

Abstract
Distinguishing between allostery and competition among modulating ligands is challenging for large target molecules. Of practical necessity, inferences are often drawn from in vitro assays on target fragments, but such inferences may belie actual mechanisms. One key example of such ambiguity concerns calcium-binding proteins (CaBPs) that tune signaling molecules regulated by calmodulin (CaM). Since CaBPs resemble CaM, CaBPs are believed to competitively replace CaM on targets. Yet, brain CaM expression far surpasses that of CaBPs, so how can CaBPs exert appreciable biological actions? Here, we devise a live-cell, holomolecule approach that reveals an allosteric mechanism for calcium channels, whose CaM-mediated inactivation is eliminated by CaBP4. Our strategy is to covalently link CaM and/or CaBP to holochannels, enabling live-cell FRET assays to resolve a cyclical allosteric binding scheme for CaM and CaBP4 to channels, thus explaining how trace CaBPs prevail. This approach may apply generally for discerning allostery in live cells.
Distinct modulatory ligands frequently act upon common target molecules1-3, supporting molecular computations important to biological signaling networks3,4. These ligands may act by competition wherein only one ligand can bind at a time, or by allostery allowing simultaneous interaction5. This contrast in classic biochemical mechanism holds crucial implications for signaling behavior, but can be notoriously difficult to establish6. This is particularly true for large target molecules resistant to reconstitution outside of live cells. If studied piecemeal as subdomain peptides, such targets often exhibit multiple potential ligand binding sites, but the relevance of these within the holomolecule often remains uncertain7.
One prime example of such mechanistic ambiguity concerns the interaction of voltage-gated Ca2+ channels (CaV) with two modulatory ligands: calmodulin (CaM) and calcium binding proteins (CaBPs). To start, CaM regulation of CaV channels plays a major role in the intracellular Ca2+ feedback orchestrating many biological processes8. This modulation frequently manifests as a Ca2+-dependent inactivation (CDI) of channel opening9,10, important for Ca2+ homeostasis11. Additionally, exciting recent discoveries identify Ca2+-binding proteins (CaBPs), a family of CaM-like brain molecules12-17 that may also bind CaV channels and other targets18,19. CaBPs are bi-lobed like CaM, with each lobe containing two EF-hand Ca2+ binding motifs12. However, while all four EF hands bind Ca2+ in CaM, one of them is non-functional in CaBPs. Coexpressing CaBPs with CaV1 channels then eliminates their CDI14,20. Indeed, modulation like this diversifies biological responses to Ca2+ signals13,14,18,21.
Based on the similarity of CaM and CaBPs, the prevailing hypothesis is that CaBPs competitively replace CaM on channels14,22,23. Indeed, these ligands exhibit competitive binding to an isolated channel IQ domain (Fig. 1a, blue oval), a principal element for CDI. The interaction of CaM and CaBPs may thus be formulated as in Fig. 1b. State 1 portrays an ‘empty’ channel without ligand and thus incapable of CaM-mediated Ca2+ regulation24,25. This empty channel may bind CaM at the IQ domain to form a complex (state 2) that can then undergo Ca2+ regulation (e.g., inactivation). Alternatively, CaBP may bind at the IQ domain, yielding a state 3 with altered Ca2+ regulatory behavior (e.g., non-inactivatable). Importantly, both CaM and CaBP cannot simultaneously bind (state 4 excluded) under competition. Hypothetical dispositions of CaBP and CaM are cartooned in Fig. 1b.
Figure 1. Potential mechanisms of CaM and CaBP interaction with calcium channels.

(a) Calcium channel landmarks. Ca2+-inactivating (CI) region (∼160 aa of proximal carboxy terminus), containing elements involved in CaM regulation. IQ domain (blue oval), harbors apoCaM pre-association and proposed site of CaBP interaction. Vestigial EF hands (squares). NSCaTE element (brown oval), N-lobe Ca2+/CaM effector site. (b) Potential mechanisms of CaM and CaBP interaction. Prevailing competitive hypothesis restricts model to states 1-3, and excludes state 4. State 1, channel lacking both CaM and CaBP. State 2, channel with single apoCaM bound (black circle), primarily to IQ domain. State 3, channel with single CaBP bound (orange square). Under competition, CaBP would occupy same site as CaM (i.e., IQ domain), allowing channel binding to either ligand, but not both. Under allostery, CaBP binds to alternate site (state 3, dashed outline square), allowing simultaneous binding of CaM and CaBP (state 4). Existence of state 4 is unknown. Far right and left, hypothetical interaction of CaBP (left) and CaM (right) with IQ element under competition. (c) Relative transcript count (RPKM, Reads Per Kilobase Million) of CaM, CaBP4, and CaBP1, obtained by deep sequencing of transcriptomes from adult mouse neocortex. (d) Serial analysis of gene expression (SAGE) reveals transcript levels from adult mouse retina. CaM expression ∼15× CaBP4, and ∼70× CaBP1. (e) Deep sequencing of transcriptomes from human prefrontal cortex. CaM transcripts ∼8× CaBP1, and ∼6000× CaBP4.(f-h) In-situ hybridization mouse brain images from Allen Brain Atlas, normalized by scaling neocortical region to match RPKM signals in c.
As with many large molecules, studies of isolated channel peptides raise the possibility of more complex scenarios. Specifically, peptide assays hint at multiple potential binding sites for CaM and CaBPs. Thus, these two ligands need not compete at the IQ domain, and the possibility of an allosteric mechanism arises (i.e., Fig. 1b, state 4). For CaM regulation itself, interaction sites extend beyond the IQ domain (Fig. 1a, blue oval). In fact, CaM regulation starts with a single apoCaM (Ca2+-free CaM) preassociated, with the C-lobe of apoCaM embracing the IQ domain24-27 (Fig. 1a, blue oval), and the N-lobe engaging upstream vestigial EF-hands24 (Fig. 1a, squares), all within an encompassing CI region9,28,29. Ca2+ binding to this ‘resident’ CaM alters channel opening by driving rearrangements that may include the binding of the N-lobe of Ca2+-bound CaM (Ca2+/CaM) to the channel amino terminus30 (Fig. 1a, tan oval). Regarding CaBPs, peptide assays also hint at interactions beyond the IQ domain31,32, but the relevance of these assays to holochannels remains unsettled. One study argues for the functional impact of such sites32, while another argues otherwise31. In all, the scheme of CaM and CaBP interaction remains ambiguous (Fig. 1b), and clear tests for concurrent ligand binding to holochannels (i.e., state 4) have proven infeasible. This state of impasse is common to many large molecules33,34.
