

Abstract
The mitochondrial electron transport chain (ETC) plays a central role in energy generation in the cell. Mitochondrial dysfunctions diminish adenosine triphosphate (ATP) production and result in insufficient energy to maintain cell function. As energy output declines, the most energetic tissues are preferentially affected. To satisfy cellular energy demands, the mitochondrial ETC needs to be able to elevate its capacity to produce ATP at times of increased metabolic demand or decreased fuel supply. This mitochondrial plasticity is reduced in many age-associated diseases. In this review, we describe the serendipitous discovery of a novel class of compounds that selectively target cardiolipin on the inner mitochondrial membrane to optimize efficiency of the ETC and thereby restore cellular bioenergetics in aging and diverse disease models, without any effect on the normal healthy organism. The first of these compounds, SS-31, is currently in multiple clinical trials.
Mitochondrial Dysfunction, Bioenergetics Failure, and Complex Diseases
A decline in bioenergetics underlies the general frailty of old age and a broad spectrum of metabolic and degenerative diseases. Mitochondria play a central role in energy generation in the cell. They are often referred to as the cell's powerhouses, generating adenosine triphosphate (ATP) to carry out essential biological functions. In major mammalian tissues, 80–90% of ATP is generated by the mitochondrial electron transport chain (ETC). Mitochondrial dysfunction results in less ATP production and insufficient energy to maintain cell function, which is followed by cell injury and even cell death. Because the ETC powers a wide range of energy-based cellular functions, genetic mitochondrial diseases can produce a broad spectrum of functional abnormalities. Every tissue and organ in the body requires mitochondrial energy, but certain tissues have especially high levels of energy demand. These include the central nervous system, heart, skeletal muscle, kidney, and liver. As energy output declines, these most energetic tissues are preferentially affected, and symptoms often include loss of motor control, muscle weakness, cardiomyopathy, and visual/hearing problems.1
Aging is associated with alterations in many components of the mitochondrial ETC. A significant decrease in coupled respiration and ATP synthesis has been reported in skeletal muscles from aged mice and humans.2,3 As mitochondria become increasingly dysfunctional over time, many age-related conditions, such as heart failure and Alzheimer's disease, set in.1 Mitochondrial dysfunction has been identified in neurodegenerative diseases, heart disease, diabetes, chronic kidney disease, retinal diseases, deafness, cancer, infertility, chronic fatigue syndrome, and skin diseases.4–7
Mitochondrial Dysfunction Caused By Therapeutic Drugs
Mitochondrial dysfunction is increasingly implicated in many drug-induced toxicities.8 Notable examples include cardiac and skeletal myopathy associated with clinical use of doxorubicin and nucleoside reverse-transcriptase inhibitors such as zidovudine. Mitochondrial toxicity has led to the withdrawal of several drugs, including troglitazone, due to hepatotoxicity. Approximately 80% of drugs with US Food and Drug Administration black box warnings have been shown to cause mitochondrial toxicity. This has led the pharmaceutical industry to screen for mitochondrial toxicity early in order to identify compounds that are too toxic to bring into clinical testing.
Mitochondria as Prime Target for Therapeutics Development
The wide-ranging impact of mitochondria in so many diseases makes them prime targets for therapeutics development. Mitochondria have been proposed as therapeutic targets for ischemic heart disease, heart failure, neurodegenerative diseases, and metabolic disorders.9 However, the development of mitoprotective drugs has been hampered by a number of challenges, and there are, at present, no approved therapies for mitochondrial diseases. Current strategies for development of therapeutics for these diseases are still driven by disease-specific approaches. Modern drug discovery tends to focus on protein targets and signaling pathways for individual disease conditions, and target identification is driven by genomics and proteomics. The discovery process is then aided by high-throughput assays and screening of chemical libraries for rapid selection of likely drug candidates that would act on the target of interest. The biggest impediment to the development of mitoprotective drugs is the lack of a specific molecular entity to be targeted in mitochondria to promote ATP synthesis. The production of ATP relies on the concerted action of a multitude of proteins and lipids within the ETC (Figure 1), indicating the futility of expecting to improve ATP production by just targeting one of the many proteins in this vast integral ETC network. Finally, the discovery of mitochondrial drug candidates is also made difficult by the requirements for their delivery to the mitochondria and to be free of mitochondrial toxicity.
Figure 1.
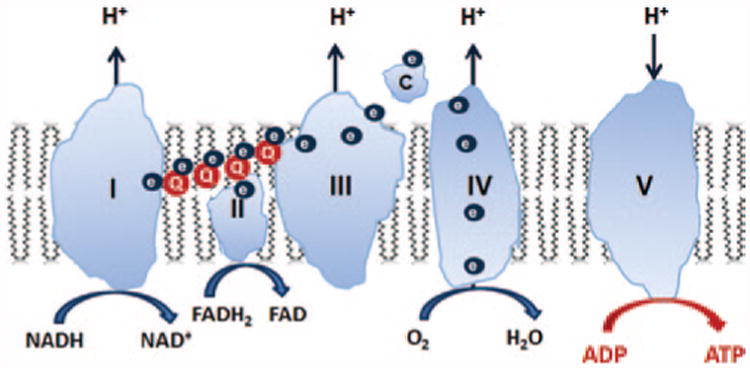
Mitochondria are composed of two membranes: an outer mitochondrial membrane and an inner mitochondrial membrane (IMM). The IMM is unique in that it has a very high concentration of proteins and is rich in cardiolipin, a very anionic phospholipid that plays a crucial role in the formation of cristae curvature and is important for efficient function of the electron transport chain (ETC). The mitochondrial matrix enclosed by the IMM contains not only mitochondrial DNA but also the major enzymes of the tricarboxylic acid cycle, which provide reducing equivalents in the form of NADH and FADH2 to the ETC. The IMM is compartmentalized into numerous cristae that greatly increase the surface area of the IMM. The four power-generating protein complexes of the ETC (complexes I–IV) reside on these cristae in the IMM, and the proton gradient generated across the IMM as a result of electron transfer from complex I to complex IV drives the production of ATP by the F0F1–ATPase (complex V). All these complexes must be assembled properly for efficient electron transfer to take place. ADP, adenosine diphosphate; ATP, adenosine triphosphate; ATPase, adenosine triphosphatase; FADH2, reduced flavin adenine dinucleotide; NADH, reduced nicotinamide adenine dinucleotide.
The Role of Serendipity in Drug Discovery
Serendipity has played a significant role throughout the history of drug discovery, and indeed, many of the most effective therapeutic agents in use today arose through serendipity. The current target-based drug discovery paradigm, using high-throughput screening of large chemical libraries, tends to remove any chance of serendipitous discoveries. In this review, we will describe the chance discovery of a family of compounds (Szeto–Schiller (SS) peptides) that selectively target the mitochondrial ETC to optimize efficiency of electron transport and restore cellular bioenergetics in aging and diverse disease models without any effect on the normal healthy organism. We will then summarize what is understood about their mechanisms of action. The first of these compounds (SS-31; also named MTP-131 and Bendavia) has entered into clinical development for a variety of complex diseases for which there is currently no satisfactory treatment. In addition, SS-31 and its analogs have served as valuable tools to further our understanding of the major role of mitochondrial dysfunction in many of the age-related complex diseases. Furthermore, these amino acid–based compounds have surprisingly remarkable drug-like properties, making us question many of the old dogmas about drug design in the pharmaceutical industry.
Serendipitous Discovery of a New Class of Small Molecules that Selectively Target Mitochondria
While working on a synthetic opioid peptide with very high affinity and selectivity for the μ-opioid receptor,10 we accidentally discovered that this highly polar, water-soluble tetrapeptide had potent central nervous system analgesic activity after s.c. administration to rodents,11 suggesting that it was capable of crossing the blood–brain barrier. It is generally thought that only highly lipophilic compounds are capable of crossing the blood–brain barrier. This was so disturbing to us that we sought confirmation of its cell permeability using cell cultures. Indeed, this peptide (SS-02 or [Dmt1]DALDA (H-Dmt-d-Arg-Phe-Lys-NH2, where Dmt = 2′,6′-dimethyl-Tyr)) (Table 1) was readily taken up by a variety of cell types without the need for specific transporters or receptors, and it even penetrated a layer of polarized epithelial cells with tight junctions.12 This was highly unexpected given its size (molecular weight = 640), the highly polar peptide backbone, and the presence of a 3+ net charge. The key discovery came after we synthesized a fluorescent-labeled analog13 and confocal fluorescence microscopy confirmed cellular uptake of this peptide, revealing highly selective mitochondrial targeting.14 Studies on isolated mitochondria subsequently confirmed rapid mitochondrial uptake of SS-02 and revealed that this peptide was selectively partitioned to the inner mitochondrial membrane (IMM), with minimal presence in the matrix.14 This was another surprise because cationic molecules are thought to penetrate the mitochondrial matrix because of the potential gradient across the IMM. We confirmed that, unlike lipophilic cations such as MitoQ, the uptake of SS-02 does not rely on mitochondrial potential and it does not cause mitochondrial depolarization.14 Recognizing the potential of a compound that can selectively target and concentrate on the IMM, where the ETC is localized, we designed other peptide analogs, SS-31 and SS-20, which also target the IMM but have negligible affinity for opioid receptors14,15 (Table 1).
