

Abstract
Structured RNA elements within messenger RNA often direct or modulate the cellular production of active proteins. As reviewed here, RNA structures have been discovered that govern nearly every step in protein production: mRNA production and stability; translation initiation, elongation, and termination; protein folding; and cellular localization. Regulatory RNA elements are common within RNAs from every domain of life. This growing body of RNA‐mediated mechanisms continues to reveal new ways in which mRNA structure regulates translation. We integrate examples from several different classes of RNA structure‐mediated regulation to present a global perspective that suggests that the secondary and tertiary structure of RNA ultimately constitutes an additional level of the genetic code that both guides and regulates protein biosynthesis.
Keywords: RNA structure, Riboswitch, Frameshifting, IRES, Translation
1. Introduction
RNA was long assumed to be a simple courier of the information contained within a DNA genome. This tidy, linear view of biology is quickly being replaced with models that emphasize the complex landscape of interactions between these macromolecules. At the center lies RNA. It is now well established that complex RNA structures are capable of functions previously thought to be the purview of proteins, including ligand binding and catalysis. These RNA structure‐mediated functions also include regulation of nearly every step of cellular protein production. Some of the regulatory mechanisms discussed below have been thoughtfully reviewed previously. Our goal in this review is not to duplicate these reviews but to present the argument that the three‐dimensional structure of messenger RNA (mRNA) constitutes an additional layer of genetic information that both guides and regulates the production of encoded proteins.
The primary sequence of an mRNA encodes the amino acid sequence of a protein, whereas structural features within mRNA molecules can determine the biological activity of the encoded protein by regulating the isoform produced, expression level, folding, localization, or stability. RNA structures that regulate biological function during translation have been identified in every kingdom of life. Consequently, developing a better understanding of how RNA structure governs protein expression and function has broad‐ranging implications. These include guiding the development of novel therapeutics for combating bacterial [1] and viral pathogens [2, 3] and extend to understanding and mitigating diverse human genetic diseases such as Huntington's disease [4], myotonic dystrophy type 1 [5], and cystic fibrosis [6].
2. mRNA as a sensor
An mRNA can govern its own translation and transcription using ligand‐binding structural elements called riboswitches. The best characterized riboswitches are located in the 5′ untranslated regions (UTRs) of bacterial mRNAs. Upon ligand binding, the RNA undergoes allosteric rearrangement that regulates transcription or translation initiation, elongation efficiency, mRNA stability, or splicing [7, 8, 9]. Riboswitches contain two domains: a metabolite‐binding region known as the “aptamer domain” and an allosteric domain termed the “expression platform” (Fig. 1 A). The expression platform enacts the regulatory function signaled by the aptamer domain. Typically one structure of the riboswitch occludes an important regulatory element such as the ribosome‐binding Shine–Dalgarno sequence. Riboswitch aptamer domains have evolved to bind diverse small molecules, and those domains that bind the same ligand tend to be highly conserved. In contrast, the expression domains vary in both sequence and function in different organisms. Thus, riboswitches are modular; a given aptamer domain has a specific target metabolite but the ultimate function depends on the linked expression platform. Riboswitches that bind ions (Mg2+, F−), carbohydrates, metabolites, proteins, and co‐enzymes have been characterized. Protein expression requires mRNA to be (i) transcribed, (ii) processed (for example, 5′ capped, spliced, polyadenylated), and (iii) translated by the ribosome. Interfering with any of these processes reduces the number of available mRNA transcripts or decreases protein production. Examples of ligand‐sensing riboswitches that influence each of these steps are discussed below.
Figure 1.
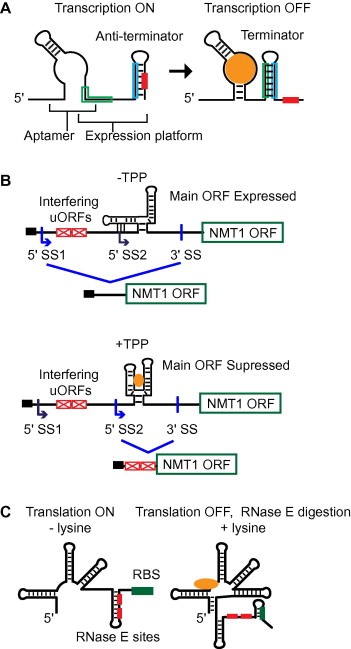
Mechanisms of riboswitch regulation. (A) Schematic example of transcription termination resulting from ligand binding to the aptamer domain of a riboswitch and subsequent stabilization of a terminator stem‐loop and exposure of a poly‐uracil stretch (red box). (B) Splicing regulation by a TPP riboswitch in the N. crassa NMT1 gene [19]. The favored 5′ splice site (SS2) is obscured in the absence of ligand. Ligand binding attenuates ORF protein expression by allowing use of SS2. (C) Combined translational and stability control by a lysine riboswitch. Ligand binding sequesters the RBS (green) and exposes two RNase E cleavage sites (red), leading to mRNA degradation.
2.1. Control of transcription elongation
In an mRNA with a riboswitch, RNA polymerase synthesizes the aptamer domain before the expression domain. This allows the nascent aptamer domain to sample the cellular environment and potentially bind a cognate ligand before the expression domain is transcribed and folded, as recently observed in a co‐transcriptional folding study of the adenine riboswitch [10]. Critically, this timing allows folding of one of the mutually exclusive structures that can be adopted by the riboswitch. For example, in many strains of bacteria, ligand binding to an aptamer domain causes an expression platform to form an intrinsic terminator stem, an RNA hairpin followed by a stretch of six or more uracil (U) residues, that inhibits RNA polymerase extension (Fig. 1A) [11, 12]. In the absence of ligand, the unbound aptamer domain structure allows the anti‐terminator stem‐loop to form, sequestering the poly‐U stretch and allowing mRNA synthesis to proceed. This type of regulation is exemplified by the T‐box mechanism: if an uncharged tRNA binds to an element in the mRNA encoding the synthetase responsible for charging the particular tRNA, an anti‐terminator helix forms that permits transcription of the message encoding the aminoacyl‐tRNA synthetase [13]. In contrast, the charged tRNA does not bind strongly to the terminator helix. In most cases, as in this example, the riboswitch mechanisms are negative feedback loops wherein a given ligand down regulates enzymes involved in ligand production. Although uncommon, there are examples of riboswitches that turn protein expression on rather than off. For example, ligand binding to the adenine riboswitch in Bacillus subtilis results in increased expression of proteins involved in adenine export [14].