Importantly, the distinctions between competitive versus allosteric mechanism concern more than academic interest; they appear to be critical to the biological activity of CaBPs. In particular, the affinity of CaV channels for apoCaM may be greater than that for CaBPs14,25,26, as the present study will substantiate. Moreover, this study will highlight a predominance of CaM over CaBP expression throughout the brain. Under competititon, then, how could CaBPs exert functional effects? By contrast, an allosteric mechanism with the right mix of parameters would readily explain how naturally occuring CaBPs could still exert modulation.
Here, we establish an allosteric mechanism for CaV1.3 Ca2+ channels, a robust prototype system where strong CaM-mediated CDI can nonetheless be completely suppressed by CaBPs. Our approach is to exploit covalent linkage of holochannels to CaM and/or CaBP, along with live-cell FRET binding assays with intact holochannel complexes. In so doing, we fully resolve a cyclical allosteric binding scheme for CaM and CaBP4 to channels (i.e., Fig. 1b). The parameters we obtain explain how trace CaBP can functionally prevail over excess CaM. This approach may furnish a general means for discriminating allosteric mechanisms in the live-cell context.
Results
Predominance of CaM over CaBPs in the CNS
To motivate exploration of competitive versus allosteric mechanisms, we first considered the relative expression of CaM and CaBPs in the CNS. Given a competitive mechanism of CaBP modulation of Ca2+ channels, CaBP must at least approach the abundance of CaM to be functionally relevant, particularly if channels were to exhibit a greater affinity for apoCaM over CaBPs. In native systems, however, CaBP expression pales in comparison to that for CaM35, even for the more prominent CaBP1 and CaBP4 isoforms. Deep sequencing in mouse neocortex36 reveals CaBP1 transcripts to be ∼5-fold rarer than those for CaM; and CaBP4 transcripts 10,000-fold rarer (Fig. 1c). Similarly, SAGE analysis of whole mouse retina37 estimates a 1:15 ratio for CaBP4 to CaM transcripts, and an even smaller ratio for CaBP1 (Fig. 1d). Moreover, deep sequencing of human prefrontal cortex38 indicates a 1:8 ratio for CaBP1 to CaM transcripts, and a 1:6,000 ratio for CaBP4 (Fig. 1e). Although transcript abundance is not equivalent to protein expression, the enormous degree of transcript imbalance here would certainly yield protein expression mismatch39. Normalized in-situ hybridization profiles of mouse brain (Figs. 1f-h) underscore the predominance of CaM35, raising a challenge for competitive mechanisms, and prompting a search for potential allostery.
As a critical prelude to this search, we sought to resolve the functional contribution of ancillary CaBP binding sites outside the IQ domain, especially within the relevant configuration of the holochannel complex. This step would be a necessary ante for considering an allosteric scenario. Thus, we undertook a two-step sequence: first, to consolidate the baseline functional effects and binding properties of CaBP to CaV1.3 channels; and second, to validate the functional relevance of potential ancillary CaBP binding sites in chimeric Ca2+ channels.
CaBP modulation and binding of CaV1.3 channels
Figure 2a displays the baseline effects of CaBPs on CaV1.3 channels. We mainly focused on the rarer CaBP4 isoform, because it faces the larger competitive challenge. Absent CaBP, CaM-mediated CDI triggers strong decay of Ca2+ current14 when compared to Ba2+ (left subpanel). Ba2+ binds CaM poorly40 and thus furnishes a baseline reference without CDI. Upon strong overexpression of CaBP4, CDI is eliminated in exemplar traces14 (right subpanel). Population data in Fig. 2b fully confirms these trends, where the fraction of peak current remaining after 300-ms depolarization (r300) gauges steady-state inactivation. When plotted versus step potential, the r300 relation obtained with Ba2+ (black) is nearly flat, corroborating weak voltage-dependent inactivation. In control, the corresponding Ca2+ r300 relation exhibits a U shape (left subpanel, red), indicating genuine CDI9. The difference between these relations (f300, at 10-mV step depolarization) then quantifies CDI in isolation. Upon coexpressing CaBP4, the Ba2+ and Ca2+ relations coalesce (right subpanel), substantiating elimination of CDI. Figure 2c portrays this effect in f300 bar-graph format, used for compactness in subsequent figures.
Figure 2. CaBP4 modulation and binding of CaV1.3 channels.
