

Abstract

Arylalkylamine N-acetyltransferase (AANAT) catalyzes the penultimate step in the biosynthesis of melatonin and other N-acetylarylalkylamides from the corresponding arylalkylamine and acetyl-CoA. The N-acetylation of arylalkylamines is a critical step in Drosophila melanogaster for the inactivation of the bioactive amines and the sclerotization of the cuticle. Two AANAT variants (AANATA and AANATB) have been identified in D. melanogaster, in which AANATA differs from AANATB by the truncation of 35 amino acids from the N-terminus. We have expressed and purified both D. melanogaster AANAT variants (AANATA and AANATB) in Escherichia coli and used the purified enzymes to demonstrate that this N-terminal truncation does not affect the activity of the enzyme. Subsequent characterization of the kinetic and chemical mechanism of AANATA identified an ordered sequential mechanism, with acetyl-CoA binding first, followed by tyramine. We used a combination of pH–activity profiling and site-directed mutagenesis to study prospective residues believed to function in AANATA catalysis. These data led to an assignment of Glu-47 as the general base in catalysis with an apparent pKa of 7.0. Using the data generated for the kinetic mechanism, structure–function relationships, pH–rate profiles, and site-directed mutagenesis, we propose a chemical mechanism for AANATA.
Biogenic amines are important neuroactive amines found in both vertebrates and invertebrates functioning as neurotransmitters, neuromodulators, or neurohormones via their binding to specific receptors. In insects, the biogenic arylalkylamines are dopamine, tyramine, serotonin, and octopamine, which function primarily as neurotransmitters.1 The pathways for biosynthesis and degradation have important roles in the function of the arylalkylamines as a balance between production and clearance is necessary to maintain the appropriate cellular concentrations of the amines. Dysfunction in either of these opposing pathways would lead to improper cellular levels of the arylalkylamines, leading to errors in the processes regulated by them.2
The biogenic arylalkylamines are all derived in vivo from the cognate aromatic amino acid (tyrosine or tryptophan) precursor. The precursor amino acid is initially hydroxylated by an aromatic amino acid hydroxylase (tyrosine hydroxylase or tryptophan hydroxylase)3,4 and then decarboxylated by aromatic l-amino acid decarboxylase (3,4-dihydroxylphenylalanine decarboxylase)5 to generate either dopamine or serotonin, respectively. Tyramine is derived from the decarboxylation of tyrosine by tyrosine decarboxylase,6,7 which can then be β-hydroxylated to generate octopamine, a reaction that is catalyzed by tyramine β-hydroxylase.8−10 One proposed inactivation reaction for the arylalkylamines is N-acetylation as catalyzed by arylalkylamine N-acetyltransferase (AANAT): acetyl-CoA + arylalkylamine → N-acetylarylalkylamine + CoA-SH.11,12 In addition to arylalkylamine inactivation, acetyl-CoA-dependent N-acetylation is involved in cuticle sclerotization13−16 and melatonin biosynthesis.17,18
AANAT (EC 2.3.1.87), also known as serotonin N-acetyltransferase and dopamine N-acetyltransferase, has been identified in Drosophila melanogaster(11) and is a member of the GCN5-related N-acetyltransferase (GNAT) superfamily.19,20 The identification of N-acetyltransferase activity for arylalkylamines in D. melanogaster was first shown in 1972 by Dewhurst et al.,11 followed by the initial characterization of AANAT in 1977 by Maranda and Hodgetts.21 In 1998, Brodbeck et al.22 identified two biologically relevant variants, variant A (AANATA) and variant B (AANATB), which differ by 35 amino acids found only at the N-terminus of the larger AANATB. Differential transcription of a single AANAT gene leads to the two different AANAT variants.22 These variants are expressed in different tissues and at different life stages, with AANATA being found in the brain, ventral nerve cord, and midgut during late stage embryogenesis and in adults. AANATB is less abundant and is found in the brain only during late pupal stages and in adults, as well.22 AANAT enzymes have been characterized from many organisms and are known to catalyze the rate-limiting penultimate step in the formation of melatonin.20,23−25 Melatonin is a hormone that is produced in a diurnal cycle24 (higher levels are found at night) and is suggested to regulate the life span of D. melanogaster.18
Eight putative AANAT-like enzymes (AANATL) have been identified in D. melanogaster(26) and are proposed to play a role in the biosynthesis of fatty acid amides.27,28 Herein, we report on the cloning, expression, purification, and characterization of D. melanogaster AANATA and AANATB from recombinant Escherichia coli. Our data show that AANATA and AANATB are catalytically similar with respect to substrate specificities and measured kinetic constants (Km,app and kcat,app). More detailed studies of AANATA were performed to define the kinetic mechanism and to elaborate a chemical mechanism that is consistent with both the pH–rate profiles and data from a set of site-directed mutant enzymes.