2.2. Regulation of splicing
Although most riboswitches identified to date exist in simple prokaryotes, a thiamine pyrophosphate (TPP) sensing riboswitch is a widely distributed element [15, 16] found in bacteria, archaea [17], and eukaryotes [16, 18, 19] including both simple [20] and complex plants [19, 21]. While bacterial TTP riboswitches typically exert control at the level of transcription [22], these elements regulate alternative splicing in eukaryotes. Eukaryotic TPP riboswitches are typically located in intronic regions of genes associated with thiamine metabolism. Differences in secondary structure between ligand‐bound and unbound forms of the precursor mRNA (pre‐mRNA) sequester, expose, or relocate splice sites resulting in alternatively spliced mRNAs. Inclusion or exclusion of upstream open reading frames (ORFs) in mRNAs affects the identity of the synthesized protein. Neurospora crassa contains several TPP riboswitches and provides the best understood examples of eukaryotic riboswitches. In the NMT1 riboswitch, ligand binding causes formation of a structure that exposes an alternative splice site that prevents production of the major ORF product (Fig. 1B) [19]. A recent study, also in N. crassa, suggests that a TPP riboswitch in the NCU01977 gene controls formation of long‐distance base pairs [23]. These base pairs do not cover or reveal splice sites, but rather place a 5′ splice donor and 3′ acceptor in favorable proximity [23]. These different forms of splicing control within a single organism highlight the functional diversity of riboswitch elements.
2.3. Regulation of mRNA stability
The glmS ribozyme, a self‐cleaving RNA that requires ligand binding for activation, is an example of an RNA structure that regulates mRNA stability. The ribozyme is located in the 5′ UTR of the mRNA encoding the glucosamine‐6‐phosphate synthetase (GlmS) enzyme, which is widely distributed in Gram positive bacteria [24]. When glucosamine‐6‐phosphate levels are high, the molecule binds to the aptamer domain, activating the ribozyme. When the glmS ribozyme self‐cleaves, a terminus recognized by RNase J1 is produced, and RNase J degrades the remaining mRNA. Another example of riboswitch modulation of mRNA stability is found in the p27 tumor suppressor mRNA [25]. A riboswitch in this mRNA binds the Pumilio RNA‐binding protein (PUM1) and causes an allosteric RNA rearrangement that reveals an miRNA target. Subsequent miRNA binding results in miRNA‐mediated gene silencing.
2.4. Translation
The initiation of translation can also be regulated by riboswitches. Two mutually exclusive RNA structures are used in this control, one of which sequesters the ribosome binding site (RBS) and diminishes translation efficiency. The second structure leaves the RBS site accessible. One example is a lysine‐responsive riboswitch in Escherichia coli (Fig. 1C) [26]. Upon ligand binding, a structure forms that inhibits translation by sequestering the RBS and that also exposes two RNase E cleavage sites, leading to subsequent mRNA degradation (Fig. 1C).
Regulation of translation by riboswitches can function in concert with other forms of riboswitch regulation to allow precise control of protein expression. For example, cellular tryptophan levels control expression of the trp operon in B. subtilis at the levels of transcription and translation through binding of a protein complex called tryptophan‐activated RNA‐binding attenuation protein (TRAP) [27, 28]. The combination of these two strategies highlights the highly sensitive regulation of gene expression achievable via RNA‐based mechanisms. In the event that metabolite concentrations do not exceed the threshold required to halt mRNA transcription, these same concentrations may trigger translational inhibition. Such dual control may also allow rapid responses to changes in metabolite concentrations.
2.5. Other mechanisms
Although most riboswitches function in cis, a trans‐acting SAM riboswitch was discovered in Listeria monocytogenes [29]. Two of the seven SAM riboswitches in L. monocytogenes (SreA and B) [29] act in trans: These RNAs bind to the 5′ UTR of the prfA virulence factor mRNA and sequester the Shine–Dalgarno sequence to inhibit prfA translation. This trans activity does not require SAM binding and therefore can be considered similar to antisense mechanisms employed by other non‐coding RNAs.
Other unique and varied riboswitch mechanisms have been characterized. For example, some riboswitches function in a cooperative manner [30]. These include the glycine and tetrahydrofolate riboswitches that contain tandem aptamer domains or bind multiple ligand molecules, respectively. These coupled binding strategies provide organisms a means to tune the degree of riboswitch response to ligand concentration. Cooperative binding of two glycine molecules, for example, likely evolved to allow precise response to cellular concentrations of glycine [31]. Another unique example found in L. monocytogenes is an RNA thermosensor. A 5′ UTR structure that inhibits translation of prfA virulence factor at temperatures below 37 °C [32]. Infection of warm host cells results in an RNA rearrangement that allows translation. This use of temperature as a ‘ligand’ demonstrates the wide range of regulation mechanisms achievable by RNA structure.
3. Hijacking the ribosome: IRES elements
3.1. Cap‐dependent translation
Under most conditions, translation initiation in eukaryotes is dependent on a 5′ 7‐methylguanylate cap on a mRNA. Initiation is mediated by eukaryotic initiation factors (eIFs) that are recruited through a series of recognition events (reviewed in [33]). This process begins with recognition and binding of the 5′ cap by the eIF4 cap‐binding complex. This event leads to formation of the 48S initiation complex, a ribonucleoprotein complex containing the 40S ribosomal subunit and several eIFs. This initiation complex scans the mRNA from 5′ to 3′ for an AUG start codon [34, 35]. At the start codon, the complex stalls, the eIFs dissociate, and the remainder of the ribosome is recruited to form an 80S unit. It is only at this point that the elongation stage of translation begins. Initiation is the rate‐limiting step of translation and regulation of this event allows tight control of protein expression. RNA structures play regulatory roles in canonical translation initiation; for example, extensive, stable mRNA structure can interfere with a scanning ribosomal complex [36].
3.2. Cap‐independent translation
Canonical translation initiation is attenuated by cellular mechanisms in response to environmental stimuli such as nutrient stress, mitosis, apoptosis, and viral infection. Even during times of stress, however, synthesis of certain proteins is required, and specific eukaryotic mRNAs use a structure‐based strategy to ‘hijack’ the ribosome and initiate translation through an internal ribosomal entry site (IRES). Viral mRNAs also co‐opt host protein synthesis machinery through cap‐independent binding. At these internal entry sites, typically in the 5′ UTR of a message, RNA structure replaces many or all of the eIF proteins required to recruit the ribosome (Fig. 2 ).
Figure 2.
