conducted an in silico analysis using portions of the Illumina data generated in this study. A series of data volumes (2, 5 and 8 Gb) were randomly selected from the total high-quality reads to simulate varied sequencing depths for the given animal sample mixture (i.e. containing 49 species of varied phylogenetic relatedness). Reads and the corresponding scaffolds/contigs that were assembled from these reads using only SOAPdenovo 2.0 were ‘BLASTed’ against the aligned protein coding genes derived from the above assembly pipeline using ‘BWA’ and ‘BLAST’, respectively. Criteria for a successful taxon recovery were defined as: 100% sequence identity, ≥90% coverage for at least one protein coding gene marker. We first calculated taxonomic recovery rates at varied sequencing depths for the standard animal CO1 barcode region, then expanded the analysis to include the full CO1 gene, CO2, CO3 and eventually all 13 MT protein coding genes.
RESULTS
Construction of mitogenomes
As shown in Figure 1, a total of 230 million raw PE reads were produced on a whole Hiseq 2000 lane (ca. 35 Gb raw data, SRA174290), while 22 million high-quality PE reads (3.3 Gb, containing candidate mitochondrial reads) were filtered out after removal of adaptors, low-quality reads and most non-mitochondrial sequences. These clean reads were used for the subsequent assembling. A total of 884 000, 208 000 and 270 000 scaffolds were obtained using SOAPdenovo, SOAPdenovo-Trans and IDBA-UD, producing 691, 383 and 416 mito-scaffolds, respectively. These three sets of mito-scaffolds were clustered and further assembled into 658 scaffolds using TGICL. A total of 649 scaffolds were retained for further analysis after manual examination.
All protein coding genes annotated from the 649 mito-scaffolds (including 118 CO1-scaffolds and 531 non-CO1-scaffolds) were blasted against the NCBI MT protein coding gene reference library using ‘megablast’. The first round of taxonomic assignment identified 47 scaffolds containing protein coding genes readily assigned to 38 input taxa, which were retained for the final mitogenome construction. An additional four CO1-scaffolds were further identified by Barcode of Life Data (BOLD) via the CO1 barcode regions and kept. After these two steps, seven (all mayflies) of the 49 input taxa were not yet associated with any mito-scaffolds. We then Sanger sequenced CO1 barcodes for these mayflies and successfully identified all mito-scaffolds for each of the mayflies using these Sanger barcodes. The failure of the initial taxonomic assignments of these mayfly mito-scaffolds was due to the poor coverage of Ephemeroptera in public sequence databases.
Protein coding genes were aligned across 49 taxa based on amino acid and nucleotide sequences using ClustalW and MEGA, where redundant Ns were removed and alignment gaps were inserted. Redundant Ns created by assembling programs, such as SOAPdenovo were not unusual, indicating the necessity of manual examination after the automated assembly procedure. Aligned protein coding genes were then placed back to the corresponding scaffolds.
Finally, a total of 60 scaffolds containing 632 MT protein coding genes were associated to the 49 input taxa, with the majority of species (44) containing a single scaffold (Figure 2, Table 1 and Supplementary Table S1). Most of the assembled mitogenomes (36) are longer than 15 kb (including 20 complete genomes), while all remaining >10 kb. The overall completeness for protein coding genes was high (99.6% of total length), where only five genes were missing from the total 637 protein coding genes (Figure 3). Thirteen out of 49 successfully assembled ATP8 genes (marked in gray color in Figure 3) failed in annotation due to incompleteness of the reference library. Annotations for protein coding and other mitochondrial genes were summarized in Figure 2 and Supplementary Figure S2 (Appendix 2).
Figure 2.
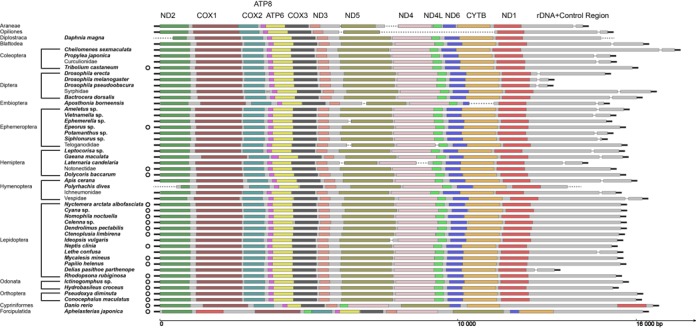
Assembled mitogenome scaffolds for all 49 taxa. Assembled mitogenomes are presented following each of the 49 taxa. Color bars represent 13 protein coding genes. Fragmented scaffolds assigned to the same taxon are connected by dash lines. To aid visualization, each mitogenome is manually broken at the beginning of ND2, with the bold black line connecting the associated scaffold containing rDNA and/or control region aligned to the right side of the super-scaffold. Complete circular mitogenomes are marked with a circle sign after the taxon name.