Experimental Procedures
Materials
The Ambion RETROscript Kit, ProBond nickel-chelating resin, and MicroPoly(A) Purist were purchased from Invitrogen. NdeI, XhoI, Antarctic Phosphatase, and T4 DNA ligase were purchased from New England Biolabs. BL21(DE3) E. coli cells, XL10 E. coli cells, and the pET-28a(+) vector were purchased from Novagen. Kanamycin monosulfate and isopropyl β-d-1-thiogalactopyranoside were purchased from Gold Biotechnology. Oligonucleotides were purchased from Eurofins MWG Operon, and PfuUltra High-Fidelity DNA polymerase was purchased from Agilent. Benzoyl-CoA, acetyl-CoA, butyryl-CoA, hexanoyl-CoA, octanoyl-CoA, decanoyl-CoA, oleoyl-CoA, and N-acetylserotonin were purchased from Sigma-Aldrich. All other reagents were of the highest quality available from either Sigma-Aldrich or Fisher Scientific.
Cloning of D. melanogaster AANATA and AANATB
A cDNA library was generated from D. melanogaster heads, using the Ambion RETROscript Kit and MicroPoly(A) Purist kits. AANATA (NCBI reference sequence NM_079115.2) and AANATB (NCBI reference sequence NM_206212.1) were amplified from the D. melanogaster head cDNA library using the following primers: 5′ GAC TCA TAT GAT GGA GGA CGC ATT GAC C 3′ (forward) and 5′ ATC CCT CGA GCT ACA GCT TGG TCT GCG C 3′ (reverse); 5′ GCT ACA TAT GAT GGA AGT GCA GAA GCT (forward) and 3′ ATC CCT CGA GCT ACA GCT TGG TCT GCG C (reverse), respectively. Polymerase chain reaction (PCR) was performed using PfuUltra High-Fidelity DNA polymerase, using the following set of conditions: initial denaturing step of 95 °C for 2 min, then 30 cycles (95 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min), and then a final extension step of 72 °C for 10 min. The AANATA product or the AANATB PCR product was inserted into a pET-28a(+) vector using the NdeI and XhoI restriction enzymes, yielding an expression vector for each enzyme, AANATA pET-28a or AANATB pET-28a, respectively. The AANATA pET-28a or AANATB pET-28a vector was then transformed into E. coli XL10 competent cells and cultured in Luria broth (LB) supplemented with 40 μg/mL kanamycin at 37 °C. The plasmids were purified using the Promega Wizard Plus SV Minipreps DNA purification kit and sequenced by Eurofins MWG operon. In separate experiments, the AANATA pET-28a or AANATB pET-28a vector was then transformed into E. coli BL21(DE3) cells for expression of AANATA or AANATB, respectively.
Expression and Purification of AANATA and AANATB
The E. coli BL21(DE3) cells harboring either the AANATA pET-28a or AANATB pET-28a expression vector were cultured in LB supplemented with 40 μg/mL kanamycin at 37 °C. The cell cultures were induced at an OD600 of 0.6 with 1 mM isopropyl β-d-1-thiogalactopyranoside for 4 h at 37 °C. The final cultures were then harvested by centrifugation at 5000g for 10 min at 4 °C, and the pellet was collected.
