

Abstract
In patients with chronic kidney disease (CKD), loss of cellular proteins increases the risks of morbidity and mortality. Persistence of muscle protein catabolism in CKD results in striking losses of muscle proteins as whole-body protein turnover is great; even small but persistent imbalances between protein synthesis and degradation cause substantial protein loss. No reliable methods to prevent CKD-induced muscle wasting currently exist, but mechanisms that control cellular protein turnover have been identified, suggesting that therapeutic strategies will be developed to suppress or block protein loss. Catabolic pathways that cause protein wasting include activation of the ubiquitin–proteasome system (UPS), caspase-3, lysosomes and myostatin (a negative regulator of skeletal muscle growth). These pathways can be initiated by complications associated with CKD, such as metabolic acidosis, defective insulin signalling, inflammation, increased angiotensin II levels, abnormal appetite regulation and impaired microRNA responses. Inflammation stimulates cellular signalling pathways that activate myostatin, which accelerates UPS-mediated catabolism. Blocking this pathway can prevent loss of muscle proteins. Myostatin inhibition could yield new therapeutic directions for blocking muscle protein wasting in CKD or disorders associated with its complications.
Introduction
A decline in the protein content of the body as a result of ageing or catabolic diseases increases the risks of morbidity and mortality.1,2 In chronic kidney disease (CKD), mortality is related to loss of muscle mass.3 These associations lead to two important questions: first, how are protein stores lost, and second, how can the losses be prevented? The excessive risks of mortality and morbidity in patients with CKD have been widely attributed to malnutrition.4,5 This conclusion is primarily based on the frequent presence of hypoalbuminaemia and reports that some patients with progressively severe CKD spontaneously restrict their dietary protein.6–9 However, epidemiological evaluations have concluded that the excessive morbidity and mortality of patients with CKD is rarely attributable to malnutrition.7,10–12 Specifically, if malnutrition was the cause of lost protein stores in these patients, then simply altering their diet should correct the excessive morbidity and mortality.10 This conclusion was examined by Ikizler and colleagues in a series of elegant experiments based on measurements of protein synthesis and degradation in patients on chronic haemodialysis before, during and 2 h after completing a dialysis session.13 The haemodialysis procedure stimulated protein degradation and reduced protein synthesis. These responses persisted for 2 h following dialysis, suggesting that a process causing protein loss was initiated by the therapy and persisted. Although increasing the intake of protein and calories improved protein turnover, it did not fully correct the responses to haemodialysis.13–16 These results indicate that uraemia or the haemodialysis process activates a mechanism of cellular protein catabolism. Increasing dietary protein will not eliminate CKD-stimulated protein loss unless the catabolic mechanism is blocked. A similar conclusion was reached following a 1-year randomized controlled trial of responses of patients on haemodialysis to intradialytic parenteral nutrition given in conjunction with oral nutritional supplements.17 This intervention did not improve 2-year mortality, BMI, laboratory markers of nutritional status or the rate of hospitalization when compared with a control group of patients who were given only the oral supplement. We do not interpret these reports as negating the importance of concentrating on dietary factors in the treatment of patients with CKD because lack of attention to diet will lead to complications, including metabolic acidosis, alterations in bone metabolism and the accumulation of uraemic toxins.18,19 However, these clinical data, in addition to measurements of muscle metabolism in experimental models of CKD, indicate that activation of cellular mechanisms that stimulate loss of protein stores contributes to CKD-induced muscle atrophy.
Regarding hypoalbuminaemia in CKD, low serum albumin levels are inversely correlated with mortality in patients on haemodialysis.6 This observation led to the proposal that malnutrition caused hypoalbuminaemia in patients with CKD. However, other mechanisms can also affect serum albumin levels.20 For example, a study of patients on haemodialysis showed that a low serum albumin level is more closely related to the presence of circulating proinflammatory markers than to changes in dietary protein.21 Moreover, young women with anorexia nervosa who had lost nearly 21% of their lean body mass had almost normal values of serum albumin.22 These results indicate that the cause of hypoalbuminaemia, as well as the loss of muscle mass, in patients with CKD involves more complex mechanisms than just provision of dietary factors. In this Review, we describe how CKD stimulates catabolic pathways that interfere with cellular protein metabolism. Knowledge of these pathways might enable the development of therapies to block muscle wasting in CKD and other catabolic conditions.
Mechanisms of muscle loss
Characteristics of normal protein turnover
Cellular proteins in the cytosol, nucleus and organelles are continually degraded and replaced by protein synthesis. Rates of these processes differ widely as some enzymes have half-lives of minutes, some proteins last for days and structural proteins are even more stable. The average rate of protein turnover also varies among tissues; human liver cells are replaced every few days, whereas replacement of proteins in muscles or the brain occurs over weeks. The amount of intracellular protein that is turned over each day is very large. In a healthy adult weighing 70 kg, about 280 g of protein is degraded and synthesized per day; the majority of this protein is intracellular. By contrast, the amount of plasma protein turned over, including albumin, is estimated at only 21 g per day.23
This considerable variability in protein turnover means that the proteolytic machinery must be highly selective and tightly regulated. Accelerated breakdown of an essential protein or a short-lived regulatory protein would irreversibly alter the function of the cell. Notably, the overall rates of protein degradation and synthesis in tissues must be equal or protein losses will occur. Even a small decrease in synthesis or degradation would cause substantial losses of body protein stores, as occurs in patients with CKD, cancer and other catabolic conditions. In addition to the requirement for precise timing of the degradation of proteins that regulate cellular function, loss of large amounts of protein from muscle might act to supply amino acids, affecting growth and energy homeostasis.23 Catabolic conditions are now recognized to stimulate loss of lean body mass by activating cellular processes and specific proteases. These catabolic processes commonly occur in experimental models of muscle wasting, so a goal of investigators is to identify strategies that will block muscle wasting in various conditions.
Mechanisms of muscle growth and repair
Loss of muscle protein stores can result from three responses: impaired growth of new muscle fibres, suppression of protein synthesis or stimulation of protein degradation. The ability to promote growth of muscles is a prime example of a highly regulated process. Mature muscles consist of muscle fibres that are surrounded by a basal lamina, which covers not only myofibrils, but also satellite cells (also known as muscle precursor cells).24,25 These cells are usually quiescent, but in response to muscle injury or to changes in levels of growth factors (for example insulin-like growth factor 1 [IGF-1]), they begin to express the myogenic regulatory factors Myf-5 and myoblast determination protein 1 (MyoD).25 The expression of these factors is followed by satellite cell proliferation and differentiation, resulting in myoblasts and, subsequently, myocytes (Figure 1). The myocytes undergo fusion and further differentiation to form myofibrils and can, therefore, repair or enlarge myofibrils.
Figure 1.

Stages of skeletal muscle regeneration. Specific biomarkers can be detected at each stage of myogenesis. Satellite cells are normally quiescent, but when stimulated by factors such as insulin-like growth factor 1, they express myogenic regulatory factors, including myoblast determination protein 1. Expression of these factors leads to proliferation and differentiation of satellite cells into myoblasts and myocytes that can fuse to form myotubes or myofibrils. Abbreviations: Myf, myogenic factor; Pax, paired box protein.
The influence of CKD on growth of muscle or the function of satellite cells has received limited attention, but some evidence supports that satellite-cell function is impaired by CKD. In a mouse model of CKD, levels of MyoD and myogenin—myogenic cellular factors released from satellite cells—were reduced.24 This reduction was associated with decreased satellite-cell proliferation and differentiation.24 In examining the mechanism underlying the CKD-induced decrease in satellite cell function, two factors were identified. Firstly, CKD impairs the ability of IGF-1 to regulate muscle protein synthesis and degradation.26 Secondly, satellite-cell function is similarly impaired in mice with partial deletion of the IGF-1 receptor and in mice with CKD. Zhang et al.24 not only identified that satellite cell dysfunction is linked to reduced IGF-1 activity, but also showed that mice with CKD or those with an inducible, partial deletion of Igf1 develop fibrosis in their muscles. As fibrosis weakens muscles, it will be interesting to determine whether patients with CKD also develop muscle fibrosis and if this contributes to their morbidity.
Protein synthesis
CKD-induced suppression of protein synthesis is a second potential mechanism causing muscle wasting. Measurements of the turnover of a labelled amino acid in patients and experimental models of CKD revealed that both leucine oxidation and nonoxidative leucine disposal (an in vivo index of protein synthesis) were significantly reduced in the setting of CKD compared with control values.27 To examine how CKD affects the turnover of muscle proteins, synthetic rates of three types of protein were measured: mixed muscle proteins, myosin and mitochondrial proteins. Compared with healthy individuals (n = 10), patients with CKD (n = 12) had a 27% lower rate of synthesis of mixed muscle proteins and a 37% lower rate of synthesis of myosin, whereas rates of synthesis of mitochondrial proteins were similar.28 Interestingly, the stimulation of protein synthesis depends on the sensing of extracellular (plasma) rather than intramuscular essential amino acid concentration.29
When mechanisms that might cause muscle wasting in a rat model of CKD were investigated, rates of protein synthesis in isolated, perfused muscles were found to be substantially lower than rates of net protein degradation.30 In the muscles of a mouse model of CKD that was subjected to exercise, levels of the markers of protein synthesis serine/threonine-protein kinase mTOR and ribosomal protein S6 kinase (p70S6K) were decreased.31 Subsequently, the development of metabolic acidosis in rats with CKD was found to cause loss of muscle protein stores, mainly because protein breakdown was stimulated whereas protein synthesis was minimally suppressed. Moreover, when the acidosis caused by CKD was corrected, muscle protein degradation decreased. Protein synthesis increased minimally, suggesting that the major effect of acidification on muscle-protein turnover is stimulation of protein degradation.32,33 Measurements of amino-acid turnover have revealed that protein synthesis is suppressed during haemodialysis,29,34 whereas acidosis increases the degradation of protein and the essential amino acid leucine.35 Notably, both responses were blocked by administering sodium bicarbonate. Acidosis quickly stimulates protein losses as shown by Ballmer et al.36 who induced acidosis by feeding ammonium chloride to healthy adults and found that albumin synthesis was suppressed and the individuals developed negative protein balance after only 48 h. On the basis of results obtained during a primed constant infusion of L-[1-13C]-leucine and L-[ring-2H5]phenylalanine, Ikizler et al.13 concluded that haemodialysis can improve whole-body protein synthesis, but as noted above, this improvement is counterbalanced by the catabolic responses to haemodialysis, resulting in proteolysis of body proteins and muscles. Thus, CKD-induced activation of protein catabolism is a more prominent cause of muscle wasting than is decreased protein synthesis.