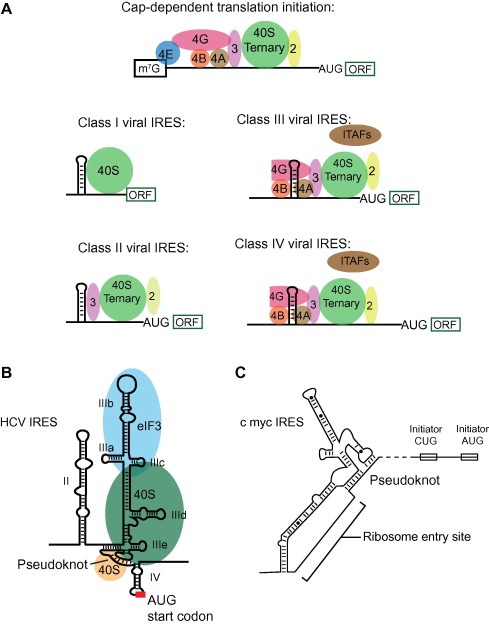
Cap‐dependent translation initiation and classes of IRES‐mediated initiation. (A) Schematic of canonical cap‐dependent initiation [102] and viral IRES classes. (B) Model of HCV IRES initiation. The pseudoknot at the base of domain III mimics a tRNA [51] and may position the AUG in the ribosome active site. (C) Simplified model of the c‐myc IRES element. The ribosome entry site is adjacent to an RNA pseudoknot [102].
Eukaryotic and viral RNAs use IRES elements in strategically different ways. Eukaryotic mRNAs with IRES elements encode proteins required for apoptosis, cell development, oncogenesis and for survival of nutrient stress, hypoxia, heat shock, and viral infection (reviewed in [37]). These conditions coincide with a decline in cap‐dependent translation initiation, and IRES mechanisms allow the cell to respond with synthesis of appropriate proteins. For example, during apoptosis, the mRNA encoding c‐myc, a transcription enhancer important to apoptosis and cancer mechanisms, switches from canonical to IRES‐mediated translation initiation and is actively produced despite inactivation of cap‐dependent translation [38]. This may be a widespread cellular strategy. In silico analyses estimate that nearly 10% of cellular mRNAs have IRES activity [39]. In addition, viruses often contain highly structured 5′ regions incompatible with traditional translation initiation, and may lack the 5′ cap or poly(A) tail required for canonical translation [40]. RNA viruses use IRES elements to proliferate in the cellular environment.
3.3. IRES structures
The first IRES elements were discovered in the positive sense RNAs of picornaviruses and encephalomyocarditis viruses [41, 42]. IRES elements in closely‐related viral species exhibit a high degree of structural conservation, despite large differences in primary sequence, emphasizing an important role for RNA structure in directing protein expression. In contrast, cellular IRES elements show much less sequence and structural conservation, suggesting these elements are more highly specialized.
IRES elements may require eIFs and/or RNA‐binding proteins called IRES trans‐acting factors (ITAFs), whereas others directly bind the ribosome to initiate translation. The number of eIF and ITAF factors required to initiate translation is roughly inversely related to the amount of compactly folded RNA structure in an IRES [43]. Viral IRES elements tend to be more structured than their cellular counterparts and bind fewer co‐factors [44]. Four classes of IRES elements have been defined for Picornaviridae (Fig. 2A). Class I viral IRES elements are self‐sufficient, RNA‐driven systems that do not require any factors for initiation. At the opposite end of the spectrum, Class IV elements require canonical eIFs as well as ITAFs [45].
Perhaps the best characterized Class I viral IRES is from a Dicistroviridae intergenic region. An RNA pseudoknot in this IRES occupies the ribosomal P‐site and mimics the structure of a bound peptidyl tRNA, allowing complete ribosomal assembly [46, 47]. A Class I IRES in Israeli acute bee paralysis virus controls levels of two proteins, one expressed from a +1 reading frame by forming an extra GU base pair in the pseudoknot occupying the P‐site, thereby shifting the frame of the codon:anticodon mimic by one nucleotide [40].
Class II viral IRES domains are less structured. The Class II HCV IRES binds directly to the ribosome [48] but requires canonical elements such as initiator tRNAMet, eIF2, and GTP for translation initiation [49]. The HCV IRES contains several major domains (II, III, and IV) that are conserved among related flaviviruses and some picornaviruses (Fig. 2B). Domain III binds eIF3 and contains a distinct 40S binding site (reviewed in [50]). A pseudoknotted element at the base of domain III is located near the AUG start codon in domain IV (Fig. 2B), and computational modeling suggests that it forms a three‐dimensional structure similar to that of tRNA [51]. This tRNA‐like structure likely interacts with the ribosome, ultimately aligning the start codon in the ribosomal P‐site [48, 49, 51, 52, 53, 54]. The use of mimicry, especially of tRNAs, appears to be a general strategy in IRES function [55].
Class III and Class IV viral IRES elements require more initiation factors than do Class II IRESs and do not directly bind the 40S ribosome. Examples of Group III IRESs are found in foot and mouth disease virus and encephalomyocarditis virus RNAs. Group IV IRES elements include those from poliovirus and hepatitis A virus. An important distinction between these two IRES classes is in their proximity to an AUG start codon. Class III IRESs immediately precede the AUG, whereas Class IV IRESs can be located at significant distance from their target ORFs.
The mRNA encoding the transcription factor c‐myc contains perhaps the best characterized cellular IRES element. Normally, proteins from the myc family control cell proliferation and apoptosis; however, a mutant form is associated with carcinogenesis [56, 57]. As discussed, c‐myc mRNA can use either canonical or IRES‐mediated translation initiation [58]. Use of the IRES element occurs under specific cellular conditions when one of four possible promoter sequences is used to produce a long carcinogenic mRNA [58] (Fig. 2C). RNA pseudoknots, similar to those important to tRNA mimicry in HCV and Dicistroviridae Class I IRESs, may form near the c‐myc ribosome landing sites in the IRES elements [59]. Understanding these RNA structures will be key to determining how these complex protein regulatory pathways function.
4. Derailing translation with programed ribosomal frameshifting
Numerous cellular and viral RNAs undergo non‐canonical translation where the genetic information within the message is “recoded” [60] by signals within the RNA (reviewed [40, 61]). We will highlight the role that RNA structure plays in inducing and regulating one example of non‐canonical translation: programed ribosomal frameshifting.
Ribosomal frameshifting occurs when elements within the RNA cause the ribosome to switch registers to either the −1 or +1 reading frame. Additionally, some studies suggest that −2 and +2 frameshifting events are possible [62, 63]. Frameshifting sites are minimally composed of two distinct parts: a “slippery sequence” and a cis‐acting element. Various cis‐acting elements, such as Shine–Dalgarno‐like sequences and stop codons [64, 65], are capable of inducing frameshifting, but most frameshifting events are mediated by highly structured RNA elements [66, 67, 68]. The cis‐element within the mRNA pauses a translating ribosome's active site over the slippery sequence (Fig. 3 A). The codon–anticodon interactions are momentarily disrupted, and the ribosome advances two or four nucleotides in the case of −1 and +1 frameshifting, respectively, before translation continues. The efficiencies of frameshifting sites vary depending on the features of the elements involved [66, 69, 70]. Even low levels of programmed frameshifting, less than 10%, can have large functional consequences.
Figure 3.