The pellet was then resuspended in 20 mM Tris (pH 7.9), 500 mM NaCl, and 5 mM imidazole; the cells were lysed by sonication, and the cellular debris was removed by centrifugation (10000g for 15 min at 4 °C). The supernatant was loaded onto a column packed with 6 mL of ProBond nickel-chelating resin. The column was first washed with 10 column volumes of 20 mM Tris-HCl (pH 7.9), 500 mM NaCl, and 5 mM imidazole and then with 10 column volumes of 20 mM Tris-HCl (pH 7.9), 500 mM NaCl, and 60 mM imidazole, and AANATA or AANATB eluted in 1 mL fractions of 20 mM Tris-HCl (pH 7.9), 500 mM NaCl, and 500 mM imidazole. Purity was assessed by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and visualized using Coomassie stain. The protein concentration was determined using the Bradford dye binding assay.
Production of Site-Directed Mutants
Each site-directed AANATA mutant was generated by the overlap extension method29 using PfuUltra High-Fidelity DNA polymerase under the following PCR conditions: initial denaturing step of 95 °C for 2 min, then 30 cycles (95 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min), and then a final extension step of 72 °C for 10 min. Primers for each mutant (Table S1 of the Supporting Information) were designed with the Agilent QuickChange Primer Design tool. The AANATA mutant PCR products were then inserted into a pET-28a(+) vector using the NdeI and XhoI restriction enzymes. The AANATA mutant pET-28a vectors were transformed into E. coli XL10 competent cells and cultured in LB supplemented with 40 μg/mL kanamycin at 37 °C. The plasmids were then purified using the Promega Wizard Plus SV Minipreps DNA purification kit and sequenced by Eurofins MWG operon. Individual mutant AANATA proteins were expressed and purified as described previously for the wild-type enzyme.
Measurement of Enzyme Activity
AANATA and AANATB activity was analyzed using Ellman’s reagent30 by measuring the release of coenzyme A at 412 nm in 300 mM Tris-HCl (pH 8.0), 150 μM DTNB, and the desired concentrations of the amino donor substrate and acyl-CoA substrate. Initial velocities of CoA-SH release were measured using a Cary 300 Bio UV–visible spectrophotometer, and the resulting initial velocity kinetic data were fit to the desired equation using SigmaPlot 12.0. Steady-state kinetic constants were obtained by a fit to eq 1
| 1 |
where vo is the initial velocity, [S] is the substrate concentration, Vmax is the maximal velocity, and Km is the Michaelis constant.
Apparent kinetic constants for each arylalkylamine substrate were determined by holding acetyl-CoA at a fixed saturating concentration, whereas those for each acyl-CoA substrate were determined by holding tyramine at a fixed saturating concentration at 22 °C. Assays were performed in triplicate, and the uncertainties for the (kcat/Km)app and relative (kcat/Km)app values were defined by using eq 2, where σ is the standard error.31
| 2 |
Kinetic Mechanism and Inhibitor Analysis
Initial velocity patterns for acetyl-CoA and tyramine were produced by varying the concentration of one substrate while holding the concentration of the other substrate at a fixed concentration and fitting the data to eq 3 for an ordered bi-bi mechanism using Igor Pro 6.34A
| 3 |
where Kia is the dissociation constant for substrate A (acetyl-CoA), Kb is the Km for substrate B (tyramine), and Ka is the Km for substrate A (acetyl-CoA). The two experimental sets were generated by holding the tyramine concentration constant (5, 10, 25, and 50 μM) and varying the concentration of acetyl-CoA, whereas the second set involved holding the concentration of acetyl-CoA constant (20, 40, 60, and 100 μM) and varying the concentration of tyramine.
The IC50 values for long-chain acyl-CoAs and tyrosol were determined at the Km concentration for the appropriate substrate, tyramine at 12 μM or acetyl-CoA at 39 μM, while varying the concentration of the respective inhibitor. The IC50 value was determined by a fit of the resulting data to eq 4 using SigmaPlot 12.0
| 4 |
where vo is the initial velocity without inhibitor, vi is the initial velocity at different inhibitor concentrations, and [I] is the inhibitor concentration. The assays were performed in triplicate.