Figure 3.
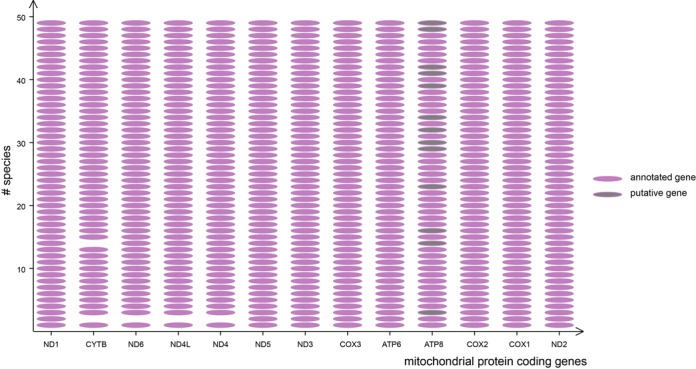
Completeness of assembled protein coding genes. Presence and absence of assembled protein coding genes are represented by pink ovals and blanks. Gray ovals represent putative genes (all ATP8) that were successfully assembled but failed in annotation due to its poor coverage in public databases. Only 5 out of 637 protein coding genes were missing from the final assemblies.
The majority of mito-scaffolds that were filtered out by the taxon assignment procedure (544, 84%) were presented at ≤10X depth, which counted for only 23% of the total assembled length (Table 2 and Figure 4, Supplementary Table S3 in Appendix 4). These low-quality scaffolds were excluded from the subsequent analysis. We then compared the remaining 45 mito-scaffolds with >10X depth (non-target scaffolds) against the BOLD system (www.boldsystems.org, ‘Species Level Barcode Records’ option) when containing CO1 or against NCBI using ‘blastn’. Sixty (green dots in Figure 4) were successfully associated to input taxa in the taxon assignment procedure (see descriptions in section S2 in Appendix 2). In addition, three scaffolds (yellow dots) potentially belonged to input taxa (CL44.Contig1 and CL213.Contig1: Opiliones; CL184.Contig1: Laternaria candelaria) but were not assigned in taxon assignments (based on ‘Megablast’) due to incompleteness of public references. Read coverage confirmed a polymorphic site in one scaffold (pink dots), which was excluded by the taxon assignment procedure because its counter part had better quality (longer in length with more protein coding genes (PCGs) and higher average depth). Seven scaffolds were identified as bacterial origin (blue dots), most of which were insect endosymbionts. A total of 23 scaffolds were identified as erroneous assemblies, where zero read coverage was discovered at ≥1 site along the scaffolds, which was likely an artefact of IDBA-UD. The taxon assignment procedure was able to successfully exclude these scaffolds from the final assembly. Finally, 10 scaffolds (gray dots) could not be identified to any of the above categories and presumably represented organisms (e.g. microbes) whose sequences were not yet available in the public reference.
Table 2. Average depth and length of assembled mito-scaffolds.
| Average depth (X) | ≤3 | 3–5 | 5–10 | >10 |
|---|---|---|---|---|
| # of scaffolds | 407 | 106 | 31 | 105 |
| Average length (bp) | 362 | 598 | 860 | 7519 |
Figure 4.
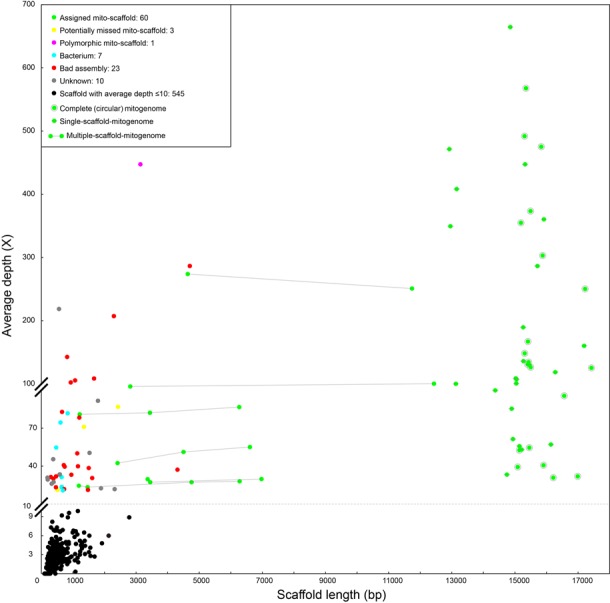
Categories of assembled mito-scaffolds. Sixty (green dots) were successfully associated to input taxa in the taxon assignment procedure; three scaffolds (yellow dots) potentially belonged to input taxa; one scaffold (pink dots) was confirmed as polymorphic nucleotide by read coverage; seven scaffolds were identified as bacterial origin (blue dots), most of which were insect endosymbionts; and 23 scaffolds were identified as erroneous assemblies as no read covered in some areas.