Table 1. Chemical structures of Szeto-Schiller (SS) peptides.
| SS-02 | H-Dmt-d-Arg-Phe-Lys-NH2 |
| SS-31 | H-d-Arg-Dmt-Lys-Phe-NH2 |
| SS-20 | H-Phe-d-Arg-Phe-Lys-NH2 |
Dmt, 2′,6′-dimethylTyr.
It was recently discovered that selective binding of SS-31 and SS-20 to cardiolipin accounts for their concentration on the IMM.16 Cardiolipin is exclusively expressed on the IMM.17 By incorporating a polarity-sensitive fluorescent amino acid (aladan) into the peptide sequences, we found that only anionic phospholipids caused a shift in the maximum wavelength (λmax) and fluorescence intensity of the peptide, with cardiolipin being the most sensitive.16 The zwitterionic phospholipids (phosphatidylcholine and phosphatidylethanolamine) had no effect, suggesting that electrostatic interaction between SS-31 and the anionic phospholipids is involved. Because phosphatidylserine is negligible in the IMM, cardiolipin appears to be the primary target for these peptides. Further nuclear magnetic resonance studies led us to propose that electrostatic interaction between the two basic amino acids (Lys and Arg) and the phosphate head groups of cardiolipin aligns the aromatic residues (Dmt and Phe) within the hydrophobic acyl chain region.18 We have evidence that SS-02 and SS-20 also interact with cardiolipin in the same manner (H.H.Z and A.V.B., unpublished data). These are the first compounds known to target specific membrane phospholipids rather than protein molecules. The selective interaction of these compounds with cardiolipin explains why they concentrate only on the IMM—cardiolipin is normally not expressed on other cellular membranes. The targeting of cardiolipin makes these peptides ideal for acting on the mitochondrial ETC and optimizing ATP production, in addition to helping to minimize of-target adverse effects.
Role of Cardiolipin in Mitochondrial Structure and Function
The IMM contains ∼10% cardiolipin.17 Unlike all other phospholipids, cardiolipin is a dimeric phospholipid with a small acidic head group and four acyl chains, thus giving it a conical structure (Figure 2). As a result of its conical shape, cardiolipin exerts lateral pressure on a membrane containing other phospholipids and promotes membrane curvature.17 The presence of cardiolipin decreases water permeability of the lipid membrane and decreases the energy required to create folds or cristae in the IMM. Cardiolipin is particularly important for cristae formation, and deficiency of cardiolipin in Barth syndrome results in loss of cristae membranes and mitochondrial failure.19 Cardiolipin rafts on cristae membranes also allow the respiratory complexes to form supercomplexes that reduce the distance between redox partners (Figure 2).20 According to the Marcus theory, the electron transfer rate between an electron donor and an acceptor decreases exponentially with increasing distance, and it has been estimated that redox partners need to be arranged at distances less than 14–20 Å.21 Besides providing a platform for the organization of the respiratory complexes, cardiolipin is also thought to serve as a proton trap on the outer leaflet of the IMM to allow rapid lateral diffusion of protons to ATP synthase with minimal changes in the bulk phase pH.22 The diffusion coefficient is at least 20 times larger along the membrane surface than within the bulk aqueous phase.
Figure 2.
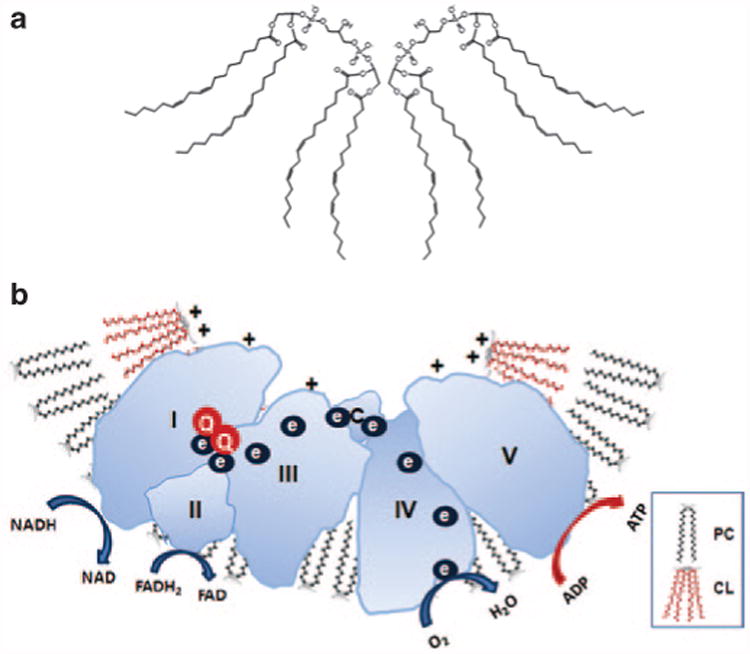
Cardiolipin promotes membrane curvature to optimize electron transport chain. (a) Cardiolipin (CL) is a dimeric phospholipid with a small acidic head group and four acyl chains, thus giving it a conical structure. As a result of its conical shape, CL exerts lateral pressure on a membrane containing other phospholipids such as phosphatidylcholine (PC), and results in membrane curvature. (b) CL rafts on cristae membranes allow the respiratory complexes to form supercomplexes that reduce the distance between redox partners. Besides providing a platform for the aggregation of the respiratory complexes, CL is also thought to serve as a proton trap on the outer leaflet of the inner mitochondrial membrane to allow rapid lateral diffusion of protons to the ATP synthase with minimal changes in the bulk phase pH. ADP, adenosine diphosphate; ATP, adenosine triphosphate; FADH2, reduced flavin adenine dinucleotide; NADH, reduced nicotinamide adenine dinucleotide.
Cardiolipin as a Drug Target
A decline in cardiolipin content has been reported with age, and this is associated with biochemical and functional alterations in many components of the mitochondrial ETC, resulting in reduced efficiency of electron transport, inhibition of oxidative phosphorylation (OXPHOS), and increased leakage of reactive oxygen species (ROS).23–25 Alterations in cardiolipin have also been reported in a variety of pathological conditions, including ischemia–reperfusion injury, heart failure, skeletal muscle weakness, neurodegenerative diseases, traumatic brain injury, diabetes, and cancer cachexia.26–29 In many of these disorders, mitochondria have been found to be swollen, with loss of cristae membranes. Thus, protection of cardiolipin may represent a common therapeutic approach to many age-associated infirmities and complex diseases.
Cardiolipin Serves as an “Address” for SS Peptides to Modulate Cytochrome C Activity
Cytochrome (cyt)c serves as an electron carrier from complex III to complex IV on the ETC. Being a soluble protein and the only nonintegral component of the respiratory chain, cytc represents a rate-limiting step in the ETC. Cardiolipin provides an anionic platform for electrostatic attraction so that this highly cationic protein remains loosely attached to the ETC and can facilitate electron transfer from complex III to complex IV (Figure 2). However, about 10–15% of cytc can also become tightly bound to cardiolipin via hydrophobic interaction. This binding changes the native conformation of cytc and disrupts the Met80–heme iron coordination so that cytc cannot participate in electron transfer and instead becomes a peroxidase that oxidizes cardiolipin.30–32 Oxidized cardiolipin disturbs cardiolipin microdomains on the IMM and causes the loss of cristae curvature. Cardiolipin oxidation also causes cytc to be detached from the IMM. All these processes result in inhibition of mitochondrial respiration and set the stage for apoptosis.33 Oxidized cardiolipin also synergizes with Ca2+ to induce opening of the mitochondrial permeability transition pore, resulting in collapse of mitochondrial potential, uncoupling of OXPHOS, and release of cytc and other proapoptotic proteins into the cytosol to trigger apoptosis. Thus, cytc is Janus-faced, having two contrasting functions, one promoting life and the other promoting death.
By binding to cardiolipin, the SS peptides modulate the hydrophobic interaction between cytc and cardiolipin and promote the electron carrier properties relative to the peroxidase activity in cytc.32 Structural studies suggest that the (SS-31/cardiolipin) complex penetrates deep into cytc to protect its heme iron and prevent peroxidase activity.16 SS-31 can also improve electron carrier function of the (cytc/cardiolipin) complex by improving π-π* interaction near the heme, and thus it is able to increase state 3 mitochondrial respiration and ATP/O ratio.16,34 Importantly, SS-31 has no effect on cytc reduction or mitochondrial O2 consumption when cytc is not bound tightly to cardiolipin.34 Thus, cardiolipin serves as an address for SS-31 to target the rate-limiting step of the ETC and thereby optimize OXPHOS.