Dead-end inhibitor analysis was conducted for both oleoyl-CoA and tyrosol. The inhibition patterns were determined by holding one substrate (acetyl-CoA or tyramine) at a fixed concentration and varying the concentration of the other substrate, with each set conducted at a different fixed concentration of the inhibitor. The initial velocities from these inhibitor experiments were fit to eqs 5–7 for competitive, noncompetitive, and uncompetitive inhibition, respectively
| 5 |
| 6 |
| 7 |
where vo is the initial velocity, Vmax is the maximal velocity, [S] is the substrate concentration, Km is the Michaelis constant, [I] is the inhibitor concentration, and Ki is the inhibition constant. The assays were performed in triplicate.
Rate versus pH Dependence
The pH dependence of the steady-state kinetic constants was determined for both acetyl-CoA and tyramine as varied substrates while holding the other substrate at a fixed concentration. Kinetic constants were determined at 0.5 pH intervals from pH 6.0 to 9.5, using the following buffers: MES (pH 6.0–7.0), Tris (pH 7.0–9.0), and 2-amino-2-methyl-1-propanol (AMeP) (pH 9.0–9.5). The resulting data were fit to eq 8 [log(kcat/Km-acetyl-CoA) or log(kcat/Km-tyramine)] and eq 9 (log kcat)
| 8 |
| 9 |
where c is the pH-independent plateau using Igor Pro 6.34A.
Intrinsic Fluorescence Measurements for the Determination of the Coenzyme A Dissociation Constant
Fluorescence spectra were generated with a JASCO FP-8300 spectrofluorometer equipped with a circulating water bath maintained at 22 °C. Emission spectra (excitation at 280 nm, emission at 290–300 nm) were measured in a 0.4 cm path length cell containing 400 μL of 300 mM Tris-HCl (pH 8.0), varying concentrations of coenzyme A, and a fixed AANATA enzyme concentration (0.08 mg/mL for the wild type, 0.1 mg/mL for R153A). Fluorescence emission spectra were acquired in triplicate, with a scan speed of 50 nm/min, an excitation bandwidth of −5 nm, and an emission bandwidth of −2.5 nm. The Kd for coenzyme A was determined by fitting the data to eq 10 with SigmaPlot 12.0
| 10 |
where ΔF is the change in intrinsic fluorescence, ΔFmax is the maximal change in fluorescence at infinite ligand concentration, Kd is the ligand dissociation constant, and [L] is the ligand concentration.
Product Characterization
Product characterization was performed using a Phenomenex Kinetex 2.6 μm C18 100 Å (50 mm × 2.1 mm) reverse phase column coupled with an Agilent 6540 liquid chromatography/quadrupole time-of-flight mass spectrometer (LC/QTOF-MS) in positive ion mode. An enzyme reaction mixture comprised of 300 mM Tris-HCl (pH 8.0), 500 μM acetyl-CoA, 1 mM serotonin, and 5 μg AANATA in a final volume of 750 μL was incubated for 30 min at room temperature. Then AANATA was removed from the reaction mixture by centrifugation using a Millipore 10 kDa filter. The resulting sample was injected on the LC/QTOF-MS, and the retention time and high-resolution mass were compared with those of a commercial standard of N-acetylserotonin. Conditions for LC/QTOF-MS analysis are described in ref (27).
Results
Overexpression and Purification of AANATA and AANATB
AANATA and AANATB were cloned from a cDNA library generated from the D. melanogaster head (Figure S1 of the Supporting Information). Both genes were inserted into a pET28a(+) vector that encodes an N-terminal His6 tag. Both AANATA and AANATB were purified using ProBond nickel-chelating resin to homogeneity, yielding 21 mg of AANATA or 3 mg of AANATB per liter of culture. Purity was assayed by SDS–PAGE (Figure S2 of the Supporting Information) and determined to be >95%.
