

Abstract
Aims: To examine the role of the enzyme methionine sulfoxide reductase A-1 (MSRA-1) in amyloid-β peptide (Aβ)-peptide aggregation and toxicity in vivo, using a Caenorhabditis elegans model of the human amyloidogenic disease inclusion body myositis. Results: MSRA-1 specifically reduces oxidized methionines in proteins. Therefore, a deletion of the msra-1 gene was introduced into transgenic C. elegans worms that express the Aβ-peptide in muscle cells to prevent the reduction of oxidized methionines in proteins. In a constitutive transgenic Aβ strain that lacks MSRA-1, the number of amyloid aggregates decreases while the number of oligomeric Aβ species increases. These results correlate with enhanced synaptic dysfunction and mislocalization of the nicotinic acetylcholine receptor ACR-16 at the neuromuscular junction (NMJ). Innovation: This approach aims at modulating the oxidation of Aβ in vivo indirectly by dismantling the methionine sulfoxide repair system. The evidence presented here shows that the absence of MSRA-1 influences Aβ aggregation and aggravates locomotor behavior and NMJ dysfunction. The results suggest that therapies which boost the activity of the Msr system could have a beneficial effect in managing amyloidogenic pathologies. Conclusion: The absence of MSRA-1 modulates Aβ-peptide aggregation and increments its deleterious effects in vivo. Antioxid. Redox Signal. 22, 48–62.
Introduction
Sporadic inclusion body myositis (IBM) is the most common muscle disease in the elderly. It affects the skeletal muscles, and its progression leads to severe muscle weakness (3, 33). Although there is also an important inflammatory component (2, 16), patients do not respond to immunosuppressor and immunomodulatory therapies (2, 3).
One of the most notable characteristics of IBM is the presence of amyloid-β peptide (Aβ) aggregates in skeletal muscle fibers (4, 6, 7, 51, 69). The accumulation of the Aβ peptide has been proposed to have a crucial role in the development of IBM, similar to what occurs in Alzheimer's disease (AD). However, in the case of AD, the buildup of Aβ occurs mainly extracellularly, while the accumulation of the peptide is intracellular in IBM (4, 6, 7, 51, 69). The prevalent theory sustains that the damage of skeletal muscle fibers is secondary to the abnormal accumulation of proteins as is the case in AD (29, 30, 67).
Innovation.
Aberrant protein aggregation is a common feature of late-onset diseases, such as Alzheimer's disease and inclusion body myositis (IBM). Using a Caenorhabditis elegans model of the amyloidogenic disease IBM that expresses the amyloid-beta peptide in muscle cells, we evaluated the role of the oxidation repair msra-1 gene in vivo.
We found that the absence of MSRA-1 is critical during early stages of IBM, being a key factor in its aggregation and toxicity. This study highlights the relevance of the methionine sulfoxide reductase system in IBM pathology.
Therapies that improve the activity of this antioxidant system could be beneficial in managing amyloidogenic diseases.
In AD, the neurotoxic effects of the Aβ peptide compromise synaptic structure and function well before the start of neurodegeneration (61, 62, 74). For instance, there is a decrease of synaptic proteins before the onset of plaque formation in murine models of AD and also in AD patients (36, 49). Similarly, there is mislocalization of neuromuscular junction (NMJ) proteins in cellular cultures from IBM biopsies (38), indicating a NMJ transmission defect (4). Current evidence shows that in AD, soluble Aβ oligomers, rather than insoluble senile plaques or insoluble Aβ fibrils, are the toxic species (50, 63, 71). Aβ oligomers are also present in muscle biopsies from IBM patients (52). We previously showed that, in a Caenorhabditis elegans model of IBM, there are structural defects in the NMJ as well as synaptic dysfunction, and that Aβ oligomeric species are the main toxic forms of the peptide (56). This IBM model expresses the human Aβ peptide in muscle cells and has been extensively used to elucidate the molecular mechanisms of AD and IBM (32, 57, 59). In addition, Aβ aggregation can be easily monitored in vivo from very early stages throughout the animals' lifespan (40, 57).
Oxidative stress is considered as playing a major role in the development of AD and also in IBM (5, 68). In vitro evidence indicates that the oxidation of Met35 in Aβ1-42 affects its aggregation and toxicity (9, 13, 23). The redox state of the methionine residue at position 35 of the Aβ peptide influences its aggregation by reducing fibril formation (23–25). There is controversy regarding whether the oxididation state of Aβ oligomeric species makes them more or less neurotoxic (10, 15, 65). Cell culture experiments show that this oxidized peptide increases the oxidative stress of the organism (9). In vitro experiments suggest that the effect of Met35 oxidation on Aβ toxicity may be influenced by experimental conditions, especially how the oligomers and fibrils were prepared and the exposure time to the cell culture (13). It appears that the formation of soluble oligomers is delayed during the first phase of aggregation when Met35 is oxidized (26). Therefore, there is a larger proportion of nontoxic Aβ monomers, making Aβ-Met35-O less toxic at the onset of short-term experiments (26). In the context of AD and Aβ peptide aggregation, an in vivo study suggests that the methionine residue at position 35 of the peptide is relevant in the generation of oxidative stress and influences its neurotoxic properties (65, 12).
The methionine sulfoxide reductase (Msr) system has been implicated in aging and protection against oxidative stress (28, 39, 43, 44, 53). This highly conserved system reverses the oxidation of methionine residues within proteins (75). In vertebrates, the enzyme methionine sulfoxide reductase is expressed in most tissues (22, 60) and specifically reduces oxidized methionines in proteins (45, 47, 48). MSRA-1 is the only Msr enzyme described in C. elegans (31). We found that the expression of the enzyme declines during aging; while its deletion increases the levels of oxidized methionines in proteins from 10% to 30% and accelerates aging (39). In rats, Msr expression decays in the kidney, brain, and liver during aging (54). Similarly, brains from AD patients show decreased MsrA expression (20). It is still unknown whether MsrA expression declines in human muscle tissue during aging, which could have relevance in the development of IBM. If this is the case, the oxidation of Aβ Met35 may remain unrepaired for longer periods of time, leading to the prevalence of more toxic oligomeric species and/or an increase in oxidative stress, as Met35 cannot act as a reactive oxygen species (ROS) scavenger (9).
To examine the role of Met35 oxidation in Aβ aggregation and toxicity in vivo, we used C. elegans models of IBM where the gene that encodes the C. elegans methionine sulfoxide reductase-1 (MSRA-1) is deleted. As a consequence, the reduction of oxidized methionines is suppressed in these strains (39). In a constitutive strain, which expresses Aβ throughout the worms' lifespan, we found that the locomotory and synaptic defects caused by Aβ are enhanced in the absence of MSRA-1. These phenotypes correlate with an increment of oligomeric species and fewer Aβ aggregates. This constitutive model resembles the course of human IBM: It is late onset, the individuals are affected progressively during their lifetime, but their lifespan is not shortened.
In an inducible Aβ strain that expresses large quantities of the peptide in a very short period of time (72), we observed a limited delay in the time course of paralysis. This model can be used only during a short window of time of Aβ aggregation, because the individuals die within hours of induction and at a very early age. Therefore, this model does not resemble human IBM, but it could, nonetheless, help us understand Aβ dynamics.
Our results suggest that the absence of the MSRA-1 enzyme could influence IBM pathology by increasing Aβ oligomeric species and synaptic dysfunction.
Results
The deletion of the msra-1 gene heightens the locomotory impairments of Aβ transgenic worms
Muscle expression of Aβ causes locomotory impairments in C. elegans (40, 56). To test the relevance of oxidation repair in this model of IBM, we first evaluated locomotion in a transgenic strain that carries a deletion of the msra-1 gene (allele tm1421) and expresses the human Aβ peptide in muscle cells. In this strain, methionine-35 of the Aβ peptide may remain oxidized. Once this modification occurs, it cannot be reduced due to the absence of the repair enzyme MSRA-1. To evaluate the general methionine oxidation state of proteins in the absence of MSRA-1 in our model, we specifically measured oxidized methionines in total protein extracts of each strain (Table 1). We found that the presence of Aβ increases the oxidation in methionines (from 17% to 23%). In msra-1 deletion mutants and in the Aβ, msra-1 strain the oxidation of methionines reaches 30% of total methionines. Therefore, we expect that around 30% of Aβ Met35 is oxidized.
Table 1.
Percentage of Oxidized Methionines in Total Protein Extracts from One-Day-Old Adult Worms
| Genotype | % Met-O/total Met | p-Value with regard to wild-type strain | p-Value with regard to Aβstrain |
|---|---|---|---|
| No Aβ | 17.2±3.5 | ||
| msra-1 | 30.8±4.4 | p≤0.1 | |
| Aβ | 23.5±1.6 | p≤0.5 | |
| Aβ; msra-1 | 31.3±2.4 | p≤0.1 | p≤0.1 |
Values are the average±(SEM) from n=3 measurements per strain. Student's t-test was used for statistical analysis. Met-O, oxidized methionines.