Validation of assemblies
We first compared six reference mitogenomes obtained from NCBI with our assemblies of the corresponding taxa. Average similarities of 99.2 and 99.6% were observed in nucleotide sequences of protein coding and rRNA genes between pairs of reference and assembly, respectively (Supplementary Figure S2 in Appendix 2). Only an average of 0.5% amino acid sequences showed difference between reference and assembly.
In addition, a total of 129 Sanger sequences of CO1, ND1 and ND5 were used to validate our assembled MT sequences, where no conflicts were observed between the Sanger and Hiseq results and no chimeras were found (Figure 5, Supplementary Tables S1 in Appendix 1, Supplementary Table S2 in Appendix 3). These Sanger sequences were not necessary for the de novo assembly of mitogenomes but were used in validation and taxonomic assignments of a few scaffolds when reference library coverage was poor.
Figure 5.
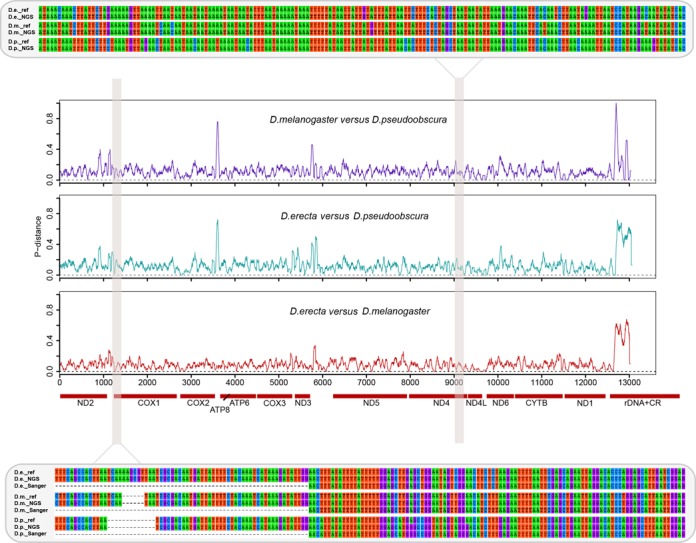
Validation of Drosophila assemblies using Sanger sequences and reference mitogenomes. Pairwise divergences are plotted along the assembled length of the mitogenomes for Drosophila melanogaster, Drosophila erecta and Drosophila pseudoobscura. P-distance values are calculated using a slide-window of 50 and 1 bp step. Protein coding genes, rDNA and the control region are marked along the mitogenome. Two regional assemblies are shown in details with the NGS assemblies aligned with reference mitogenomes obtained from GenBank and Sanger sequences (only available here for the 5′ end of the CO1 barcode region as shown in the bottom panel). NGS assemblies are confirmed by both reference mitogenomes and Sanger sequences and successfully recover the characteristic gap regions for D. melanogaster and D. pseudoobscura.
A close examination of the assembly results for the three Drosophila species demonstrated the robustness of our assembly pipeline (Figure 5). Pairwise divergences between the three congeners showed significant variations along the length of the genome, where the phylogenetically more closely related pair—D. erecta and D. melanogaster exhibited smaller interspecific variations. Examination of regions showing low divergences revealed that the assembly pipeline was able to correctly reconstruct homologous fragments of high similarity for each of the three mitogenomes. Where reference genomes obtained from GenBank showed intraspecific variations compared with our assembly (they were sequenced from different individuals), Sanger sequences confirmed the NGS results.
Therefore the accuracy of our Hiseq assembly results have been confirmed by three lines of evidence: (i) the validation via 129 Sanger sequences, (ii) the conservativeness of amino acid sequences and length in protein coding genes across taxa and (iii) confirmation by nucleotide and amino acid sequences from six model organisms.
The gene order in protein coding genes is highly conservative among all arthropods we sequenced, including species from Araneae, Cladocera and 11 insect orders. Rearrangements in gene order are also clear in the zebrafish and starfish (Figure 2).