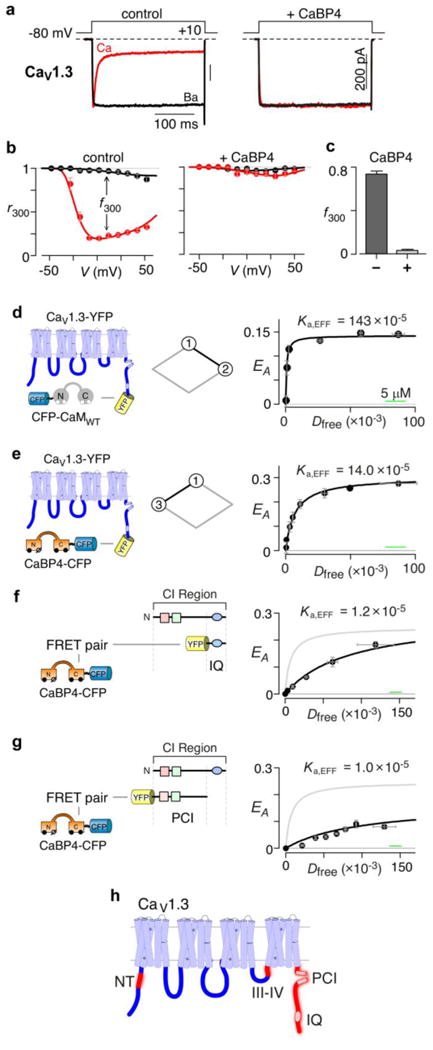
(a) Functional CaBP4 effects on whole-cell CaV1.3 current traces. Left, CaM-mediated CDI without CaBP4. Scale bar pertains to Ca2+ currents (red). Ba2+ currents (black) scaled down ∼3× for comparison of decay kinetics. Format utilized throughout. Right, coexpressing CaBP4 eliminates CDI. (b) Population data confirming CaBP4 elimination of CDI. Left, fraction of peak current after 300-msec depolarization (r300), versus step voltage for control cells. Black relation for Ba2+ currents; red for Ca2+ (mean ± SEM, n = 7 cells). Difference between relations at +10 mV (f300) reports steady-state CDI14. Right, elimination of CDI by CaBP4 (n = 7). (c) Bar-graph summary for relations in b (n = 7). (d) Left, cartoon of FRET 2-hybrid pairs: CaV1.3 fused with YFP on carboxy terminus (CaV1.3–YFP), and CFP–CaMWT. Middle, diamond schematic (cf., Fig. 1b) showing predominant transition (black segment) probed by FRET experiment. Right, FRET efficiency (EA) for binned groups of cells (∼4-5 cells per symbol) plotted versus Dfree, relative free donor concentration26,41 (CFP–CaMWT), forming a binding isotherm26,41. Dfree, microscopic-specific units are calibrated to μM by horizontal green scale bar24 (here and in e-g). Half-maximal EA reached at Kd,EFF = 700 Dfree units yielding Ka,EFF = (1 / Kd,EFF) = 143 × 10-5 (Dfree-1 units), equivalent to Ka ∼44 μM-1. (e) FRET analysis for CaBP4–CFP versus CaV1.3– YFP. Format as in d. Black symbols bin ∼7-8 cells. Ka,EFF ∼14 × 10-5 (Dfree-1 units), or Ka ∼4.4 μM-1. (f) FRET 2-hybrid analysis for CaBP4–CFP versus YFP–IQ. Left, cartoon of FRET partners. Right, corresponding binding curve (black); symbols average ∼5-6 cells. Gray, fit to holochannel relation from e. (g) FRET 2-hybrid analysis for CaBP4–CFP versus YFP–PCI peptide. Format as in e. Black symbols average ∼7-8 cells. (h) Cartoon of main α1D subunit of CaV1.3, comprising four homologous domains, with intracellular loops (dark blue) facing downwards. Red, potential CaBP binding regions, based on peptide FRET 2-hybrid analysis.
Regarding binding, CaBP4 might outcompete excess CaM, if CaBP4 were to exhibit a higher affinity for channels than CaM. Prior peptide assays conflict on this point14,27, and quantification of CaM and CaBP4 binding to functional holochannel complexes has seldom been achieved. We previously developed a holochannel FRET 2-hybrid assay26,41 enabling characterization of CaM binding to functional CaV1.2 in live cells. Here, we utilize this approach to quantify interaction between YFP-tagged CaV1.3 channels and CFP-tagged CaM or CaBP4 (Figs. 2d, e). Within the scheme in Fig. 1b, these assays measure the association constants between states 1 and 2 (Fig. 2d, diamond schematic), and between states 1 and 3 (Fig. 2e). The rightmost subpanel in Fig. 2d plots FRET efficiency (EA) versus the relative free concentration of apoCaM (Dfree), yielding a binding curve with a large association constant of K12 = 44 ± 9 μM-1 (Ka,eff = 143×10-5 microscope-specific units). By contrast, though CaBP4 does bind well to the channel (Fig. 2e), the affinity is ten-fold lower with a Ka = 4.4 ± 0.7 μM-1. Likewise, CaBP1 binding was 2.6 ± 0.4 μM-1 (Supplementary Results, Supplementary Fig. 1) Of note, interaction affinity (i.e., Ka,EFF) is the reciprocal of the free concentration of donor-tagged molecules (Dfree) at half-maximal EA, whereas the distance/orientation of donor and acceptor fluorophores in the bound complex specifies maximal EA (EA/max) at large Dfree values26,41.
To identify candidate segments for CaBP4 binding with holochannels (Fig. 2e), we scanned the intracellular loops of CaV1.3 for CaBP4 interaction (Fig. 2e, left subpanel, dark blue segments), using a peptide version of FRET 2-hybrid assays26,41. CaBP4-ECFP was the donor, and EYFP fused to various channel segments the acceptor. Though peptide affinities may not translate quantitatively to the intact context, they serve to identify regions meriting full analysis via integrative holochannel assays. Figure 2f confirms CaBP4 binding to the isolated IQ domain of CaV1.3 (black), as observed previously14,27. Notably, the interaction affinity is lower than for the holochannel (gray fit), suggesting that other segments contribute. Indeed, CaBP4 also interacts with the upstream PCI region shown in Fig. 2g (black data), and further results identify other candidate interaction sites at the channel amino terminus (NT) and the C-terminal third of the loop between domains III and IV (III-IV loop) (Fig. 2h, red regions; Supplementary Fig. 2). These findings largely agree with those in homologous CaV1.2 channels20,22.
Donating CaBP4 modulation to chimeric channels
To test for functional relevance of candidate CaBP4 binding segments (Fig. 2h, red) at the holochannel level, we sought to confer CaBP4 sensitivity to CaBP-insensitive channels via donation of essential CaV1.3 modules. We thus turned to the CaV2.3 isoform (Fig. 3a, left; Supplementary Fig. 3a), which readily forms functional chimeras with CaV1 channels28,42. FRET between CaBP4-CFP and CaV2.3 fused to YFP revealed little binding (Fig. 3a, right). Moreover, CaBP4 also did not influence CDI (Figs. 3b, c). Thus, CaV2.3 channels served as a ‘blank slate’ lacking both binding and modulation by CaBP4. First, we replaced the CI region of CaV2.3 with the corresponding segment of CaV1.3 (Fig. 3d, left, eeeed). CaBP4 now bound appreciably to this chimeric channel (Fig. 3d, right). More importantly, CDI in these chimeric channels could now also be attenuated, though not eliminated by CaBP4 (Figs. 3e, f; Supplementary Fig. 3b). Further chimeric-channel experiments indicated that adding both the NT and III-IV loops of CaV1.3 were required for full CaBP4 sensitivity (Supplementary Fig. 3-4). The final outcome is demonstrated by the chimeric channel (Fig. 3g, left, deedd) containing all three candidate binding sites (NT, III-IV loop, and CI region) of CaV1.3. CaBP4 binding to this chimera shows the highest affinity (Fig. 3g, right; Supplementary Fig. 3e), and CDI is now completely eliminated by CaBP4 (Figs. 3h, i; Supplementary Fig. 3d). Direct deletions and mutagenesis performed on the CaV1.3 channel backbone further confirmed the functional contribution of all these segments to CaBP4 sensitivity (Supplementary Figs. 5-7). Overall, the CaV1.3 CI region appeared necessary, and all three segments (NT, III-IV, PCI, and IQ) together were required for full functional sensitivity to CaBP4.