Mitochondrial Plasticity
Mitochondrial OXPHOS is primarily determined by adenosine diphosphate (ADP) concentration and cellular demand for ATP synthesis (Figure 3). Mitochondrial OXPHOS capacity is defined as maximal ADP-stimulated OXPHOS elicited by a high ADP:ATP ratio at saturating concentrations of substrates and under unlimited O2 supply.4 Only substrate availability lower than the energetic demand or low O2 concentrations will limit ATP generation by mitochondria. The mitochondrial ETC needs to be able to elevate its capacity to produce ATP at times of increased metabolic demand or decreased fuel supply. Its ability to do so has been called “spare respiratory capacity” or “mitochondrial plasticity.”4 Mitochondrial plasticity is determined by the efficacy of mitochondrial coupling between O2 consumption and the rate of ATP synthesis (P/O ratio). Figure 3 shows that state 3 respiration is dependent on substrate concentration and O2 supply. Mitochondrial plasticity provides an increase in respiratory capacity and a shift in the Michaelis–Menten constant Km so that higher mitochondrial respiration can be achieved with lower substrate or O2 concentrations. The effect is less pronounced at high concentrations of substrates. The respiratory control ratio shows that state 4 respiration is not changed, thus implying increased P/O coupling. This concept is most relevant for ischemic injury, in which there is a paucity of fuel and O2 supply; it is also important when glucose uptake into skeletal muscle is reduced because of insulin resistance. Mitochondrial plasticity is also relevant in situations of increased energy demand, such as during endurance exercise, heart failure, sepsis, and after severe trauma. Under these conditions, the ability of the mitochondrial ETC to increase P/O coupling is critical for maintaining cell function and cell survival. On the other hand, increasing mitochondrial coupling may even be beneficial in the context of metabolic overload and low energy demand (low ADP), in order to reduce electron leak and ROS generation.
Figure 3.
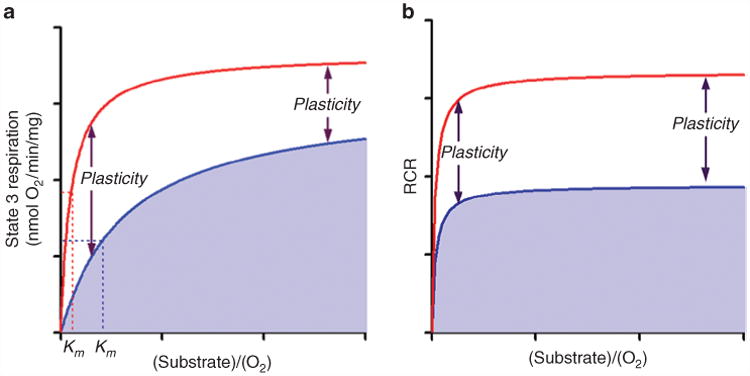
The mitochondrial electron transport chain needs to be able to elevate its capacity to produce ATP at times of increased metabolic demand or decreased fuel supply. Its ability to do so has been called “spare respiratory capacity” or “mitochondrial plasticity.” (a) Mitochondrial state 3 respiration (ADP-stimulated respiration) is dependent on substrate concentration and O2 supply (blue line). Mitochondrial plasticity provides a shift in the Michaelis–Menten constant (Km) so that higher mitochondrial respiration can be achieved with lower substrate or O2 concentration (red line). The effect is less at high concentrations of substrates. (b) The respiratory control ratio (RCR) shows that state 4 respiration is not changed. Mitochondrial plasticity is determined by the efficacy of mitochondrial coupling between O2 consumption and the rate of ATP synthesis (P/O ratio). ADP, adenosine diphosphate; ATP, adenosine triphosphate.
Resting mitochondrial activity and mitochondrial plasticity are reduced in the elderly and in insulin-resistant patients. In the long term, mitochondrial capacity may be increased by increasing mitochondrial content via transcriptional regulators such as peroxisome proliferator-activated receptor (PPAR)-γ coactivators 1-α and 1-β or through AMP-activated protein kinase. However, short-term mitochondrial plasticity requires improving the efficiency of the ETC. Uncouplers increase mitochondrial electron transfer and inhibit ROS formation, but at the price of diminished ATP synthesis. Furthermore, promoting electron transfer by increasing electron entry will only increase electron leak and ROS generation. In general, mitochondrial plasticity requires increased coupling of electron transfer to ATP synthesis in the ETC.
Recent evidence suggests that the SS peptides can improve mitochondrial capacity and coupling, thereby increasing mitochondrial plasticity. By targeting the rate-limiting step (cytc) in the ETC, the SS peptides promote electron flux through the ETC, increase state 3 respiration, increase P/O coupling, increase ATP synthesis, and reduce electron leak.34,35 Both SS-31 and SS-20 can increase state 3 respiration when substrate concentration is low, without changing state 4 respiration, thereby increasing respiratory control ratio, suggesting increased mitochondrial coupling.34,35 This increase in mitochondrial plasticity may account for the beneficial effects of SS peptides reported in many preclinical disease models. Furthermore, we recently found that these peptides can also enhance OXPHOS coupling by reducing proton leak (A.V.B. and H.H.S., unpublished data). Proton leak accounts for ∼25% of resting O2 consumption; this futile cycle represents an energy sink and may therefore serve as a target for improving mitochondrial plasticity.36 Proton leak appears to be regulated by membrane phospholipids, especially cardiolipin.37 Cardiolipin serves as a proton trap on the IMM, and by protecting cardiolipin content and cristae density, SS-31 can help to minimize proton leak and improve mitochondrial coupling. In the following section, we will summarize the evidence for SS peptides protecting cell function and survival under conditions of low substrate availability, increased energy demand, high fuel supply, or decreased metabolic demand. The results suggest that the SS peptides can promote mitochondrial plasticity in order to appropriately meet metabolic changes. It should be noted that SS-31 was used as either a trifluoroacetate salt (SS-31) or an acetate salt (MTP-131).
Decline in Bioenergetics Associated with Normal Aging
Sarcopenia, loss of skeletal muscle function with age, is a growing public health crisis in terms of both quality of life and economic cost to society. Reduced exercise efficiency in aged humans and mice has been associated with mild mitochondrial uncoupling in skeletal muscle, significant decrease in P/O ratio, maximal ATP synthesis, and increased ROS production.2,3 The traditional view is that the accumulation of oxidative damage to DNA, proteins, and lipids results in mitochondrial dysfunction with age. Therefore, the finding that 1 h after a single dose of SS-31 to aged mice the decline in resting and maximal mitochondrial ATP production, P/O ratio, and ROS emission were all reversed was quite unexpected.38 Skeletal muscle from aged mice was also more fatigue resistant 1 h after treatment, and 8 days of treatment led to increased endurance on the treadmill.38 Importantly, SS-31 had no effect on skeletal muscle bioenergetics in healthy 5-month-old mice. By acting on the rate-limiting step of the ETC, SS-31 improved skeletal muscle performance and exercise capacity only in aged mice.
Disuse Muscle Atrophy
Skeletal muscle weakness and atrophy commonly occur during prolonged periods of inactivity due to limb immobility or prolonged bed rest, and these are especially a problem in the aging population. Hind-limb casting in rats resulted in decreased state 3 respiration and decreased respiratory control ratio in the soleus muscle, suggesting both diminished activity of the ETC and uncoupling.39 Swollen mitochondria lacking cristae membranes were observed in the rat soleus muscle after hind-limb suspension, suggestive of cardiolipin peroxidation and depletion. SS-31 treatment prevented the uncoupling seen following immobilization and prevented skeletal muscle atrophy.39,40 The reduction in muscle atrophy was due to both inhibition of cell death pathways and protein degradation, as well as to an increase in anabolic signaling pathways.39,40 It should be noted that SS-31 had no effect on mitochondrial function in ambulatory animals.39 These studies suggest that SS-31 may protect against muscle loss from prolonged bed rest, especially in aged populations. Similarly, prolonged mechanical ventilation is associated with diaphragmatic weakness and atrophy, which result in difficulty in weaning off the ventilator. Treatment with SS-31 preserved OXPHOS, reduced mitochondrial ROS emission, and protected against mechanical ventilation–induced diaphragmatic atrophy and contractile dysfunction.41 These findings suggest that SS-31 may have therapeutic potential in preventing “failure to wean” in patients on mechanical ventilation.
Hypermetabolic Response to Severe Burn Trauma
Severe burn injury causes a major hypermetabolic response that is associated with increased energy expenditure and substrate release from protein stores, resulting in loss of lean body mass and muscle wasting that can persist for 9 months after injury. Burn patients develop the classic signs of insulin resistance, including hyperglycemia, increased protein catabolism, and impaired insulin-stimulated glucose uptake in skeletal muscle.42 This catabolic response is associated with decreased ATP synthesis and increased mitochondrial ROS in skeletal muscle. Treatment of mice with a single dose of SS-31 immediately after burn injury increased the ATP synthesis rate fivefold, reduced oxidative stress, and prevented apoptosis in skeletal muscle.43,44 SS-31 also prevented insulin resistance in these burn-injured mice.43,45 These studies suggest that SS-31 may be very helpful in minimizing the catabolic response after severe burn trauma and may also prevent insulin resistance in critically ill patients, especially those with sepsis.