In silico simulation of whole mitogenome metagenomics
At any given sequencing volume, longer mitochondrial sequence reads (e.g. barcode versus full CO1) or more gene markers tended to deliver higher taxonomic recovery rates (Figure 6). For example, with 2 Gb clean reads (dashed curve with hollow diamond points, Figure 6), two more species were detected when using full-length CO1 gene compared to the standard CO1 barcode region (39 versus 37) and an additional three species were discovered when CO2 and CO3 were also included in the reference (42 versus 39). The total number of detectable taxa at a given sequencing volume eventually reached a plateau when all mito-scaffolds of the target species had been identified from the DNA mixture. The curves tended to flatten out sooner with increased sequencing volume.
Figure 6.
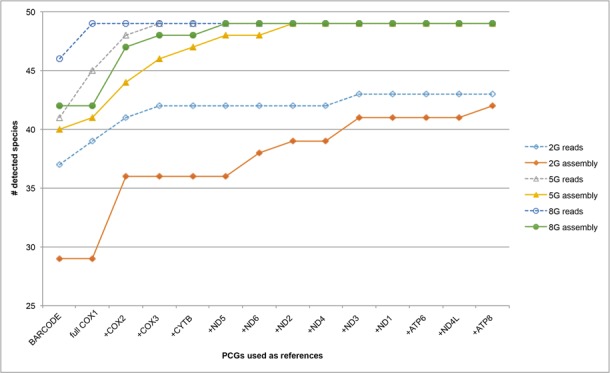
In silico simulation for taxon detection using multiple loci. Varied proportions of the clean raw reads (2G, 5G and 8G) were randomly selected as the in silico datasets to test efficiency of a multi-locus identification approach. The selected reads and assembled contigs using these reads via SOAPdenovo 2.0were blasted against the 632 protein coding gene references extracted from the assembled mitogenomes. A taxon was considered detected when the sequence coverage reached 90% at 100% sequence identity for at least one reference protein coding gene. This was tested for the standard CO1 barcode region first, and then extended to include the full CO1 gene and other protein coding genes one by one. Taxonomic recovery rates were improved with increased sequencing volume and inclusion of multiple loci.
Raw reads apparently always provided better taxonomic discoveries when compared with assembled scaffolds at all sequencing volumes, when fewer gene markers were considered. Not surprisingly, with a given set of MT gene markers, increased sequencing tended to deliver better taxonomic recovery for the pooled sample until species curves had reached plateau. When all 13 protein coding genes were used as references, a total of 43 (88%), 49 (100%) and 49 (100%) species were discovered by 2, 5 and 8 Gb clean Hiseq data, respectively. Our findings suggest that a mito-metagenomics pipeline using a multi-locus approach can improve species discovery from bulk samples at a given sequencing volume.
DISCUSSION
High throughput construction of multiplex mitogenomes
Overall, mitochondrial reads only accounted for a small fraction of the total genomic DNA (0.5%) in the pooled sample. However, this minute trace of MT DNA produced impressive coverage for all pooled species (ranging from ∼10X to 660X, Table 1 and Supplementary Table S1 in Appendix 1), assuring high-quality genome assemblies. Among many factors that may influence assembly results for pooled mitogenomes, we address a few key features: DNA quantity and quality, sequencing depth and phylogenetic distance.
DNA quantity and quality
Although we normalized the total DNA quantity for each of the 49 taxa before pooling (i.e. 100 ng of each genomic DNA was mixed), it is important to remember that the total molar weight of the mitochondrial DNA remains largely unknown and uneven for pooled taxa. Both genome size (which subsequently determines the nuclear to mitochondrial DNA ratio) and non-target DNA (e.g. microbial symbionts, gut contents) may significantly affect the proportion of MT DNA in total DNA extracts. Given that studies on many of these specifics are lacking for the animals with regards to proportion of mitochondrial DNA, it is impossible to completely eliminate MT DNA molar bias from pooled samples. However, some adjustment can be made for certain taxa. For instance, species with large genome sizes (e.g. most orthopterans and stick insects) should be pooled with more genomic DNA. Similarly, those known to maintain large amount of symbionts (e.g. termites, some hemipterans and wasps) should be processed with more DNA or the gut could be removed before DNA extraction.