Figure 3. Chimeric channels reveal importance of CaBP sites beyond the IQ domain.

(a-c) No CaBP4 binding or functional modulation of wild-type CaV2.3. (a) FRET experiment for CaV2.3 fused to YFP, versus CaBP4-CFP. Little binding. Each symbol bins ∼4-5 cells. (b) Exemplar whole-cell current traces illustrating absence of CaBP4 effect on CDI. (c) Population data confirm lack of CaBP4 effects on CDI. Bars average n = 5 cells each. (d) Left, cartoon of holochannel FRET experiment, pitting CaBP4-CFP versus YFP-tagged chimeric CaV2.3 with CaV1.3 CI substitution (eeeed). Right, significantly increased binding affinity of eeeed with CaBP4 (symbols bin ∼3-4 cells each) compared to the control in gray (from a). (e) Exemplar whole-cell currents now exhibit partial CDI suppression by CaBP4. Control on left, with strong CaBP4 coexpression on right. (f) Population data confirms partial CDI suppression by CaBP4 in eeeed channels. Control bar averages n = 5 cells. Light gray bar for CaBP4 condition averages n = 7 cells. (g, h, i) Chimeric CaV2.3 channels that include CaV1.3 amino terminus, III-IV loop, and CI module (deedd) show strongest CaBP4 binding (g, symbols average ∼5 cells each), and complete CDI elimination by CaBP4 (h, i). (i) Control bar graph averages n = 8 cells, and light gray bar for CaBP4 condition averages n = 5 cells.
CaM and CaBP4 concurrently bind to holochannels
With ample evidence for the functional contribution of CaBP binding beyond the IQ module, we pursued definitive tests for competitive versus allosteric interaction between CaM and CaBP4 (Fig. 1b). A crucial discriminator would be the presence or absence of simultaneous ligand binding and modulation of holochannels. Such a test would establish or exclude the existence of state 4 in Fig. 1b, thus furnishing a decisive mechanistic distinction.
To this end, we covalently fused CaM or CaBP4 to CaV1.3 channels, a strategy we used previously with functional assays alone to probe CaM interactions with CaV1.2 channels43. Figures 4a, b introduce the approach, here applied anew to CaV1.3 channels. To start, we considered CaV1.3 channels without covalently linked CaM, which again demonstrate robust CDI (Fig. 4a, control), just as observed earlier. Upon strong overexpression of a dominant-negative mutant CaM9,14 (CaM1234), which is Ca2+ insensitive, CDI is completely eliminated14 (Fig. 4a, traces labeled +CaM1234 and bargraph). This outcome occurs because CaM1234 has competitively replaced endogenous functional CaM on the channel IQ domain24. By contrast, when the analogous experiment is performed with CaV1.3 channels fused to a single CaM (Fig. 4b, far left, CaV1.3–CaM construct), a completely different outcome results. Before coexpressing CaM1234 (Fig. 4b, control), these channels undergo robust CDI indicating unfettered functionality of the covalently attached CaM. Importantly, however, the covalent linkage of a single CaM would enrich enormously the local concentration of CaM to levels extending into the millimolar range43. This local concentration would far surpass the concentrations of CaM1234 achievable by expression as a separate molecule24 (∼10 μM). One would thereby predict that separately coexpressing CaM1234 in this context would have little or no effect on CDI. Indeed, we observed exactly this preservation of CDI (Fig. 4b, +CaM1234 traces, and bar graph; Supplementary Figs. 8a, b). This outcome demonstrates that covalently attached CaM effectively occludes the IQ module in CaV1.3.
Figure 4. CaBP4 and CaM simultaneously bind CaV1.3 channels.

(a) Effect of Ca2+-insensitive mutant CaM (CaM1234) to completely eliminate CDI. Left, schematic of wild-type CaV1.3 channel. Control exemplar traces illustrating baseline CDI for reference. +CaM1234 traces illustrating complete loss of CDI upon CaM1234 coexpression. Bar graphs, confirming complete elimination of CDI by CaM1234 in population, where bars average n = 5 cells each. (b) Complete occlusion of CaM1234 effects in CaV1.3 channels attached at carboxy terminus to wild-type CaM via 12-glycine linker (CaV1.3–CaM in left schematic). Format as in a. Control traces show fullbore CDI, indistinguishable from that in CaV1.3 channels lacking covalent linkage. By contrast, +CaM1234 exemplar traces illustrate complete sparing of CDI. Bar-graph summaries confirm this trend, where each bar averages n = 6 cells each. (c) CaBP4 effects on CaV1.3–CaM. Format as in b. Control whole-cell current traces reproduced from b for reference. Remarkably, coexpressing CaBP4 with CaV1.3–CaM here completely eliminates CDI in both exemplar traces (+CaBP4), and population bar-graph data, where the dark control bar averages n = 6 cells, and the light gray bar for the CaBP4 condition averages n = 7 cells. (d) CaBP4 can still bind with CaV1.3–CaM, where black symbols bin ∼5-6 cells each. As reference, gray curve plots amplitude-normalized fit obtained without CaM fusion from Fig. 3e.