We performed thrashing assays (number of body bends per minute) in liquid medium at day 1 (Fig. 1A) and day 3 of adulthood (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/ars). The locomotory defect of the Aβ strain (40% reduction with regard to the control strain) is enhanced in the absence of msra-1 (Fig. 1A and Table 2). The motility impairments of the Aβ; msra-1 strain (65% total decrease in thrashing rates) are more severe than the sum of the defects shown by each independent strain (Aβ and msra-1 strains) in 1-day-old adults (Table 2). Therefore, the absence of the MSRA-1 enzyme aggravates the locomotory defects caused by Aβ and the impairments persist for at least during the first 3 days of adulthood (Supplementary Fig. S1). When we rescued the Aβ; msra-1 strain with the wild-type msra-1 gene, the motility improves to reach that of the Aβ strain (Fig. 1A, Supplementary Fig. S1 and Table 2).
FIG. 1.

The deletion of the msra-1 gene worsens the locomotory impairments of amyloid-beta peptide (Aβ) transgenic worms. (A) The graph shows the quantification of movement in liquid medium (thrashes per minute) of 1-day-old adult worms. Aβ transgenic worms (black bar; strain CL2120) move slower than the controls that do not express Aβ (white bar; strain CL2122). In an msra-1 mutant background, the transgenic Aβ worms (dark gray bar; strain ANM19) show a significant decrease in motility compared with the Aβ worms (black bar). Deletion of the msra-1 gene (light gray bar) causes a slight decrease in motility compared with the control worms at this age. Transformation rescue of the wild-type msra-1 gene into the Aβ; msra-1 strain (lined bar, strain ANM45) restores motility to reach that of the Aβ transgenic worms. Data are means±SEM from at least four independent experiments. n≥50 animals per bar. One-way ANOVA followed by Bonferroni's post hoc comparisons test were used for statistical analysis. ***p≤0.001. (B) The graph shows the quantification of movement in liquid medium (thrashes per minute) of 1-day-old adult worms. Aβ transgenic worms (black bar; strain CL2120) move slower than the controls that do not express Aβ (white bar; strain CL2122). In a sod-3 mutant background, the transgenic Aβ worms (dark gray bar; strain ANM48) show a moderate decrease in motility compared with the Aβ worms (black bar). Deletion of the sod-3 gene (light gray bar; strain VC433) causes a significant decrease in motility compared with the control worms at this age. Data are means±SEM from at least four independent experiments. n≥30 animals per bar. One-way ANOVA followed by Bonferroni's post hoc comparisons tests were used for statistical analysis. ***p≤0.001, **p≤0.01 (C) The graph shows motility in liquid medium (thrashes per minute) of 1-day-old adult worms. Control worms (no Aβ; strain CL2122) and Aβ transgenic worms (strain CL2120) were subjected to msra-1 RNAi (black bars). The msra-1 knocked-down worms show decreased motility in the presence of Aβ. Data are means±SEM n≥50 animals per strain. Unpaired Student's t-test was used for statistical analysis. ***p≤0.001 (D) The fluorescence images show worms that express MSRA-1::GFP under the control of the endogenous msra-1 promoter. The top image shows that MSRA-1::GFP is expressed in several tissues, including the intestine, muscle cells, and neurons (strain ANM57). The bottom image shows a worm from the same strain described earlier that has been subjected to msra-1 RNAi. Expression of MSRA-1::GFP remains only in the neurons.
Table 2.
Quantification of Movement in Liquid Medium (Thrashes per Minute) of One-Day-Old Adult Worms
| Genotype | Thrashes/min | % with regard to wild-type control | % decrease with regard to wild-type control | % decrease with regard to Aβ |
|---|---|---|---|---|
| No Aβ Control (n=30) | 201±3.9 | 100 | 0 | |
| Aβ (n=50) | 119±5.8 | 59 | 41 | |
| msra-1 (n=52) | 178±4.3 | 89 | 11 | |
| Aβ; msra-1 (n=50) | 70±2.9 | 35 | 65 | 41 |
| Rescued strain (n=62) | 99±2.5 | 49 | 51 | |
| sod-3 (n=30) | 116±2.8 | 58 | 42 | |
| Aβ; sod-3 (n=30) | 82±2.7 | 41 | 59 | 31 |
Values are the average±(SEM) from n≥30 worms per strain.
In order to test the effect of another antioxidant enzyme in motility impairments caused by Aβ, we constructed a strain that carries a mutation in the iron/manganese superoxide dismutase gene sod-3 and which also overexpresses the Aβ peptide in muscle cells. The superoxide dismutase system acts directly on ROS levels, but it is unable to reduce oxidized proteins. We found that a mutation in this enzyme worsens the Aβ phenotype by 31% (Fig. 1B and Table 2). However, the lack of MSRA-1 reduces Aβ worms' motility by 41% (Fig. 1A and Table 2), even though in a wild-type background the msra-1 phenotype is milder than that of sod-3 (Fig. 1A, B and Table 2).
We also knocked down msra-1 expression in the Aβ strain using RNA interference (RNAi). In these experiments, we obtained similar effects as those observed with the null msra-1 mutant. The absence of MSRA-1 exacerbates the motility defects of the Aβ worms (Fig. 1C). We corroborated msra-1 knockdown in a strain that expresses MSRA-1::GFP under the control of the endogenous msra-1 promoter. MSRA-1 expression is undetectable in all tissues, including muscle cells where Aβ is expressed, but remains in the nervous system, as RNAi is not effective in this type of cells (Fig. 1D).
The absence of MSRA-1 exacerbates synaptic dysfunction in the Aβ transgenic worms
Previously, we showed that the constitutive Aβ transgenic strain develops functional defects in the NMJ, displaying resistance to nicotine but not to levamisole (both drugs are acetylcholine receptor agonists). Therefore, Aβ seems to specifically affect the subtype of nicotinic acetylcholine receptors that are sensitive to nicotine but insensitive to levamisole (40, 56). These receptors are closely related to the α7-nicotinic acetylcholine receptor, proposed to be associated with Aβ1-42 neurotoxic effects in mammals (55, 59). Paralysis assays on nicotine performed at day 1 of adulthood show that the absence of MSRA-1 enhances the resistance of the Aβ worms to this drug (Fig. 2A and Supplementary Fig. S2). This enhanced resistance to nicotine is abolished when the wild-type msra-1 gene under the control of its own promoter is transformed into the Aβ; msra-1 strain (Fig. 2A). No difference in behavior between these strains is observed when the worms are exposed to levamisole (Fig. 2B). These results suggest that the absence of MSRA-1 aggravates the functional defects of a NMJ already compromised by Aβ, probably through the same targets.
FIG. 2.
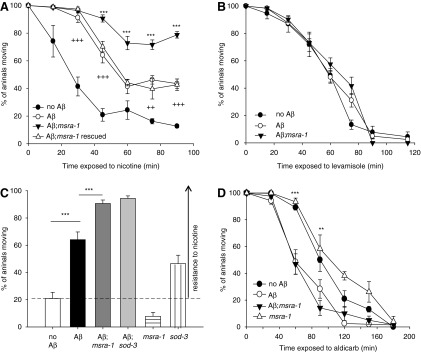
The absence of MSRA-1 exacerbates synaptic dysfunction in the Aβ transgenic worms. (A) The graph shows the time course of paralysis in response to nicotine (31 mM). One-day-old control worms (CL2122), Aβ transgenic worms (CL2120), Aβ; msra-1 worms (ANM19), and Aβ; msra-1 worms rescued with the wild-type msra-1 gene (ANM45) were assayed on nicotine plates. The absence of MSRA-1 increases the nicotine resistance of the Aβ strain. Aβ; msra-1 rescued strain ANM45 shows the same behavior as the Aβ strain. Data are means±SEM from at least four independent experiments. n≥45 animals per strain in each experiment. Repeated-measures two-way ANOVA followed by Bonferroni's post hoc comparisons tests were used for statistical analysis. ***p≤0.001 comparing the Aβ transgenic worms with Aβ; msra-1 worms. ++p≤0.01 comparing Aβ transgenic worms with the control worms. +++p≤0.001 comparing Aβ transgenic worms with the control worms. (B) One-day-old control worms (CL2122), Aβ transgenic worms (CL2120), and Aβ; msra-1 animals (ANM19) were assayed on levamisole plates (0.25 mM). Data are means±SEM from at least four independent experiments. n≥45 animals per strain in each experiment. The paralysis curves show no differences between the strains. (C) The graph compares the paralysis of the Aβ expressing worms in different backgrounds (msra-1 or sod-3) after a 45-min exposure to nicotine 31 mM. The absence of both msra-1 and sod-3 worsens the nicotine resistance of the Aβ worms. Contrary to what we observe in the msra-1 mutant, the sod-3 mutants show nicotine resistance even in the absence of Aβ. Data are means±SEM from at least four independent experiments. n≥45 animals per strain in each experiment. One-way ANOVA followed by Bonferroni's post hoc comparisons tests were used for statistical analysis. ***p≤0.001 (D) The graph shows the time course of paralysis in response to aldicarb (1 mM). One-day-old control worms (CL2122), Aβ transgenic worms (CL2120), Aβ; msra-1 animals (ANM19), and msra-1 mutant worms were assayed on aldicarb plates. The absence of MSRA-1 does not change the hypersensitivity of the Aβ worms to aldicarb. Data are means±SEM from at least four independent experiments, n≥45 animals per strain in each experiment. Repeated-measures two-way ANOVA followed by Bonferroni's post hoc comparisons tests were used for statistical analysis. **p≤0.01, ***p≤0.001 comparing Aβ transgenic worms with the control worms.