Protein degradation by specific proteases
A third mechanism resulting in a gain or loss of muscle proteins is a change in the rate of protein degradation. The major mechanism that degrades proteins in muscle (and in other tissues) is the ubiquitin–proteasome system (UPS; Figure 2).23,37 As expected, the degradation of protein by the UPS is remarkably specific.38 The importance of the UPS in degrading proteins that impact cellular functions was recognized when the 2004 Nobel Prize in Chemistry was awarded to Hershko, Ciechanover and Rose.23,39,40
Figure 2.
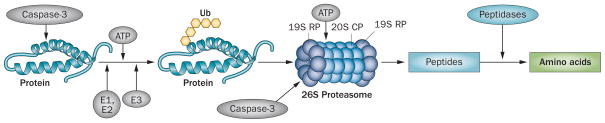
Chronic kidney disease-induced protein degradation by the UPS. An initial step in the degradation cascade involves cleavage of the complex structure of muscle protein by caspase-3, which produces substrates for degradation. Protein substrates are conjugated to Ub by an ATP-dependent process involving the enzymes E1, E2 and E3. The selectivity of protein substrates principally depends on recognition of the protein to be degraded by specific E3 Ub-ligases (for example, TRIM63 for muscle proteins). After five Ub proteins are attached to the protein substrate, the complex can be recognized by the 26S proteasome, which releases Ubs, unfolds the protein substrate and ‘injects’ it into the 20S CP in which proteins are degraded to peptides. At this stage, caspase-3 also cleaves the 26S protease regulatory subunit 4 and 26S protease regulatory subunit 8, which are specific subunit proteins of the 19S proteasome RP. This reaction stimulates degradation of proteins in the 20S proteasome CP. Peptides released into the cytoplasm are degraded into amino acids. Abbreviations: CP, core particle; RP, regulatory particle; Ub, ubiquitin; UPS, ubiquitin–proteasome system.
The ubiquitin–proteasome system
Protein degradation must be tightly regulated because proteins that regulate transcription or metabolic pathways and those that participate in protein scaffolding have vastly different half-lives. If removal of a protein is not precise, cell function and even survival could be jeopardized. It is somewhat surprising, therefore, that most intracellular proteins are degraded by the same ATP-dependent system—the UPS. The activity of this system is regulated at several steps and begins by marking proteins to be degraded (Figure 2).37 For example, a protein destined for degradation is first marked by the addition of ubiquitin, a small member of the heat shock protein family. This reaction requires ATP and is initiated by a single E1 ubiquitin-activating enzyme followed by interaction with one of about 20 E2 ubiquitin-carrier proteins that can interact with an E3 ubiquitin-ligase enzyme. In the presence of ATP, the activated ubiquitin is transferred to a lysine of the substrate protein or to lysines that are contained in ubiquitin. The process is repeated until a chain of five ubiquitin proteins forms a complex that is recognized by the 26S proteasome and the marked protein is subsequently degraded. Specificity in these reactions is possible because >1,000 E3 ubiquitin-ligases exist and each recognizes specific proteins or a specific class of proteins and marks them for destruction with ubiquitin. Specific sequences or structures of the E3 ubiquitin-ligases, including HECT, CHIP and RING finger domains, are responsible for this recognition and conjugation.37
Degradation of a marked protein takes place in the 26S proteasome, a complex consisting of >60 subunit proteins.32,37 The complex is composed of a 20S barrel-shaped core and 19S regulatory particles at either or both ends of the 20S proteasome. Notably, the 19S particles consist of many subunits, which recognize the polyubiquitin chains that are bound to proteins. Using ATP, the 19S particles cleave the polyubiquitin chain from the substrate protein to release ubiquitin, which can be recycled to mark other substrate proteins. The 19S particles use ATP to unfold proteins so the protein can then be ‘injected’ into the 20S particle where it is converted to peptides, which are released into the cytoplasm and degraded by cytoplasmic peptidases.41 The 20S proteasome is a cylinder-like structure composed of four stacked rings. The ringed structure forms a narrow, gated pore through which substrates that have been unfolded can enter and the proteolytic products exit.42 Another unique feature of protein degradation in the 26S proteasome is that peptide-bond cleavage occurs via a hydroxyl moiety present in threonines at each proteolytic site. Because threonine-based proteolysis is so unusual, highly specific inhibitors (for example MG132, lactacystin and epoxomicin) have been synthesized. One proteasome inhibitor, bortezomib, has become a successful chemotherapeutic drug for the treatment of multiple myeloma.
Caspase-3
The discovery that the activities of caspase-3 and the UPS work together to stimulate muscle wasting indicates how complex proteolytic processes can achieve increased proteolysis while maintaining specificity. Caspase-3 has two types of involvement. Firstly, caspase-3 is activated by catabolic conditions (such as CKD) and it acts to cleave the complex structure of muscle proteins, yielding substrates for the UPS.43,44 This activity of caspase-3 is needed because the UPS digests the complex structure of muscle (that is, actomyosin or myofibrils) slowly whereas it degrades monomeric myosin or actin rapidly. Interestingly, caspase-3 function has been identified in muscle biopsy samples from animal models and patients with catabolic conditions.43,44 Specifically, stimulation of caspase-3 proteolytic activity leaves behind a marker of proteolysis in the insoluble fraction of muscle tissue, a 14-kDa fragment of actin. As this actin fragment is rapidly degraded when it is in solution with components of the UPS, it can only be found in the insoluble fraction of muscle where it is protected from degradation. Measurement of the 14-kDa actin fragment in muscles was used to investigate the cleavage of muscle proteins by caspase-3 in three groups: patients with muscle atrophy as a result of osteoarthritis who were undergoing hip replacement surgery; patients with severe burn injuries; and patients on haemodialysis who were participating in an 18-week trial of the ability of exercise to improve muscle metabolism.44 Results from patients in the osteoarthritis group revealed that the rate of protein degradation (measured by the turnover of labelled amino acids) was closely related to the level of the 14-kDa actin fragment in muscle biopsy samples (r = 0.7; P <0.01). In biopsy samples of muscle from patients with burn injuries, an increase in the level of the 14-kDa actin fragment was observed even in muscles that were uninjured and distant from the burn, whereas in patients on haemodialysis, endurance training reduced the levels of the 14-kDa actin fragment to values similar to those found in muscles of healthy adults. These results suggest that the 14-kDa actin fragment could be developed as a marker of muscle wasting.
Secondly, caspase-3 activity can stimulate UPS-mediated protein degradation in muscle by directly stimulating the proteolytic activity of the proteasome.45 The biochemical mechanism involves cleavage of specific subunit proteins of the 19S particle, namely 26S protease regulatory subunit 4 and 26S protease regulatory subunit 8.45 These subunits are located near the opening of the central channel of the 20S proteasome and their cleavage could, therefore, increase the amount of unfolded proteins being inserted into the channel of the 20S proteasome (the site of proteolysis). This ‘feed forward’ response would result in increased degradation of muscle proteins and also provides another level of proteolytic control of UPS-mediated protein degradation.
Notably, evidence exists for coordinated proteolytic mechanisms besides changes in caspase-3 activity. For example, mRNAs of components of the UPS increase simultaneously in the muscles of rodent models of various catabolic disorders (such as cancer, diabetes, CKD or starvation); this finding suggests that a genetic programme is activated in response to catabolic conditions.46
Autophagy and lysosomal proteolysis
Autophagy is a process that results in lysosomal degradation of damaged organelles (for example, mitochondria) or aggregates of abnormal proteins. During autophagy, double membranes form around defective organelles or cytoplasmic proteins that are disrupted or damaged. The complex creates autophagosomes that can fuse with lysosomes containing DNAases, RNAases, lipases and proteases, which break down proteins and organelles.47,48 Importantly, the formation of autophagosomes is stimulated by decreased levels of phosphatidylinositol 3-kinase (PI3K) together with activation of the autophagy-related gene BECN1. Autophagy is triggered in response to a decrease in PI3K activity, so it is not surprising that an increase in the levels of mTOR (a marker of protein synthesis and part of the IGF-1/PI3K/Akt signalling pathway) is critical for suppression of the process.48 The influence of CKD on the activation of autophagy has not been rigorously evaluated and the magnitude of autophagy-mediated proteolysis in relation to total protein degradation is unclear.