Insulin Resistance and Type 2 Diabetes
Maintenance of normal blood glucose depends on insulin responsiveness of skeletal muscles and glucose-stimulated insulin secretion by pancreatic β-cells. Insulin resistance generally precedes the onset of diabetes in most cases of type 2 diabetes. Under excess lipid supply, β-oxidation of fatty acids exceeds the capacity of the tricarboxylic acid cycle and the ETC, resulting in incomplete fat oxidation, accumulation of intramyocellular lipids, and increased ROS production.46 Rats fed a high-fat diet for as little as 3 days showed increased emission of mitochondrial H2O2 in their skeletal muscles.47 Six weeks of high-fat diet led to insulin resistance and was associated with elevated mitochondrial H2O2 emission and a decrease in the ratio of reduced to oxidized glutathione, but no change in mitochondrial state 3 respiration was observed.47 Similar findings were reported in young insulin-resistant obese humans. Reduction of oxidative stress by SS-31 treatment or by overexpression of catalase targeted to mitochondria prevented insulin resistance in mice but had no effect on weight gain.47,48 Thus, mitochondrial oxidative stress appears to play a role in the initiation of insulin resistance, even though mitochondrial respiratory capacity remains normal in the early phase of insulin resistance.
Patients with type 2 diabetes not only have reduced secretion of insulin by pancreatic β-cells, they also fail to increase their OXPHOS capacity in response to insulin. This impaired mitochondrial plasticity is due not only to lower availability of substrates but also to impaired submaximal ADP-stimulated OXPHOS.4 Diabetic retinopathy is a major complication in type 2 diabetes, and insulin signaling is compromised in the retina.49 We recently found dramatically swollen mitochondria and loss of cristae membranes in the retinal pigment epithelium of diabetic mice, and this was associated with progressive decline in visual acuity (Alam N.M., Mills W.C., Szeto H.H., Prusky G.T., personal communication). Treatment with MTP-131 (either systemically or topically to the eye) protected mitochondrial structure in the retinal pigment epithelium (Figure 4) and restored vision, without any effect on blood glucose or glucose tolerance. These findings suggest that impaired mitochondrial plasticity may underlie diabetic complications and may be independent of hyperglycemia.
Figure 4.
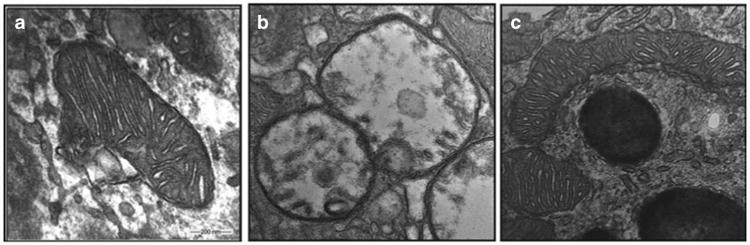
SS-31 protects the mitochondrial structure in retinal pigment epithelium (RPE) cells in diabetic mice. Mice were fed either a normal diet (ND) or a diabetic diet (DD) starting at 4 weeks of age. The DD mice also received streptozotocin (STZ) at 8 weeks to reduce insulin secretion (DD+STZ). These mice then received saline or SS-31 applied as eye drops daily, starting at 12 weeks. Representative electron microscopic images of RPE mitochondria at 32 weeks in (a) ND, (b) DD+STZ plus saline, and (c) DD+STZ plus SS-31 mice. Mitochondria are swollen and lack cristae in DD+STZ mice. SS-31-treated mitochondria retained their normal structure and cristae architecture.
Heart Failure
Heart failure is a mismatch between supply and demand of ATP. The heart consumes 20–30 times its own weight in ATP every day, and 90% of the ATP is derived from mitochondrial OXPHOS. This mismatch may result from decreased mitochondrial production of ATP postischemia or increased workload to the myocardium following hypertension. There is a significant decrease in myocardial ATP and phosphocreatine content in heart failure.50 However, current treatments all rely on “energy sparing” by decreasing workload. Targeting mitochondrial plasticity to improve ATP supply may provide an alternative approach to the treatment of heart failure. Cardiac mitochondria in failing hearts have structural abnormalities and loss of cristae, suggesting loss of cardiolipin with alterations in cardiolipin biosynthesis and remodeling.51,52 SS-31 has been found to be beneficial in several experimental heart failure models.
The first study was in a mouse model of hypertensive cardiomyopathy induced by angiotensin.53 Continuous administration of angiotensin for 4 weeks significantly increased both systolic and diastolic pressure. Echocardiography showed increase in left-ventricular mass and decline in diastolic function. Treatment with SS-31 ameliorated the cardiac hypertrophy and diastolic dysfunction without reducing blood pressure.
SS-31 was also tested in a pressure-overload heart failure model (transverse aortic constriction (TAC)). In mice, TAC causes increase in left-ventricular mass, dilation of the lef ventricle, and a 50% decline in fractional shortening within 4 weeks. Treatment with SS-31 completely ameliorated the cardiac hypertrophy, systolic failure, and myocardial fibrosis.54 The loss of mitochondrial cristae and increase in mitochondrial oxidative damage were both mitigated by SS-31. A direct action of SS-31 on mitochondria was confirmed by cardiac proteomic analysis. Of the 538 proteins that significantly changed after TAC, 30% were mitochondrial proteins and 25% were involved in metabolism. Most of the mitochondrial proteins declined in abundance after TAC, but SS-31 attenuated this decline in 84% of these proteins. The major pathways affected in TAC were mitochondrial dysfunction/OXPHOS and the citrate cycle, and these pathways were prevented by SS-31 treatment. This study supports the idea of targeting mitochondrial metabolism as a treatment approach.
SS-31 was recently evaluated in a canine myocardial infarct model induced by repeated intracoronary embolizations with microspheres. Sustained depression of cardiac function is seen in this model after embolization is discontinued. In dogs with advanced heart failure, acute MTP-131 treatment significantly increased ejection fraction, stroke volume, cardiac output, and left-ventricular contractility index without changing heart rate or blood pressure.55 Chronic administration of MTP-131 daily for 3 months significantly improved ejection fraction and left-ventricular end-diastolic pressure.56 These functional improvements were associated with improved mitochondrial potential, improved mitochondrial state 3 respiration, and increased ATP synthesis.57 Importantly, MTP-131 normalized the expression of cardiolipin-remodeling genes and proteins, suggesting that MTP-131 can improve mitochondrial function by protecting mitochondrial cardiolipin.58 Because MTP-131 has no effect on blood pressure or heart rate, it may be used to complement current therapies for heart failure.
Ischemia–Reperfusion Injury
Ischemia contributes to morbidity and mortality in a wide range of pathologies, including myocardial infarction, ischemic stroke, and acute kidney injury. The most effective therapeutic intervention for reducing ischemic damage and limiting the size of the infarct is timely reperfusion. However, ATP recovery is often delayed on reperfusion because ischemia causes mitochondrial swelling and unfolding of cristae membranes.16 By targeting mitochondrial cardiolipin and inhibiting cardiolipin peroxidation, the SS peptides protect mitochondrial cristae and facilitate ATP recovery in ischemic tissues.16 These peptides have been shown to be highly effective against cerebral, myocardial, and renal ischemia–reperfusion injury.
Administration of SS-31 30 min after ligation of the middle cerebral artery in mice significantly reduced infarct volume after transient, but not permanent, ischemic stroke.59,60 SS-31 also significantly reduced hemispheric swelling and inflammation as measured by monocyte chemoattractant protein 1 expression in transient stroke.60 These results suggest that SS-31 may serve as a potential therapeutic for minimizing cerebral ischemia– reperfusion injury, especially in stroke patients who receive tissue plasminogen activator.
The efficacy of the SS peptides in minimizing myocardial ischemia–reperfusion injury has been demonstrated in a number of studies. Treatment with SS-02 or SS-31 before coronary artery ligation significantly reduced infarct size and decreased severity of arrhythmias in rats.61 SS-31 administered after the onset of ischemia reduced infarct size in rabbits and sheep after coronary artery ligation, attenuated the extent of no-reflow in rabbits, and reduced infarct size in isolated perfused guinea pig hearts.62 Importantly, the investigators reported that SS-31 is rapidly taken up by the myocardium even after ischemia.
The benefit of mitochondrial protection in minimizing ischemia–reperfusion injury has been studied extensively in the kidney. Treatment with SS-31 30 min before ligation significantly improved renal function after both 30 and 45 min ischemia in a dose-dependent manner.63 SS-20 pretreatment was able to increase warm ischemia tolerance time from 30 to 45 min.35 Treatment with SS-31 or SS-20 significantly protected mitochondrial structure and preserved cristae architecture in tubular cells.16,35,63,64 Mitochondrial state 3 respiration was significantly improved with treatment using either SS-31 or SS-20, and tissue ATP levels were restored to control levels in 1 h, resulting in significant reduction in apoptosis, necrosis, oxidative stress, and inflammation, along with promotion of regeneration of tubular epithelial cells.
In renal tubular epithelial cells, as in other cells with high metabolic demand, cristae membrane makes up most of the IMM, with deep invaginations that extend into the matrix (Figure 5, left panel). Ischemia-induced mitochondrial ATP depletion compromises osmotic regulation in mitochondria and leads to matrix swelling (Figure 5, middle panel). Because mitochondria remain swollen after ischemia, ATP synthesis remains compromised despite restoration of blood flow on reperfusion. Treatment with SS-20 or SS-31 before ischemia prevented mitochondrial swelling and preserved cristae membrane (Figure 5, right panel). Because swelling is caused by failure of energy-driven water efflux mechanisms, these findings suggest that renal tubular cells treated with the SS peptides managed to produce sufficient ATP during ischemia to maintain fluid regulation in mitochondria. Enhancing mitochondrial bioenergetics may be an important target for improving ischemia tolerance during surgical procedures such as partial nephrectomy or organ transplantation.