In order to establish whether the lack of other antioxidant response enzymes also increases the resistance of Aβ transgenic worms to nicotine, we performed nicotine paralysis assays on Aβ; sod-3 worms. Figure 2C shows that the absence of SOD-3 makes the Aβ transgenic worms more resistant to nicotine, similar to what we described for msra-1 mutants in the Aβ background. However, sod-3 mutant worms (in the absence of Aβ) show resistance to nicotine, while msra-1 mutant worms do not. In fact, msra-1 mutants seem to be slightly sensitive to the drug (Fig. 2C). These results would indicate that sod-3 mutants have additional phenotypes which might not be related to oxidative stress. In fact, it was reported that sod-3 mutants show axon guidance defects (www.wormabase.org).
To further test the influence of MSRA-1 on the synaptic function of the Aβ transgenic strain, we determined the effects of aldicarb (acetylcholinesterase inhibitor). Our results show that the Aβ strain is sensitive to aldicarb. However, this behavior is not altered in the msra-1 mutant background (Fig. 2D). Therefore, the absence of MSRA-1 in the IBM model does not seem to affect the function of the presynaptic terminal.
Taken together, these results suggest that in our animal model, MSRA-1 function is relevant at the postsynaptic terminal and may affect nAChRs which are insensitive to levamisole through the Aβ peptide.
MSRA-1 knockdown enhances the mislocalization of the nAChR ACR-16 in the NMJ of Aβ transgenic worms
Previously, we showed that the nicotinic acetylcholine receptor ACR-16 is mislocalized in the NMJ of Aβ transgenic worms, while the levamisole sensitive nAChRs are not altered (1, 56). ACR-16 mislocalization in these worms correlates with nicotine resistance. Figure 3Ad and B show that in worms that express Aβ and lack MSRA-1, the intensity of ACR-16::GFP fluorescence is greatly reduced. Aβ expression by itself does not change ACR-16 expression levels (56). Besides, msra-1 knockdown by RNAi in a wild-type background does not affect the clustering of ACR-16 or the intensity of ACR-16::GFP fluorescence (Fig. 3Aa,b, B). Western blot analysis indicates that ACR-16::GFP is present at similar levels in all our experimental conditions (Fig. 3C, D). Therefore, the diminished fluorescence could be a consequence of ACR-16 inability to form clusters at the NMJ in the presence of permanently oxidized Aβ.
FIG. 3.

MSRA-1 knockdown enhances the mislocalization of the nAChR ACR-16 in the NMJ of Aβ transgenic worms. (A) Control worms expressing ACR-16::GFP show fluorescent puncta over the ventral nerve cord (a). msra-1 knockdown does not alter the localization of ACR-16::GFP in muscle cells (b). Worms that express Aβ show a markedly altered localization of ACR-16::GFP puncta which can be seen in several muscle arms (arrows) (c). In the presence of Aβ, msra-1 knockdown significantly reduces ACR-16 fluorescence (d). (B) The graph shows the quantification of ACR-16::GFP fluorescence as integrated density of the puncta. Data are means±SEM from n≥30 worms per strain. Student's t-test was used for statistical analysis. ***p≤0.001. (C) Western blot of total protein extracts from strains shown in (A). ACR-16::GFP detected with anti-GFP antibodies. Anti-α-tubulin antibodies were used as a loading control. (D) The graph shows quantification of the Western blot in (C). Data are means±SEM of three independent Western blots. Student's t-test was used for statistical analysis.
MSRA-1 modulates Aβ aggregation in vivo
In vitro evidence shows that the oxidation state of Met35 of the Aβ peptide influences its aggregation by reducing fibril formation (23–25). Therefore, we tested aggregation in vivo in our Aβ, msra-1 strain. C. elegans strains that express Aβ present high levels of oxidative stress (18, 73). Consequently, in the absence of the repair enzyme MSRA-1, the amino acid Met35 of the peptide will likely remain permanently oxidized (Table 1). First, we stained the worms with ThS, a dye that binds to Aβ aggregates. We found that in the absence of MSRA-1, the area and number of ThS positive deposits decreases compared with the control strain [Aβ, msra-1 (+/+)] (Fig. 4A–C). We also observed that, while the area of ThS-positive amyloid aggregates decreases, the percentage area that stains with (Trans, trans)-1-bromo-2,5-bis-(3-hydroxycarbonyl-4-hydroxy) styrylbenzene (BSB) (a dye with higher affinity for Aβ oligomeric species) increases (Fig. 4D, E). However, the number of BSB-positive aggregates does not change when the MSRA-1 enzyme is absent (Fig. 4F). We have already demonstrated that the number of ThS-positive aggregates increases with age (40). In this work, we also show that the BSB-positive areas increase with age (Supplementary Fig. S3), and this process is exacerbated in the absence of MSRA-1.
FIG. 4.

MSRA-1 modulates Aβ aggregation in vivo. (A) ThS staining of 1-day-old Aβ (top image) and Aβ; msra-1 (bottom image) worms show amyloid deposits in the head muscles. Very few ThS-positive amyloid deposits are detected in the absence of MSRA-1 (arrows). (B) The graph shows the quantification of the area of ThS-positive amyloid aggregates in the head region. Data are means±SEM from n≥30 worms per strain. Student's t-test was used for statistical analysis. ***p≤0.001. (C) Quantification of the total number of amyloid deposits in the head area. Data are means±SEM from n≥30 worms per strain. Student's t-test was used for statistical analysis. ***p≤0.001. (D) BSB staining of 1-day-old Aβ (top image) and Aβ; msra-1 (bottom image) worms. The arrows show BSB-positive areas that are increased in the absence of msra-1. (E) The graph shows quantification of the BSB-positive areas in the head of the animal. Data are means±SEM from n≥30 worms per strain. Student's t-test was used for statistical analysis. ***p≤0.001. (F) Quantification of the total number of BSB-positive areas in the animal's head. Data are means±SEM from n≥30 worms per strain. Student's t-test was used for statistical analysis. ***p≤0.001. (G) Western blot from Wt, Aβ, and Aβ; msra-1 worms. Bands of higher-molecular-weight (MW) decrease in intensity in the absence of msra-1. A representative blot from four independent experiments is shown. (H) The graph shows the quantification of the bands in (G) by densitometry. In the absence of msra-1, the fraction of high MW Aβ species decreases with regard to the lower MW Aβ oligomers. Data are means±SEM of three independent Western blots. *p≤0.01. Student's t-test was used for statistical analysis.
Analysis of Aβ oligomeric species through Western blot indicates that the relative levels of low-molecular-weight (MW) oligomers (8–24 kDa) with regard to higher MW species (26–50 kDa) increase in the absence of MSRA-1 (Fig. 4G, H and Supplementary Fig. S4).
The formation of fewer and smaller amyloid aggregates in the absence of oxidation repair of methionines correlates with an increase in the relative levels of oligomeric species and with enhanced motility impairments and synaptic defects.
The absence of MSRA-1 delays paralysis in an inducible Aβ strain
In vitro experiments suggest that the effect of Met35 oxidation on Aβ toxicity may be influenced by experimental conditions, especially how the oligomers and fibrils are prepared and the length of exposure to the cell culture (13). It appears that initial Aβ aggregation (formation of soluble oligomers) is delayed during the first phase of aggregation when Met35 is oxidized (26). Therefore, there is a larger proportion of nontoxic Aβ monomers during the first phase of aggregation at the beginning of short-term experiments. In order to test Aβ-Met35-O toxicity in a shorter time frame in vivo, we used the inducible strain CL4176. This strain produces large quantities of Aβ in a very short period of time (18, 72). The worms paralyze very quickly, and most worms cannot move 24 h after induction. In contrast to the constitutive Aβ worms and to IBM pathological features, they do not form ThS-positive amyloid aggregates (18). The graph in Figure 5A compares the percentage of worms paralyzed over time in the inducible strain and in strains that express Aβ constitutively. Most worms from the inducible strain (CL4176) are paralyzed 24 h after induction as previously described (17); while the worms that express Aβ constitutively remain motile, as we have already shown (40, 56). Another constitutive strain (CL2006) that forms fewer Aβ aggregates than CL2120 presents an intermediate paralysis curve as already described (72). We introduced the msra-1 deletion into the inducible strain through standard genetic crosses and then, we analyzed paralysis over time after Aβ induction. The graph in Figure 5B shows the paralysis time course of inducible Aβ transgenic worms compared with inducible Aβ worms that lack MSRA-1. The absence of MSRA-1 causes a small delay in the time course of paralysis, suggesting reduced Aβ toxicity. However, this protection is temporary, as both strains are completely paralyzed 29 h after induction (Fig. 5B).