Surprisingly, DNA quality had no obvious influence on either sequencing coverage or assembling quality. The level of DNA degradation was scored as levels A to D, where A represented the best DNA quality for genome sequencing and D was typically considered as ‘junk’ DNA (Supplementary Table S1 in Appendix 1) according to the standard BGI protocol. Given most of the pooled taxa were collected and preserved in ethanol, DNA degradation was expected. Therefore, much of the DNA used in this study was highly degraded (rated as C or D level). We categorized pooled taxa based on DNA quality levels and summarized the average numbers of assembled protein coding genes for each category. No significant difference was observed among DNA quality levels (Supplementary Figure S3 in Appendix 2). Similarly, there was no correlation between the average sequencing depth and DNA quality level (Supplementary Table S1 in Appendix 1). The independence of mitogenome assembly quality from DNA quality is unexpected, but not unreasonable. Typical DNA degradation should still provide sufficient DNA of acceptable quality (e.g. 100–500 bp in fragment size) for NGS shotgun sequencing. Our result suggests that DNA extracted from degraded samples will likely produce a large portion of full mitochondrial genomes. An additional advantage of multiplex sequencing is that the required DNA quantity for each of the mixed species is significantly reduced when compared with routine protocols (i.e. one library per species). The new pipeline only requires 1–2 μg of total pooled DNA for library construction on the Illumina Hiseq platform, reducing DNA quantity requirements for each species to 100 ng or less, which can be effectively obtained from animal tissues (e.g. insect legs). These discoveries are very encouraging for the construction of a comprehensive reference library for mitogenomes, considering that a significant portion of the biodiversity belongs to rare taxa, many of which can only be found in museum collections.
How closely related the taxa to be pooled?
The annotation and subsequent concatenation of assemblies from three different assembler programs significantly reduced the number of scaffolds from 1 362 000 (884 000 + 208 000 + 270 000) to 649, therefore alleviating the needs for taxonomic assignment for fragmented scaffolds. Consequently, this improvement led to a much more flexible taxon pooling strategy. The fact that all 49 taxa were successfully assembled suggests that species from different families, subfamilies or even congeners (e.g. three Drosophila spp. were each assembled into a single scaffold) can be pool-sequenced using our pipeline.
A major concern for pool-sequencing of multiplex mitogenomes is the accuracy of sequence assembly and the risk in creating chimeras. The issue can become more serious when closely related taxa are mixed. Our results demonstrate such an NGS pipeline can produce mitogenome sequences at an equivalent quality comparable to Sanger sequencing (Figure 5, Supplementary Table S2 in Appendix 3), which has been considered as the gold standard.
Main research fields that may benefit from the current work include phylogenetics and reference genome construction for biodiversity studies. Fulfilling needs in investigating various levels of evolutionary history, taxa from different families, genera and even within the same genus now can be pool-sequenced for mitogenomes. Both gene sequences (including protein coding genes, rRNA, tRNA genes and control region) and gene order can be used in phylogenetic inference. Similarly, when closely related taxa co-occur in natural communities, our pipeline is expected to successfully assemble mitogenomes of most, if not all, taxa for the entire community. These genomes will serve as critical references for a PCR-free based mito-metagenomics approach.
Technical improvements (wet-lab sequencing or informatics) allowing for longer assembly of mito-scaffolds will enable us to pool more closely related taxa. Furthermore, the growing databases for both whole mitogenomes and MT protein coding genes will improve our ability to assign fragmented scaffolds to finer taxonomic levels, therefore creating further flexibility in taxon pooling.
How many species can be pooled and how much does it cost?
Of all the 49 species that were successfully assembled for at least a large portion of the mitogenomes, the sequencing coverages range from ca. 10 to 660X. Because the lowest coverage (10–20X) produced good-quality assemblies, we expect this sequencing depth should be sufficient for multiplex mitogenome assembly as the minimum coverage. Considering the average mitochondrial proportion of total DNA of the 49 pooled species is 0.5% and the current sequencing capacity, we predict that it will be possible to sequence as many as 1000 taxa in a single lane on an Illumina Hiseq 2000. Obviously, given the expected heterogeneous DNA quantity, different taxa will gain varied assembling qualities. Nevertheless, the current strategy should allow for generating most of the MT protein coding genes for a vast number of animal taxa at a single effort.
The biggest advantage of the new NGS pipeline is the significant reduction of library construction cost for each taxon. Although the final cost will obviously vary from one laboratory to another (e.g. research-oriented versus commercial), it is possible to provide a general understanding based on publically available information. We calculated the average cost for each mitogenome based on published chemistry cost of the Hiseq 2000 (41), current sequencing capacity, average cost for Hiseq library construction, average MT DNA proportion from the present study and minimum sequencing coverage required for good-quality assembly (10–20X, from this study). The chemistry cost for producing a single mitogenome ranges from ca. 30 to <2 USD, assuming 50 to 1000 species can be pool-sequenced in a single lane of Hiseq 2000, respectively. Therefore, even the current cost is already at the same level of classic Sanger sequencing of a single gene, yet providing >10-fold increase in genetic information. Occasionally, Sanger sequences are needed to assure taxon assignment for assembled scaffolds (not mandatory for de novo assembly) when public databases are not well represented taxonomically. For example, in the current work, 7 CO1, 2 ND5 and 1 ND1 Sanger sequences were applied to link 10 scaffolds to the corresponding species. The reliance on these Sanger sequences slightly increased the overall analytical cost (ca. 10 USD/Sanger sequence). However, we expect such reliance shall be rapidly removed once comprehensive mitogenome reference can be built—the major goal of the current study.