Figure 5.
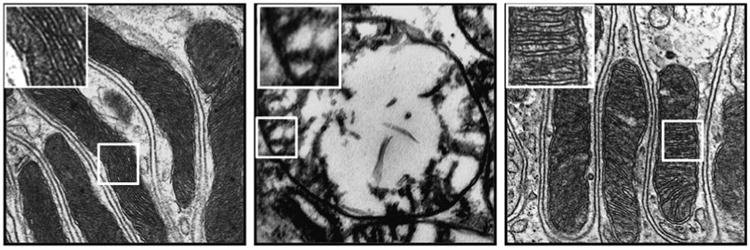
SS-20 protects kidney mitochondria during ischemia. Rats were treated with SS-20 or saline 30 min before bilateral occlusion of renal blood flow for 45 min. Electron microscopic images were taken from kidney sections 5 min after onset of reperfusion. Left panel: representative section from sham animals shows elongated mitochondria with dense cristae membranes. Middle panel: saline-treated ischemic kidneys show matrix swelling and loss of cristae membranes. Right panel: SS-20-treated ischemic kidneys show normal mitochondrial structure with preserved cristae architecture. All images are presented at ×80,000 original magnification.
Postischemic Tissue Remodeling
Most tissues—including the heart, kidney, and brain—undergo some kind of tissue remodeling (wound healing or scar formation) after acute ischemic injury. Myocardial infarctions result in extensive fibrosis that enhances myocardial stiffness and hampers both diastolic and systolic function. Likewise, recovery from acute kidney injury is marked by progressive structural damage, including glomerulosclerosis, tubular atrophy, and interstitial fibrosis. Although the focus has primarily been on minimizing death of cardiomyocytes, neurons, and renal tubular cells, little attention has been paid to the loss of endothelial cells during ischemia. The loss of the microvasculature results in chronic tissue hypoxia, causing further capillary and tubular damage, inflammation, and fibrosis.65,66 SS-31 protects cristae architecture of endothelial mitochondria during ischemia, maintains endothelial viability after ischemia, and prevents inflammation and fibrosis 4 weeks later.64
Renal artery stenosis is a major cause of hypertension and chronic kidney disease. Revascularization by angioplasty can successfully lower blood pressure but does not necessarily improve renal blood flow or glomerular filtration rate. Correction of renal artery stenosis does not correct microvascular remodeling in the kidney.67 In an animal model of renal artery stenosis, a single dose of MTP-131 at the time of angioplasty restored cortical microvasculature 4 weeks later, in addition to improving renal blood flow and glomerular filtration rate.67 This was accompanied by protection of mitochondrial biogenesis and significant mitigation of apoptosis, oxidative stress, interstitial inflammation, and fibrosis.67 A recent study has now shown that renal dysfunction secondary to renal artery stenosis is associated with loss of cardiolipin.68 Treatment with MTP-131 for 4 weeks in this model, without revascularization, managed to restore cardiolipin content and improve vascular density, tissue oxygenation, renal blood flow, and glomerular filtration rate. Oxidative stress and fibrosis were also ameliorated. These studies implicate cardiolipin loss and mitochondrial damage in postischemia tissue remodeling and position the mitochondria as a central therapeutic target.
Neurodegenerative Diseases
Mitochondrial impairment and oxidative stress are intimately involved in the pathogenesis of neurodegenerative diseases. Age-associated loss of mitochondrial plasticity may play a role in Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis. Synaptic mitochondria and ATP production are critical for proper synaptic function. It has been estimated that half of the ATP within neurons is used for maintenance of ionic gradients and another 30% is dedicated to synaptic transmission. In the transgenic amyloid-β protein precursor mouse model of Alzheimer's disease, SS-31 reduced broken cristae in mitochondria and restored both axonal transport of mitochondria and synaptic viability.69,70 These studies suggest that, by these actions, SS-31 can provide adequate ATP supply at the synapse to improve neuronal function.
In a model of Parkinson's disease caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, both SS-31 and SS-20 prevented loss of dopamine neurons in the substantia nigra and preserved dopamine levels in the striatum.71 Studies with isolated mitochondria showed that both peptides prevented MPP+-induced inhibition of mitochondrial respiration and ATP production, as well as mitochondrial swelling. In the G93A superoxide dismutase-1 (SOD1) mutant mouse, a model for amyotrophic lateral sclerosis, SS-31 treatment delayed the onset of resting tremors, improved rotarod performance, reduced motor neuron cell loss, and prolonged survival.72
Protection against Herbicide- and Drug-Induced Mitochondrial Toxicity
In line with their origin from bacteria, mitochondria are susceptible to a number of herbicides and pesticides. Picloram and triclopyr, two of the most commonly used herbicides in the world, are known to have severe toxic effects in humans. SS-31 prevented inhibition of mitochondrial respiration and protected primary neurons from the toxicity of picloram and triclopyr.73
Many therapeutic drugs can cause mitochondrial toxicity, and some have been withdrawn from the market. Doxorubicin is one of the most effective antineoplastic agents, but its cumulative cardiac toxicity restrains its broad clinical use. Doxorubicin can undergo one-electron reduction at complex I and generate superoxide, but it is also known to interact with cardiolipin and can promote cardiolipin peroxidation. SS-31 has been reported to reduce the increase in cytosolic ROS and prevent catabolism in myotubes treated with doxorubicin.74 Acute and chronic neuropathy is observed with the use of oxaliplatin and other platinum-based chemotherapeutics. Concomitant use of SS-31 with oxaliplatin prevented hypersensitivity to both cold and mechanical pain, and this was associated with reduced oxidative stress in the dorsal root ganglion.75 In addition, iodinated contrast agents such as diatrizoate are the third most important cause of acute kidney injury. In a recent report, both SS-31 and SS-20 significantly reduced oxidative stress and renal injury in rats given diatrizoate.76 These studies highlight the number of drug-induced toxicities that appear to be mediated by mitochondrial impairment and suggest that SS-31 can significantly protect against such toxicities caused by diverse chemical compounds.
SS Peptides Promote Mitochondrial Plasticity
It has been recognized only recently that aging and many complex diseases are characterized by impaired mitochondrial plasticity, in that mitochondria are unable to increase P/O coupling to meet metabolic demands. Mitochondrial plasticity plays an important role in both conditions of “famine” and “feast.” When substrates and O2 supply are low, an increase in P/O coupling provides more ATP per O2 consumed. On the other hand, in the setting of low energy demand, e.g., a sedentary lifestyle, a metabolic overload stress on the mitochondria would result in excess electron leak unless the mitochondria are able to simultaneously increase efficiency of electron transport coupled to ATP synthesis. Such an increase in mitochondrial capacity may act as a buffer to reduce this metabolic stress and maintain insulin sensitivity. The SS peptides can increase mitochondrial capacity and improve P/O coupling, thereby producing more ATP and reducing ROS formation. ADP-stimulated O2 consumption depends on substrate concentration when it is low but is saturated at higher substrate concentrations. Mitochondrial plasticity may be represented as an increase in state 3 respiration as a function of substrate concentration, with an upward shift of the normal curve and a greater than fourfold decrease in Km (Figure 3). The effect of SS peptides on state 3 respiration is much more pronounced at low substrate concentrations, and this may explain why SS-31 had no effect on skeletal muscle P/O ratios in young mice but was able to restore mitochondrial capacity and coupling in aged mice.38 An improved rate of electron transfer, together with increased P/O coupling, would reduce ROS generation, and this may contribute to the ability of SS-31 to mitigate high ROS production in rats fed a high-fat diet.47
Although increased mitochondrial biogenesis may improve mitochondrial plasticity in the long term, it cannot explain the effects that are observed with acute use of SS-31. The ability of a single dose of SS-31 to improve maximal ATP synthesis in aged skeletal muscles suggests that SS-31 must have a direct effect on the ETC. We believe that this is accomplished by promoting electron transfer at the rate-limiting step of the ETC, where cytc must transfer electrons efficiently from complex III to complex IV in order to prevent electron leak at complex III. For cytc to transfer electrons efficiently, it needs to be in close proximity to the two respiratory complexes, and this is accomplished by its affinity for cardiolipin. Under conditions of low ATP, as seen in ischemia, cytc tends to be hydrophobically bound to cardiolipin, and this inhibits its ability to accept electrons. By modulating the interaction between cardiolipin and the heme iron of cytc, the SS peptides promote π-π* interaction and electron transfer.34 This allows more ATP production per available O2 and thereby increases the ischemia time that can be tolerated before cell death occurs. These peptides also inhibit cytc peroxidase activity by protecting the reactive heme iron from H2O2, thus preventing cardiolipin peroxidation and destruction of cristae membranes.