FIG. 5.

The absence of MSRA-1 delays paralysis in an inducible strain. (A) In the graph, each curve is the result of at least five independent experiments and it shows the progression of Aβ-induced paralysis in three different Aβ transgenic strains. CL2120 forms numerous Aβ aggregates and does not paralyze over the course of the experiment. CL2120 is the constitutive transgenic Aβ strain we used in all previous experiments. CL2006 is another constitutive Aβ transgenic strain that forms fewer Aβ aggregates than CL2120, and it paralyzes over time. CL4176 is an inducible strain that does not form Aβ aggregates but accumulates large amounts of Aβ. All worms are completely paralyzed 26 h after temperature shift. (B) The graph compares the progression of paralysis in the Aβ-inducible strain in the presence and absence of MSRA-1. In the absence of MSRA-1, paralysis is delayed between 22 and 26 h after temperature shift. By 28 h, there is no difference in the fractions of animals paralyzed in the presence and absence of MSRA-1. Data are means±SEM from at least four independent experiments. Repeated-measures two-way ANOVA followed by Bonferroni's post hoc comparisons tests were used for statistical analysis. **p≤0.01. (C) The Western blot shows oxidative state of Aβ Met35. Aβ was immunoprecipitated from protein extracts of synchronized strains CL4176 (Aβ) and ANM12 (Aβ; msra-1) at 24 h after induction. For the Western blot, the anti-methionine sulfoxide antibody was used.
To test Met35 oxidation state in these strains, we immunoprecipitated Aβ from protein extracts and then, we detected the presence of oxidized methionines using an anti-methionine sulfoxide antibody. The Aβ peptide has only one methionine; therefore, we can only detect the oxidized Met35. As expected from the results shown in Table 1, we found that there is methionine oxidation in both strains. Unfortunately, this assay is only qualitative, and, therefore, we cannot establish a quantitative difference between the strains (Fig. 5C and Supplementary Fig. S5).
Discussion
We postulate that the absence of MSRA-1 in the transgenic C. elegans model of IBM triggers changes in the composition and toxicity of amyloid aggregates in muscle cells. These alterations may be related to the oxidation in Met35 of the Aβ peptide. Based on the evidence reported by several research groups, there is general consensus on the role of Met35 in controlling Aβ oligomerization and aggregation (24, 25, 35). On the other hand, it is unclear whether the oxidation state of Met35 influences the peptide's toxicity in vivo (13, 14).
Oxidative stress induced by Aβ is central in the pathological cascade that leads to neurodegeneration in AD (12–14, 65). Therefore, one of the key questions is whether the oxidation in Met35 increases the peptide's toxicity. As stated earlier, this point has not been settled, as there are numerous reports showing conflicting results depending on the selected experimental strategy (13). The available evidence on the oxidized peptide toxicity is largely based on cell culture experiments where the peptide is prepared in vitro and then used to treat the cells. The results depend on how the peptide is prepared and on the exposure times (13, 58). In IBM, the peptide aggregates intracellularly in muscle cells (3, 4, 6); therefore, the previous in vitro evidence based on the peptide added to the extracellular space is less relevant in studying this disease. In our approach, we used a transgenic C. elegans strain that constitutively expresses the human Aβ peptide in the body wall muscle cells and also carries a deletion of the msra-1 gene, the only Msr enzyme described in C. elegans (31, 39) to avoid reduction of oxidized methionines. This model enables us to focus on the aggregation and toxicity of the peptide itself, as there is no APP processing involved or changes in the peptide's expression. It was originally demonstrated that in mammals, the absence of MsrA causes an increase in the oxidation of proteins and elevated levels of brain pathologies (42, 46 and references therein). In C. elegans, we showed that there is an increment in the percentage of oxidized methionines in the null msra-1 mutants (39). In our constitutive transgenic C. elegans Aβ model, we can recapitulate in vivo the natural assembly of Aβ over time, as it happens in IBM; we can measure its toxicity at different stages of Aβ aggregation, and we can establish the effects of the absence of MSRA-1. Moreover, we were able to measure the oxidation state of methionines in total proteins and, indirectly, in the Aβ peptide (Table 1, Fig. 5C and Supplementary Fig. S5). These results support earlier evidence showing that Aβ expression in C. elegans models of IBM triggers an increase in the oxidation state of proteins which correlates with the paralysis phenotype (18, 73). In terms of toxicity, our evidence shows that in the constitutive Aβ in vivo model, which resembles the human disease, the accumulation of oxidized peptide in adult individuals leads to increased toxicity. The toxicity translates into decreased synaptic function, compromised synaptic structure, and diminished locomotory capacity. Our results support previous reports showing that the oxidized oligomers are more toxic in long-term experiments (58). Moreover, there is evidence showing that the MsrA system has a role in protecting cultured neurons from Aβ1-42 toxicity. The oxidation of Met35 to Met-sulfoxide induces the activity of this oxidation repair system to increase cell viability in cortical and hippocampal neurons (41, 46). In C. elegans, the absence of SOD-3, an enzyme that contributes to the redox balance of the organism, triggers serious motility impairments even in the absence of Aβ. This phenotype worsens only slightly in the presence of Aβ in contrast to what occurs in msra-1 mutants. Therefore, the lack of SOD-3 may influence Aβ toxicity as a consequence of a general increase in oxidative stress. Instead, the inability to specifically repair oxidized methionines (lack of MSRA-1) seems to have a greater impact in increasing Aβ toxicity.
In our previous work, we found that the NMJ functional defects (increased nicotine resistance) caused by Aβ correlate with the specific mislocalization of the nicotinic acetylcholine receptor ACR-16 in the postsynaptic region (56). This receptor shows a high identity with the α7 subunit of the vertebrate nicotinic AChR expressed in neurons (21). The evidence shows that Aβ can affect the function of nAChRs. For example, Aβ1–42 binds with a high affinity to α7 and α4β2 nAChRs (70). In this work, we show that in the absence of MSRA-1, which makes oxidation repair of methionines impossible, the clustering of ACR-16 is further compromised in the Aβ transgenic worms. This result is in agreement with the phenotype of increased nicotine resistance observed in this strain (Fig. 2A). There is no clear evidence indicating whether the Aβ oligomers and protofibrils interact directly or indirectly with the nicotinic receptors (8). However, Pym 2007 demonstrated that the subtype-specific actions on neuronal nAChRs, displayed by the Aβ peptide, are dependent on Met35 of the Aβ1–42 sequence (55). Interestingly, the effects observed on neuronal nAChR subtypes are strongly dependent on the presence of a sulfoxidized amino acid in position 35 of Aβ, suggesting that an interaction between Aβ and nAChRs may play a role in these effects.
It is expected that the absence of the MSRA-1 enzyme will result in the permanent oxidation of many other proteins, and this could be the real cause of the aggravated phenotypes we observe in the Aβ, msra-1 strain. This is a caveat that is very difficult to resolve completely. However, based on our previous and thorough analysis of the msra-1 mutants (39), we performed our experiments in developmental stages where the msra-1 deletion does not cause any apparent phenotypes in the absence of Aβ.
In this work, we also show that, in the absence of MSRA-1, the aggregation kinetic of intracellular Aβ-peptide in vivo is similar to what has already been shown in vitro. Zagorsky and collaborators showed that the oxidation of Met35 in the Aβ peptide hinders the formation of Aβ aggregates in vitro (23–25). We show that there is a significant decrease in the formation of Aβ aggregates when we prevent the oxidation repair of methionines in vivo. Our results suggest that while the number and area of Aβ aggregates decrease, the oligomeric species increase. Our experimental design enables us to indirectly address whether the oxidation in Met35 has a role in Aβ assembly and toxicity (12, 34, 73). In contrast, other experimental approaches substitute Met35 by a redox-unreactive amino acid, which can alter the aggregation of the peptide itself, independent of the oxidation in Met35 (11, 13).
Interestingly, the toxicity results we obtained with the inducible Aβ strain in the msra-1 background are in conflict with those obtained with the constitutive Aβ strain. The worms from the inducible strain generate large amounts of oligomers in a very short period of time, they never form amyloid aggregates, and they paralyze and die in a matter of hours. This inducible model does not resemble the time course and the pathological features of IBM. However, we believe that it can give some insights into the very early stages of Aβ aggregation. Worms from the constitutive strain express Aβ during the animal's lifetime, they form numerous amyloid aggregates and show a wild-type lifespan (40), similar to what happens in the human disease. In the absence of MSRA-1, the worms from the inducible Aβ strain show a mild and transient improvement in the phenotype (the paralysis is delayed) that lasts for only a few hours. We could explain these results based on the aggregation kinetics of the oxidized peptide in vitro where the oxidized monomers require longer times to form soluble oligomers and fibrils (23–26). We could hypothesize that since the Aβ monomers are less toxic than the oligomers, toxicity and paralysis will be temporarily delayed. However, since this is a very dynamic process, the monomers will quickly form toxic oligomers that trigger paralysis. This approach only enables us to look at a very specific time point on the kinetics of Aβ aggregation in a situation where large quantities of peptide are produced in a very short time frame.