Recommended strategy
Based on our pilot study, we make the following observations for multiplex sequencing and assembly of mitochondrial genomes:
Species as close as congeners can be pooled;
At least 50 species can be pool-sequenced in a single lane of Hiseq 2000, while hundreds or even thousands taxa can be expected to produce good-quality assemblies with most of the mitogenomes recovered;
Degraded voucher specimens may be suitable for sequencing;
On average, 100 ng or less genomic DNA from each species is sufficient for pooling;
Species with large genome size and those carrying abundant microbes should be compensated for with more DNA;
Multiple assembler programs should be applied; the concatenated results will produce better genomes.
Expanding single-gene metabarcoding to whole-mitochondrion metagenomics: prospects
The rapid developments of NGS technologies have created new possibilities to utilize genomic information in various fields of biological studies. Our pilot work represents one of the first empirical data to quickly and cost-efficiently build a reference library for whole mitochondrial genomes for a wide range of animal lineages. Although the accumulation of whole mitogenomes in the public domain has been expedited over the past years, taxonomic representation has been largely biased (23). For instance, only ca. 20% of the extant insect families are represented by a mitogenome sequence. A comprehensive mitogenome library covering the Tree of Animal Life will improve our knowledge on evolutionary history of animals and global patterns in genomic features of mitochondria (AT/CG ratio, gene order, evolutionary rates, etc.)
Furthermore, the new pipeline opens up a new venue for biodiversity research based on HTS. Although the current PCR-free metabarcoding method (9) has the advantage in reducing taxonomic biases, it relies exclusively on a specific gene (e.g. CO1 barcode for animals). It not only leaves out the majority of informative mitochondrial sequences, but also faces the possibility of missing the CO1 barcode target due to insufficient sequencing or DNA degradation. Our in silico simulation results suggest that by recruiting more informative mitochondrial markers, species detection for bulk samples can be more effective as it can be achieved at lower sequencing depths (hence more cost efficient). Apparently, the feasibility of expanding the current CO1 metabarcoding approach to whole-mitochondrion metagenomics (‘mito-metagenomics’) relies on the comprehensiveness of mitogenome references. The new multiplex mitogenome sequencing and assembly pipeline allows for high-throughput and cost-efficient construction of mitochondria for many animal taxa. Furthermore, when dealing with a specific eco-system, the new pipeline can rapidly build a full set of mitogenomes for the common, if not all, animals found in a given community, e.g. a stream, a pond or a crop field being investigated. Such a well-represented mitogenome reference for the focused locality will enable studies integrating phylogenetic history and ecological niches (e.g. phylogenetic diversity) and improve performance of NGS-based biodiversity surveys, especially those using a PCR-free shotgun sequencing approach.
ACCESSION NUMBERS
SRA174290, KM207019–KM207147 and KM244654–KM244713.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank the following collaborators for their contribution of specimens: Prof. Zhongli Sha of the Institute of Oceanology, Chinese Academy of Sciences (starfish), Prof. Xiaoli Tong and Miss Weifang Shi of South China Agricultural University (Ephemeroptera and Embioptera) and Prof. Boping Han of Jinan University (Daphnia). Colleagues from BGI-Shenzhen helped to optimize the laboratory and informatics pipelines.
Footnotes
Present Address: Yiyuan Li, Department of Biological Sciences, Galvin Life Science Center, University of Notre Dame, IN 46556, USA.
FUNDING
Ministry of Science and Technology of the People's Republic of China through the National High-tech Research and Development Project (863) of China [2012AA021601]; National Science and Technology Support Program of China [2012BAK11B06-4]. Funding for open access charge: BGI-Shenzhen.
Conflict of interest statement. None declared.