Characterization of the Amino Acceptor Substrates
Amino acceptors are defined herein as any acyl-CoA or aryl-CoA substrate. Our data for AANATA and AANATB (Table 1) show that the amino acceptor specificities are approximately the same for both enzymes. Both catalyze the formation of N-acyltyramines, utilizing a set of straight-chain saturated acyl-CoAs with little variation in the Km,app values from acetyl-CoA to hexanoyl-CoA for the two enzymes. A two-carbon increase in acyl-chain length to decanoyl-CoA has a dramatic and negative effect on catalysis. The Km,app is ∼10-fold higher and the kcat,app ∼10-fold lower than the values for octanoyl-CoA, such that the (kcat/Km)app,decanoyl-CoA is ∼1% of the (kcat/Km)app,octanoyl-CoA and only ∼0.04% of the (kcat/Km)app,acetyl-CoA (Table 1). Acyl-CoA thioesters possessing an acyl chain of ≥12 carbon atoms were not substrates for AANATA. Although lauroyl-CoA and the longer-chain acyl-CoA thioesters were not AANATA substrates, all were inhibitors of the enzyme with IC50 values of ≤1.1 μM. We find a slight increase in the apparent binding affinity as the acyl-chain length increases, with the IC50 values decreasing from 1.1 μM for lauroyl-CoA to 0.4 μM for oleoyl-CoA (Table 2).
Table 1. Steady-State Kinetic Constants for AANATA and AANATB with Different Acyl-CoAs.
| substratea | Km,app (μM) | kcat,app (s–1) | (kcat/Km)app (M–1 s–1) | relative (kcat/Km)app |
|---|---|---|---|---|
| AANATAb,c | ||||
| benzoyl-CoA | 65 ± 13 | 0.37 ± 0.03 | (5.7 ± 1.2) × 103 | 1.0 ± 0.3 |
| acetyl-CoA | 39 ± 12 | 16 ± 1 | (4.1 ± 1.3) × 105 | 72 ± 27 |
| butyryl-CoA | 36 ± 2 | 9.9 ± 0.4 | (2.8 ± 0.2) × 105 | 49 ± 11 |
| hexanoyl-CoA | 23 ± 3 | 1.6 ± 0.1 | (6.8 ± 0.9) × 104 | 12 ± 3 |
| octanoyl-CoA | 18 ± 3 | 0.26 ± 0.01 | (1.4 ± 0.2) × 104 | 2.5 ± 0.6 |
| decanoyl-CoA | 220 ± 60 | 0.04 ± 0.01 | (1.7 ± 0.5) × 102 | 0.04 ± 0.01 |
| AANATBc,d | ||||
| benzoyl-CoA | 80 ± 6 | 0.12 ± 0.04 | (1.6 ± 0.5) × 103 | 1.0 ± 0.4 |
| acetyl-CoA | 64 ± 9 | 8.3 ± 0.4 | (1.3 ± 0.2) × 105 | 81 ± 31 |
| butyryl-CoA | 19 ± 3 | 4.4 ± 0.2 | (2.3 ± 0.3) × 105 | 150 ± 55 |
| hexanoyl-CoA | 23 ± 6 | 0.88 ± 0.04 | (3.8 ± 1.0) × 104 | 25 ± 10 |
Reaction condition: 300 mM Tris-HCl (pH 8.0), 150 μM DTNB, 1.0 mM tyramine, and varying acyl-CoA concentrations.
Relative (kcat/Km)app values for AANATA amino acceptor substrates were indexed to benzoyl-CoA.
Kinetic constants are reported with the standard error (n = 3).
Relative (kcat/Km)app values for AANATB amino acceptor substrates were indexed to benzoyl-CoA.