The C. elegans model is a powerful tool for genetic analysis of biological processes. In this work, we have taken advantage of this characteristic to examine the role of MSRA-1 in Aβ-peptide aggregation and toxicity in vivo. Our analyses enabled us to gain insights into the unsolved problem of Aβ redox state by correlating phenotypic analyses, the aggregation state of Aβ, and the absence of the system for Met-O reduction. It is clear that additional biochemical analyses are needed to achieve a quantitative evaluation of Met35 redox state in the Aβ peptide. However, C. elegans has limitations in this regard, and it is, therefore, necessary to look for more suitable animal models (mouse models of IBM and AD).
Our proposed model of the pathological events in a C. elegans model of IBM states that the accumulation of specific Aβ species in muscle cells causes motility impairments, and structural and functional defects in the neuromuscular synapse (Fig. 6A). The redox state of Aβ is a key component in the modulation of Aβ aggregation. In our IBM model that constitutively expresses Aβ in muscle cells and lacks MSRA-1, the toxic Aβ oligomeric species will accumulate because the oxidation in Met35 inhibits the formation of less toxic fibrils and aggregates (Fig. 6B). This toxicity is associated with motility defects and early synaptic dysfunction at the NMJ. In future, it will be important to explore other processes that are being affected directly or indirectly by the oxidized oligomers, perhaps later in the development of the disease, such as mitochondrial function and protein homeostasis.
FIG. 6.

Proposed model. The cartoon shows the early pathological events in the IBM C. elegans model. (A) The accumulation of Aβ species in different states of oligomerization at the neuromuscular synapse causes an alteration of specific postsynaptic molecules, including nAChRs. The oxidative state of Aβ Met35 alters Aβ aggregation kinetics. (B) During the development of IBM, the oxidation of Aβ-Met35 may shift the aggregation kinetics toward the formation of more toxic oligomeric species. The oxidation of the toxic-soluble oligomers prevents the formation of fibrils and, ultimately, of less toxic amyloid aggregates. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Materials and Methods
Nematode strains and culture
Nematodes were raised at 20°C (unless otherwise indicated) under standard laboratory conditions on agar plates cultured with Escherichia coli (OP50) (64).
The following strains were used: N2 (Bristol) as the wild type, msra-1(tm1421)II, VC433 sod-3(gk235) X. Transgenic strains CL2120 (dvIs14[unc-54/Aβ1-42(pCL12)+mtl-2::GFP(pCL26)]) and CL2122 (dvIs15[pPD30.38(unc-54vector)+mtl-2::GFP(pCL26)]) and CL2006 (dvIs2[pCL12(unc-54/hAβ1–42)+pRF4(rol-6(su1006)] were previously described (19). Briefly, CL2120 predominantly expresses the human Aβ3-42 peptide under the control of the unc-54 (myosin heavy chain) promoter that drives expression in the body wall muscle cells. These animals form intracellular amyloid deposits constitutively in their muscle cells (37). CL2122 is the control strain for CL2120, without Aβ expression. Strain CL4176smg-1(cc546)I;dvIs27[pAF29(Pmyo-3::Aβ1-42)+pRF4(rol-6(su1006)] expresses human Aβ3-42 in the body wall muscle cells when induced at 23°C, and has the roller phenotype. The Aβ transgenic strains and their controls were a gift of Dr. Christopher Link, University of Colorado, Boulder, CO.
Strains carrying GFP fusion proteins Pmyo3::ACR-16::GFP were a gift from Dr. M. Francis, University of Massachusetts, Worcester, MA.
We constructed the following strains by standard genetic crosses:
ANM19 dvIs14[unc-54/Aβ1-42(pCL12)+mtl-2::GFP(pCL26)];msra-1(tm1421)II, ANM48dvIs14[unc-54/Aβ1-42(pCL12)+mtl-2::GFP(pCL26);sod-3(gk235)X, ANM30(dvIs2[pCL12(unc-54/hAβ1–42)+pRF4(rol-6(su1006)];Pmyo3::ACR-16::GFP and ANM31[pCL12(unc-54/Aβ1-42)pRF4(su1006)], [Punc-54::Aβ1-42;pRF4(rol-6(su1006)];Pmyo3::ACR-16::GFP), ANM12smg-1(cc546)I; dvIs27[pAF29(Pmyo-3::Aβ1-42)+pRF4(rol-6(su1006)];msra-1(tm1421)II.
Transgenic strains were generated by germline transformation using standard microinjection techniques (64). Multiple independent transgenic lines were generated for each transgenic strain. For (dvIs14[unc-54/Aβ1-42(pCL12)+mtl-2::GFP(pCL26)]);msra-1(tm1421) rescue experiments, ANM19 strain was injected with the pTTX3::cherry plasmid (70 ng/μl) along with plasmid pRA1006 carrying the complete wild-type msra-1 gene (100 ng/μl) to generate strain ANM45. For RNAi assays, we constructed strain ANM57 uccEx17[rol-6(su2006)pmsra-1::MSRA-1::GFP] that enables us to confirm the specific interference of the MSRA-1 expression in all tissues except the nervous system.
Plasmid construction
pRA1006: the plasmid carries the complete msra-1 gene plus a −1403 bp 5′UTR. The DNA was obtained by polymerase chain reaction-mediated amplification of cosmid F43E2.5 using oligonucleotide primers 5′AAACAATAgTgATgTACTgTg3′ and 5′TAAAATTCATTgAAAACCTgATAAA3′. The polymerase chain reaction (PCR) fragment was cloned into pGEM-T easy vector (Promega).
pRA1045: the plasmid carries the complete msra-1 gene plus a −1403 bp 5′UTR. The DNA was obtained by polymerase chain reaction-mediated amplification of cosmid F43E2.5 using oligonucleotide primers 5′AAACAATAgTgATgTACTgTg3′ and 5′TAAAATTCATTgAAAACCTgATAAA3′. The PCR fragment was cloned into the pPD95_75GFP vector (Addgene).
Thrashing assays
Individual 1- and 3-day-old adult animals (between 15–25 animals per experiment) were placed on a 30 μl drop of M9 buffer (64). After a 2-min recovery period, each individual worm was recorded for 1.0 min and the thrashes were counted.
Microscopy
Animals were anesthetized in 20 μM NaAzide and mounted on slides for epifluorescence. Fluorescence images were acquired using the same exposure parameters for all experimental conditions, with 40×or 100×objectives in an Olympus BX51 microscope (Shinjuku) equipped with a digital camera Micropublisher 3.3 RTV (JH Technologies).
Thioflavine-S staining
Thioflavine-S (ThS) staining was performed as previously described (19). Briefly, the worms washed from the plates were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) pH 7.4 for 24 h at 4°C. The fixative solution was removed, replaced by permeabilization solution (125 mM Tris, pH 7.4, 1% Triton X-100, and 5% β-mercaptoethanol), and incubated at 37°C for 24 h. The animals were washed thrice in PBS-T (PBS+Triton X-100 0.1%), stained in 0.125% Th-S (Sigma) in 50% ethanol for 2 min, and destained for another 2 min in 50% ethanol. The stained samples were resuspended in PBS, mounted in fluorescence mounting medium (DAKO), observed with epifluorescence microscopy (Olympus BX51), and photographed with a digital camera (Micropublisher 3.3RTV; JH Technologies).
BSB staining
BSB was purchased from AnaSpec, Inc. and prepared according to the manufacturer's instructions. The assay was performed according to the protocol established by Toledo and Inestrosa (66) with the following modifications. Briefly, synchronized worms were cultured and collected at different stages, fixed with 4% paraformaldehide, and permeabilized as indicated by Fay et al. (19). Then, the worms were incubated with BSB (0.02% resuspended in ethanol 50%), for 4 h at 37° C. The worms were then washed with lithium carbonate (1 M) with water and finally mounted for microscopy observation. BSB staining intensity was estimated using the WCIF Image J software.
Image fluorescence quantification
Digital quantification of areas positive for ThS, BSB, or ACR-16::GFP fluorescence was done using the ImageJ software. For ThS or BSB, the analysis revealed the total average area of the deposits. The total area of deposits was divided by the total head area to standardize it to the worm's size. The images were transformed to 8-bit and after subtraction of background fluorescence, a region of interest was drawn around individual areas of fluorescence in the worm's head. In the case of ACR16::GFP puncta, quantification of fluorescence was performed along the ventral nerve cord. Pictures were always obtained from the anterior or posterior regions nearest to the vulva. Data were estimated as an integrated density value using the same threshold parameters for control and treatment situations.