REFERENCES
- 1.Taberlet P., Coissac E., Pompanon F., Brochmann C., Willerslev E. Towards next-generation biodiversity assessment using DNA metabarcoding. Mol. Ecol. 2012;21:2045–2050. doi: 10.1111/j.1365-294X.2012.05470.x. [DOI] [PubMed] [Google Scholar]
- 2.Hebert P.D., Cywinska A., Ball S.L. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. B Biol. Sci. 2003;270:313–321. doi: 10.1098/rspb.2002.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bellemain E., Carlsen T., Brochmann C., Coissac E., Taberlet P., Kauserud H. ITS as an environmental DNA barcode for fungi: an in silico approach reveals potential PCR biases. BMC Microbiol. 2010;10:189. doi: 10.1186/1471-2180-10-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arif I.A., Khan H.A., Al Sadoon M., Shobrak M. Limited efficiency of universal mini-barcode primers for DNA amplification from desert reptiles, birds and mammals. Genet. Mol. Res. 2011;10:3559–3564. doi: 10.4238/2011.October.31.3. [DOI] [PubMed] [Google Scholar]
- 5.Haas B.J., Gevers D., Earl A.M., Feldgarden M., Ward D.V., Giannoukos G., Ciulla D., Tabbaa D., Highlander S.K., Sodergren E., et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011;21:494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan M.J., Chariton A.A., Hartley D.M., Court L.N., Hardy C.M. Improved inference of taxonomic richness from environmental DNA. PLoS One. 2013;8:e71974. doi: 10.1371/journal.pone.0071974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu S., Li Y., Lu J., Su X., Tang M., Zhang R., Zhou L., Zhou C., Yang Q., Ji Y. SOAPBarcode: revealing arthropod biodiversity through assembly of Illumina shotgun sequences of PCR amplicons. Methods Ecol. Evol. 2013;4:1142–1150. [Google Scholar]
- 8.Yu D.W., Ji Y., Emerson B.C., Wang X., Ye C., Yang C., Ding Z. Biodiversity soup: metabarcoding of arthropods for rapid biodiversity assessment and biomonitoring. Methods Ecol. Evol. 2012;3:613–623. [Google Scholar]
- 9.Zhou X., Li Y., Liu S., Yang Q., Su X., Zhou L., Tang M., Fu R., Li J., Huang Q. Ultra-deep sequencing enables high-fidelity recovery of biodiversity for bulk arthropod samples without PCR amplification. Gigascience. 2013;2:4. doi: 10.1186/2047-217X-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.CBOL Plant Working Group. A DNA barcode for land plants. Proc. Natl. Acad. Sci. U.S.A. 2009;106:12794–12797. doi: 10.1073/pnas.0905845106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krawczyk K., Szczecińska M., Sawicki J. Evaluation of 11 single-locus and seven multilocus DNA barcodes in Lamium L. (Lamiaceae) Mol. Ecol. Resour. 2013;14:272–285. doi: 10.1111/1755-0998.12175. [DOI] [PubMed] [Google Scholar]
- 12.Bourke B.P., Oliveira T.P., Suesdek L., Bergo E.S., Sallum M.A. A multi-locus approach to barcoding in the Anopheles strodei subgroup (Diptera: Culicidae) Parasit. Vectors. 2013;6:111. doi: 10.1186/1756-3305-6-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roe A.D., Rice A.V., Bromilow S.E., Cooke J.E., Sperling F.A. Multilocus species identification and fungal DNA barcoding: insights from blue stain fungal symbionts of the mountain pine beetle. Mol. Ecol. Resour. 2010;10:946–959. doi: 10.1111/j.1755-0998.2010.02844.x. [DOI] [PubMed] [Google Scholar]
- 14.Dupuis J.R., Roe A.D., Sperling F.A. Multi‐locus species delimitation in closely related animals and fungi: one marker is not enough. Mol. Ecol. 2012;21:4422–4436. doi: 10.1111/j.1365-294X.2012.05642.x. [DOI] [PubMed] [Google Scholar]
- 15.Zhan A., Bailey S.A., Heath D.D., Macisaac H.J. Performance comparison of genetic markers for high-throughput sequencing-based biodiversity assessment in complex communities. 2014;14:1049–1059. doi: 10.1111/1755-0998.12254. [DOI] [PubMed] [Google Scholar]
- 16.David O., Laredo C., Leblois R., Schaeffer B., Vergne N. Coalescent-based DNA barcoding: multilocus analysis and robustness. J. Comput. Biol. 2012;19:271–278. doi: 10.1089/cmb.2011.0122. [DOI] [PubMed] [Google Scholar]
- 17.Chesters D., Yu F., Cao H., Dai Q., Wu Q., Shi W., Zheng W., Zhu C. Heuristic optimization for global species clustering of DNA sequence data from multiple loci. Methods Ecol. Evol. 2013;4:961–970. [Google Scholar]