In one study, the rate of proteolysis in isolated muscles changed minimally when lysosomal functions were inhibited with weak bases, such as methylamine.32,49 However, phagocytosis–lysosomal proteolysis might have a role in CKD-induced muscle wasting because CKD causes insulin resistance and suppresses IGF-1/PI3K/Akt signalling, which would also stimulate the autophagy–lysosome system.48,50,51
Myostatin/activin pathway and muscle wasting
Although the expression of several proinflammatory cytokines (tumour necrosis factor [TNF], IL-6, IL-1 and IFN-γ) is associated with lost muscle mass, the precise contribution of inflammation to muscle wasting in various catabolic conditions remains uncertain. However, abundant evidence indicates that transforming growth factor β (TGF-β) and members of the TGF-β family (myostatin [also known as growth/differentiation factor 8] and activin A) are closely associated with muscle-protein loss in catabolic conditions.52 Binding of TGF-β ligands to surface receptors on muscle activates a Smad2/Smad3-mediated signalling pathway that leads to stimulation of proteolysis and muscle atrophy.53 An important member of the TGF-β family, myostatin, is predominantly expressed in skeletal muscle, but low levels are present in heart muscle and adipocytes. Myostatin suppresses growth of skeletal muscles and in animals lacking myostatin, very large increases in muscle mass and strength have been observed.52,54 The mechanism for decreased muscle growth in response to myostatin includes inhibition of satellite cell function and proliferation.55 The signalling pathway following binding of myostatin to its receptor involves activation of Smad2/Smad3 and phosphorylation of Akt in muscle. The latter causes muscle protein catabolism because a low level of phosphorylated (p)Akt reduces phosphorylation of the Forkhead box O (FoxO) family of transcription factors.52,55,56 This observation is important because dephosphorylated FoxO transcription factors enter the nucleus to increase the expression of the E3 Ub-ligases TRIM63 (also known as MuRF1) and F-box only protein 32 (MAFbx, also known as atrogin-1). These E3 Ub-ligases stimulate UPS-mediated degradation of muscle proteins, including contractile proteins. We have added myostatin to the list of mechanisms that cause CKD-stimulated muscle protein wasting because inhibition of myostatin prevents muscle atrophy by improving satellite cell function and suppressing protein degradation.55
Pathogenesis of muscle wasting in CKD
Metabolic acidosis
Developing an appropriate animal model to study how CKD influences muscle metabolism is difficult because of variability in the degree of kidney function impairment and responses to dietary factors (for example, variation in food intake and, therefore, blood urea nitrogen and metabolic acidosis). A rodent model of CKD similar to that published in the 1930s,30,57 namely subtotal nephrectomy and feeding a high protein diet, is useful because these animals develop markers of uraemia and metabolic acidosis similar to that found in patients with CKD (serum bicarbonate ~16 mM and blood urea nitrogen >28.5 mmol/l [>80 mg/dl]).30,57 A study was carried out to examine whether CKD mainly suppresses protein synthesis or stimulates protein degradation and how metabolic acidosis affects responses. The experiment was possible because acidosis can be neutralized by adding sodium bicarbonate to the rodent chow. In this case, normal rats responded to metabolic acidosis by stimulating protein catabolism in muscle to a greater degree than the decrease in protein synthesis.30,58 In a rat model of CKD, adding sodium bicarbonate to the chow largely eliminated the increase in protein degradation in muscles. These results were confirmed clinically; in infants, children, adults and elderly patients, preventing the consequences of metabolic acidosis improves growth and estimates of protein stores.19 Clinical investigations have also confirmed that protein metabolism and indices of nutritional status that are associated with prevention of metabolic acidosis are improved in patients with CKD.35,59
How are the catabolic responses to metabolic acidosis mediated? To address this question, two strategies were used: firstly, the effects of metabolic acidosis on changes in gene expression or circulating hormones that could affect muscle protein metabolism were examined; and secondly, whether metabolic acidosis lowers the intracellular pH in muscle to trigger muscle protein catabolism was investigated. In studies of cultured muscle cells and isolated muscles, metabolic acidosis increased the expression of ubiquitin mRNA and subunits of the proteasome, indicating activation of proteolysis in the UPS.49,60 To determine whether changes in hormone levels induced by metabolic acidosis act as a stimulus for muscle proteolysis, adrenalectomized rats or mice were studied because the influence of glucocorticoids is eliminated in these animals. Glucocorticoids were necessary to activate the UPS, but this activation occurred only if there was a second stimulus, namely metabolic acidosis. The presence of either acidosis or a physiologically high dose of glucocorticoids alone did not stimulate activity of the UPS.58,61
To determine if metabolic acidosis stimulates the UPS by decreasing muscle cell pH, intracellular pH in the muscles of three groups of rats was measured using NMR.62 The first group included normal rats infused with hydrochloric acid, leading to a plasma bicarbonate concentration of 5 mM. The second group included normal rats that were gavage-fed ammonium chloride to produce an average intracellular pH of 7.09. The third group included rats with CKD that exhibited increased gene transcription and UPS-mediated protein degradation. No intracellular pH-dependent mechanisms that stimulate muscle protein losses were detected. This finding led to the identification of another potential mediator, namely defects in insulin responses, because metabolic acidosis can induce insulin resistance and defects in insulin signalling are associated with loss of muscle mass.63,64
Insulin/IGF-1 signalling and muscle wasting
An intact insulin/IGF-1 intracellular signalling pathway is critical for the regulation of muscle proteins. This pathway is initiated when IGF-1 or insulin interact with their cell membrane receptors, resulting in a series of phosphorylations of intermediate mediators of insulin/IGF-1 signalling, such as insulin receptor substrate 1 (IRS-1), PI3K and Akt. Phosphorylation of Akt stimulates metabolic responses, including an increase in protein synthesis and suppression of protein degradation (Figure 3). In a mouse model of CKD, insulin/IGF-1 signalling was suppressed, leading to muscle atrophy, whereas upregulation of the PI3K/Akt pathway prevented muscle atrophy.24,31,56,65,66 Impaired insulin signalling in cultured muscle cells or mouse models of CKD or insulin deficiency (streptozotocin treatment) stimulated proteolysis by the UPS and caspase-3, causing muscle atrophy.32,33,43,56
Figure 3.

Insulin/IGF-1 signalling stimulates protein synthesis and suppresses protein degradation. Binding of insulin or IGF-1 to the IGF-1 receptor stimulates tyrosine phosphorylation of IRS-1. Activation of IRS-1 stimulates the PI3K/Akt cascade, leading to phosphorylation of Akt, which activates mTOR (leading to protein synthesis) and phosphorylates (inactivates) Fox0. As pFoxO cannot enter the nucleus and, therefore, does not stimulate muscle protein losses, this process limits muscle protein wasting. In the absence of pAkt, active Fox0 increases transcription of the E3 ubiquitin ligases MAFbx and TRIM63, resulting in muscle wasting. MicroRNAs regulate IGF-1/PI3K/Akt signalling; miR-486 downregulates PTEN, which results in an increase in pAkt levels, whereas miR-23a suppresses translation of MAFbx and TRIM63 by interacting with their 3′-untranslated regions. The result is an inhibition of muscle atrophy. Abbreviations: FoxO, forkhead box protein O; IGF, insulin-like growth factor; IRS-1, insulin receptor substrate 1; MAFbx, F-box only protein 32; mTOR, mammalian target of rapamycin; p, phosphorylated; PI3K, phosphatidylinositol 3-kinase; PTEN, phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase and dual-specificity protein phosphatase; TRIM63, E3 ubiquitin-protein ligase TRIM63.
A major factor that mediates the regulation of muscle protein metabolism is the phosphorylation of Akt (pAkt); pAkt phosphorylates and downregulates FoxO function. In muscles, FoxO1 and FoxO3 regulate the expression of TRIM63 and MAFbx, which are critical for muscle-protein breakdown.67,68,71 Thus, when insulin resistance occurs, the levels of pAkt, phosphorylated FoxO1 and FoxO3 are low. Activated (dephosphorylated) FoxO3 or FoxO1 can enter the nucleus to stimulate the expression of TRIM63 and MAFbx.56,69,70 As noted above, when the expression of TRIM63 and MAFbx is increased, muscle-specific proteins are degraded to peptides and ultimately to amino acids.23,37,71 The expression of MAFbx simulates the degradation of MyoD, thereby impairing muscle regeneration.
Other potential regulatory mechanisms are involved when intracellular signalling of the IGF-1/insulin/PI3K/Akt pathway is activated. For example, mice fed a high-fat diet develop insulin resistance and increased expression of phosphatidylinositol 3,4,5-trisphosphate 3 phosphatase and dual-specificity protein phosphatase (PTEN).72 This finding is relevant because PTEN dephosphorylates phosphatidylinositol 3,4,5-trisphosphate (PIP3) to form the inactive phosphatidylinositol 4,5-bisphosphate (PIP2). Thus, an increase in PTEN expression has the same negative impact on insulin/IGF-1 signalling as a decrease in PI3K activity and inhibition of PTEN can prevent muscle-protein loss.72–75
Another phosphatase, tyrosine-protein phosphatase nonreceptor type substrate 1 (SIRP-α), also contributes to CKD-induced muscle-protein wasting.76 The presence of CKD was found to markedly induce the expression of SIRP-α, resulting in two important outcomes: firstly, tyrosine phosphorylation of the insulin receptor and IRS-1 was reduced, resulting in the development of insulin resistance; and secondly, muscle wasting was stimulated.76 Thus, regulation of IGF-1/insulin/PI3K/Akt intracellular signalling can occur at several steps.
Inflammation and muscle wasting
Levels of circulating proinflammatory cytokines, including IL-6, TNF-α, serum amyloid A and C-reactive protein, are increased in patients with CKD.55,77,78 The latter increase is notable because high circulating levels of C-reactive protein predict all-cause mortality, especially in patients with hypoalbuminaemia, which presumably reflects the impact of inflammation on albumin synthesis.8,79–81 Injecting IL-6 into mice was reported to cause muscle wasting but the mechanism was not identified.78
Two mechanisms linking IL-6-related cytokines to muscle wasting have been identified (Figure 4). Firstly, a mouse model of inflammation was created by infusing angiotensin II, which resulted in accelerated muscle-protein degradation.82 In this model, circulating and muscle levels of IL-6, as well as levels of circulating serum amyloid A, were increased. These changes were associated with an increase in the expression of the suppressor of cytokine signalling 3 (SOCS-3) in muscle.78 This finding is interesting because increased SOCS-3 expression in muscle contributes to the loss of muscle proteins. A high level of SOCS-3 reduces IRS-1 levels and suppresses intracellular insulin signalling, leading to activation of caspase-3 and the UPS.83,84 Notably, these responses were blocked in mice that had a genetic deletion of Il6. Thus, proinflammatory conditions that increase IL-6 levels stimulate muscle-protein degradation by interfering with insulin/PI3K/Akt signalling.
Figure 4.

A simplified mechanism of myostatin-induced muscle atrophy. Inflammation increases levels of circulating or local proinflammatory cytokines, such as IL-6. These cytokines stimulate phosphorylation of Stat3, which in turn stimulates SOCS-3 expression. The result is suppression of insulin/IGF-1 signalling, leading to muscle atrophy. IL-6-induced activation of pStat3 also stimulates C/EBPδ, which activates myostatin and, therefore, results in muscle loss. Activation of Stat3 can be blocked by the small-molecule inhibitor C188–9, which suppresses CKD-induced muscle wasting. Myostatin can also be blocked with either an anti-myostatin peptibody or miR-27 to prevent muscle wasting. Abbreviations: C/EBPδ, CCAAT/enhancer-binding protein δ; CKD, chronic kidney disease; IGF, insulin-like growth factor; IRS-1, insulin receptor substrate 1; SOCS-3, suppressor of cytokine signalling 3.