Neuromuscular synaptic transmission assay
Worms were cultured on regular nematode growth medium (NGM) agar plates (64). Synchronized worms of the same age (25–30) were placed on assay plates (NGM supplemented with 0.25 mM Levamisole (Sigma)), 31 mM Nicotine, or 1 mM Aldicarb (Sigma). We then analyzed the drug-induced paralysis over time (56).
Protein extraction
Worms were collected from the plates with M9 buffer, transferred to tubes, centrifuged, and washed twice to eliminate bacteria. The final worm pellet was resuspended in 200 μl lysis buffer in the presence of protease inhibitors (50 mM HEPES pH=7.5; 6 mM MgCl2; 1 mM EDTA; 75 mM sucrose; 25 mM benzamidine; and 1%Triton X-100) and frozen at 80°C. The samples were sonicated three times on ice for 15 s and centrifuged at 12,000 g for 15 min; the supernatants were removed and use for Western blot analysis.
Western blot analyses
The crude protein extracts were centrifuged for 5 min at 9500 g to eliminate the cuticles. The supernatant was transferred to a new tube, and total protein content was quantified using the Bradford method. Equal protein quantities were suspended in loading buffer (50 mM Tris-HCl pH 6.8; 5% SDS; 10% glycerol; bromophenol blue; and 5% β-mercaptoethanol) and boiled for 3 min adapted from (72). The samples were subjected to electrophoresis on 10%–20% Tris-Tricine Precast Gel (BioRad) at a constant voltage (100 V) of 4°C. The proteins were then transferred to polyvinylidene difluoride membranes for 1 h at 4°C (300 mA). The membranes were boiled in PBS for 3 min before blocking with a solution of PBT (PBS, 0.1% Tween-20) plus 5% milk for 1 h at room temperature. We used the mouse anti-Aβ monoclonal antibody 6E10 (Chemicon, Millipore Corporation) 1:1000 dilution or mouse anti-α-tubulin monoclonal antibody (Sigma) at 1:5000 dilution overnight at 4°C. We used goat anti-mouse HRP as a secondary antibody. For GFP Western blot, we use the A11121 monoclonal antibody 11E5 (LifeTechnologies, Invitrogen) following the manufacturer's recommendations.
Immunoprecipitation assay
Briefly, crude protein extracts were diluted to 1 μg/μl in the lysis buffer in the presence of protease inhibitors. From this dilution, 200 μg of protein were precleared with 20 μl of Protein G PLUS-agarose beads (Santa Cruz Biotechnology) for 30 min at 4°C, with rotation. Samples were centrifuged at 2370 g for 5 min, and the supernatant was incubated with 1 μg of mouse anti-Aβ antibody (6E10; Millipore) for 1 h at 4°C. 25 μl of new Protein G beads were added for 3 h at 4°C, with rotation. The immune-complexes were finally pelleted at 590 g for 5 min, washed thrice with cold PBS, and resuspended with 2X loading buffer. Immnunoblots were performed with the rabbit anti-methionine sulfoxide polyclonal antibody (Novus Biologicals) to detect oxidized methionine, and the loading buffer used had no dithiothreitol or β-mercaptoethanol.
Methionine oxidation quantification
Met-O was measured by high-performance liquid chromatography (HPLC)-fluorometric detection, using a modification of the method reported by Minniti et al. and references therein (39). Briefly, total protein extracts were hydrolyzed using methanesulfonic acid and then derivatized with orto-phthaldialdehyde. L-Methionine sulfone (Met-O2) was used as an internal standard. Analyses were performed by reverse-phase HPLC using a Merck-Hitachi L-6200 Intelligent pump engaged to an F-1000 fluorescence spectrophotometer detector and a column oven L-7350 (Merck LaChrom). The excitation wavelength was set at 340 nm, and the emission was set at 455 nm. Amino acids were separated on an Inertsil ODS-3 (C18) 150 mm×4.6 mm×5 μm GLSciences column. Gradient elution was performed with solution A, composed of 75 mM sodium acetate (pH 4.5), 19% methanol, and 1% tetrahydrofuran and solution B, composed of 75 mM sodium acetate (pH 4.5) and 80% methanol, delivered at a flow rate of 0.9 ml/min, as follows: initially and during the first 1.5 min with 100% A; for the next 11.5 min, with 96% A; next 5 min with 86% A; next 7 with 60% A; and finally, 13 min with 0% A. The injection volume for the extract was 20 μl. All measurements were performed in the “Centro de Nutrición Molecular y Enfermedades Crónicas, Pontificia Universidad Católica de Chile (CNMEC-UC).”
RNAi experiments
These experiments were performed using standard RNAi feeding protocols (27) and the corresponding E. coli strain from the Ahringer RNAi feeding library (F43E2.5clone). We acknowledge the originator of the library, Dr. Julie Ahringer, and Source BioScience LifeSciences for providing the library. The exposure of worms to the msra-1 RNAi clone was done from the embryo stage. Verification of interference was performed for every assay using strain ANM57 (MSRA::GFP).
Data and statistical analysis
The statistical analyses were performed using GraphPadPrism5 software. The specific statistical tests used in each experiment are indicated in the corresponding Figure Legends.
Supplementary Material
Abbreviations Used
- ACR-16
acetylcholine receptor16
- AD
Alzheimer's disease
- Aβ
amyloid-beta peptide
- BSB
(Trans, trans)-1-bromo-2,5-bis-(3-hydroxycarbonyl-4-hydroxy) styrylbenzene
- EDTA
ethylene-diamine-tetraacetic acid
- GFP
green fluorescent protein
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HPLC
high-performance liquid chromatography
- IBM
inclusion body myositis
- Met35
methionine at position 35 of the Aβ peptide
- Met-O
methionine sulfoxide
- Msr
methionine sulfoxide reductase
- MSRA-1
methionine sulfoxide reductase A-1 enzyme
- msra-1
methionine sulfoxide reductase A-1 gene
- mtl-2
metallothionein2 gene
- MW
molecular weight
- NaAzide
sodium azide
- nAChR
nicotinic acetylcholine receptor
- NGM
nematode growth medium
- NMJ
neuromuscular junction
- PBS
phosphate-buffer saline
- PCR
polymerase chain reaction
- RNAi
RNA interference
- rol-6
roller-6 gene
- ROS
reactive oxygen species
- SDS
sodium dodecyl sulfate
- SOD-3
superoxide dismutase-3 enzyme
- sod-3
superoxide dismutase-3 gene
- ThS
thioflavine-S
- unc-54
uncoordinated 54 gen
- UTR
untranslated region
- Wt
wild type
Acknowledgments
This research was supported by FONDECYT 1120213 to RA, the Center for Aging and Regeneration (CARE), PFB12/2007, Base Financing Program for Scientific and Technological Centers of Excellence, CONICYT. Some nematode strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources (NCRR). The tm1421 allele was obtained from the National Bioresource Project. The authors thank Dr. Chris Link for providing the Aβ strains, Dr. Michael Francis for the ACR-16::GFP strain, and Dr. Olivia Casanueva for the msra-1 RNAi clone.
Author Disclosure Statement
No conflicting financial interests exist for any of the authors.