Table 2. AANATA IC50 Values for Long-Chain Acyl-CoAs.
| inhibitora | carbon skeleton | IC50 (nM) |
|---|---|---|
| oleoyl-CoA | 18:1 (Δ9) | 380 ± 5 |
| palmitoleoyl-CoA | 16:1 (Δ9) | 430 ± 40 |
| myristoyl-CoA | 14:0 | 580 ± 70 |
| lauroyl-CoA | 12:0 | 1100 ± 130 |
Reaction was fixed at the Km,app values for both acetyl-CoA (39 μM) and tyramine (12 μM) while varying the concentration of the long-chain acyl-CoA inhibitor. Kinetic constants are reported with the standard error (n = 3).
Benzoyl-CoA was also included in our analysis of amino acceptor substrates to facilitate the comparison of our substrate specificity data with those published for other N-acyltransferases.32,33 The (Km,app)benzoyl-CoA is similar to the values we measured for short-chain acyl-CoA thioesters, but the kcat,app is significantly lower, such (kcat/Km)app,benzoyl-CoA is ∼1.5% of (kcat/Km)app,acetyl-CoA.
Characterization of the Amino Donor Substrates
Amino donors are defined herein as compounds that contain a free amino moiety. The substrate specificity of AANATA was evaluated using a set of arylalkylamine amino donors; these data show that tyramine is the amino donor with the highest (kcat/Km)app value at saturating acetyl-CoA (Table 3). The kinetic constants for tyramine, with both AANATA and AANATB, were very similar at saturating acetyl-CoA concentrations [for AANATB, (Km,app)tyramine = 20 ± 3 μM and (kcat,app)tyramine = 16 ± 1 s–1 compared with the AANATA data in Table 3].
Table 3. AANATA Steady-State Kinetic Constants for Different Arylalkylamines.
Reaction condition: 300 mM Tris-HCl pH 8.0, 150 μM DTNB, 500 μM acetyl-CoA, and varying concentration of arylalkylamine.
Kinetic constants are reported ± standard error (n = 3).
A group of other arylalkylamines was evaluated as AANATA amino donor substrates to better understand the general structural features that affect binding and catalysis (Table 3). The (kcat/Km)app values ranged over 2–3 orders of magnitude for the amino donors we included in our study, with tyramine as the best amino donor with the highest (kcat/Km)app value and 3,4-dimethoxyphenethylamine as the worst amino donor substrate with the lowest (kcat/Km)app value. Tyramine and tryptamine exhibit relatively high (kcat/Km)app values (Table 3); therefore, modification at the α-position was explored to determine if their cognate amino acids could serve as substrates. No activity was observed above the baseline rate of acetyl-CoA hydrolysis for tyrosine, tyrosine methyl ester, or tryptophan. Next, we assessed tyrosine, tyrosine methyl ester, and tryptophan as AANATA inhibitors at a concentration of 1.0 mM, with the acetyl-CoA (39 μM) and tyramine (12 μM) concentrations fixed at their respective Km,app values. No inhibition was observed for these three compounds.
The amino donor substrate specificity for AANATA was further evaluated to determine if non-arylalkylamines could serve as substrates. The decarboxylated amino acids, histamine (histidine) and ethanolamine (serine), were not AANATA substrates. Isoniazid, a first-line drug for the treatment of tuberculosis,34−36 was evaluated for AANATA binding. There was no observed decrease in the rate of reaction in the presence of 1.0 mM isoniazid.
Product Characterization
The product formed from the AANATA reaction using acetyl-CoA and serotonin as substrates was analyzed by LC/QTOF-MS in positive ion mode. Identification of the product formed from this reaction was compared with the commercial standard, N-acetylserotonin. The AANATA product, N-acetylserotonin, was successfully identified by comparison of both the retention time on a Kinetex 2.6 μm C18 100 Å (50 mm × 2.1 mm) reverse phase column and the [M + H]+ (m/z) high-resolution mass spectroscopy peak with those of the commercial standard (Table S2 of the Supporting Information).