Secondly, the relationship between IL-6 and the signal transducer and activator of transcription 3 (Stat3) and its target genes were examined. These experiments were planned because CKD was found to be associated with a marked increase in the expression of Stat3 in the muscles of patients and rodents.85 To determine whether blocking Stat3 would improve protein balance, mice with muscle-specific knockout of Stat3 were examined; Stat3 knockout ameliorated loss of body and muscle weight in a mouse model of CKD. Changes in the expression of Stat3 produced similar changes in the expression of myostatin. IL-6 also stimulated Stat3 to increase the expression of CCAAT/enhancer-binding protein δ (C/EBPδ). To assess whether C/EBPδ was influencing myostatin expression, mice with a genetic absence of Cebpd were studied. In these mice, myostatin expression was reduced and CKD-induced loss of muscle mass was prevented. Moreover, mice lacking C/EBPδ exhibited an increase in their survival compared with wild-type mice with intact C/EBPδ. These results, taken together with those associated with inhibition of myostatin, indicate the potential for developing translational strategies that could suppress the muscle wasting associated with CKD.55,85
Defective hypothalamic appetite regulation
As discussed above, CKD can cause loss of muscle and adipose tissue, possibly because patients with catabolic disease have poor appetites, leading to true malnutrition. Using genetic and pharmacological approaches, Cheung et al.86 demonstrated that CKD is indeed associated with defects in the hypothalamic regulation of appetite. These investigators found that injecting mediators of appetite directly into regions of the hypothalamus in mice with CKD led to an improvement in muscle growth. As leptin and the central melanocortin systems in the hypothalamus are not only regulators of metabolism, but are also targets of cytokine actions, this report provides information about the impact of inflammation on appetite and metabolism.
The investigators also examined specific pathways affecting appetite, including how the propeptide precursor pro-opiomelanocortin inhibits food intake and energy storage. The complicated responses they identified included production and release of α-melanocyte-stimulating hormone, which binds to melanocortin receptor 3 (MC3-R) and melanocortin receptor 4 (MC4-R) and inhibits food intake. Interestingly, genetic or pharmacological blockade of MC4-R reduced the development of cachexia, which is associated with CKD.86,87 Injections of the neurotransmitter agouti-related protein, an antagonist to MC4-R, increased food intake and reduced the muscle loss associated with CKD. These investigators have since reported that loss of muscle mass in CKD depends on a peripheral mechanism that results from an imbalance in levels of myostatin and IGF-1.88,89
MicroRNAs and muscle wasting
MicroRNAs are capable of regulating biological processes under diverse physiological and pathological conditions. They act by binding to complementary sequences in the 3′-untranslated regions (UTRs) of mRNAs, resulting in inhibition of mRNA translation, which causes changes in protein expression. Notably, a single microRNA can target multiple mRNAs and one mRNA frequently serves as a target for regulation by multiple microRNAs.
To identify whether microRNAs influence CKD-induced muscle wasting and muscle levels of microRNAs in mice with CKD or sham-operated, pair-fed controls were studied using a TaqMan® Low Density Array (Life Technologies, USA).90 In this study, 12 significant changes in microRNA levels were associated with CKD, including decreases in the levels of miR-29a and miR-29b (Table 1). Upon investigation, miR-29 was found to contain a complementary sequence to the 3′-UTR of transcriptional repressor protein YY1 (also known as Yin and Yang 1). A decrease in miR-29 level resulted in an upregulation of YY1 protein, which inhibited myogenesis. This finding is relevant because inhibition of myogenesis contributes to CKD-induced muscle atrophy.90
Table 1.
CKD-induced changes in microRNA levels that affect muscle metabolism
| microRNA | Fold increase | Fold decrease | Target | P value (CKD vs control) | Reference(s) |
|---|---|---|---|---|---|
| miR-181d | 5.08 | – | Hox-A11 | P <0.05 | 90,132 |
| miR-434-5p | 6.624 | – | Unknown | P <0.05 | 90 |
| miR-455-3p | 2.848 | – | Unknown | P <0.05 | 90,133 |
| miR-496 | 3.669 | – | Unknown | P <0.05 | 90 |
| miR-668 | 4.088 | – | Unknown | P <0.05 | 90 |
| miR-23a | – | −3.726 | TRIM63, MAFbx, myosin-1, myosin-2, myosin-4 | P <0.05 | 90,91,134 |
| miR-29a | – | −4.181 | YY1, HDAC4, DBT | P <0.05 | 90,135,136 |
| miR-29b | – | −3.732 | YY1, HDAC4, DBT | P <0.05 | 90,136,137 |
| miR-124a | – | −4.181 | Unknown | P <0.05 | 90 |
| miR-187 | – | −43.93 | Unknown | P <0.05 | 90 |
| miR-199b | – | −3.732 | Unknown | P <0.05 | 90 |
| miR-376a | – | −10.204 | Unknown | P <0.05 | 90 |
| miR-29c | 1.058 | – | YY1, HDAC4, DBT | P >0.05 | 90,135,136 |
| miR-486 | – | −0.223 | FoxO1, PTEN, Pax-7 | P >0.05 | 93,95 |
Abbreviations: CKD, chronic kidney disease; DBT, lipoamide acyltransferase component of branched-chain α-keto acid dehydrogenase complex, mitochondrial; FoxO1, forkhead box protein O1; HDAC4, histone deacetylase 4; Hox-A11, homeobox protein Hox-A11; MAFbx, F-box only protein 32; Pax-7, paired box protein Pax-7; PTEN, phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase and dual-specificity protein phosphatase; TRIM63, E3 ubiquitin-protein ligase TRIM63; YY1, transcriptional repressor protein YY1.
The level of miR-23a was also decreased in muscles of mice with CKD.90 This observation is of interest because miR-23a suppresses the translation of both MAFbx and/or TRIM63 in a 3′-UTR-dependent manner.91,92 Expression of miR-23a can protect muscles from glucocorticoid-induced skeletal muscle atrophy so a decrease in miR-23a expression might be involved in CKD-induced muscle wasting.
In another example of microRNA-induced responses, miR-486 was found to have therapeutic potential, at least in terms of the suppression of muscle wasting in CKD.93 In mice with CKD, the miR-486 mimic blocked dexamethasone-stimulated muscle-protein degradation without influencing protein synthesis. The response to miR-486 could be explained by a downregulation of PTEN. As noted above, downregulation of PTEN increases the level of pAkt, leading to phosphorylation of FoxO1 and FoxO3, and hence reduced expression of TRIM63 and MAFbx, which stimulates proteolysis by the UPS (Figure 3). Another study found that an increase in miR-486 expression leads to a decrease in FoxO1 expression.94 Regarding the influence of miR-486 on muscle regeneration, Dey et al.95 documented that miR-486 expression was associated with an increase in satellite cell differentiation. By contrast, Alexander et al.96 reported that miR-486 decreased the muscle mass of patients with Duchenne muscular dystrophy and that overexpressing miR-486 in vivo impaired muscle regeneration. This result could be attributable to the fact that miR-486 alters cell cycle kinetics that are activated during muscle fibre regeneration.96
To illustrate the complexity of responses to microRNAs, consider the following reports: one group determined that muscle-specific microRNAs comprise a series that includes miR-1, miR-133 and miR-206, each of which can modulate muscle-cell growth and differentiation.97 However, their action is a secondary level of response, because each of these microRNAs target IGF-1. For example, miR-1 was shown to directly inhibit the 3′-UTR of IGF-1 mRNA in C2C12 cells (mouse muscle cells).97 A different study showed that miR-133 could bind to the 3′-UTR of the IGF-1 receptor in C2C12 cells and an increase in miR-133 expression actually suppressed the levels of the IGF-1 receptor by posttranscriptional mechanisms.98 Another study demonstrated that miR-206 directly downregulates IGF-1 mRNA in 293T cells, whereas a low level of miR-206 substantially increased IGF-1 mRNA in vivo.99
MicroRNAs can also affect muscle metabolism by interacting with myostatin (Figure 4). For example, miR-27a stimulates muscle-cell proliferation by directly inhibiting myostatin expression through an interaction between miR-27a and a target sequence in the myostatin 3′-UTR.100 miR-27b expression can also attenuate myostatin activity; mutating the miR-27 recognition sequence in the 3′-UTR of myostatin led to a sharp decrease in the degradation of myostatin mRNA.101 This finding is relevant to inflammation-induced muscle loss because TNF ligand superfamily member 12 induced muscle atrophy by downregulating miR-27a and miR-27b. The miR-27 microRNAs are sensitive to individual amino acids. Leucine is known to increase protein synthesis in muscle, but how this response is mediated has not yet been determined. Chen et al.102 showed that treating C2C12 cells with leucine increased miR-27a expression with downregulation of myostatin, leading to cell proliferation. These results offer an explanation for leucine-induced changes in muscle metabolism.102 In summary, microRNAs are a fascinating area for research and should suggest beneficial approaches for preventing the loss of muscle mass that is induced by CKD and possibly other catabolic conditions.
Strategies for blocking muscle atrophy
Diet and control of acidosis
The first-line therapy for prevention or treatment of CKD-induced muscle atrophy is to ameliorate the many complications of CKD, which include metabolic acidosis, hypertension, increased angiotensin II levels, poorly controlled diabetes, oedema and the accumulation of unexcreted waste products of metabolism.103 Preventing these complications can improve the physical status and well-being of patients. Unfortunately, patients with stages 1–4 CKD eat substantially more protein than is recommended.103,104 This finding is relevant because one complication of eating too much protein is the development of metabolic acidosis and correcting acidosis in patients with CKD will suppress protein wasting.59
Exercise to prevent muscle wasting
Considering the problems associated with CKD, such as anaemia, hypertension and bone and muscle losses, as well as the many medicines required by patients, it is not difficult to understand why these patients frequently avoid exercise. This outcome is unfortunate not only because exercise is beneficial for cardiovascular health, but also because progressive loss of muscle mass adversely affects overall health. An extreme model of resistance exercise (muscle overloading) in rats with CKD increased the expression of IGF-1 as well as downstream mediators of IGF-1, including IRS-1, PI3K, pAkt and myostatin.105,106 A single session of resistance exercise was found to stimulate protein anabolism in muscles of patients on haemodialysis, whereas 18 weeks of endurance exercise improved markers of protein metabolism, including IGF-1 and myostatin.107,108 In this study, proteolysis in muscle was sharply suppressed.
To determine which types of exercise can counteract muscle-protein losses most efficiently, two mouse models of exercise (muscle resistance or overloading and treadmill running) were evaluated.31 In mice with CKD, resistance exercise not only reduced muscle protein breakdown, but also increased muscle weight and improved intracellular signalling mediators that regulate protein synthesis and progenitor cell functions. The treadmill running exercise improved the excessive rates of muscle proteolysis, but did not improve protein synthesis or functions of progenitor cells.
For bedridden patients or those in wheelchairs, novel therapies that can safely suppress muscle wasting are needed. Electrical stimulation or acupuncture have been suggested as two such therapies. Acupuncture was reported to ameliorate skeletal muscle atrophy in mice that were subjected to hind-limb suspension (a model of muscle wasting).109 Other investigators concluded that electrical acupuncture can suppress myostatin expression, leading to proliferation of satellite cells and improved repair of muscle injury.110 One attractive possibility for therapy is low-frequency electrical stimulation, which mimics acupuncture therapy or exercise in mice with CKD-induced muscle atrophy. Low-frequency electrical stimulation significantly improved (P <0.05) weights of the red-fibre, soleus and white-fibre extensor digitorum longus muscles in mice with CKD.111 The mechanism for these observations involve a suppression of CKD-induced muscle proteolysis and an increase in muscle IGF-1 levels. Further work is needed to confirm these preliminary observations.