References
- 1.Aldunate R, Minniti AN, Rebolledo D, and Inestrosa NC. Synaptic defects associated with s-inclusion body myositis are prevented by copper. Biometals 25: 815–824, 2012 [DOI] [PubMed] [Google Scholar]
- 2.Amato AA. and Barohn RJ. Inclusion body myositis: old and new concepts. J Neurol Neurosurg Psychiatry 80: 1186–1193, 2009 [DOI] [PubMed] [Google Scholar]
- 3.Askanas V. and Engel WK. Inclusion-body myositis, a multifactorial muscle disease associated with aging: current concepts of pathogenesis. Curr Opin Rheumatol 19: 550–559, 2007 [DOI] [PubMed] [Google Scholar]
- 4.Askanas V. and Engel WK. Inclusion-body myositis: a myodegenerative conformational disorder associated with Abeta, protein misfolding, and proteasome inhibition. Neurology 66: S39–S48, 2006 [DOI] [PubMed] [Google Scholar]
- 5.Askanas V. and Engel WK. Inclusion-body myositis: muscle-fiber molecular pathology and possible pathogenic significance of its similarity to Alzheimer's and Parkinson's disease brains. Acta Neuropathol 116: 583–595, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Askanas V. and Engel WK. Sporadic inclusion-body myositis: conformational multifactorial ageing-related degenerative muscle disease associated with proteasomal and lysosomal inhibition, endoplasmic reticulum stress, and accumulation of amyloid-beta42 oligomers and phosphorylated tau. Presse Med 40: e219–e235, 2011 [DOI] [PubMed] [Google Scholar]
- 7.Askanas V, Engel WK, and Nogalska A. Pathogenic considerations in sporadic inclusion-body myositis, a degenerative muscle disease associated with aging and abnormalities of myoproteostasis. J Neuropathol Exp Neurol 71: 680–693, 2012 [DOI] [PubMed] [Google Scholar]
- 8.Buckingham SD, Jones AK, Brown LA, and Sattelle DB. Nicotinic acetylcholine receptor signalling: roles in Alzheimer's disease and amyloid neuroprotection. Pharmacol Rev 61: 39–61, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Butterfield DA. Amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer's disease brain. A review. Free Radic Res 36: 1307–1313, 2002 [DOI] [PubMed] [Google Scholar]
- 10.Butterfield DA. and Boyd-Kimball D. The critical role of methionine 35 in Alzheimer's amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity. Biochim Biophys Acta 1703: 149–156, 2005 [DOI] [PubMed] [Google Scholar]
- 11.Butterfield DA. and Bush AI. Alzheimer's amyloid beta-peptide (1–42): involvement of methionine residue 35 in the oxidative stress and neurotoxicity properties of this peptide. Neurobiol Aging 25: 563–568, 2004 [DOI] [PubMed] [Google Scholar]
- 12.Butterfield DA, Galvan V, Lange MB, Tang H, Sowell RA, Spilman P, Fombonne J, Gorostiza O, Zhang J, Sultana R, and Bredesen DE. In vivo oxidative stress in brain of Alzheimer disease transgenic mice: Requirement for methionine 35 in amyloid beta-peptide of APP. Free Radic Biol Med 48: 136–144, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butterfield DA. and Sultana R. Methionine-35 of abeta(1–42): importance for oxidative stress in Alzheimer disease. J Amino Acids 2011: 198430, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Butterfield DA, Swomley AM, and Sultana R. Amyloid beta-peptide (1–42)-induced oxidative stress in Alzheimer disease: importance in disease pathogenesis and progression. Antioxid Redox Signal 19: 823–835, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clementi ME, Pezzotti M, Orsini F, Sampaolese B, Mezzogori D, Grassi C, Giardina B, and Misiti F. Alzheimer's amyloid beta-peptide (1–42) induces cell death in human neuroblastoma via bax/bcl-2 ratio increase: an intriguing role for methionine 35. Biochem Biophys Res Commun 342: 206–213, 2006 [DOI] [PubMed] [Google Scholar]
- 16.Dalakas MC. Inflammatory muscle diseases: a critical review on pathogenesis and therapies. Curr Opin Pharmacol 10: 346–352, 2010 [DOI] [PubMed] [Google Scholar]
- 17.Dostal V, Roberts CM, and Link CD. Genetic mechanisms of coffee extract protection in a Caenorhabditis elegans model of beta-amyloid peptide toxicity. Genetics 186: 857–866, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drake J, Link CD, and Butterfield DA. Oxidative stress precedes fibrillar deposition of Alzheimer's disease amyloid beta-peptide (1–42) in a transgenic Caenorhabditis elegans model. Neurobiol Aging 24: 415–420, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Fay DS, Fluet A, Johnson CJ, and Link CD. In vivo aggregation of beta-amyloid peptide variants. J Neurochem 71: 1616–1625, 1998 [DOI] [PubMed] [Google Scholar]
- 20.Gabbita SP, Aksenov MY, Lovell MA, and Markesbery WR. Decrease in peptide methionine sulfoxide reductase in Alzheimer's disease brain. J Neurochem 73: 1660–1666, 1999 [DOI] [PubMed] [Google Scholar]
- 21.Grassi F, Palma E, Tonini R, Amici M, Ballivet M, and Eusebi F. Amyloid beta(1–42) peptide alters the gating of human and mouse alpha-bungarotoxin-sensitive nicotinic receptors. J Physiol 547: 147–157, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansel A, Heinemann SH, and Hoshi T. Heterogeneity and function of mammalian MSRs: enzymes for repair, protection and regulation. Biochim Biophys Acta 1703: 239–247, 2005 [DOI] [PubMed] [Google Scholar]
- 23.Hou L, Kang I, Marchant RE, and Zagorski MG. Methionine 35 oxidation reduces fibril assembly of the amyloid abeta-(1–42) peptide of Alzheimer's disease. J Biol Chem 277: 40173–40176, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Hou L, Lee HG, Han F, Tedesco JM, Perry G, Smith MA, and Zagorski MG. Modification of amyloid-beta1-42 fibril structure by methionine-35 oxidation. J Alzheimers Dis 37: 9–18, 2013 [DOI] [PubMed] [Google Scholar]
- 25.Hou L, Shao H, Zhang Y, Li H, Menon NK, Neuhaus EB, Brewer JM, Byeon IJ, Ray DG, Vitek MP, Iwashita T, Makula RA, Przybyla AB, and Zagorski MG. Solution NMR studies of the A beta(1–40) and A beta(1–42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J Am Chem Soc 126: 1992–2005, 2004 [DOI] [PubMed] [Google Scholar]
- 26.Johansson AS, Bergquist J, Volbracht C, Paivio A, Leist M, Lannfelt L, and Westlind-Danielsson A. Attenuated amyloid-beta aggregation and neurotoxicity owing to methionine oxidation. Neuroreport 18: 559–563, 2007 [DOI] [PubMed] [Google Scholar]
- 27.Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, Welchman DP, Zipperlen P, and Ahringer J. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421: 231–237, 2003 [DOI] [PubMed] [Google Scholar]
- 28.Kim HY. and Gladyshev VN. Methionine sulfoxide reductases: selenoprotein forms and roles in antioxidant protein repair in mammals. Biochem J 407: 321–329, 2007 [DOI] [PubMed] [Google Scholar]
- 29.LaFerla FM, Green KN, and Oddo S. Intracellular amyloid-beta in Alzheimer's disease. Nat Rev 8: 499–509, 2007 [DOI] [PubMed] [Google Scholar]
- 30.LaFerla FM. and Oddo S. Alzheimer's disease: Abeta, tau and synaptic dysfunction. Trends Mol Med 11: 170–176, 2005 [DOI] [PubMed] [Google Scholar]
- 31.Lee BC, Lee YK, Lee HJ, Stadtman ER, Lee KH, and Chung N. Cloning and characterization of antioxidant enzyme methionine sulfoxide-S-reductase from Caenorhabditis elegans. Arch Biochem Biophys 434: 275–281, 2005 [DOI] [PubMed] [Google Scholar]
- 32.Link CD. C. elegans models of age-associated neurodegenerative diseases: lessons from transgenic worm models of Alzheimer's disease. Exp Gerontol 41: 1007–1013, 2006 [DOI] [PubMed] [Google Scholar]
- 33.Lotz BP, Engel AG, Nishino H, Stevens JC, and Litchy WJ. Inclusion body myositis. Observations in 40 patients. Brain 112 (Pt 3): 727–747, 1989 [DOI] [PubMed] [Google Scholar]
- 34.Maiti P, Lomakin A, Benedek GB, and Bitan G. Despite its role in assembly, methionine 35 is not necessary for amyloid beta-protein toxicity. J Neurochem 113: 1252–1262, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martinez J, Lisa S, Sanchez R, Kowalczyk W, Zurita E, Teixido M, Giralt E, Andreu D, Avila J, and Gasset M. Selenomethionine incorporation into amyloid sequences regulates fibrillogenesis and toxicity. PloS One 6: e27999, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW, Jr., and Morris JC. Altered expression of synaptic proteins occurs early during progression of Alzheimer's disease. Neurology 56: 127–129, 2001 [DOI] [PubMed] [Google Scholar]
- 37.McColl G, Roberts BR, Gunn AP, Perez KA, Tew DJ, Masters CL, Barnham KJ, Cherny RA, and Bush AI. The Caenorhabditis elegans A beta 1–42 model of Alzheimer disease predominantly expresses A beta 3–42. J Biol Chem 284: 22697–22702, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McFerrin J, Engel WK, and Askanas V. Impaired innervation of cultured human muscle overexpressing betaAPP experimentally and genetically: relevance to inclusion-body myopathies. Neuroreport 9: 3201–3205, 1998 [DOI] [PubMed] [Google Scholar]
- 39.Minniti AN, Cataldo R, Trigo C, Vasquez L, Mujica P, Leighton F, Inestrosa NC, and Aldunate R. Methionine sulfoxide reductase A expression is regulated by the DAF-16/FOXO pathway in Caenorhabditis elegans. Aging cell 8: 690–705, 2009 [DOI] [PubMed] [Google Scholar]