Clinical Development of SS-31
In addition to the very promising preclinical results summarized herein, SS-31 has excellent “drug-like” properties and a promising safety profile. The absorption, distribution, metabolism, and excretion profile and the pharmacokinetics of SS-31 have been reported in preclinical studies.15,32 Being a very water-soluble compound, SS-31 has a very low apparent volume of distribution. Distribution to the major organs—kidney, heart, liver, skeletal muscle, and lung—occurred within 30 min after s.c. administration, with the highest concentration found in the kidney and little distribution to the adipose tissue.16,38 Plasma levels and total body exposure, as measured by the area under the concentration–time curve, are dose proportional within the dose range used in preclinical efficacy studies.15 In the isolated perfused heart, ∼25% of the dose was extracted per minute.62 When applied topically to the eyes of rabbits, the concentration of SS-31 is very high in the anterior chamber (conjunctiva, cornea, and sclera) and then is distributed via periocular penetration to the retina, with very low levels in the vitreous humor (Szeto HH., unpublished data). SS-31 undergoes some hydrolysis starting at the carboxyl terminus to form tripeptide and dipeptide fragments. Both the parent drug and its metabolites are excreted entirely by the kidneys within 48 h, and their clearances are directly proportional to creatinine clearance. The elimination half-life of ∼2 h in rats, dogs, and monkeys is sufficient for once-daily dosing to achieve pharmacological efficacy in these species.
SS-31 entered into clinical development with a commercial sponsor (Stealth Peptides, Newton, MA) in 2010 using the acetate salt form (MTP-131). Several phase I studies have assessed its safety, tolerability, and pharmacokinetics in healthy male and female subjects after i.v. and p.o. dosing. The i.v. formulation was well tolerated when infused over a wide dose range (0.01– 0.25 mg/kg/h), achieving potentially effective plasma drug levels even with the lowest dose and demonstrating predictable linear pharmacokinetics. The pharmacokinetic results from humans were compatible with the pharmacokinetics from several animal models, with an elimination half-life of ∼4 h and a very small apparent volume of distribution. The p.o. formulation provided plasma concentrations known to be cardioprotective in preclinicalcal studies and was also well tolerated.
On the basis of the preclinical effectiveness of SS-31and MTP-131 for reducing myocardial ischemia–reperfusion injury,61,62,77 the first multinational clinical phase II trial was focused on ischemia–reperfusion injury and microvascular injuries in patients experiencing acute ST-segment elevation myocardial infarction (STEMI). The rationale and design of the EMBRACE-STEMI trial has been published.78 This is a randomized, double-blind, placebo-controlled trial enrolling patients with first-time anterior STEMI undergoing primary percutaneous coronary intervention. Patients are randomized to receive MTP-131 at 0.05 mg/kg/h or placebo as an i.v. infusion. This dose is based on the pharmacokinetic/pharmacodynamic relationship in several animal models and human pharmacokinetic data from phase I studies.78 The primary end point is infarct size as measured by creatine kinase release and cardiac magnetic resonance imaging with gadolinium enhancement. The EMBRACE-STEMI trial, begun in June 2012, enrolled more than 300 patients across 40 sites within the United States and Europe (NCT01572909). Enrollment has been completed, and the results of this trial are forthcoming.
A second phase II study involves the treatment of acute kidney injury and renal microvascular dysfunction in hypertension. This study is based on the demonstrated effectiveness of MTP-131 in improving renal microvascular blood flow and glomerular filtration rate after angioplasty in pigs with atherosclerotic renal artery stenosis.67 Previous studies have shown that angioplasty alone fails to reverse structural and functional deterioration in stenotic kidneys.79 This clinical study is also supported by other animal studies showing the effectiveness of MTP-131 in preventing acute ischemic kidney injury and in mitigating microvascular rarefaction.16,63,64 The phase II clinical study is intended to assess improvement of renal function in patients after angioplasty for severe unilateral renal artery stenosis. This is a randomized, placebo-controlled, single-center study, and patients will receive either MTP-131 (0.05 mg/kg//h) or saline for a maximum duration of 4 h (NCT01755858). The primary outcome measure is glomerular filtration rate at 8 weeks after angioplasty. Secondary outcome measures include renal volume, regional renal blood flow, renal oxygenation, and a number of inflammatory and oxidative biomarkers.
Other phase II studies that are scheduled to begin in 2014 include one examining acute treatment of heart failure, which is based on preclinical studies described earlier in dogs with severe postischemic heart failure.55 In another study, an eye drop formulation of MTP-131 (Ocuvia) will be used to treat diabetic macular edema. This trial is based on the ability of MTP-131 to reach the retina after topical application and on the preclinical results obtained in diabetic mice showing mitochondrial protection in the retinal pigment epithelium and restoration of visual function.
SS-31 represents the first of a class of new chemical entities that selectively target mitochondrial cardiolipin to improve mitochondrial plasticity and restore optimal bioenergetics.32 These compounds provide an entirely novel approach to the treatment of complex diseases that at first glance appear to be totally unrelated. Common to all of them, however, is the loss of cellular energy that accounts for their failure to function properly, and these peptides act by recharging the powerhouse of all cells. Importantly, they have no effect on normal mitochondria, which accounts for their excellent safety profile. SS-31 has been studied extensively in preclinical disease models by many independent investigators, and together we have found that restoring mitochondrial bioenergetics can improve these diverse clinical disorders. The ongoing clinical studies will allow this idea to be validated and hopefully bring relief to patients suffering from these chronic diseases with unmet needs.
Reflections
Serendipity played a central role in the initial discovery of the SS peptides. This platform would never have been discovered by rational design because we simply do not know enough about the targets for improving mitochondrial plasticity or the structural requirements for mitochondria targeting. This chance discovery was followed by rational peptide design rather than high-throughput screening of chemical libraries. Rational design was involved in the design of peptide analogs that have suitable chemical, pharmacokinetic, and pharmacodynamic properties. For many years, although numerous pharmacological studies demonstrated the efficacy of these peptides in protecting mitochondrial structure and function, the “target” of these peptides remained elusive. Even as these compounds went into clinical development, their mechanism of action was not entirely understood.
The ETC on the IMM is one of the most difficult targets for drug development. Targeting any one protein in this highly elaborate electron transfer system is unlikely to improve ATP production. SS-31 and its analogs have shown that it is possible to influence OXPHOS outcome by targeting cardiolipin that serves as a structural platform for organizing the respiratory complexes into supercomplexes for highly efficient electron transfer. Furthermore, these compounds have demonstrated that they can interfere with the interaction between cardiolipin and the heme iron of the Janus-faced cytc to favor enhancement of the electron carrier function as opposed to peroxidase generation.
The numerous studies demonstrating the effectiveness of these peptides in remarkably diverse animal disease models, and in famine or feast, support the hypothesis that these peptides act by promoting mitochondrial plasticity. With insufficient substrates and/or O2 during famine, these peptides can increase mitochondrial respiration and ATP production, and prolong survival. With excess substrates during feast, they can promote OXPHOS coupling, reduce electron leak, and prevent ROS production. By selectively targeting the IMM—and by promoting mitochondrial plasticity rather than activating/inhibiting a specific target—these peptides have no effect on normal healthy mitochondria and have great safety profiles. Long-term treatment for 8 weeks revealed no safety issues in pigs,67,68,80 and no adverse effects were reported after 8 months of daily treatment in mice (Alam et al., personal communication).
These amino acid–based compounds have surprising drug-like properties, making us question many of the old dogmas about drug design. Although peptides are generally considered poor drug candidates, SS-31 shows many advantages relative to other small molecules, in terms of (i) the ease of synthesis and formulation and (ii) highly favorable linear pharmacokinetics. The most unusual aspect is their ability to cross the blood–brain barrier and be efficacious against central nervous system disorders. We have learned from structural activity studies that their alternating aromatic–cationic motif (see Table 1) is what allows them to permeate cell membranes because the aromatic rings provide an electronic cage for the cautions. Most peptide designs have tended toward increasing lipophilicity, but they have not been particularly successful. Our experience is certainly not the first reminder that serendipity can triumph over rational design when it comes to developing drugs.
Acknowledgments
The authors' research was supported by the National Institutes of Health (DA-08924, NS-048295, AG-35844, HL-101186, AG-0001751, and AG-042637) and the Research Program in Mitochondrial Therapeutics at the Weill Cornell Medical College. We are most grateful to all of our collaborators who have contributed so much to the understanding of the SS peptides. Special thanks go to the following members of the Szeto laboratory for their creativity and devotion to the study of SS peptides: Shaoyi Liu, Yi Soong, William C. Mills, and Wesley Chao.
Footnotes
Conflict of Interest: The SS peptides described in this article have been licensed for commercial research and development to Stealth Peptides, a clinical-stage biopharmaceutical company in which H.H.S., A.V.B., and the Cornell Research Foundation have financial interests. H.H.S. is the scientific founder of Stealth Peptides International and a consultant to Stealth Peptides. The Research Program in Mitochondrial Therapeutics was established at the Weill Cornell Medical College with a gift from Stealth Peptides International.