- 18.Nicolas V., Schaeffer B., Missoup A.D., Kennis J., Colyn M., Denys C., Tatard C., Cruaud C., Laredo C. Assessment of three mitochondrial genes (16S, Cytb, CO1) for identifying species in the Praomyini tribe (Rodentia: Muridae) PLoS One. 2012;7:e36586. doi: 10.1371/journal.pone.0036586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Z., Li H., Bin S., Ma J., He H., Li X., Gong F., Lin J. Sequence analysis of mitochondrial ND1 gene can reveal the genetic structure and origin of Bactrocera dorsalis s.s. BMC Evol. Biol. 2014;14:55. doi: 10.1186/1471-2148-14-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Botero-Castro F., Tilak M.K., Justy F., Catzeflis F., Delsuc F., Douzery E.J. Next-generation sequencing and phylogenetic signal of complete mitochondrial genomes for resolving the evolutionary history of leaf-nosed bats (Phyllostomidae) Mol. Phylogenet. Evol. 2013;69:728–739. doi: 10.1016/j.ympev.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 21.Cameron S.L. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 2014;59:95–117. doi: 10.1146/annurev-ento-011613-162007. [DOI] [PubMed] [Google Scholar]
- 22.Timmermans M.J., Dodsworth S., Culverwell C.L., Bocak L., Ahrens D., Littlewood D.T., Pons J., Vogler A.P. Why barcode? High-throughput multiplex sequencing of mitochondrial genomes for molecular systematics. Nucleic Acids Res. 2010;38:e197. doi: 10.1093/nar/gkq807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cameron S.L. How to sequence and annotate insect mitochondrial genomes for systematic and comparative genomics research. 2014;39:400–411. [Google Scholar]
- 24.Williams S., Foster P., Littlewood D. The complete mitochondrial genome of a turbinid vetigastropod from MiSeq Illumina sequencing of genomic DNA and steps towards a resolved gastropod phylogeny. Gene. 2014;533:38–47. doi: 10.1016/j.gene.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Groenenberg D.S., Pirovano W., Gittenberger E., Schilthuizen M. The complete mitogenome of Cylindrus obtusus (Helicidae, Ariantinae) using Illumina next generation sequencing. BMC Genomics. 2012;13:114. doi: 10.1186/1471-2164-13-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma P., Guo Z., Li D. Rapid sequencing of the bamboo mitochondrial genome using Illumina technology and parallel episodic evolution of organelle genomes in grasses. PLoS One. 2012;7:e30297. doi: 10.1371/journal.pone.0030297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hahn C., Bachmann L., Chevreux B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic Acids Res. 2013;41:e129-e129. doi: 10.1093/nar/gkt371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rubinstein N.D., Feldstein T., Shenkar N., Botero-Castro F., Griggio F., Mastrototaro F., Delsuc F., Douzery E.J., Gissi C., Huchon D. Deep sequencing of mixed total DNA without barcodes allows efficient assembly of highly plastic ascidian mitochondrial genomes. Genome Biol. Evol. 2013;5:1185–1199. doi: 10.1093/gbe/evt081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dettai A., Gallut C., Brouillet S., Pothier J., LeCO1ntre G., Debruyne R. Conveniently pre-tagged and pre-packaged: extended molecular identification and metagenomics using complete metazoan mitochondrial genomes. PLoS One. 2012;7:e51263. doi: 10.1371/journal.pone.0051263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ivanova N.V., Dewaard J.R., Hebert P.D. An inexpensive, automation-friendly protocol for recovering high-quality DNA. Mol. Ecol. Notes. 2006;6:998–1002. [Google Scholar]
- 31.Luo R., Liu B., Xie Y., Li Z., Huang W., Yuan J., He G., Chen Y., Pan Q., Liu Y., et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1:18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li R., Zhu H., Ruan J., Qian W., Fang X., Shi Z., Li Y., Li S., Shan G., Kristiansen K., et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20:265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie Y., Wu G., Tang J., Luo R., Patterson J., Liu S., Huang W., He G., Gu S., Li S., et al. SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics. 2014;30:1660–1666. doi: 10.1093/bioinformatics/btu077. [DOI] [PubMed] [Google Scholar]
- 34.Peng Y., Leung H.C., Yiu S.M., Chin F.Y. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28:1420–1428. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- 35.Pertea G., Huang X., Liang F., Antonescu V., Sultana R., Karamycheva S., Lee Y., White J., Cheung F., Parvizi B., et al. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003;19:651–652. doi: 10.1093/bioinformatics/btg034. [DOI] [PubMed] [Google Scholar]
- 36.Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., Buxton S., Cooper A., Markowitz S., Duran C., et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quail M.A., Smith M., Coupland P., Otto T.D., Harris S.R., Connor T.R., Bertoni A., Swerdlow H.P., Gu Y. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics. 2012;13:341. doi: 10.1186/1471-2164-13-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.