Strategies to inhibit myostatin or Stat3
CKD is associated with increased expression of myostatin, a negative regulator of skeletal muscle growth. An increase in myostatin expression in the muscles of mice with CKD suppressed protein synthesis, increased protein degradation and impaired satellite-cell myogenesis.55 Whether inhibition of myostatin could improve muscle mass was explored using an anti-myostatin peptibody. Treatment with this humanized, neutralizing antibody to myostatin (injected every second day for 28 days) led to an increase in the weights of the mice and their individual muscles compared with results from pair-fed mice with CKD injected with an inactive, control antibody. The inhibition of myostatin led to an increase in IGF-1/insulin/PI3K/Akt intracellular signalling, providing a mechanism for muscle growth. Improvements in satellite-cell functions and an increase in myogenesis were also noted.
An unexpected benefit of myostatin inhibition was a decrease in the circulating levels of TNF-α, IL-6, INF-γ and macrophage colony-stimulating factor 1.55 This reduction in the level of inflammation could at least partially explain why suppressing myostatin ameliorated the insulin resistance that occurs frequently in CKD.78,112 We emphasize these positive responses to myostatin inhibition because preliminary results from a clinical trial indicate that the anti-myostatin peptibody exhibited an acceptable safety profile in patients with muscular dystrophy.113 Inhibition of myostatin might, therefore, be beneficial for patients with CKD and muscle-protein losses.
Other potential methods of suppressing myostatin and other members of the TGF-β superfamily exist. For example, a circulating, locally acting protein, activin A, interacts with the activin receptor type-2B that is present on muscle membranes. This receptor stimulates the intracellular signalling events that cause muscle atrophy. The effectiveness of blocking activin receptor type-2B was tested by Zhou et al.114 who infused cancer-bearing mice with soluble activin receptor type-2B. The treatment neutralized activin A and myostatin and resulted in remarkable sparing of muscle proteins despite the catabolic influence of cancer. Surprisingly, mice treated with the soluble receptor showed improved survival compared with cancer-bearing mice that were treated with a control protein. Taken together, the studies discussed above suggest that treatments inhibiting myostatin and/or activin A could be clinically useful in catabolic conditions.
Another treatment strategy for blocking muscle-protein losses in catabolic diseases has been tested.85 A small molecule, C188–9, was identified that can block the Stat3 signalling pathway and prevent activation of myostatin. C188–9 renders Stat3 inactive and does not cross-react with upstream JAK or Src kinases.85,115 In mice with or without CKD, C188–9 was well-tolerated and CKD-induced muscle wasting was blocked during 1 month of treatment.85 The mechanism underlying the anticatabolic influence of C188–9 depends on blocking Stat3 activity in muscle followed by inhibition of C/EBPδ expression. In this case, suppression of myostatin leads to growth of the mouse and its muscles despite the presence of CKD. These results indicate that an inhibitor of Stat3 could be used to uncover how aberrant intracellular signalling causes muscle proteolysis, and might lead to a therapeutic strategy that prevents the muscle atrophy induced by CKD and potentially other catabolic conditions (Figure 4).85
Pharmacological methods to stop muscle loss
Androgenic steroids such as nandrolone decanoate (ND), a synthetic derivative of testosterone, increase muscle mass in healthy adults and in patients with CKD. In a phase II clinical trial that included 54 patients with stage 5 CKD, an ND-induced increase in appendicular mass with no evidence of fluid overloading was reported.116 However, no consistent effect of the therapy on physical function was found and the highest dose of ND used (100 mg per week) was intolerable in females because of virilization.
In catabolic conditions, an increase in IGF-1 in muscle could presumably exert beneficial effects on intracellular signalling.117 Clinical studies of the long-term effects of administering IGF-1 and its analogues are not available and concern has been raised that prolonged inhibition of protein degradation could have a negative impact on protein metabolic balance.118 CKD-induced defects in response to growth hormone would interfere with muscle protein metabolism in a similar fashion to IGF-1. In fact, CKD causes an almost 50% decrease in the activation of JAK2 and STAT5 transcription factors by growth hormone.119–121 How these effects impair muscle metabolism was not identified.
Angiotensin II induces loss of muscle mass by a mechanism that involves inhibition of IGF-1/insulin/PI3K/Akt intracellular signalling.82 Not surprisingly, the angiotensin II receptor inhibitor, losartan, has been used to promote muscle regeneration associated with inhibition of TGF-β-initiated cellular signalling in muscle.122 Moreover, in mice with hind-limb immobilization, losartan was associated with protection against loss of muscle mass.123 As with angiotensin II infusion, the protective mechanism that prevented muscle wasting involved increased activation of the IGF-1/insulin/Akt/mTOR pathway. The long-term consequences of blocking angiotensin II in patients with CKD have not been reported.82,123
Evidence suggests that treatment with the gastric hormone ghrelin can modulate systemic inflammation and insulin action, leading to suppression of CKD-induced muscle atrophy.124 In mice, ghrelin can inhibit dexamethasone-induced atrophy of myotubes (a model of skeletal muscle atrophy).125 Caution is warranted, however, because mice with muscle atrophy from denervation or fasting that were treated with ghrelin developed impaired muscle atrophy without stimulating muscle hypertrophy.125 A randomized, placebo-controlled study in malnourished patients receiving peritoneal dialysis yielded evidence that subcutaneous injections of ghrelin increased food intake.126 More information is needed to document whether ghrelin will chronically improve nutritional status and outcome in patients with CKD. Cheung et al.127 found that pegylated leptin antagonist (7 mg/kg per day) might represent a viable therapeutic strategy for cachexia in mice with CKD.
Inhibitors of the UPS
As the UPS is the major pathway for degrading muscle proteins, a UPS inhibitor could prevent muscle atrophy.128,129 However, caution is required; the UPS is critical for the regulation of cell functions, so prolonged testing would be needed to understand the consequences of UPS inhibition in patients with CKD. Some patients with multiple myeloma who received >12 weeks of treatment with the proteasome inhibitor bortezomib displayed cardiac complications.130 New inhibitors might be more specific in terms of which proteins can and cannot be targeted for degradation. For example, the small-molecule inhibitor of TRIM63 might have sufficient specificity for muscle that it could be used to avoid losses of muscle proteins being degraded by the UPS. To identify such an inhibitor, Eddins et al.131 screened a small-molecule library and identified P013222, an inhibitor of TRIM63 activation. The specificity for TRIM63 was documented by showing degradation of myosin in a cell model.131
Conclusions
Several catabolic conditions, including CKD, result in loss of muscle mass, but no readily available treatments for successfully preventing muscle wasting in these conditions currently exist. Over the past 15 years, much has been learned about the mechanisms of protein metabolism and turnover, and how these processes regulate cellular functions. Some of these mechanisms stimulate protein synthesis and suppress protein degradation whereas others do the opposite. A major cause of muscle loss is impairment in IGF-1/insulin/PI3K/Akt intracellular signalling. Acidosis, inflammation, myostatin, autophagy and (potentially) activation of microRNAs act to suppress intracellular IGF-1/insulin signalling in muscle. Reports from the past few years based on unravelling the biochemical disorders that cause muscle wasting have suggested potential methods for preventing or suppressing losses of muscle protein in CKD. An obvious candidate is to correct metabolic acidosis. Fortunately, exciting preclinical reports indicate that therapeutic strategies could be developed to combat muscle wasting in CKD and perhaps in other catabolic conditions. These include treatment with anti-myostatin peptibody, a strategy that has only been transiently tried in patients with muscle-protein loss. Promising results have been reported with inhibitors of myostatin-induced responses and potential molecular techniques based on microRNAs or inhibitors of autophagy pathways. Much more information is needed, including well-controlled clinical trials, but there is at least reason to hope that treatments for muscle wasting are being developed.
Key points.
Muscle atrophy frequently complicates the course of chronic kidney disease (CKD) and is associated with excess morbidity and mortality
CKD-induced mechanisms that cause muscle atrophy include activation of the ubiquitin–proteasome system, caspase-3 and myostatin
Triggers that activate proteolysis in CKD include metabolic acidosis, defects in insulin/insulin-like growth factor 1 (IGF-1) intracellular signalling, inflammation and catabolic responses to microRNAs
Current strategies aimed at preventing muscle atrophy include correction of acidosis plus exercise; specific therapies that inhibit myostatin or signal transducer and activator of transcription 3 (Stat3) are being developed
Review criteria.
We searched MEDLINE and PubMed for original articles focusing on chronic kidney disease-induced muscle atrophy published between 1996 and 2014. The search terms used were “chronic kidney disease”, “muscle wasting” and “malnutrition”. All papers identified were English-language, full-text papers. We also searched the reference lists of identified articles for further papers.
Acknowledgments
This work was supported by NIAMS R01 AR060268 (X.H.W.) and NIDDK R37 DK037175 (W.E.M.).
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
Both authors researched the data for the article, provided substantial contributions to discussions of its content, wrote the article and edited the manuscript before submission.
Contributor Information
Xiaonan H. Wang, Renal Division, Department of Medicine, Emory University, 1639 Pierce Drive, WMB 338, Atlanta, GA 30322, USA
William E. Mitch, Nephrology Division, Department of Medicine, Baylor College of Medicine, One Baylor Plaza, ABBR R705, Houston, TX 77030, USA
References
- 1.Griffiths RD. Muscle mass, survival, and the elderly ICU patient. Nutrition. 1996;12:456–458. doi: 10.1016/s0899-9007(96)00141-4. [DOI] [PubMed] [Google Scholar]
- 2.Windsor JA, Hill GL. Risk factors for postoperative pneumonia. The importance of protein depletion. Ann Surg. 1988;208:209–214. doi: 10.1097/00000658-198808000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gracia-Iguacel C, et al. Prevalence of protein-energy wasting syndrome and its association with mortality in haemodialysis patients in a centre in Spain. Nefrologia. 2013;33:495–505. doi: 10.3265/Nefrologia.pre2013.Apr.11979. [DOI] [PubMed] [Google Scholar]
- 4.Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351:1296–1305. doi: 10.1056/NEJMoa041031. [DOI] [PubMed] [Google Scholar]
- 5.United States Renal Data System. USRDS 2009 Annual Data Report: Atlas of End-Stage Renal Disease in the United States. 2009 online http://www.usrds.org/atlas09.aspx.