Thus armed, we could perform a pivotal experiment—to test the effect of coexpressing CaBP4 with the CaV1.3–CaM construct. If CaBP4 and CaM compete at the IQ module, then CaBP4 should hardly influence CDI of CaV1.3–CaM, just as observed for CaM1234 (Fig. 4b). In striking contrast, we observed complete elimination of CDI (Fig. 4c, control versus +CaBP4 traces), as confirmed by population data (Fig. 4c, bar graph; Supplementary Fig. 8c). We also observed similar outcomes with CaBP1 acting on CaV1.3–CaM (Supplementary Fig. 9), as well as with CaBP modulation of analogous CaV1.2–CaM channels (Supplementary Fig. 10). These remarkable results strongly argue that CaBP and CaM can bind to distinct sites on the channel, in contradiction to a competitive mechanism (Fig. 1b).
We advanced the approach of tethered CaM even further, and directly corroborated the functional result above with holochannel binding assays involving CaV1.3–CaM. Holochannel FRET 2-hybrid analysis demonstrated that CaBP4 still binds to CaV1.3–CaM (Fig. 4d, black data and curve), with only moderately reduced affinity compared to channels without covalently linked CaM (Fig. 4d, unlinked gray fit reproduced for reference). Interestingly, owing to occlusion of the CaM binding site by tethered CaM, the binding of CaBP4 to CaV1.3–CaM directly probes transitions between states 2 and 4 (Fig. 4d, schematic). This yields an association constant K24 = 1.3 ± 0.2 μM-1. Thus, we would argue that state 4 in Fig. 1b exists.
A key remaining issue was the possibility of more than one CaBP4 molecule working together to modulate channels. Accordingly, we extended our linkage strategy still further, and fused a single CaBP4 to the amino terminus of a channel that already had a carboxy-tail linkage to CaM. This created the dual-linkage construct in Fig. 5a (left subpanel, CaBP4–CaV1.3–CaM). Remarkably, this single CaBP4 entirely eliminated CDI, demonstrating that only one CaBP4 molecule suffices for full modulation (Fig. 5a, other subpanels; Supplementary Fig. 8d). Furthermore, FRET 2-hybrid assays between CaBP4–CaV1.3–YFP holochannels and CaBP4– CFP resulted in negligible binding (Fig. 5b), demonstrating explicitly that only a single CaBP4 can bind to the channel. In this dual-linkage construct, the fused CaBP4 must certainly be interacting with the channel, otherwise CDI would not have been eliminated. Yet, the tethered CaM might conceivably be dislodged, retaining affiliation only via its linker. This possibility was excluded, however, because holochannel FRET assays confirmed that CaM, as a separate molecule, can indeed bind holochannels fused to CaBP4 (Fig. 5c, black data and fit). Because of occlusion of the CaBP4 binding pocket by linked CaBP4, binding of CaM to CaBP4–CaV1.3 directly probes transitions between states 3 and 4 (Fig. 5c, schematic), with an association constant K34 = 10 ± 2 μM-1. Moreover, the maximal FRET efficiency at large Dfree values (EA/max) is indistinguishable from that for the corresponding experiment with CaV1.3 channels without fusion (as reference, gray trace for unlinked channel experiment in Fig. 3d). Thus, apoCaM appears to bind to holochannels in largely the same configuration, whether CaBP is present or not.
Figure 5. Only one CaBP4 can bind per CaV1.3 channel.

(a) Fusion of both CaBP4 (by 8-glycine linker) and CaM (by 12-glycine linker) to CaV1.3 (CaBP4–CaV1.3–CaM, left cartoon) completely eliminates CDI, as seen from exemplar traces (middle left), overlay of Ba2+ (black) and Ca2+ (red) r300 relations (middle right, n = 5 cells per condition), and bar-graph f300, averaging n = 5 cells for each bar. (b) CaBP4-CFP does not bind CaBP4–CaV1.3–YFP (left schematic showing CaV1.3 channel with CaBP4 fused to its amino terminus, and YFP to its carboxy terminus), suggesting only one CaBP4 can bind to CaV1.3. Middle diamond schematic indicates that CaBP4–CaV1.3–YFP cannot be perturbed from state 3 by overexpressing CaBP4-CFP as a separate molecule. Black symbols average ∼3-4 cells each. Reference gray fit, CaM binding CaV1.3 channels without fusion to CaBP4 from Fig. 3e. (c) Holochannel FRET indicates that CaM can still bind to CaBP4–CaV1.3–YFP. Right, corresponding CFP–CaM binding curve (black symbols bin ∼3-4 cells each, black fit). Amplitude-normalized reference gray fit, CaM binding CaV1.3 channels without fusion to CaBP4, from Fig. 3e.
In all, these results argue that CaBP4 and CaM can bind concurrently to CaV1.3 channels. While apoCaM likely interacts with the IQ domain24, CaBP4 modulation may rely on binding to elements outside the IQ domain.
Modulation by trace CaBP levels explained
That shown, how could trace concentrations of CaBPs modulate channels in excess CaM (Figs. 1c-h)? Accordingly, we elaborated the four-state scheme of Fig. 1b, by including relevant association constants obtained above (Figs. 2, e, 4d, and 5c). Figure 6a displays the result, which comprises a thermodynamic cycle of CaM and CaBP interaction with channels. Thus viewed, it was reassuring that (K12 · K24) / (K13 · K34) ∼ 1, in accord with thermodyanmic constraints44. We thus proceeded to in-depth analysis of this scheme. If CaBP4 were to interact in a purely competitive manner, state 4 would not exist. Given a rough estimate of free endogenous CaM at 10 μM25, the remaining 3-state system would predict that a CaBP concentration of 107 μM would be required for half block of CDI (Fig. 6b, Competitive Model, black). This concentration of CaBP4 far exceeds that generally present in the brain (Figs. 1c-h). If state 4 is included, so that an allosteric mechanism pertains, a very different outcome arises. In particular, if the binding of CaM and CaBP4 were completely independent, then transitions into state 4 would be governed by association constants K34 and K24, which would be respectively identical to K12 and K13. Alternatively, if the transitions were to interact cooperatively, then K34 and K24 would be equal to K12 and K23 multiplied by a common factor λ that derives from thermodynamic mandates44. Here, λ > 1 would signify positive cooperativity in the binding of CaBP4 and CaM to the channel, λ < 1 would denote negative cooperativity, and λ = 0 would correspond to purely competitive binding of CaM and CaBP4. Intriguingly, our experiments permitted direct determination of the λ factor. K34 is lower than K12 by about 4 fold, as is K24 compared to K13. Thus, we estimate λ ∼ 0.23, confirming a modest degree of negative coooperativity in the binding of both CaBP4 and apoCaM to channels. Importantly, given this precise diamond-shaped system, the projected effects of CaBP4 on CDI corresponds to the red-dashed curve in Fig. 6b, with a half CDI block at a CaBP4 concentration of ∼0.8 μM. This concentration is plausibly consistent with levels in the brain. Moreover, accounting for the effects of endogenous CaM on our binding analysis produced little change in this prediction (Supplementary Fig. 11; Supplementary Notes 1-2) Thus, physiological CaBP4 may be able to exert modulation as a direct consequence of the allosteric mechanism in Fig. 6a.