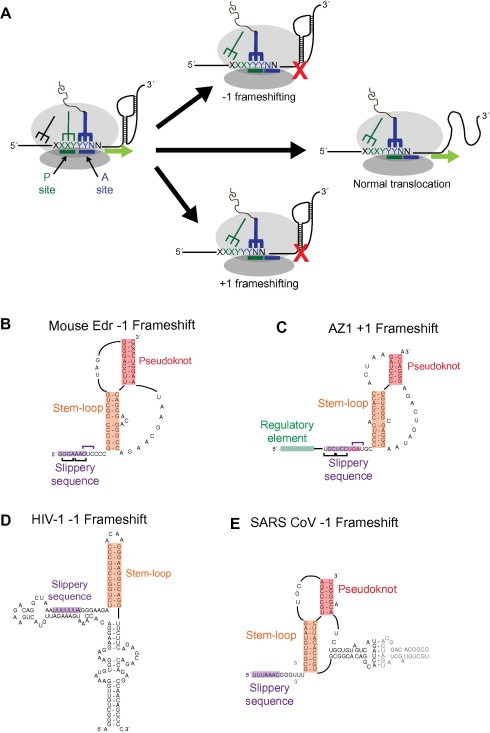
Structural diversity within programmed ribosomal frameshifting elements. (A) Schematic diagram of −1 and +1 programmed frameshifting events. A translating ribosome encounters a slippery sequence upstream of a stable RNA structure. The post‐translocation position of the peptide‐linked tRNA (blue structure) is shown for normal translocation (middle) and −1 and +1 frameshifting events. Proposed secondary structures for (B) the −1 frameshift element for the mouse Edr gene [73], (C) the +1 frameshifting region of the human AZ1 gene [76, 79] (D) the gag‐pol frameshift in HIV‐1 [82], and (E) the replicase‐transcriptase frameshift of SARS‐CoV [84]. In each example, the “slippery sequence” (purple box) is located upstream of a stable structure (orange and red boxes).
RNA pseudoknots are structural motifs that are often found at both −1 and +1 ribosomal frameshifting sites, presumably due to their ability to resist the helicase activity of the ribosome [71]. Pseudoknots contain a minimum of two base‐paired helices; nucleotides within the loop of one helix base pair with nucleotides outside the stem region to form the “pseudoknotted” structure [72]. An example of pseudoknot within a −1 frameshifting element in mammalian cells is found in the mouse embryonal carcinoma differentiation regulated (Edr) gene [73] (Fig. 3B). Edr and the human ortholog PEG10 are highly expressed during development and encode proteins with two overlapping reading frames [74, 75]. A pseudoknot structure also can be found in the +1 frameshifting sites within eukaryotic antizyme genes [76, 77], which encode inhibitors of ornithine decarboxylase [78]. Translation of functional proteins from these genes requires a +1 frameshift [79]. The AZ1 frameshifting site is complex and is composed of a (UGC UCC UGA) slippery sequence flanked by a 5′ regulatory element and pseudoknot (Fig. 3C) [76, 79]. The slippery sequence contains a UGA stop‐codon that will terminate protein translation unless a frameshift occurs. Increased levels of polyamines, the downstream products of ornithine decarboxylase, up regulate levels of frameshifting through action of the 5′ regulatory element [80].
One of the first −1 frameshifting elements identified occurs at the end of the Gag reading frame in HIV‐1 [70] (Fig. 3D). This frameshifting element induces a −1 frameshift for 5–10% of translating ribosomes, allowing for the translation of a gag‐pol fusion protein that contains gag protein fused to the viral protease and reverse transcriptase enzymes [70]. The frameshifting element was initially suggested to consist of the slippery sequence (UUUUUUA) and a simple stem‐loop structure [70]; however, this simple model has been revised as studies of this element suggested that additional sequences are required for biological activity [81]. Studies of the frameshifting element in the context of the intact HIV‐1 genomic RNA suggest that the stem‐loop is part of a larger structured RNA domain (Fig. 3D) [82]. The surrounding structural domain functions to attenuate the frameshifting efficiency [83].
A complex −1 frameshifting element exists within the genome of the SARS corona‐virus (SARS‐CoV) at the end of the first ORF. Frameshifting results in the translation of the SARS‐CoV the replicase‐transcriptase peptide, which is cleaved by proteases to generate up to 16 individual proteins [68]. The programed frameshift in SARS‐CoV appears to regulate the ratios of SARS viral proteins. The frameshifting element in SARS‐CoV contains a stem‐loop in one of the pseudoknot loops (Fig. 3E) [84]. The end of this internal stem‐loop structure contains a palindromic sequence that allows the element to dimerize (Fig. 3E) [85]. Mutations that interfered with dimerization of this domain, both reduced frameshifting efficiency and inhibited viral replication [85]. These data suggest that the frameshifting efficiency within SARS‐CoV is regulated by the ability of the genome to dimerize, connecting two critical steps in the replication cycle of the virus.
The frameshifting elements described here are just a few examples of a rapidly expanding list of frameshifting elements that function in diverse organisms and viruses. RNA structures within these frameshifting elements often have modular properties, and stable stem‐loop structures can substitute for pseudoknots in some elements [86]. The frameshifting efficiency of the core elements can be regulated by the surrounding RNA sequence and structure [83], tertiary interactions [85], and ligands [80] that ultimately create dynamic elements capable of both precisely controlling frameshifting efficiency and responding to conditions within the cellular environment.
5. RNA structure and setting ribosomal speed limits
Folding of a nascent chain of amino acids into functional protein domains begins immediately after the peptide emerges from the ribosome (reviewed in [87, 88]). The rate of translation can significantly impact protein folding [89]. The most extensively studied examples of regulated translational pausing in cells are at rare codons. Rare codons, those codons with low abundance tRNA partners, often occur at the junctions between independently folding protein domains [90]. Ribosomal pausing at these rare codons is thought to facilitate the folding of native proteins in cells [91]. The correlation between translational pausing and rare codon frequency is imperfect, and there is also evidence that mRNA structure modulates translation rates. For example, when individual ribosomes translating an RNA hairpin were monitored using optical tweezers, the length of time that the ribosome paused during translation was dependent on the stability of the RNA structure [92]; stable structures are eventually unwound by the mechanical forces exerted by the ribosome [93].