- 6.Lowrie EG, Lew NL. Death risk in hemodialysis patients: the predictive value of commonly measured variables and an evaluation of death rate differences between facilities. Am J Kidney Dis. 1990;15:458–482. doi: 10.1016/s0272-6386(12)70364-5. [DOI] [PubMed] [Google Scholar]
- 7.Stenvinkel P, Heimbürger O, Lindholm B. Wasting, but not malnutrition, predicts cardiovascular mortality in end-stage renal disease. Nephrol Dial Transplant. 2004;19:2181–2183. doi: 10.1093/ndt/gfh296. [DOI] [PubMed] [Google Scholar]
- 8.Carrero JJ, et al. Muscle atrophy, inflammation and clinical outcome in incident and prevalent dialysis patients. Clin Nutr. 2008;27:557–564. doi: 10.1016/j.clnu.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 9.Hakim RM, Lazarus JM. Initiation of dialysis. J Am Soc Nephrol. 1995;6:1319–1328. doi: 10.1681/ASN.V651319. [DOI] [PubMed] [Google Scholar]
- 10.Mitch WE. Malnutrition: a frequent misdiagnosis for hemodialysis patients. J Clin Invest. 2002;110:437–439. doi: 10.1172/JCI16494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fouque D, et al. A proposed nomenclature and diagnostic criteria for protein-energy wasting in acute and chronic kidney disease. Kidney Int. 2008;73:391–398. doi: 10.1038/sj.ki.5002585. [DOI] [PubMed] [Google Scholar]
- 12.Abrass CK. Overview: obesity: what does it have to do with kidney disease? J Am Soc Nephrol. 2004;15:2768–2772. doi: 10.1097/01.ASN.0000141963.04540.3E. [DOI] [PubMed] [Google Scholar]
- 13.Ikizler TA, et al. Hemodialysis stimulates muscle and whole body protein loss and alters substrate oxidation. Am J Physiol Endocrinol Metab. 2002;282:E107–E116. doi: 10.1152/ajpendo.2002.282.1.E107. [DOI] [PubMed] [Google Scholar]
- 14.Pupim LB, et al. Intradialytic parenteral nutrition improves protein and energy homeostasis in chronic hemodialysis patients. J Clin Invest. 2002;110:483–492. doi: 10.1172/JCI15449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pupim LB, Majchrzak KM, Flakoll PJ, Ikizler TA. Intradialytic oral nutrition improves protein homeostasis in chronic hemodialysis patients with deranged nutritional status. J Am Soc Nephrol. 2006;17:3149–3157. doi: 10.1681/ASN.2006040413. [DOI] [PubMed] [Google Scholar]
- 16.Carrero JJ, et al. Etiology of the protein-energy wasting syndrome in chronic kidney disease: a consensus statement from the International Society of Renal Nutrition and Metabolism (ISRNM) J Ren Nutr. 2013;23:77–90. doi: 10.1053/j.jrn.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 17.Cano NJ, et al. Intradialytic parenteral nutrition does not improve survival in malnourished hemodialysis patients: a 2-year multicenter, prospective, randomized study. J Am Soc Nephrol. 2007;18:2583–2591. doi: 10.1681/ASN.2007020184. [DOI] [PubMed] [Google Scholar]
- 18.Dobre M, Meyer TW, Hostetter TH. Searching for uremic toxins. Clin J Am Soc Nephrol. 2013;8:322–327. doi: 10.2215/CJN.04260412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitch WE, Ikizler TA, editors. Handbook of Nutrition and the Kidney. Lippincott Williams & Wilkins; 2010. [Google Scholar]
- 20.Tripathy K, Klahr S, Lotero H. Utilization of exogenous urea nitrogen in malnourished adults. Metabolism. 1970;19:253–262. doi: 10.1016/0026-0495(70)90060-0. [DOI] [PubMed] [Google Scholar]
- 21.Kaysen GA, et al. Inflammation and reduced albumin synthesis associated with stable decline in serum albumin in hemodialysis patients. Kidney Int. 2004;65:1408–1415. doi: 10.1111/j.1523-1755.2004.00520.x. [DOI] [PubMed] [Google Scholar]
- 22.Smith G, Robinson PH, Fleck A. Serum albumin distribution in early treated anorexia nervosa. Nutrition. 1996;12:677–684. doi: 10.1016/s0899-9007(96)00170-0. [DOI] [PubMed] [Google Scholar]
- 23.Mitch WE, Goldberg AL. Mechanisms of muscle wasting. The role of the ubiquitin-proteasome pathway. N Engl J Med. 1996;335:1897–1905. doi: 10.1056/NEJM199612193352507. [DOI] [PubMed] [Google Scholar]
- 24.Zhang L, Wang XH, Wang H, Du J, Mitch WE. Satellite cell dysfunction and impaired IGF-1 signaling cause CKD-induced muscle atrophy. J Am Soc Nephrol. 2010;21:419–427. doi: 10.1681/ASN.2009060571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grefte S, Kuijpers-Jagtman AM, Torensma R, Von den Hoff JW. Skeletal muscle development and regeneration. Stem Cells Dev. 2007;16:857–868. doi: 10.1089/scd.2007.0058. [DOI] [PubMed] [Google Scholar]
- 26.Ding H, Gao XL, Hirschberg R, Vadgama JV, Kopple JD. Impaired actions of insulin-like growth factor 1 on protein synthesis and degradation in skeletal muscle of rats with chronic renal failure. Evidence for a postreceptor defect. J Clin Invest. 1996;97:1064–1075. doi: 10.1172/JCI118499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castellino P, et al. Glucose and amino acid metabolism in chronic renal failure: effect of insulin and amino acids. Am J Physiol. 1992;262:F168–F176. doi: 10.1152/ajprenal.1992.262.2.F168. [DOI] [PubMed] [Google Scholar]
- 28.Adey D, Kumar R, McCarthy JT, Nair KS. Reduced synthesis of muscle proteins in chronic renal failure. Am J Physiol Endocrinol Metab. 2000;278:E219–E225. doi: 10.1152/ajpendo.2000.278.2.E219. [DOI] [PubMed] [Google Scholar]
- 29.Bohé J, Low A, Wolfe RR, Rennie MJ. Human muscle protein synthesis is modulated by extracellular, not intramuscular amino acid availability: a dose-response study. J Physiol. 2003;552:315–324. doi: 10.1113/jphysiol.2003.050674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.May RC, Kelly RA, Mitch WE. Mechanisms for defects in muscle protein metabolism in rats with chronic uremia. Influence of metabolic acidosis. J Clin Invest. 1987;79:1099–1103. doi: 10.1172/JCI112924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang XH, Du J, Klein JD, Bailey JL, Mitch WE. Exercise ameliorates chronic kidney disease-induced defects in muscle protein metabolism and progenitor cell function. Kidney Int. 2009;76:751–759. doi: 10.1038/ki.2009.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bailey JL, et al. The acidosis of chronic renal failure activates muscle proteolysis in rats by augmenting transcription of genes encoding proteins of the ATP-dependent ubiquitin-proteasome pathway. J Clin Invest. 1996;97:1447–1453. doi: 10.1172/JCI118566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bailey JL, Zheng B, Hu Z, Price SR, Mitch WE. Chronic kidney disease causes defects in signaling through the insulin receptor substrate/phosphatidylinositol 3-kinase/Akt pathway: implications for muscle atrophy. J Am Soc Nephrol. 2006;17:1388–1394. doi: 10.1681/ASN.2004100842. [DOI] [PubMed] [Google Scholar]
- 34.Raj DS, et al. Protein turnover and amino acid transport kinetics in end-stage renal disease. Am J Physiol Endocrinol Metab. 2004;286:E136–E143. doi: 10.1152/ajpendo.00352.2003. [DOI] [PubMed] [Google Scholar]
- 35.Reaich D, et al. Correction of acidosis in humans with CRF decreases protein degradation and amino acid oxidation. Am J Physiol. 1993;265:E230–E235. doi: 10.1152/ajpendo.1993.265.2.E230. [DOI] [PubMed] [Google Scholar]
- 36.Ballmer PE, et al. Chronic metabolic acidosis decreases albumin synthesis and induces negative nitrogen balance in humans. J Clin Invest. 1995;95:39–45. doi: 10.1172/JCI117668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lecker SH, Goldberg AL, Mitch WE. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol. 2006;17:1807–1819. doi: 10.1681/ASN.2006010083. [DOI] [PubMed] [Google Scholar]
- 38.Rock KL, et al. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 39.Lecker SH, Mitch WE. Proteolysis by the ubiquitin-proteasome system and kidney disease. J Am Soc Nephrol. 2011;22:821–824. doi: 10.1681/ASN.2010090958. [DOI] [PubMed] [Google Scholar]
- 40.Wang XH, Mitch WE. Muscle wasting from kidney failure—a model for catabolic conditions. Int J Biochem Cell Biol. 2013;45:2230–2238. doi: 10.1016/j.biocel.2013.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saric T, Graef CI, Goldberg AL. Pathway for degradation of peptides generated by proteasomes: a key role for thimet oligopeptidase and other metallopeptidases. J Biol Chem. 2004;279:46723–46732. doi: 10.1074/jbc.M406537200. [DOI] [PubMed] [Google Scholar]
- 42.Groll M, et al. A gated channel into the proteasome core particle. Nat Struct Biol. 2000;7:1062–1067. doi: 10.1038/80992. [DOI] [PubMed] [Google Scholar]
- 43.Du J, et al. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J Clin Invest. 2004;113:115–123. doi: 10.1172/JCI200418330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Workeneh BT, et al. Development of a diagnostic method for detecting increased muscle protein degradation in patients with catabolic conditions. J Am Soc Nephrol. 2006;17:3233–3239. doi: 10.1681/ASN.2006020131. [DOI] [PubMed] [Google Scholar]
- 45.Wang XH, et al. Caspase-3 cleaves specific 19 S proteasome subunits in skeletal muscle stimulating proteasome activity. J Biol Chem. 2010;285:21249–21257. doi: 10.1074/jbc.M109.041707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lecker SH, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004;18:39–51. doi: 10.1096/fj.03-0610com. [DOI] [PubMed] [Google Scholar]