Figure 6. Mechanism of CaV1.3 channel modulation by modest levels of CaBP4.

(a) Allosteric model of CaM and CaBP4 binding to CaV1.3 channels, based on Fig. 1b, but now with experimentally determined association constants as listed and drawn from Figs. 2d, e, 4d, and 5c. (b) Model predictions for CaBP4 dependence of CDI. If dual-bound state 4 does not exist (i.e., pure competition), then black relation results under estimated free CaM of 10 μM.. Entire 4-state allosteric scheme, with experimentally determined association constants predicts dashed curve for CDI dependence upon free CaBP concentration. Solid red curve fits experimental data from approach outlined in c, where symbols average n = 5 cells each. (c) Schematic of patch fluorometry experiments. (d) Top, schematic for increasing CaBP4 concentration. Bottom, Ca2+ traces corresponding to various symbols in b, as labeled.
This notable prediction nonetheless relies on the accuracy of holochannel FRET assays. We thus utilized patch fluorometry to directly determine the sensitivity of CaV1.3 CDI to CaBP4 (Fig. 6c). Specifically, whole-cell electrophysiology measured CDI, and concurrent whole-cell fluorescence measurements estimated the concentration of fluorescently labeled CaBP4 molecules. CaM was fused to CaV1.3 channels to ensure a stable and high local concentration of CaM. Data thus obtained should decorate the predicted dashed-red curve in Fig. 6b. Exemplar calcium traces (Fig. 6d) obtained under various CaBP4 concentrations illustrate variable CDI attenuation. Absent CaBP4 (point a in Figs. 6b, d), the full measure of wild-type CDI is apparent. Notably, at estimated CaBP concentrations of less than 2.5 μM, CDI already exhibits substantial attenuation (points b and c in Figs. 6b, d). Indeed, CDI becomes virtually absent at [CaBP4] of 4 μM and higher (point d in Figs. 6b, d). Population data corroborate these trends (Fig. 6b, symbols, solid-red curve) and essentially overlays the predicted outcome from holochannel FRET assays (Fig. 6b, dashed-red curve). This correspondence substantiates our allosteric model (Fig. 6a), and its explanation for how trace amounts of CaBPs may exert their action on channels despite an abundance of CaM. As these experiments employed CaV1.3 fused covalently to CaM, the CaBP sensitivity here is a lower-limit estimate, making the outcome still more compelling.
Discussion
Calcium-binding proteins (CaBPs) tune the Ca2+ responsiveness of numerous Ca2+ signaling molecules regulated by CaM, thereby projecting influence over diverse biological processes13,18,19,21. Channel peptide studies and the homology between CaBPs and CaM support the view that CaBPs act by competitively replacing CaM on target molecules14,22,27. However, CaM expression far exceeds that of CaBPs in the brain, raising questions about the ability of CaBPs to exert appreciable effects. To overcome the interpretive limitations of peptide studies, we devised functional and live-cell FRET interaction assays on CaV1.3 holochannels covalently linked to CaM and/or CaBP4. Notably, CaV1.3 channels fused to CaM are still strongly modulated by CaBP4, and holochannel FRET assays directly confirm CaBP4 binding to such channels. These results firmly establish that apoCaM and CaBP4 can bind simultaneously, implicating an allosteric scheme that predicts and explains how trace CaBP4 can modulate channels in excess CaM. We directly confirm this prediction using patch fluorometry.
These results project a new view of CaBP modulation of CaV1.3 channels that incorporates our recent molecular model of CDI24 (Supplementary Fig. 12). The channel/apoCaM complex in Supplementary Fig. 12a corresponds to configuration 2 in Fig. 6a. The C-terminal lobe of apoCaM interacts strongly with the IQ domain, and the N-lobe engages more weakly with EF-hand regions. CDI occurs upon Ca2+/CaM switching interactions to its effector sites (Supplementary Fig. 12b), with the N-lobe of Ca2+/CaM embracing an NSCaTE module in the channel amino terminus, and the C-lobe forming a tripartite complex with the channel IQ module and upstream EF-hand region. To overlay CaBP4, we positioned CaBP4 with major contacts on the amino terminus, III-IV loop, and EF-hand regions (Supplementary Fig. S12c, corresponding to Fig 6a, state 4). During rest, CaBP and apoCaM may bind simultaneously because principal interaction sites appear distinct for the two ligands. This proposed arrangement retains strong interaction of the IQ domain with the C-lobe of apoCaM, based on interpreting the equivalent EA/max values in Figs. 2d and 5c as evidence that apoCaM adopts a similar bound configuration, whether CaBP is absent or also bound. As this equivalence could conceivably accomodate different apoCaM arrangements, the suggested configuration is provisional. That said, the proposed layout (Supplementary Fig. 12c) does project a potential steric clash between the N-lobe of apoCaM and CaBP4 at the EF-hand region, which nicely rationalizes the modest negative cooperativity factor of λ ∼0.23. On Ca2+ elevation, access of Ca2+/CaM to its effector sites may be inhibited by CaBP4 preoccupying nearby or partially overlapping sites, thus prohibiting CDI. In this regard, the mechanism of CDI inhibition may be a nuanced combination of allosteric and competitive mechanisms. Under resting Ca2+, CaBP and apoCaM posture for baseline position via an allosteric mechanism (i.e., Fig. 6a); however, upon Ca2+ elevation, CDI fails to occur because CaBP occludes Ca2+/CaM effector sites via a potentially competitive regime. This class of mechanism may generalize to other CaBP target molecules that share distinctions between sites for apoCaM preassociation and Ca2+/CaM effectuation. CaV1.2 channels are homologous to CaV1.345, and likely to subscribe to an analogous mechanism. Ryanodine receptors are CaM regulated and exhibit movement of CaM between distinct apoCaM and Ca2+/CaM sites46. Closely related IP3 receptors host both apoCaM47 and Ca2+/CaM48,49 binding sites, and are modulated by CaBP18,19.