References
- 1.Wallace DC. A mitochondrial bioenergetic etiology of disease. J Clin Invest. 2013;123:1405–1412. doi: 10.1172/JCI61398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marcinek DJ, Schenkman KA, Ciesielski WA, Lee D, Conley KE. Reduced mitochondrial coupling in vivo alters cellular energetics in aged mouse skeletal muscle. J Physiol. 2005;569:467–473. doi: 10.1113/jphysiol.2005.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conley KE, Jubrias SA, Cress ME, Esselman P. Exercise efficiency is reduced by mitochondrial uncoupling in the elderly. Exp Physiol. 2013;98:768–777. doi: 10.1113/expphysiol.2012.067314. [DOI] [PubMed] [Google Scholar]
- 4.Szendroedi J, Phielix E, Roden M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol. 2012;8:92–103. doi: 10.1038/nrendo.2011.138. [DOI] [PubMed] [Google Scholar]
- 5.Rosca MG, Tandler B, Hoppel CL. Mitochondria in cardiac hypertrophy and heart failure. J Mol Cell Cardiol. 2013;55:31–41. doi: 10.1016/j.yjmcc.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barot M, Gokulgandhi MR, Mitra AK. Mitochondrial dysfunction in retinal diseases. Curr Eye Res. 2011;36:1069–1077. doi: 10.3109/02713683.2011.607536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Che R, Yuan Y, Huang S, Zhang A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am J Physiol Renal Physiol. 2014;306:F367–F378. doi: 10.1152/ajprenal.00571.2013. [DOI] [PubMed] [Google Scholar]
- 8.Dykens JA, Will Y. The significance of mitochondrial toxicity testing in drug development. Drug Discov Today. 2007;12:777–785. doi: 10.1016/j.drudis.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 9.Hersh SP. Mitochondria: an emerging target for therapeutics. Clin Pharmacol Ther. 2010;87:630–632. doi: 10.1038/clpt.2010.22. [DOI] [PubMed] [Google Scholar]
- 10.Shimoyama M, Shimoyama N, Zhao GM, Schiller PW, Szeto HH. Antinociceptive and respiratory effects of intrathecal H-Tyr-D-Arg-Phe-Lys-NH2 (DALDA) and [Dmt1] DALDA. J Pharmacol Exp Ther. 2001;297:364–371. [PubMed] [Google Scholar]
- 11.Zhao GM, et al. Profound spinal tolerance after repeated exposure to a highly selective mu-opioid peptide agonist: role of delta-opioid receptors. J Pharmacol Exp Ther. 2002;302:188–196. doi: 10.1124/jpet.302.1.188. [DOI] [PubMed] [Google Scholar]
- 12.Zhao K, Luo G, Zhao GM, Schiller PW, Szeto HH. Transcellular transport of a highly polar 3+ net charge opioid tetrapeptide. J Pharmacol Exp Ther. 2003;304:425–432. doi: 10.1124/jpet.102.040147. [DOI] [PubMed] [Google Scholar]
- 13.Szeto HH, Schiller PW, Zhao K, Luo G. Fluorescent dyes alter intracellular targeting and function of cell-penetrating tetrapeptides. FASEB J. 2005;19:118–120. doi: 10.1096/fj.04-1982fje. [DOI] [PubMed] [Google Scholar]
- 14.Zhao K, et al. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem. 2004;279:34682–34690. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- 15.Szeto HH, Schiller PW. Novel therapies targeting inner mitochondrial membrane–from discovery to clinical development. Pharm Res. 2011;28:2669–2679. doi: 10.1007/s11095-011-0476-8. [DOI] [PubMed] [Google Scholar]
- 16.Birk AV, et al. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol. 2013;24:1250–1261. doi: 10.1681/ASN.2012121216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Osman C, Voelker DR, Langer T. Making heads or tails of phospholipids in mitochondria. J Cell Biol. 2011;192:7–16. doi: 10.1083/jcb.201006159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Birk AV, Chao WM, Bracken C, Warren JD, Szeto HH. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol. 2014;171:2017–2028. doi: 10.1111/bph.12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schlame M, Ren M. The role of cardiolipin in the structural organization of mitochondrial membranes. Biochim Biophys Acta. 2009;1788:2080–2083. doi: 10.1016/j.bbamem.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bazán S, Mileykovskaya E, Mallampalli VK, Heacock P, Sparagna GC, Dowhan W. Cardiolipin-dependent reconstitution of respiratory supercomplexes from purified Saccharomyces cerevisiae complexes III and IV. J Biol Chem. 2013;288:401–411. doi: 10.1074/jbc.M112.425876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cordes M, Giese B. Electron transfer in peptides and proteins. Chem Soc Rev. 2009;38:892–901. doi: 10.1039/b805743p. [DOI] [PubMed] [Google Scholar]
- 22.Arnarez C, Marrink SJ, Periole X. Identification of cardiolipin binding sites on cytochrome c oxidase at the entrance of proton channels. Sci Rep. 2013;3:1263. doi: 10.1038/srep01263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paradies G, Ruggiero FM, Petrosillo G, Quagliariello E. Age-dependent decline in the cytochrome c oxidase activity in rat heart mitochondria: role of cardiolipin. FEBS Lett. 1997;406:136–138. doi: 10.1016/s0014-5793(97)00264-0. [DOI] [PubMed] [Google Scholar]
- 24.Lesnefsky EJ, et al. Aging decreases electron transport complex III activity in heart interfibrillar mitochondria by alteration of the cytochrome c binding site. J Mol Cell Cardiol. 2001;33:37–47. doi: 10.1006/jmcc.2000.1273. [DOI] [PubMed] [Google Scholar]
- 25.Petrosillo G, Matera M, Casanova G, Ruggiero FM, Paradies G. Mitochondrial dysfunction in rat brain with aging Involvement of complex I, reactive oxygen species and cardiolipin. Neurochem Int. 2008;53:126–131. doi: 10.1016/j.neuint.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 26.Han X, Yang J, Yang K, Zhao Z, Abendschein DR, Gross RW. Alterations in myocardial cardiolipin content and composition occur at the very earliest stages of diabetes: a shotgun lipidomics study. Biochemistry. 2007;46:6417–6428. doi: 10.1021/bi7004015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Julienne CM, et al. Cardiolipin content is involved in liver mitochondrial energy wasting associated with cancer-induced cachexia without the involvement of adenine nucleotide translocase. Biochim Biophys Acta. 2014;1842:726–733. doi: 10.1016/j.bbadis.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 28.Ji J, et al. Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nat Neurosci. 2012;15:1407–1413. doi: 10.1038/nn.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sparagna GC, et al. Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. J Lipid Res. 2007;48:1559–1570. doi: 10.1194/jlr.M600551-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Hanske J, Tofey JR, Morenz AM, Bonilla AJ, Schiavoni KH, Pletneva EV. Conformational properties of cardiolipin-bound cytochrome c. Proc Natl Acad Sci USA. 2012;109:125–130. doi: 10.1073/pnas.1112312108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Basova LV, et al. Cardiolipin switch in mitochondria: shutting of the reduction of cytochrome c and turning on the peroxidase activity. Biochemistry. 2007;46:3423–3434. doi: 10.1021/bi061854k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol. 2014;171:2029–2050. doi: 10.1111/bph.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonzalvez F, Gottlieb E. Cardiolipin: setting the beat of apoptosis. Apoptosis. 2007;12:877–885. doi: 10.1007/s10495-007-0718-8. [DOI] [PubMed] [Google Scholar]
- 34.Birk AV, Chao WM, Bracken C, Warren JD, Szeto HH. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol. 2014;171:2017–2028. doi: 10.1111/bph.12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szeto HH, Liu S, Soong Y, Birk AV. Improving mitochondrial bioenergetics under ischemic conditions increases warm ischemia tolerance in the kidney. Am J Physiol Heart Renal Physiol. doi: 10.1152/ajprenal.00366.2014. in press. [DOI] [PubMed] [Google Scholar]
- 36.Rolfe DF, Brand MD. The physiological significance of mitochondrial proton leak in animal cells and tissues. Biosci Rep. 1997;17:9–16. doi: 10.1023/a:1027327015957. [DOI] [PubMed] [Google Scholar]
- 37.Porter RK, Hulbert AJ, Brand MD. Allometry of mitochondrial proton leak: influence of membrane surface area and fatty acid composition. Am J Physiol. 1996;271:R1550–R1560. doi: 10.1152/ajpregu.1996.271.6.R1550. [DOI] [PubMed] [Google Scholar]
- 38.Siegel MP, et al. Mitochondrial-targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell. 2013;12:763–771. doi: 10.1111/acel.12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Min K, Smuder AJ, Kwon OS, Kavazis AN, Szeto HH, Powers SK. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J Appl Physiol (1985) 2011;111:1459–1466. doi: 10.1152/japplphysiol.00591.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Talbert EE, Smuder AJ, Min K, Kwon OS, Szeto HH, Powers SK. Immobilization-induced activation of key proteolytic systems in skeletal muscles is prevented by a mitochondria-targeted antioxidant. J Appl Physiol (1985) 2013;115:529–538. doi: 10.1152/japplphysiol.00471.2013. [DOI] [PubMed] [Google Scholar]