- 47.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 48.Sandri M. Protein breakdown in muscle wasting: role of autophagy-lysosome and ubiquitin-proteasome. Int J Biochem Cell Biol. 2013;45:2121–2129. doi: 10.1016/j.biocel.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mitch WE, et al. Metabolic acidosis stimulates muscle protein degradation by activating the adenosine triphosphate-dependent pathway involving ubiquitin and proteasomes. J Clin Invest. 1994;93:2127–2133. doi: 10.1172/JCI117208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Price SR, et al. Muscle wasting in insulinopenic rats results from activation of the ATP-dependent, ubiquitin-proteasome proteolytic pathway by a mechanism including gene transcription. J Clin Invest. 1996;98:1703–1708. doi: 10.1172/JCI118968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu Z, Wang H, Lee IH, Du J, Mitch WE. Endogenous glucocorticoids and impaired insulin signaling are both required to stimulate muscle wasting under pathophysiological conditions in mice. J Clin Invest. 2009;119:3059–3069. doi: 10.1172/JCI38770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Han HQ, Zhou X, Mitch WE, Goldberg AL. Myostatin/activin pathway antagonism: molecular basis and therapeutic potential. Int J Biochem Cell Biol. 2013;45:2333–2347. doi: 10.1016/j.biocel.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 53.Sartori R, et al. Smad2 and 3 transcription factors control muscle mass in adulthood. Am J Physiol Cell Physiol. 2009;296:C1248–C1257. doi: 10.1152/ajpcell.00104.2009. [DOI] [PubMed] [Google Scholar]
- 54.McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-β superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 55.Zhang L, et al. Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J. 2011;25:1653–1663. doi: 10.1096/fj.10-176917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee SW, et al. Regulation of muscle protein degradation: coordinated control of apoptotic and ubiquitin-proteasome systems by phosphatidylinositol 3 kinase. J Am Soc Nephrol. 2004;15:1537–1545. doi: 10.1097/01.asn.0000127211.86206.e1. [DOI] [PubMed] [Google Scholar]
- 57.Chanutin A, Ludewig S. Experimental renal insufficiency produced by partial nephrectomy. V Diets containing whole dried meat. Arch Intern Med. 1936;58:60–80. [Google Scholar]
- 58.May RC, Kelly RA, Mitch WE. Metabolic acidosis stimulates protein degradation in rat muscle by a glucocorticoid-dependent mechanism. J Clin Invest. 1986;77:614–621. doi: 10.1172/JCI112344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de Brito-Ashurst I, Varagunam M, Raftery MJ, Yaqoob MM. Bicarbonate supplementation slows progression of CKD and improves nutritional status. J Am Soc Nephrol. 2009;20:2075–2084. doi: 10.1681/ASN.2008111205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Isozaki U, Mitch WE, England BK, Price SR. Protein degradation and increased mRNAs encoding proteins of the ubiquitin-proteasome proteolytic pathway in BC3H1 myocytes require an interaction between glucocorticoids and acidification. Proc Natl Acad Sci USA. 1996;93:1967–1971. doi: 10.1073/pnas.93.5.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Price SR, England BK, Bailey JL, Van Vreede K, Mitch WE. Acidosis and glucocorticoids concomitantly increase ubiquitin and proteasome subunit mRNAs in rat muscle. Am J Physiol. 1994;267:C955–C960. doi: 10.1152/ajpcell.1994.267.4.C955. [DOI] [PubMed] [Google Scholar]
- 62.Bailey JL, England BK, Long RC, Jr, Weissman J, Mitch WE. Experimental acidemia and muscle cell pH in chronic acidosis and renal failure. Am J Physiol. 1995;269:C706–C712. doi: 10.1152/ajpcell.1995.269.3.C706. [DOI] [PubMed] [Google Scholar]
- 63.Folli F, Saad MJ, Kahn CR. Insulin receptor/IRS-1/PI 3-kinase signaling system in corticosteroid-induced insulin resistance. Acta Diabetol. 1996;33:185–192. doi: 10.1007/BF02048541. [DOI] [PubMed] [Google Scholar]
- 64.DeFronzo RA, Beckles AD. Glucose intolerance following chronic metabolic acidosis in man. Am J Physiol. 1979;236:E328–E334. doi: 10.1152/ajpendo.1979.236.4.E328. [DOI] [PubMed] [Google Scholar]
- 65.Wang X, Hu Z, Hu J, Du J, Mitch WE. Insulin resistance accelerates muscle protein degradation: activation of the ubiquitin-proteasome pathway by defects in muscle cell signaling. Endocrinology. 2006;147:4160–4168. doi: 10.1210/en.2006-0251. [DOI] [PubMed] [Google Scholar]
- 66.Zhou Q, Du J, Hu Z, Walsh K, Wang XH. Evidence for adipose-muscle cross talk: opposing regulation of muscle proteolysis by adiponectin and fatty acids. Endocrinology. 2007;148:5696–5705. doi: 10.1210/en.2007-0183. [DOI] [PubMed] [Google Scholar]
- 67.Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22:8983–8998. doi: 10.1038/sj.onc.1207115. [DOI] [PubMed] [Google Scholar]
- 68.Katso R, et al. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17:615–675. doi: 10.1146/annurev.cellbio.17.1.615. [DOI] [PubMed] [Google Scholar]
- 69.Musarò A, et al. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet. 2001;27:195–200. doi: 10.1038/84839. [DOI] [PubMed] [Google Scholar]
- 70.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 71.Sandri M, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hu Z, et al. PTEN inhibition improves muscle regeneration in mice fed a high-fat diet. Diabetes. 2010;59:1312–1320. doi: 10.2337/db09-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 74.Dupont J, Renou JP, Shani M, Hennighausen L, LeRoith D. PTEN overexpression suppresses proliferation and differentiation and enhances apoptosis of the mouse mammary epithelium. J Clin Invest. 2002;110:815–825. doi: 10.1172/JCI13829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hu Z, et al. PTEN expression contributes to the regulation of muscle protein degradation in diabetes. Diabetes. 2007;56:2449–2456. doi: 10.2337/db06-1731. [DOI] [PubMed] [Google Scholar]
- 76.Thomas SS, Dong Y, Zhang L, Mitch WE. Signal regulatory protein-α interacts with the insulin receptor contributing to muscle wasting in chronic kidney disease. Kidney Int. 2013;84:308–316. doi: 10.1038/ki.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cheung WW, Paik KH, Mak RH. Inflammation and cachexia in chronic kidney disease. Pediatr Nephrol. 2010;25:711–724. doi: 10.1007/s00467-009-1427-z. [DOI] [PubMed] [Google Scholar]
- 78.Zhang L, et al. IL-6 and serum amyloid A synergy mediates angiotensin II-induced muscle wasting. J Am Soc Nephrol. 2009;20:604–612. doi: 10.1681/ASN.2008060628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Menon V, et al. C-reactive protein and albumin as predictors of all-cause and cardiovascular mortality in chronic kidney disease. Kidney Int. 2005;68:766–772. doi: 10.1111/j.1523-1755.2005.00455.x. [DOI] [PubMed] [Google Scholar]
- 80.Knight EL, et al. Kidney dysfunction, inflammation, and coronary events: a prospective study. J Am Soc Nephrol. 2004;15:1897–1903. doi: 10.1097/01.asn.0000128966.55133.69. [DOI] [PubMed] [Google Scholar]
- 81.Hung AM, Ellis CD, Shintani A, Booker C, Ikizler TA. IL-1β receptor antagonist reduces inflammation in hemodialysis patients. J Am Soc Nephrol. 2011;22:437–442. doi: 10.1681/ASN.2010070760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Song YH, et al. Muscle-specific expression of IGF-1 blocks angiotensin II-induced skeletal muscle wasting. J Clin Invest. 2005;115:451–458. doi: 10.1172/JCI22324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem. 2002;277:42394–42398. doi: 10.1074/jbc.C200444200. [DOI] [PubMed] [Google Scholar]
- 84.Zhang L, et al. Chemokine CXCL16 regulates neutrophil and macrophage infiltration into injured muscle, promoting muscle regeneration. Am J Pathol. 2009;175:2518–2527. doi: 10.2353/ajpath.2009.090275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang L, et al. Stat3 activation links a C/EBPδ to myostatin pathway to stimulate loss of muscle mass. Cell Metab. 2013;18:368–379. doi: 10.1016/j.cmet.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cheung W, et al. Role of leptin and melanocortin signaling in uremia-associated cachexia. J Clin Invest. 2005;115:1659–1665. doi: 10.1172/JCI22521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cheung WW, et al. Peripheral administration of the melanocortin-4 receptor antagonist NBI-12i ameliorates uremia-associated cachexia in mice. J Am Soc Nephrol. 2007;18:2517–2524. doi: 10.1681/ASN.2006091024. [DOI] [PubMed] [Google Scholar]
- 88.Cheung WW, Rosengren S, Boyle DL, Mak RH. Modulation of melanocortin signaling ameliorates uremic cachexia. Kidney Int. 2008;74:180–186. doi: 10.1038/ki.2008.150. [DOI] [PubMed] [Google Scholar]
- 89.Cheung WW, Mak RH. Melanocortin antagonism ameliorates muscle wasting and inflammation in chronic kidney disease. Am J Physiol Renal Physiol. 2012;303:F1315–F1324. doi: 10.1152/ajprenal.00341.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang XH, et al. Decreased miR-29 suppresses myogenesis in CKD. J Am Soc Nephrol. 2011;22:2068–2076. doi: 10.1681/ASN.2010121278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wada S, et al. Translational suppression of atrophic regulators by microRNA-23a integrates resistance to skeletal muscle atrophy. J Biol Chem. 2011;286:38456–38465. doi: 10.1074/jbc.M111.271270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lin Z, et al. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc Natl Acad Sci USA. 2009;106:12103–12108. doi: 10.1073/pnas.0811371106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xu J, et al. Transcription factor FoxO1, the dominant mediator of muscle wasting in chronic kidney disease, is inhibited by microRNA-486. Kidney Int. 2012;82:401–411. doi: 10.1038/ki.2012.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Small EM, et al. Regulation of PI3-kinase/Akt signaling by muscle-enriched microRNA-486. Proc Natl Acad Sci USA. 2010;107:4218–4223. doi: 10.1073/pnas.1000300107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dey BK, Gagan J, Dutta A. miR-206 and -486 induce myoblast differentiation by downregulating Pax7. Mol Cell Biol. 2011;31:203–214. doi: 10.1128/MCB.01009-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Alexander MS, et al. Regulation of DMD pathology by an ankyrin-encoded miRNA. Skelet Muscle. 2011;1:27. doi: 10.1186/2044-5040-1-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Elia L, et al. Reciprocal regulation of microRNA-1 and insulin-like growth factor-1 signal transduction cascade in cardiac and skeletal muscle in physiological and pathological conditions. Circulation. 2009;120:2377–2385. doi: 10.1161/CIRCULATIONAHA.109.879429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Huang MB, Xu H, Xie SJ, Zhou H, Qu LH. Insulin-like growth factor-1 receptor is regulated by microRNA-133 during skeletal myogenesis. PLoS One. 2011;6:e29173. doi: 10.1371/journal.pone.0029173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yan B, Zhu CD, Guo JT, Zhao LH, Zhao J. L miR-206 regulates the growth of the teleost tilapia (Oreochromis niloticus) through the modulation of IGF-1 gene expression. J Exp Biol. 2013;216:1265–1269. doi: 10.1242/jeb.079590. [DOI] [PubMed] [Google Scholar]