An extensive body of evidence demonstrating the effect of mRNA structure on translation comes from studies of the yeast ASH1 gene. Ash1p is a transcription factor that is localized to newly formed daughter cells during mitosis and regulates mating‐type switching. During mitosis, four structured elements within the coding sequence of the ASH1 mRNA function both to localize the mRNA to newly forming cells and also to translationally silence the mRNA in the mother cell [94, 95]. These two activities are separable: When the structured RNA elements are moved from the coding sequence to the 3′ UTR, the mRNA is correctly localized, but localization of the Ash1p protein is impaired [96]. The structured elements within the ASH1 mRNA appear to function as ribosome stop‐lights, preventing protein translation until proper localization of the message. Another example of the effect of mRNA structure on protein translation comes from analysis of single nucleotide polymorphisms in the human pain regulator gene catechol‐O‐methyltransferase (COMT) [97]. Polymorphisms within the COMT gene are associated with increased pain sensitivity in humans [98]. The increased pain sensitivity phenotypes are caused by dramatic increases in COMT protein levels due to changes in the global structure of the mRNA [97, 99]. These differences in COMT protein expression are attributed, in part, to structural changes near the start codon that affect translational efficiency of the mRNA [99].
Recent innovations in RNA structure probing technologies have facilitated systematic analyses of RNA structure on genome‐wide scales and examination of relationships between RNA structure and the organization of the encoded protein. The RNA probing technology SHAPE was used to analyze the structure of an HIV‐1 genomic RNA [82]. The regions of the genomic RNA that encode polyprotein linkers and interdomain loops within HIV‐1 proteins are highly structured (Fig. 4 A and B). This direct correlation between the structure of the mRNA and the organization of protein domains suggests that these structured regions of the mRNA function as ribosomal pause sites and facilitate the folding of active viral proteins. The hypothesis that structures within coding regions of mRNAs play an important role in protein folding is also supported by analysis of the mRNA transcriptome in yeast [100]. A technology termed PARS was used to probe the structures of over 3000 yeast mRNAs. There is a correlation between mRNA structure and the organization of coding sequences such that both the start and stop codons generally occur in regions that lack stable structures (Fig. 4C). The internal coding regions of most mRNAs appear to have a significantly higher level of structure than the 5′ and 3′ UTRs. Taken together, these recent studies suggest that RNA structure represents a second layer of information, an RNA structurome [101], that regulates the kinetics of protein production to ensure proper expression and folding.
Figure 4.

Relationships between RNA structure and protein domain organization. (A) Median SHAPE reactivities over 75‐nucleotide windows, which provide a metric for overall RNA structure, for the 5′ end of the HIV‐1 genome [82]. (B) Low SHAPE reactivities, reflecting higher levels of RNA structure, correlate with unstructured protein linkers in the gag polyprotein. Interdomain linkers (green) and the intra‐domain loops (yellow) are illustrated between the independently folding domains of gag (blue, red, pink, and grey structures). (C) PARS scores averaged over 3000 yeast mRNAs for 5′ UTR (green), coding region (blue), and 3′ UTR (red) regions [100].
Protein coding regions within mRNAs are often depicted as long rectangles, as if they were translation highways. The underlying assumption is that once the ribosome clears the obstacles imposed by initiation, translation proceeds uninterrupted until a stop codon is encountered. Current evidence indicates, however, that coding regions within mRNAs are complex landscapes defined by both codon usage and the physical structure of the mRNA.
6. Conclusions
We have presented an overview of strategies by which RNA structure controls and modulates the synthesis and function of proteins. These examples emphasize that structured elements in RNA function as an additional level of information to govern protein expression. Conversely, in the absence of structured RNA regulatory elements, many transcriptional and translational control functions would be lost. Proteins carry out the vast majority of cellular catalytic, signaling, regulatory, and replicative functions. Ultimately, the three‐dimensional structure of an mRNA exerts a great deal of influence over the function of its protein product. The influence and complexity of RNA structure in governing protein expression, synthesis and function can appropriately be considered another level of the genetic code, one that we are likely to have only glimpsed thus far.
Acknowledgements
We are indebted to Liz Dethoff and Andy Lavender for discussions and critical readings of the manuscript. Work in our lab focused on understanding RNA‐based genetic codes is supported by the (AI068462) and NSF (MCB‐1121024). D.M.M. is supported by postdoctoral fellowship from the American Cancer Society (PF‐11‐172‐01‐RMC). N.A.S. is supported by a Ruth L. Kirschstein National Research Service Award (F32 GM 101696‐1).
Mauger David M.,Siegfried Nathan A. and Weeks Kevin M.(2013), The genetic code as expressed through relationships between mRNA structure and protein function, FEBS Letters, 587, doi: 10.1016/j.febslet.2013.03.002
References
- 1. Deigan K.E., Ferre-D‧Amare A.R., Riboswitches: discovery of drugs that target bacterial gene-regulatory RNAs. Acc. Chem. Res., 44, (2011), 1329– 1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ahn D.G., Interference of ribosomal frameshifting by antisense peptide nucleic acids suppresses SARS coronavirus replication. Antiviral Res., 91, (2011), 1– 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marcheschi R.J., Tonelli M., Kumar A., Butcher S.E., Structure of the HIV-1 frameshift site RNA bound to a small molecule inhibitor of viral replication. ACS Chem. Biol., 6, (2011), 857– 864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yu D., Single-stranded RNAs use RNAi to potently and allele-selectively inhibit mutant huntingtin expression. Cell, 150, (2012), 895– 908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Parkesh R., Design of a bioactive small molecule that targets the myotonic dystrophy type 1 RNA via an RNA motif-ligand database and chemical similarity searching. J. Am. Chem. Soc., 134, (2012), 4731– 4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu X., Partial correction of endogenous DeltaF508 CFTR in human cystic fibrosis airway epithelia by spliceosome-mediated RNA trans-splicing. Nat. Biotechnol., 20, (2002), 47– 52. [DOI] [PubMed] [Google Scholar]