More broadly, measuring binding and function in holomolecules fused to modulatory ligands may apply to many systems. Particular advantages include the ability to readily isolate individual transitions within cyclical binding schemes, and the interpretive relevance of results obtained on holomolecules in live cells. Indeed, the approaches developed here enrich a burgeoning toolkit for pursuing quantitative biochemistry in functional assemblies within live cells. This endeavor promises rapid progress in understanding native molecular mechanisms50.
Methods
Methods and any associated references are available in the online version of the paper.
Supplementary Material
Acknowledgments
We thank Wanjun Yang for dedicated technical support, and other members of the Ca2+ signals lab for valuable comments. Hojjat Bazzazi generously made available the β2a–CaMWT construct. Dr. Yuan Gao contributed the data analysis in Fig. 1d from human prefrontal cortex. Supported by grants from the NHLBI MERIT Award (to D.T.Y.), NIDCD (P.S.Y. and Paul Fuchs), and NIMH (M.B.J.).
Footnotes
Author contributions: P.S.Y created mutant, chimeric, and engineered channels. P.S.Y. and M.B.J. performed electrophysiology and FRET experiments; and undertook extensive data analysis. M.B.J. performed molecular modeling. D.T.Y. supervised and helped conceive the project. All authors refined hypotheses, wrote the paper, and created figures.
Competing financial interests: The authors declare no competing financial interests.
References
- 1.Wang L, Martin B, Brenneman R, Luttrell LM, Maudsley S. Allosteric modulators of g protein-coupled receptors: future therapeutics for complex physiological disorders. The Journal of pharmacology and experimental therapeutics. 2009;331:340–348. doi: 10.1124/jpet.109.156380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ritter SL, Hall RA. Fine-tuning of GPCR activity by receptor-interacting proteins. Nature reviews Molecular cell biology. 2009;10:819–830. doi: 10.1038/nrm2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Y, Krupa B, Kang JS, Bolshakov VY, Liu G. Glycine site of NMDA receptor serves as a spatiotemporal detector of synaptic activity patterns. Journal of neurophysiology. 2009;102:578–589. doi: 10.1152/jn.91342.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 5.Alberts B, et al. Molecular biology of the cell. 3rd. Garland: 1996. pp. 195–222. [Google Scholar]
- 6.Goodey NM, Benkovic SJ. Allosteric regulation and catalysis emerge via a common route. Nature chemical biology. 2008;4:474–482. doi: 10.1038/nchembio.98. [DOI] [PubMed] [Google Scholar]
- 7.Grunwald ME, Zhong H, Lai J, Yau KW. Molecular determinants of the modulation of cyclic nucleotide-activated channels by calmodulin. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:13444–13449. doi: 10.1073/pnas.96.23.13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dunlap K. Calcium channels are models of self-control. J Gen Physiol. 2007;129:379–383. doi: 10.1085/jgp.200709786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+ -dependent inactivation of L- type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 10.Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- 11.Alseikhan BA, DeMaria CD, Colecraft HM, Yue DT. Engineered calmodulins reveal the unexpected eminence of Ca2+ channel inactivation in controlling heart excitation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:17185–17190. doi: 10.1073/pnas.262372999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haeseleer F, et al. Five members of a novel Ca(2+)-binding protein (CABP) subfamily with similarity to calmodulin. The Journal of biological chemistry. 2000;275:1247–1260. doi: 10.1074/jbc.275.2.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haeseleer F, et al. Essential role of Ca2+-binding protein 4, a Cav1.4 channel regulator, in photoreceptor synaptic function. Nat Neurosci. 2004;7:1079–1087. doi: 10.1038/nn1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang PS, et al. Switching of Ca2+-dependent inactivation of CaV1.3 channels by calcium binding proteins of auditory hair cells. J Neurosci. 2006;26:10677–10689. doi: 10.1523/JNEUROSCI.3236-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cui G, et al. Ca2+-binding proteins tune Ca2+-feedback to Cav1.3 channels in mouse auditory hair cells. The Journal of physiology. 2007;585:791–803. doi: 10.1113/jphysiol.2007.142307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee S, Briklin O, Hiel H, Fuchs P. Calcium-dependent inactivation of calcium channels in cochlear hair cells of the chicken. The Journal of physiology. 2007;583:909–922. doi: 10.1113/jphysiol.2007.135582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee A, et al. Differential modulation of Ca(v)2.1 channels by calmodulin and Ca2+-binding protein 1. Nat Neurosci. 2002;5:210–217. doi: 10.1038/nn805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang J, et al. Identification of a family of calcium sensors as protein ligands of inositol trisphosphate receptor Ca(2+) release channels. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:7711–7716. doi: 10.1073/pnas.102006299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kasri NN, et al. Regulation of InsP3 receptor activity by neuronal Ca2+-binding proteins. The EMBO journal. 2004;23:312–321. doi: 10.1038/sj.emboj.7600037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou H, et al. Ca2+-binding protein-1 facilitates and forms a postsynaptic complex with Cav1.2 (L-type) Ca2+ channels. J Neurosci. 2004;24:4698–4708. doi: 10.1523/JNEUROSCI.5523-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haeseleer F, Palczewski K. Calmodulin and Ca2+-binding proteins (CaBPs): variations on a theme. Advances in experimental medicine and biology. 2002;514:303–317. doi: 10.1007/978-1-4615-0121-3_18. [DOI] [PubMed] [Google Scholar]