- 100.Huang Z, Chen X, Yu B, He J, Chen D. MicroRNA-27a promotes myoblast proliferation by targeting myostatin. Biochem Biophys Res Commun. 2012;423:265–269. doi: 10.1016/j.bbrc.2012.05.106. [DOI] [PubMed] [Google Scholar]
- 101.Allen DL, Loh AS. Posttranscriptional mechanisms involving microRNA-27a and b contribute to fast-specific and glucocorticoid-mediated myostatin expression in skeletal muscle. Am J Physiol Cell Physiol. 2011;300:C124–C137. doi: 10.1152/ajpcell.00142.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen X, Huang Z, Chen D, Yang T, Liu G. MicroRNA-27a is induced by leucine and contributes to leucine-induced proliferation promotion in C2C12 cells. Int J Mol Sci. 2013;14:14076–14084. doi: 10.3390/ijms140714076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fouque D, Mitch WE. In: Brenner and Rector’s The Kidney. 9. Taal MW, et al., editors. Ch 60. Elsevier; 2012. pp. 2170–2204. [Google Scholar]
- 104.Moore LW, et al. The mean dietary protein intake at different stages of chronic kidney disease is higher than current guidelines. Kidney Int. 2013;83:724–732. doi: 10.1038/ki.2012.420. [DOI] [PubMed] [Google Scholar]
- 105.Chen Y, Sood S, Biada J, Roth R, Rabkin R. Increased workload fully activates the blunted IRS-1/PI3-kinase/Akt signaling pathway in atrophied uremic muscle. Kidney Int. 2008;73:848–855. doi: 10.1038/sj.ki.5002801. [DOI] [PubMed] [Google Scholar]
- 106.Sun DF, Chen Y, Rabkin R. Work-induced changes in skeletal muscle IGF-1 and myostatin gene expression in uremia. Kidney Int. 2006;70:453–459. doi: 10.1038/sj.ki.5001532. [DOI] [PubMed] [Google Scholar]
- 107.Majchrzak KM, Pupim LB, Flakoll PJ, Ikizler TA. Resistance exercise augments the acute anabolic effects of intradialytic oral nutritional supplementation. Nephrol Dial Transplant. 2008;23:1362–1369. doi: 10.1093/ndt/gfm773. [DOI] [PubMed] [Google Scholar]
- 108.Kopple JD, et al. Exercise in maintenance hemodialysis patients induces transcriptional changes in genes favoring anabolic muscle. J Am Soc Nephrol. 2007;18:2975–2986. doi: 10.1681/ASN.2006070794. [DOI] [PubMed] [Google Scholar]
- 109.Onda A, et al. Acupuncture ameliorated skeletal muscle atrophy induced by hindlimb suspension in mice. Biochem Biophys Res Commun. 2011;410:434–439. doi: 10.1016/j.bbrc.2011.05.152. [DOI] [PubMed] [Google Scholar]
- 110.Takaoka Y, et al. Electroacupuncture suppresses myostatin gene expression: cell proliferative reaction in mouse skeletal muscle. Physiol Genomics. 2007;30:102–110. doi: 10.1152/physiolgenomics.00057.2006. [DOI] [PubMed] [Google Scholar]
- 111.Hu L, et al. Low-frequency electrical stimulation attenuates muscle atrophy in CKD—a potential treatment strategy. J Am Soc Nephrol. doi: 10.1681/ASN.2014020144. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kobayashi S, Maesato K, Moriya H, Ohtake T, Ikeda T. Insulin resistance in patients with chronic kidney disease. Am J Kidney Dis. 2005;45:275–280. doi: 10.1053/j.ajkd.2004.09.034. [DOI] [PubMed] [Google Scholar]
- 113.Wagner KR, et al. A phase I/II trial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol. 2008;63:561–571. doi: 10.1002/ana.21338. [DOI] [PubMed] [Google Scholar]
- 114.Zhou X, et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell. 2010;142:531–543. doi: 10.1016/j.cell.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 115.Redell MS, Ruiz MJ, Alonzo TA, Gerbing RB, Tweardy DJ. Stat3 signaling in acute myeloid leukemia: ligand-dependent and -independent activation and induction of apoptosis by a novel small-molecule Stat3 inhibitor. Blood. 2011;117:5701–5709. doi: 10.1182/blood-2010-04-280123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Macdonald JH, et al. Nandrolone decanoate as anabolic therapy in chronic kidney disease: a randomized phase II dose-finding study. Nephron Clin Pract. 2007;106:c125–c135. doi: 10.1159/000103000. [DOI] [PubMed] [Google Scholar]
- 117.Butterfield GE, et al. Effect of rhGH and rhIGF-I treatment on protein utilization in elderly women. Am J Physiol. 1997;272:E94–E99. doi: 10.1152/ajpendo.1997.272.1.E94. [DOI] [PubMed] [Google Scholar]
- 118.Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech. 2013;6:25–39. doi: 10.1242/dmm.010389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Garibotto G, et al. Effects of recombinant human growth hormone on muscle protein turnover in malnourished hemodialysis patients. J Clin Invest. 1997;99:97–105. doi: 10.1172/JCI119139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sun DF, et al. Chronic uremia attenuates growth hormone-induced signal transduction in skeletal muscle. J Am Soc Nephrol. 2004;15:2630–2636. doi: 10.1097/01.ASN.0000139492.36400.6C. [DOI] [PubMed] [Google Scholar]
- 121.Mehls O, Ritz E, Hunziker EB, Tönshoff B, Heinrich U. Role of growth hormone in growth failure of uraemia—perspectives for application of recombinant growth hormone. Acta Paediatr Scand Suppl. 1988;343:118–126. doi: 10.1111/j.1651-2227.1988.tb10811.x. [DOI] [PubMed] [Google Scholar]
- 122.Cohn RD, et al. Angiotensin II type 1 receptor blockade attenuates TGF-β-induced failure of muscle regeneration in multiple myopathic states. Nat Med. 2007;13:204–210. doi: 10.1038/nm1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Burks TN, et al. Losartan restores skeletal muscle remodeling and protects against disuse atrophy in sarcopenia. Sci Transl Med. 2011;3:82ra37. doi: 10.1126/scitranslmed.3002227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Barazzoni R, Gortan Cappellari G, Zanetti M, Guarnieri G. Ghrelin and muscle metabolism in chronic uremia. J Ren Nutr. 2012;22:171–175. doi: 10.1053/j.jrn.2011.10.017. [DOI] [PubMed] [Google Scholar]
- 125.Porporato PE, et al. Acylated and unacylated ghrelin impair skeletal muscle atrophy in mice. J Clin Invest. 2013;123:611–622. doi: 10.1172/JCI39920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wynne K, et al. Subcutaneous ghrelin enhances acute food intake in malnourished patients who receive maintenance peritoneal dialysis: a randomized, placebo-controlled trial. J Am Soc Nephrol. 2005;16:2111–2118. doi: 10.1681/ASN.2005010039. [DOI] [PubMed] [Google Scholar]
- 127.Cheung WW, et al. A pegylated leptin antagonist ameliorates CKD-associated cachexia in mice. J Am Soc Nephrol. 2014;25:119–128. doi: 10.1681/ASN.2013040432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Caron AZ, et al. The proteasome inhibitor MG132 reduces immobilization-induced skeletal muscle atrophy in mice. BMC Musculoskelet Disord. 2011;12:185. doi: 10.1186/1471-2474-12-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jamart C, Raymackers JM, Li An G, Deldicque L, Francaux M. Prevention of muscle disuse atrophy by MG132 proteasome inhibitor. Muscle Nerve. 2011;43:708–716. doi: 10.1002/mus.21949. [DOI] [PubMed] [Google Scholar]
- 130.Enrico O, et al. Unexpected cardiotoxicity in haematological bortezomib treated patients. Br J Haematol. 2007;138:396–397. doi: 10.1111/j.1365-2141.2007.06659.x. [DOI] [PubMed] [Google Scholar]
- 131.Eddins MJ, et al. Targeting the ubiquitin E3 ligase MuRF1 to inhibit muscle atrophy. Cell Biochem Biophys. 2011;60:113–118. doi: 10.1007/s12013-011-9175-7. [DOI] [PubMed] [Google Scholar]
- 132.Naguibneva I, et al. The microRNA miR-181 targets the homeobox protein Hox-A11 during mammalian myoblast differentiation. Nat Cell Biol. 2006;8:278–284. doi: 10.1038/ncb1373. [DOI] [PubMed] [Google Scholar]
- 133.Russell AP, et al. Disruption of skeletal muscle mitochondrial network genes and miRNAs in amyotrophic lateral sclerosis. Neurobiol Dis. 2012;49C:107–117. doi: 10.1016/j.nbd.2012.08.015. [DOI] [PubMed] [Google Scholar]
- 134.Wang L, et al. MiR-23a inhibits myogenic differentiation through down regulation of fast myosin heavy chain isoforms. Exp Cell Res. 2012;318:2324–2334. doi: 10.1016/j.yexcr.2012.06.018. [DOI] [PubMed] [Google Scholar]
- 135.Wang H, et al. NF-κB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell. 2008;14:369–381. doi: 10.1016/j.ccr.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Winbanks CE, et al. TGF-β regulates miR-206 and miR-29 to control myogenic differentiation through regulation of HDAC4. J Biol Chem. 2011;286:13805–13814. doi: 10.1074/jbc.M110.192625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mersey BD, Jin P, Danner DJ. Human microRNA (miR29b) expression controls the amount of branched chain α-ketoacid dehydrogenase complex in a cell. Hum Mol Genet. 2005;14:3371–3377. doi: 10.1093/hmg/ddi368. [DOI] [PubMed] [Google Scholar]