- 7. Mandal M., Breaker R.R., Gene regulation by riboswitches. Nat. Rev. Mol. Cell Biol., 5, (2004), 451– 463. [DOI] [PubMed] [Google Scholar]
- 8. Smith A.M., Fuchs R.T., Grundy F.J., Henkin T.M., Riboswitch RNAs: regulation of gene expression by direct monitoring of a physiological signal. RNA Biol., 7, (2010), 104– 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Breaker R.R., Ancient, giant riboswitches at atomic resolution. Nat. Struct. Mol. Biol., 19, (2012), 1208– 1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Frieda K.L., Block S.M., Direct observation of cotranscriptional folding in an adenine riboswitch. Science, 338, (2012), 397– 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gusarov I., Nudler E., The mechanism of intrinsic transcription termination. Mol. Cell, 3, (1999), 495– 504. [DOI] [PubMed] [Google Scholar]
- 12. Yarnell W.S., Roberts J.W., Mechanism of intrinsic transcription termination and antitermination. Science, 284, (1999), 611– 615. [DOI] [PubMed] [Google Scholar]
- 13. Grundy F.J., Winkler W.C., Henkin T.M., tRNA-mediated transcription antitermination in vitro: codon–anticodon pairing independent of the ribosome. Proc. Natl. Acad. Sci. USA, 99, (2002), 11121– 11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mandal M., Breaker R.R., Adenine riboswitches and gene activation by disruption of a transcription terminator. Nat. Struct. Mol. Biol., 11, (2004), 29– 35. [DOI] [PubMed] [Google Scholar]
- 15. Rodionov D.A., Vitreschak A.G., Mironov A.A., Gelfand M.S., Comparative genomics of thiamin biosynthesis in procaryotes. New genes and regulatory mechanisms. J. Biol. Chem., 277, (2002), 48949– 48959. [DOI] [PubMed] [Google Scholar]
- 16. Sudarsan N., Barrick J.E., Breaker R.R., Metabolite-binding RNA domains are present in the genes of eukaryotes. RNA, 9, (2003), 644– 647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miranda-Rios J., Navarro M., Soberon M., A conserved RNA structure (thi box) is involved in regulation of thiamin biosynthetic gene expression in bacteria. Proc. Natl. Acad. Sci. USA, 98, (2001), 9736– 9741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kubodera T., Watanabe M., Yoshiuchi K., Yamashita N., Nishimura A., Nakai S., Gomi K., Hanamoto H., Thiamine-regulated gene expression of Aspergillus oryzae thiA requires splicing of the intron containing a riboswitch-like domain in the 5′-UTR. FEBS Lett., 555, (2003), 516– 520. [DOI] [PubMed] [Google Scholar]
- 19. Cheah M.T., Wachter A., Sudarsan N., Breaker R.R., Control of alternative RNA splicing and gene expression by eukaryotic riboswitches. Nature, 447, (2007), 497– 500. [DOI] [PubMed] [Google Scholar]
- 20. Croft M.T., Moulin M., Webb M.E., Smith A.G., Thiamine biosynthesis in algae is regulated by riboswitches. Proc. Natl. Acad. Sci. USA, 104, (2007), 20770– 20775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bocobza S., Adato A., Mandel T., Shapira M., Nudler E., Aharoni A., Riboswitch-dependent gene regulation and its evolution in the plant kingdom. Genes Dev., 21, (2007), 2874– 2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barrick J.E., Breaker R.R., The distributions, mechanisms, and structures of metabolite-binding riboswitches. Genome Biol., 8, (2007), R239– [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li S., Breaker R.R., Eukaryotic TPP riboswitch regulation of alternative splicing involving long-distance base pairing. Nucleic Acids Res., 41, (2013), 3022– 3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ferre-D‧Amare A.R., The glmS ribozyme: use of a small molecule coenzyme by a gene-regulatory RNA. Q. Rev. Biophys., 43, (2010), 423– 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kedde M., van Kouwenhove M., Zwart W., Oude Vrielink J.A., Elkon R., Agami R., A Pumilio-induced RNA structure switch in p27-3′ UTR controls miR-221 and miR-222 accessibility. Nat. Cell Biol., 12, (2010), 1014– 1020. [DOI] [PubMed] [Google Scholar]
- 26. Caron M.P., Bastet L., Lussier A., Simoneau-Roy M., Masse E., Lafontaine D.A., Dual-acting riboswitch control of translation initiation and mRNA decay. Proc. Natl. Acad. Sci. USA, 109, (2012), E3444– E3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Babitzke P., Stults J.T., Shire S.J., Yanofsky C., TRAP, the trp RNA-binding attenuation protein of Bacillus subtilis, is a multisubunit complex that appears to recognize G/UAG repeats in the trpEDCFBA and trpG transcripts. J. Biol. Chem., 269, (1994), 16597– 16604. [PubMed] [Google Scholar]
- 28. Babitzke P., Yanofsky C., Reconstitution of Bacillus subtilis trp attenuation in vitro with TRAP, the trp RNA-binding attenuation protein. Proc. Natl. Acad. Sci. USA, 90, (1993), 133– 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Loh E., A trans-acting riboswitch controls expression of the virulence regulator PrfA in Listeria monocytogenes . Cell, 139, (2009), 770– 779. [DOI] [PubMed] [Google Scholar]
- 30. Serganov A., Patel D.J., Molecular recognition and function of riboswitches. Curr. Opin. Struct. Biol., 22, (2012), 279– 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mandal M., Lee M., Barrick J.E., Weinberg Z., Emilsson G.M., Ruzzo W.L., Breaker R.R., A glycine-dependent riboswitch that uses cooperative binding to control gene expression. Science, 306, (2004), 275– 279. [DOI] [PubMed] [Google Scholar]
- 32. Johansson J., Mandin P., Renzoni A., Chiaruttini C., Springer M., Cossart P., An RNA thermosensor controls expression of virulence genes in Listeria monocytogenes . Cell, 110, (2002), 551– 561. [DOI] [PubMed] [Google Scholar]
- 33. Jackson R.J., Hellen C.U., Pestova T.V., The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol., 11, (2010), 113– 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hinnebusch A.G., Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol. Mol. Biol. Rev., 75, (2011), 434– 436. first page of table of contents [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kozak M., An analysis of 5′-non-coding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res., 15, (1987), 8125– 8148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kozak M., Circumstances and mechanisms of inhibition of translation by secondary structure in eucaryotic mRNAs. Mol. Cell. Biol., 9, (1989), 5134– 5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fitzgerald K.D., Semler B.L., Bridging IRES elements in mRNAs to the eukaryotic translation apparatus. Biochim. Biophys. Acta, 2009, (1789), 518– 528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stoneley M., Paulin F.E., Le Quesne J.P., Chappell S.A., Willis A.E., C-Myc 5′ untranslated region contains an internal ribosome entry segment. Oncogene, 16, (1998), 423– 428. [DOI] [PubMed] [Google Scholar]