- 41.Powers SK, et al. Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit Care Med. 2011;39:1749–1759. doi: 10.1097/CCM.0b013e3182190b62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cree MG, et al. Human mitochondrial oxidative capacity is acutely impaired after burn trauma. Am J Surg. 2008;196:234–239. doi: 10.1016/j.amjsurg.2007.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee HY, Kaneki M, Andreas J, Tompkins RG, Martyn JA. Novel mitochondria-targeted antioxidant peptide ameliorates burn-induced apoptosis and endoplasmic reticulum stress in the skeletal muscle of mice. Shock. 2011;36:580–585. doi: 10.1097/SHK.0b013e3182366872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Righi V, et al. Mitochondria-targeted antioxidant promotes recovery of skeletal muscle mitochondrial function after burn trauma assessed by in vivo 31P nuclear magnetic resonance and electron paramagnetic resonance spectroscopy. FASEB J. 2013;27:2521–2530. doi: 10.1096/fj.12-220764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carter EA, et al. Evaluation of the antioxidant peptide SS31 for treatment of burn-induced insulin resistance. Int J Mol Med. 2011;28:589–594. doi: 10.3892/ijmm.2011.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goodpaster BH. Mitochondrial deficiency is associated with insulin resistance. Diabetes. 2013;62:1032–1035. doi: 10.2337/db12-1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anderson EJ, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009;119:573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee HY, et al. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab. 2010;12:668–674. doi: 10.1016/j.cmet.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kondo T, Kahn CR. Altered insulin signaling in retinal tissue in diabetic states. J Biol Chem. 2004;279:37997–38006. doi: 10.1074/jbc.M401339200. [DOI] [PubMed] [Google Scholar]
- 50.Beer M, et al. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J Am Coll Cardiol. 2002;40:1267–1274. doi: 10.1016/s0735-1097(02)02160-5. [DOI] [PubMed] [Google Scholar]
- 51.Sabbah HN, Sharov V, Riddle JM, Kono T, Lesch M, Goldstein S. Mitochondrial abnormalities in myocardium of dogs with chronic heart failure. J Mol Cell Cardiol. 1992;24:1333–1347. doi: 10.1016/0022-2828(92)93098-5. [DOI] [PubMed] [Google Scholar]
- 52.Karamanlidis G, Bautista-Hernandez V, Fynn-Thompson F, Del Nido P, Tian R. Impaired mitochondrial biogenesis precedes heart failure in right ventricular hypertrophy in congenital heart disease. Circ Heart Fail. 2011;4:707–713. doi: 10.1161/CIRCHEARTFAILURE.111.961474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dai DF, et al. Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol. 2011;58:73–82. doi: 10.1016/j.jacc.2010.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dai DF, et al. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ Heart Fail. 2013;6:1067–1076. doi: 10.1161/CIRCHEARTFAILURE.113.000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sabbah HN, Wang M, Zhang K, Gupta RC, Rastogi S. Acute intravenous infusion of Bendavia (MTP-131), a novel mitochondria-targeting peptide, improves left ventricular systolic function in dogs with advanced heart failure. Circulation. 2012;126:A15385. [Google Scholar]
- 56.Sabbah HN, Wang M, Zhang K, Gupta RC, Rastogi S. Long-term therapy with Bendavia (MTP-131), a novel mitochondria-targeting peptide, increases myocardial ATP synthesis and improves left ventricular systolic function in dogs with chronic heart failure. J Am Coll Cardiol. 2013;61:E709. [Google Scholar]
- 57.Sabbah HN, Gupta RC, Rastogi S, Wang M, Zhang K. Long-term therapy with Bendavia (MTP-131), a novel mitochondria-targeting peptide, normalizes functional mitochondrial abnormalities in left ventricular myocardium of dogs with heart failure. Mitochondrion. 2013;13:912. [Google Scholar]
- 58.Sabbah HN, et al. Bendavia (MTP-131), a movel mitochondria-targeting peptide, normalizes expression of cardiolipin remodeling genes and proteins in left ventricular myocardium of dogs with advanced heart failure. J Am Coll Cardiol. 2014;63:A758. [Google Scholar]
- 59.Cho S, Szeto HH, Kim E, Kim H, Tolhurst AT, Pinto JT. A novel cell-permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down-regulating CD36. J Biol Chem. 2007;282:4634–4642. doi: 10.1074/jbc.M609388200. [DOI] [PubMed] [Google Scholar]
- 60.Kim E, Tolhurst AT, Szeto HH, Cho S. Targeting CD36-mediated inflammation reduces acute brain injury in transient, but not permanent, ischemic stroke. CNS Neurosci Ther. 2014 Sep 12; doi: 10.1111/cns.12326. e-pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cho J, et al. Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coron Artery Dis. 2007;18:215–220. doi: 10.1097/01.mca.0000236285.71683.b6. [DOI] [PubMed] [Google Scholar]
- 62.Kloner RA, et al. Reduction of ischemia/reperfusion injury with bendavia, a mitochondria-targeting cytoprotective Peptide. J Am Heart Assoc. 2012;1:e001644. doi: 10.1161/JAHA.112.001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Szeto HH, et al. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol. 2011;22:1041–1052. doi: 10.1681/ASN.2010080808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu S, Soong Y, Seshan SV, Szeto HH. Novel cardiolipin therapeutic protects endothelial mitochondria during renal ischemia and mitigates microvascular rarefaction, inflammation, and fibrosis. Am J Physiol Renal Physiol. 2014;306:F970–F980. doi: 10.1152/ajprenal.00697.2013. [DOI] [PubMed] [Google Scholar]
- 65.Eardley KS, et al. The role of capillary density, macrophage infiltrations and interstitial scarring in the pathogenesis of human chronic kidney disease. Kidney Int. 2008;74:495–504. doi: 10.1038/ki.2008.183. [DOI] [PubMed] [Google Scholar]
- 66.Sutton TA, Fisher CJ, Molitoris BA. Microvascular endothelial injury and dysfunction during ischemic acute renal failure. Kidney Int. 2002;62:1539–1549. doi: 10.1046/j.1523-1755.2002.00631.x. [DOI] [PubMed] [Google Scholar]
- 67.Eirin A, et al. A mitochondrial permeability transition pore inhibitor improves renal outcomes after revascularization in experimental atherosclerotic renal artery stenosis. Hypertension. 2012;60:1242–1249. doi: 10.1161/HYPERTENSIONAHA.112.199919. [DOI] [PubMed] [Google Scholar]
- 68.Eirin A, et al. Mitochondrial protection restores renal function in swine atherosclerotic renovascular disease. Cardiovasc Res. 2014;103:461–472. doi: 10.1093/cvr/cvu157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Manczak M, et al. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer's disease neurons. J Alzheimers Dis. 2010;20(suppl 2):S609–S631. doi: 10.3233/JAD-2010-100564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum Mol Genet. 2011;20:4515–4529. doi: 10.1093/hmg/ddr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang L, Zhao K, Calingasan NY, Luo G, Szeto HH, Beal MF. Mitochondria targeted peptides protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Antioxid Redox Signal. 2009;11:2095–2104. doi: 10.1089/ars.2009.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Petri S, et al. Cell-permeable peptide antioxidants as a novel therapeutic approach in a mouse model of amyotrophic lateral sclerosis. J Neurochem. 2006;98:1141–1148. doi: 10.1111/j.1471-4159.2006.04018.x. [DOI] [PubMed] [Google Scholar]
- 73.Reddy TP, et al. Toxicity of neurons treated with herbicides and neuroprotection by mitochondria-targeted antioxidant SS31. Int J Environ Res Public Health. 2011;8:203–221. doi: 10.3390/ijerph8010203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gilliam LA, et al. Doxorubicin acts via mitochondrial ROS to stimulate catabolism in C2C12 myotubes. Am J Physiol Cell Physiol. 2012;302:C195–C202. doi: 10.1152/ajpcell.00217.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Toyama S, Shimoyama N, Ishida Y, Koyasu T, Szeto HH, Shimoyama M. Characterization of acute and chronic neuropathies induced by oxaliplatin in mice and differential effects of a novel mitochondria-targeted antioxidant on the neuropathies. Anesthesiology. 2014;120:459–473. doi: 10.1097/01.anes.0000435634.34709.65. [DOI] [PubMed] [Google Scholar]
- 76.Duan SB, et al. Mitochondria-targeted peptides prevent on contrast-induced acute kidney injury in the rats with hypercholesterolemia. Ren Fail. 2013;35:1124–1129. doi: 10.3109/0886022X.2013.815107. [DOI] [PubMed] [Google Scholar]
- 77.Sloan RC, et al. Mitochondrial permeability transition in the diabetic heart: contributions of thiol redox state and mitochondrial calcium to augmented reperfusion injury. J Mol Cell Cardiol. 2012;52:1009–1018. doi: 10.1016/j.yjmcc.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 78.Chakrabarti AK, et al. Rationale and design of the EMBRACE STEMI study: a phase 2a, randomized, double-blind, placebo-controlled trial to evaluate the safety, tolerability and efficacy of intravenous Bendavia on reperfusion injury in patients treated with standard therapy including primary percutaneous coronary intervention and stenting for ST-segment elevation myocardial infarction. Am Heart J. 2013;165:509–514.e7. doi: 10.1016/j.ahj.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 79.Eirin A, et al. Persistent kidney dysfunction in swine renal artery stenosis correlates with outer cortical microvascular remodeling. Am J Physiol Renal Physiol. 2011;300:F1394–F1401. doi: 10.1152/ajprenal.00697.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Eirin A, et al. Mitochondrial targeted peptides attenuate residual myocardial damage after reversal of experimental renovascular hypertension. J Hypertens. 2014;32:154–165. doi: 10.1097/HJH.0b013e3283658a53. [DOI] [PMC free article] [PubMed] [Google Scholar]