- 40.Minniti AN, Rebolledo DL, Grez PM, Fadic R, Aldunate R, Volitakis I, Cherny RA, Opazo C, Masters C, Bush AI, and Inestrosa NC. Intracellular amyloid formation in muscle cells of Abeta-transgenic Caenorhabditis elegans: determinants and physiological role in copper detoxification. Mol Neurodegeneration 4: 2, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Misiti F, Clementi ME, and Giardina B. Oxidation of methionine 35 reduces toxicity of the amyloid beta-peptide(1–42) in neuroblastoma cells (IMR-32) via enzyme methionine sulfoxide reductase A expression and function. Neurochem Int 56: 597–602, 2010 [DOI] [PubMed] [Google Scholar]
- 42.Moskovitz J, Bar-Noy S, Williams WM, Requena J, Berlett BS, and Stadtman ER. Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals. Proc Natl Acad Sci U S A 98: 12920–12925, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moskovitz J, Berlett BS, Poston JM, and Stadtman ER. The yeast peptide-methionine sulfoxide reductase functions as an antioxidant in vivo. Proc Natl Acad Sci U S A 94: 9585–9589, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moskovitz J, Flescher E, Berlett BS, Azare J, Poston JM, and Stadtman ER. Overexpression of peptide-methionine sulfoxide reductase in Saccharomyces cerevisiae and human T cells provides them with high resistance to oxidative stress. Proc Natl Acad Sci U S A 95: 14071–14075, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moskovitz J, Jenkins NA, Gilbert DJ, Copeland NG, Jursky F, Weissbach H, and Brot N. Chromosomal localization of the mammalian peptide-methionine sulfoxide reductase gene and its differential expression in various tissues. Proc Natl Acad Sci U S A 93: 3205–3208, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moskovitz J, Maiti P, Lopes DH, Oien DB, Attar A, Liu T, Mittal S, Hayes J, and Bitan G. Induction of methionine-sulfoxide reductases protects neurons from amyloid beta-protein insults in vitro and in vivo. Biochemistry 50: 10687–10697, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moskovitz J, Poston JM, Berlett BS, Nosworthy NJ, Szczepanowski R, and Stadtman ER. Identification and characterization of a putative active site for peptide methionine sulfoxide reductase (MsrA) and its substrate stereospecificity. J Biol Chem 275: 14167–14172, 2000 [DOI] [PubMed] [Google Scholar]
- 48.Moskovitz J, Weissbach H, and Brot N. Cloning the expression of a mammalian gene involved in the reduction of methionine sulfoxide residues in proteins. Proc Natl Acad Sci U S A 93: 2095–2099, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, and McConlogue L. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 20: 4050–4058, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nimmrich V. and Ebert U. Is Alzheimer's disease a result of presynaptic failure? Synaptic dysfunctions induced by oligomeric beta-amyloid. Rev Neurosci 20: 1–12, 2009 [DOI] [PubMed] [Google Scholar]
- 51.Nogalska A, D'Agostino C, Engel WK, and Askanas V. Activation of the gamma-secretase complex and presence of gamma-secretase-activating protein may contribute to Abeta42 production in sporadic inclusion-body myositis muscle fibers. Neurobiol Dis 48: 141–149, 2012 [DOI] [PubMed] [Google Scholar]
- 52.Nogalska A, D'Agostino C, Engel WK, Klein WL, and Askanas V. Novel demonstration of amyloid-beta oligomers in sporadic inclusion-body myositis muscle fibers. Acta Neuropathol 120: 661–666, 2010 [DOI] [PubMed] [Google Scholar]
- 53.Oien DB. and Moskovitz J. Substrates of the methionine sulfoxide reductase system and their physiological relevance. Curr Top Dev Biol 80: 93–133, 2008 [DOI] [PubMed] [Google Scholar]
- 54.Petropoulos I, Mary J, Perichon M, and Friguet B. Rat peptide methionine sulphoxide reductase: cloning of the cDNA, and down-regulation of gene expression and enzyme activity during aging. Biochem J 355: 819–825, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pym LJ, Buckingham SD, Tsetlin V, Boyd CA, and Sattelle DB. The Abeta1-42M35C mutated amyloid peptide Abeta1-42 and the 25–35 fragment fail to mimic the subtype-specificity of actions on recombinant human nicotinic acetylcholine receptors (alpha7, alpha4beta2, alpha3beta4). Neurosci Lett 427: 28–33, 2007 [DOI] [PubMed] [Google Scholar]
- 56.Rebolledo DL, Aldunate R, Kohn R, Neira I, Minniti AN, and Inestrosa NC. Copper reduces Abeta oligomeric species and ameliorates neuromuscular synaptic defects in a C. elegans model of inclusion body myositis. J Neurosci 31: 10149–10158, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rebolledo DL, Minniti AN, Grez PM, Fadic R, Kohn R, and Inestrosa NC. Inclusion body myositis: a view from the Caenorhabditis elegans muscle. Mol Neurobiol 38: 178–198, 2008 [DOI] [PubMed] [Google Scholar]
- 58.Ripoli C, Piacentini R, Riccardi E, Leone L, Li Puma DD, Bitan G, and Grassi C. Effects of different amyloid beta-protein analogues on synaptic function. Neurobiol Aging 34: 1032–1044, 2013 [DOI] [PubMed] [Google Scholar]
- 59.Sattelle DB. and Buckingham SD. Invertebrate studies and their ongoing contributions to neuroscience. Invert Neurosci 6: 1–3, 2006 [DOI] [PubMed] [Google Scholar]
- 60.Schallreuter KU, Rubsam K, Chavan B, Zothner C, Gillbro JM, Spencer JD, and Wood JM. Functioning methionine sulfoxide reductases A and B are present in human epidermal melanocytes in the cytosol and in the nucleus. Biochem Biophys Res Commun 342: 145–152, 2006 [DOI] [PubMed] [Google Scholar]
- 61.Selkoe DJ. Alzheimer's disease is a synaptic failure. Science (New York, NY: 298: 789–791, 2002 [DOI] [PubMed] [Google Scholar]
- 62.Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res 192: 106–113, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sivanesan S, Tan A, and Rajadas J. Pathogenesis of Abeta oligomers in synaptic failure. Curr Alzheimer Res 10: 316–323, 2013 [DOI] [PubMed] [Google Scholar]
- 64.Stiernagle T. Maintenance of C. elegans. WormBook e: The C. elegans Research Community, WormBook, 2006. DOI: 10.1895/wormbook.1.101.1, http://www.wormbook.org [DOI] [PMC free article] [PubMed]
- 65.Sultana R, Robinson RA, Lange MB, Fiorini A, Galvan V, Fombonne J, Baker A, Gorostiza O, Zhang J, Cai J, Pierce WM, Bredesen DE, and Butterfield DA. Do proteomics analyses provide insights into reduced oxidative stress in the brain of an Alzheimer disease transgenic mouse model with an M631L amyloid precursor protein substitution and thereby the importance of amyloid-beta-resident methionine 35 in Alzheimer disease pathogenesis? Antioxid Redox Signal 17: 1507–1514, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Toledo EM. and Inestrosa NC. Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1DeltaE9 mouse model of Alzheimer's disease. Mol Psychiatry 15: 272–285, 228, 2009 [DOI] [PubMed] [Google Scholar]
- 67.Tseng BP, Kitazawa M, and LaFerla FM. Amyloid beta-peptide: the inside story. Curr Alzheimer Res 1: 231–239, 2004 [DOI] [PubMed] [Google Scholar]
- 68.Varadarajan S, Yatin S, Aksenova M, and Butterfield DA. Review: Alzheimer's amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. J Struct Biol 130: 184–208, 2000 [DOI] [PubMed] [Google Scholar]
- 69.Vattemi G, Nogalska A, King Engel W, D'Agostino C, Checler F, and Askanas V. Amyloid-beta42 is preferentially accumulated in muscle fibers of patients with sporadic inclusion-body myositis. Acta Neuropathol 117: 569–574, 2009 [DOI] [PubMed] [Google Scholar]
- 70.Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, and Reitz AB. beta-Amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer's disease pathology. J Biol Chem 275: 5626–5632, 2000 [DOI] [PubMed] [Google Scholar]
- 71.Wilcox KC, Lacor PN, Pitt J, and Klein WL. Abeta oligomer-induced synapse degeneration in Alzheimer's disease. Cell Mol Neurobiol 31: 939–948, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu Y, Wu Z, Butko P, Christen Y, Lambert MP, Klein WL, Link CD, and Luo Y. Amyloid-beta-induced pathological behaviors are suppressed by Ginkgo biloba extract EGb 761 and ginkgolides in transgenic Caenorhabditis elegans. J Neurosci 26: 13102–13113, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yatin SM, Varadarajan S, Link CD, and Butterfield DA. In vitro and in vivo oxidative stress associated with Alzheimer's amyloid beta-peptide (1–42). Neurobiol Aging 20: 325–330; discussion 339–342, 1999 [DOI] [PubMed] [Google Scholar]
- 74.Ye CP, Selkoe DJ, and Hartley DM. Protofibrils of amyloid beta-protein inhibit specific K+ currents in neocortical cultures. Neurobiol Dis 13: 177–190, 2003 [DOI] [PubMed] [Google Scholar]
- 75.Zhang XH. and Weissbach H. Origin and evolution of the protein-repairing enzymes methionine sulphoxide reductases. Biol Rev Camb Philos Soc 83: 249–257, 2008 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.