- 39. Spriggs K.A., Stoneley M., Bushell M., Willis A.E., Re-programming of translation following cell stress allows IRES-mediated translation to predominate. Biol. Cell, 100, (2008), 27– 38. [DOI] [PubMed] [Google Scholar]
- 40. Firth A.E., Brierley I., Non-canonical translation in RNA viruses. J. Gen. Virol., 93, (2012), 1385– 1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pelletier J., Sonenberg N., Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature, 334, (1988), 320– 325. [DOI] [PubMed] [Google Scholar]
- 42. Jang S.K., Krausslich H.G., Nicklin M.J., Duke G.M., Palmenberg A.C., Wimmer E., A segment of the 5′ non-translated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J. Virol., 62, (1988), 2636– 2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Filbin M.E., Kieft J.S., Toward a structural understanding of IRES RNA function. Curr. Opin. Struct. Biol., 19, (2009), 267– 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hellen C.U., Sarnow P., Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev., 15, (2001), 1593– 1612. [DOI] [PubMed] [Google Scholar]
- 45. Kieft J.S., Viral IRES RNA structures and ribosome interactions. Trends Biochem. Sci., 33, (2008), 274– 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schuler M., Structure of the ribosome-bound cricket paralysis virus IRES RNA. Nat. Struct. Mol. Biol., 13, (2006), 1092– 1096. [DOI] [PubMed] [Google Scholar]
- 47. Spahn C.M., Jan E., Mulder A., Grassucci R.A., Sarnow P., Frank J., Cryo-EM visualization of a viral internal ribosome entry site bound to human ribosomes: the IRES functions as an RNA-based translation factor. Cell, 118, (2004), 465– 475. [DOI] [PubMed] [Google Scholar]
- 48. Spahn C.M., Kieft J.S., Grassucci R.A., Penczek P.A., Zhou K., Doudna J.A., Frank J., Hepatitis C virus IRES RNA-induced changes in the conformation of the 40s ribosomal subunit. Science, 291, (2001), 1959– 1962. [DOI] [PubMed] [Google Scholar]
- 49. Pestova T.V., Shatsky I.N., Fletcher S.P., Jackson R.J., Hellen C.U., A prokaryotic-like mode of cytoplasmic eukaryotic ribosome binding to the initiation codon during internal translation initiation of hepatitis C and classical swine fever virus RNAs. Genes Dev., 12, (1998), 67– 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lukavsky P.J., Structure and function of HCV IRES domains. Virus Res., 139, (2009), 166– 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lavender C.A., Ding F., Dokholyan N.V., Weeks K.M., Robust and generic RNA modeling using inferred constraints: a structure for the hepatitis C virus IRES pseudoknot domain. Biochemistry, 49, (2010), 4931– 4933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Boehringer D., Thermann R., Ostareck-Lederer A., Lewis J.D., Stark H., Structure of the hepatitis C virus IRES bound to the human 80S ribosome: remodeling of the HCV IRES. Structure, 13, (2005), 1695– 1706. [DOI] [PubMed] [Google Scholar]
- 53. Otto G.A., Puglisi J.D., The pathway of HCV IRES-mediated translation initiation. Cell, 119, (2004), 369– 380. [DOI] [PubMed] [Google Scholar]
- 54. Fraser C.S., Hershey J.W., Doudna J.A., The pathway of hepatitis C virus mRNA recruitment to the human ribosome. Nat. Struct. Mol. Biol., 16, (2009), 397– 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hammond J.A., Rambo R.P., Filbin M.E., Kieft J.S., Comparison and functional implications of the 3D architectures of viral tRNA-like structures. RNA, 15, (2009), 294– 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Paulin F.E., West M.J., Sullivan N.F., Whitney R.L., Lyne L., Willis A.E., Aberrant translational control of the c-myc gene in multiple myeloma. Oncogene, 13, (1996), 505– 513. [PubMed] [Google Scholar]
- 57. Pelengaris S., Khan M., Evan G., C-Myc: more than just a matter of life and death. Nat. Rev. Cancer, 2, (2002), 764– 776. [DOI] [PubMed] [Google Scholar]
- 58. Nanbru C., Lafon I., Audigier S., Gensac M.C., Vagner S., Huez G., Prats A.C., Alternative translation of the proto-oncogene c-myc by an internal ribosome entry site. J. Biol. Chem., 272, (1997), 32061– 32066. [DOI] [PubMed] [Google Scholar]
- 59. Jopling C.L., Spriggs K.A., Mitchell S.A., Stoneley M., Willis A.E., L-Myc protein synthesis is initiated by internal ribosome entry. RNA, 10, (2004), 287– 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gesteland R.F., Weiss R.B., Atkins J.F., Recoding: reprogrammed genetic decoding. Science, 257, (1992), 1640– 1641. [DOI] [PubMed] [Google Scholar]
- 61. Namy O., Rousset J.P., Napthine S., Brierley I., Reprogrammed genetic decoding in cellular gene expression. Mol. Cell, 13, (2004), 157– 168. [DOI] [PubMed] [Google Scholar]
- 62. Fang Y., Efficient -2 frameshifting by mammalian ribosomes to synthesize an additional arterivirus protein. Proc. Natl. Acad. Sci. USA, 109, (2012), E2920– E2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chung B.Y., Miller W.A., Atkins J.F., Firth A.E., An overlapping essential gene in the Potyviridae. Proc. Natl. Acad. Sci. USA, 105, (2008), 5897– 5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Craigen W.J., Caskey C.T., Expression of peptide chain release factor 2 requires high-efficiency frameshift. Nature, 322, (1986), 273– 275. [DOI] [PubMed] [Google Scholar]
- 65. Mejlhede N., Atkins J.F., Neuhard J., Ribosomal −1 frameshifting during decoding of Bacillus subtilis cdd occurs at the sequence CGA AAG. J. Bacteriol., 181, (1999), 2930– 2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jacks T., Varmus H.E., Expression of the Rous sarcoma virus pol gene by ribosomal frameshifting. Science, 230, (1985), 1237– 1242. [DOI] [PubMed] [Google Scholar]
- 67. Jacks T., Madhani H.D., Masiarz F.R., Varmus H.E., Signals for ribosomal frameshifting in the Rous sarcoma virus gag-pol region. Cell, 55, (1988), 447– 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Marra M.A., The genome sequence of the SARS-associated coronavirus. Science, 300, (2003), 1399– 1404. [DOI] [PubMed] [Google Scholar]
- 69. Cao S., Chen S.J., Predicting ribosomal frameshifting efficiency. Phys. Biol., 5, (2008), 016002– [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jacks T., Power M.D., Masiarz F.R., Luciw P.A., Barr P.J., Varmus H.E., Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature, 331, (1988), 280– 283. [DOI] [PubMed] [Google Scholar]