- 22.Oz S, et al. Competitive and non-competitive regulation of calcium-dependent inactivation in CaV1.2 L-type Ca2+channels by calmodulin and Ca2+-binding protein 1. The Journal of biological chemistry. 2013;288:12680–12691. doi: 10.1074/jbc.M113.460949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Findeisen F, Minor DL., Jr Structural basis for the differential effects of CaBP1 and calmodulin on Ca(V)1.2 calcium-dependent inactivation. Structure. 2010;18:1617–1631. doi: 10.1016/j.str.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ben Johny M, Yang PS, Bazzazi HX, Yue DT. Dynamic switching of calmodulin interactions underlies Ca2+ regulation of CaV1.3 channels. Nature communications. 2013;4:2370. doi: 10.1038/ncomms2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu X, Yang PS, Yang W, Yue DT. Enzyme-inhibitor-like tuning of Ca2+ channel connectivity with calmodulin. Nature. 2010;463:968–972. doi: 10.1038/nature08766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Erickson MG, Liang H, Mori MX, Yue DT. FRET two-hybrid mapping reveals function and location of L-type Ca2+ channel CaM preassociation. Neuron. 2003;39:97–107. doi: 10.1016/s0896-6273(03)00395-7. [DOI] [PubMed] [Google Scholar]
- 27.Findeisen F, Rumpf C, Minor DL. Apo states of calmodulin and CaBP1 control CaV1 voltage-gated calcium channel function through direct competition for the IQ domain. Journal of Molecular Biology. 2013;425:3217–3234. doi: 10.1016/j.jmb.2013.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Leon M, et al. Essential Ca(2+)-binding motif for Ca(2+)-sensitive inactivation of L-type Ca2+ channels. Science. 1995;270:1502–1506. doi: 10.1126/science.270.5241.1502. [DOI] [PubMed] [Google Scholar]
- 29.Kim J, Ghosh S, Nunziato DA, Pitt GS. Identification of the components controlling inactivation of voltage-gated Ca2+ channels. Neuron. 2004;41:745–754. doi: 10.1016/s0896-6273(04)00081-9. [DOI] [PubMed] [Google Scholar]
- 30.Dick IE, et al. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature. 2008;451:830–834. doi: 10.1038/nature06529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oz S, Tsemakhovich V, Christel CJ, Lee A, Dascal N. CaBP1 regulates voltage-dependent inactivation and activation of Ca(V)1.2 (L-type) calcium channels. The Journal of biological chemistry. 2011;286:13945–13953. doi: 10.1074/jbc.M110.198424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou H, Yu K, McCoy KL, Lee A. Molecular mechanism for divergent regulation of Cav1.2 Ca2+ channels by calmodulin and Ca2+-binding protein-1. The Journal of biological chemistry. 2005;280:29612–29619. doi: 10.1074/jbc.M504167200. [DOI] [PubMed] [Google Scholar]
- 33.Lorenzen A, Schwabe V. In: Purinergic and pyrimidinergic signalling I. Abbracchio MP, Williams M, editors. Springer-Verlag; 2001. pp. 19–34. [Google Scholar]
- 34.Logothetis DE, Lupyan D, Rosenhouse-Dantsker A. Diverse Kir modulators act in close proximity to residues implicated in phosphoinositide binding. The Journal of physiology. 2007;582:953–965. doi: 10.1113/jphysiol.2007.133157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lein ES, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 36.Belgard TG, et al. A transcriptomic atlas of mouse neocortical layers. Neuron. 2011;71:605–616. doi: 10.1016/j.neuron.2011.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blackshaw S, et al. Genomic analysis of mouse retinal development. PLoS Biol. 2004;2:E247. doi: 10.1371/journal.pbio.0020247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Colantuoni C, et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature. 2011;478:519–523. doi: 10.1038/nature10524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nature reviews Genetics. 2012;13:227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chao SH, Suzuki Y, Zysk JR, Cheung WY. Activation of calmodulin by various metal cations as a function of ionic radius. Mol Pharmacol. 1984;26:75–82. [PubMed] [Google Scholar]
- 41.Erickson MG, Alseikhan BA, Peterson BZ, Yue DT. Preassociation of calmodulin with voltage-gated Ca(2+) channels revealed by FRET in single living cells. Neuron. 2001;31:973–985. doi: 10.1016/s0896-6273(01)00438-x. [DOI] [PubMed] [Google Scholar]
- 42.Mori MX, Vander Kooi CW, Leahy DJ, Yue DT. Crystal structure of the CaV2 IQ domain in complex with Ca2+/calmodulin: high-resolution mechanistic implications for channel regulation by Ca2+ Structure. 2008;16:607–620. doi: 10.1016/j.str.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mori MX, Erickson MG, Yue DT. Functional stoichiometry and local enrichment of calmodulin interacting with Ca2+ channels. Science. 2004;304:432–435. doi: 10.1126/science.1093490. [DOI] [PubMed] [Google Scholar]
- 44.Hill TL. Free energy transduction in biology: the steady-state kinetic and thermodynamic formalism. Vol. 64. Academic Press; 1977. [Google Scholar]
- 45.Xu W, Lipscombe D. Neuronal Ca(V)1.3alpha(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci. 2001;21:5944–5951. doi: 10.1523/JNEUROSCI.21-16-05944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Samso M, Wagenknecht T. Apocalmodulin and Ca2+-calmodulin bind to neighboring locations on the ryanodine receptor. The Journal of biological chemistry. 2002;277:1349–1353. doi: 10.1074/jbc.M109196200. [DOI] [PubMed] [Google Scholar]
- 47.Bosanac I, et al. Crystal structure of the ligand binding suppressor domain of type 1 inositol 1,4,5-trisphosphate receptor. Molecular cell. 2005;17:193–203. doi: 10.1016/j.molcel.2004.11.047. [DOI] [PubMed] [Google Scholar]
- 48.Lin C, Widjaja J, Joseph SK. The interaction of calmodulin with alternatively spliced isoforms of the type-I inositol trisphosphate receptor. The Journal of biological chemistry. 2000;275:2305–2311. doi: 10.1074/jbc.275.4.2305. [DOI] [PubMed] [Google Scholar]
- 49.Yamada M, et al. The calmodulin-binding domain in the mouse type 1 inositol 1,4,5-trisphosphate receptor. The Biochemical journal. 1995;308(Pt 1):83–88. doi: 10.1042/bj3080083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gough NR. Biochemistry: Quantifying interactions. Science Signaling. 2010;3:ec62. [Google Scholar]
- 51.Eaves HL, Gao Y. MOM: maximum oligonucleotide mapping. Bioinformatics. 2009;25:969–970. doi: 10.1093/bioinformatics/btp092. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.