- 71. Tholstrup J., Oddershede L.B., Sorensen M.A., MRNA pseudoknot structures can act as ribosomal roadblocks. Nucleic Acids Res., 40, (2012), 303– 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pleij C.W., Rietveld K., Bosch L., A new principle of RNA folding based on pseudoknotting. Nucleic Acids Res., 13, (1985), 1717– 1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Manktelow E., Shigemoto K., Brierley I., Characterization of the frameshift signal of Edr, a mammalian example of programmed −1 ribosomal frameshifting. Nucleic Acids Res., 33, (2005), 1553– 1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shigemoto K., Brennan J., Walls E., Watson C.J., Stott D., Rigby P.W., Reith A.D., Identification and characterisation of a developmentally regulated mammalian gene that utilises −1 programmed ribosomal frameshifting. Nucleic Acids Res., 29, (2001), 4079– 4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ono R., Kobayashi S., Wagatsuma H., Aisaka K., Kohda T., Kaneko-Ishino T., Ishino F., A retrotransposon-derived gene, PEG10, is a novel imprinted gene located on human chromosome 7q21. Genomics, 73, (2001), 232– 237. [DOI] [PubMed] [Google Scholar]
- 76. Ivanov I.P., Gesteland R.F., Atkins J.F., Antizyme expression: a subversion of triplet decoding, which is remarkably conserved by evolution, is a sensor for an autoregulatory circuit. Nucleic Acids Res., 28, (2000), 3185– 3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bekaert M., Ivanov I.P., Atkins J.F., Baranov P.V., Ornithine decarboxylase antizyme finder (OAF): fast and reliable detection of antizymes with frameshifts in mRNAs. BMC Bioinf., 9, (2008), 178– [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Murakami Y., Matsufuji S., Kameji T., Hayashi S., Igarashi K., Tamura T., Tanaka K., Ichihara A., Ornithine decarboxylase is degraded by the 26S proteasome without ubiquitination. Nature, 360, (1992), 597– 599. [DOI] [PubMed] [Google Scholar]
- 79. Matsufuji S., Matsufuji T., Miyazaki Y., Murakami Y., Atkins J.F., Gesteland R.F., Hayashi S., Autoregulatory frameshifting in decoding mammalian ornithine decarboxylase antizyme. Cell, 80, (1995), 51– 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Petros L.M., Howard M.T., Gesteland R.F., Atkins J.F., Polyamine sensing during antizyme mRNA programmed frameshifting. Biochem. Biophys. Res. Commun., 338, (2005), 1478– 1489. [DOI] [PubMed] [Google Scholar]
- 81. Dulude D., Baril M., Brakier-Gingras L., Characterization of the frameshift stimulatory signal controlling a programmed −1 ribosomal frameshift in the human immunodeficiency virus type 1. Nucleic Acids Res., 30, (2002), 5094– 5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Watts J.M., Dang K.K., Gorelick R.J., Leonard C.W., Bess J.W. Jr., Swanstrom R., Burch C.L., Weeks K.M., Architecture and secondary structure of an entire HIV-1 RNA genome. Nature, 460, (2009), 711– 716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mouzakis K.D., Lang A.L., Vander Meulen K.A., Easterday P.D., Butcher S.E., HIV-1 frameshift efficiency is primarily determined by the stability of base pairs positioned at the mRNA entrance channel of the ribosome. Nucleic Acids Res., 41, (2013), 1901– 1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Baranov P.V., Henderson C.M., Anderson C.B., Gesteland R.F., Atkins J.F., Howard M.T., Programmed ribosomal frameshifting in decoding the SARS-CoV genome. Virology, 332, (2005), 498– 510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ishimaru D., RNA dimerization plays a role in ribosomal frameshifting of the SARS coronavirus. Nucleic Acids Res., 41, (2012), 2594– 2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Yu C.H., Noteborn M.H., Pleij C.W., Olsthoorn R.C., Stem-loop structures can effectively substitute for an RNA pseudoknot in −1 ribosomal frameshifting. Nucleic Acids Res., 39, (2011), 8952– 8959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Cabrita L.D., Dobson C.M., Christodoulou J., Protein folding on the ribosome. Curr. Opin. Struct. Biol., 20, (2010), 33– 45. [DOI] [PubMed] [Google Scholar]
- 88. Komar A.A., A pause for thought along the co-translational folding pathway. Trends Biochem. Sci., 34, (2009), 16– 24. [DOI] [PubMed] [Google Scholar]
- 89. Huard F.P., Deane C.M., Wood G.R., Modelling sequential protein folding under kinetic control. Bioinformatics, 22, (2006), e203– e210. [DOI] [PubMed] [Google Scholar]
- 90. Purvis I.J., Bettany A.J., Santiago T.C., Coggins J.R., Duncan K., Eason R., Brown A.J., The efficiency of folding of some proteins is increased by controlled rates of translation in vivo. A hypothesis. J. Mol. Biol., 193, (1987), 413– 417. [DOI] [PubMed] [Google Scholar]
- 91. Kimchi-Sarfaty C., Oh J.M., Kim I.W., Sauna Z.E., Calcagno A.M., Ambudkar S.V., Gottesman M.M., A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science, 315, (2007), 525– 528. [DOI] [PubMed] [Google Scholar]
- 92. Wen J.D., Lancaster L., Hodges C., Zeri A.C., Yoshimura S.H., Noller H.F., Bustamante C., Tinoco I., Following translation by single ribosomes one codon at a time. Nature, 452, (2008), 598– 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Qu X., Wen J.D., Lancaster L., Noller H.F., Bustamante C., Tinoco I. Jr., The ribosome uses two active mechanisms to unwind messenger RNA during translation. Nature, 475, (2011), 118– 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chartrand P., Meng X.H., Singer R.H., Long R.M., Structural elements required for the localization of ASH1 mRNA and of a green fluorescent protein reporter particle in vivo. Curr. Biol., 9, (1999), 333– 336. [DOI] [PubMed] [Google Scholar]
- 95. Gonzalez I., Buonomo S.B., Nasmyth K., von Ahsen U., ASH1 mRNA localization in yeast involves multiple secondary structural elements and Ash1 protein translation. Curr. Biol., 9, (1999), 337– 340. [DOI] [PubMed] [Google Scholar]
- 96. Chartrand P., Meng X.H., Huttelmaier S., Donato D., Singer R.H., Asymmetric sorting of ash1p in yeast results from inhibition of translation by localization elements in the mRNA. Mol. Cell, 10, (2002), 1319– 1330. [DOI] [PubMed] [Google Scholar]
- 97. Nackley A.G., Shabalina S.A., Tchivileva I.E., Satterfield K., Korchynskyi O., Makarov S.S., Maixner W., Diatchenko L., Human catechol-O-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science, 314, (2006), 1930– 1933. [DOI] [PubMed] [Google Scholar]
- 98. Diatchenko L., Genetic basis for individual variations in pain perception and the development of a chronic pain condition. Hum. Mol. Genet., 14, (2005), 135– 143. [DOI] [PubMed] [Google Scholar]
- 99. Tsao D., Shabalina S.A., Gauthier J., Dokholyan N.V., Diatchenko L., Disruptive mRNA folding increases translational efficiency of catechol-O-methyltransferase variant. Nucleic Acids Res., 39, (2011), 6201– 6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kertesz M., Wan Y., Mazor E., Rinn J.L., Nutter R.C., Chang H.Y., Segal E., Genome-wide measurement of RNA secondary structure in yeast. Nature, 467, (2010), 103– 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Westhof E., Romby P., The RNA structurome: high-throughput probing. Nat. Methods, 7, (2010), 965– 967. [DOI] [PubMed] [Google Scholar]
- 102. Le Quesne J.P., Stoneley M., Fraser G.A., Willis A.E., Derivation of a structural model for the c-myc IRES. J. Mol. Biol., 310, (2001), 111– 126. [DOI] [PubMed] [Google Scholar]