Kinetic Mechanism and Inhibitor Analysis
Initial velocity plots were generated using tyramine and acetyl-CoA, by varying the concentration of one substrate while holding the other substrate at a fixed saturated concentration. The resulting double-reciprocal plots (Figure 1) reveal a pattern of intersecting lines, suggesting a sequential kinetic mechanism in which both substrates must be bound before catalysis can occur. Inhibition studies were used to differentiate between a random and an ordered sequential mechanism for AANATA, using oleoyl-CoA and tyrosol as the dead-end inhibitors. Oleoyl-CoA is an analogue of acetyl-CoA, whereas tyrosol is an analogue of tyramine; neither is a substrate for AANATA. Oleoyl-CoA is a competitive inhibitor (Figure 2) versus acetyl-CoA and a pure noncompetitive inhibitor versus tyramine, with inhibition constants of 78 ± 8 nM and 150 ± 9 nM, respectively. Tyrosol is uncompetitive versus acetyl-CoA and competitive versus tyramine, with inhibition constants of 260 ± 10 μM and 302 ± 34 μM, respectively.
Figure 1.

Double-reciprocal plot of initial velocities for acetyl-CoA and tyramine. (A) Velocities measured at a fixed concentration of tyramine: 50 μM (●), 25 μM (○), 10 μM (▼), and 5 μM (△). (B) Velocities measured at a fixed concentration of acetyl-CoA: 100 μM (●), 60 μM (○), 40 μM (▼), and 20 μM (△).
Figure 2.

Dead-end inhibition analysis of AANATA. (A) Velocities measured at a fixed concentration of tyramine (12 μM), varying the concentration of acetyl-CoA, and varying the concentration of the inhibitor, oleoyl-CoA: 0 nM (●), 200 nM (○), and 375 nM (▼) (Ki = 78 ± 8 nM). (B) Velocities measured at a fixed concentration of acetyl-CoA (39 μM), varying the concentration of tyramine, and varying the concentration of the inhibitor, oleoyl-CoA: 0 nM (●), 200 nM (○), and 375 nM (▼) (Ki = 150 ± 7 nM). (C) Velocities measured at a fixed concentration of tyramine (12 μM), varying the concentration of acetyl-CoA, and varying the concentration of the inhibitor, tyrosol: 0 μM (●), 100 μM (○), and 580 μM (▼) (Ki = 260 ± 10 μM). (D) Velocities measured at a fixed concentration of acetyl-CoA (39 μM), varying the concentration of tyramine, and varying the concentration of the inhibitor, tyrosol: 0 μM (●), 100 μM (○), and 580 μM (▼) (Ki = 302 ± 34 μM).
pH Dependence of the Initial Rates
We studied the pH dependence on the kcat,app and (kcat/Km)app for both acetyl-CoA and tyramine. A rising pH profile was observed for both (kcat/Km)app-acetyl-CoA (Figure 3C) and (kcat/Km)app-tyramine (Figure 3D). The data for both are best fit using a single pKa,app value, yielding a pKa,app value of 7.1 ± 0.2 for the (kcat/Km)app-acetyl-CoA profile and a pKa,app value of 7.0 ± 0.3 for the (kcat/Km)app-tyramine profile. The kcat,app profile is bell-shaped (Figure 3A,B) for acetyl-CoA and tyramine, yielding two ionizable groups with the same apparent pKa values, consisting of an apparent pKa of 7.0 ± 0.1 and an apparent pKa of 9.8 ± 0.2.
Figure 3.

AANATA pH–rate profile: (A) kcat,app-acetyl-CoA, (B) kcat,app-tyramine, (C) (kcat/Km)app-acetyl-CoA, and (D) (kcat/Km)app-tyramine.
Site-Directed Mutagenesis of Amino Acids Proposed To Function in Acid–Base Catalysis
Our pH–rate data indicate that AANATA catalysis involves one species with a pKa,app of 7.0 and a second species with a pKa,app of 9.8. The pH–rate profile data provide the apparent pKa values for the entities involved in catalysis but do not provide definitive information concerning the identity of these chemical species. Identification of these species is critical for a more complete understanding of the catalytic mechanism. One method we used to pinpoint residues important for catalysis was the alignment of the primary sequences of known AANAT-like (AANATL) enzymes from D. melanogaster (Figure S3 of the Supporting Information).26
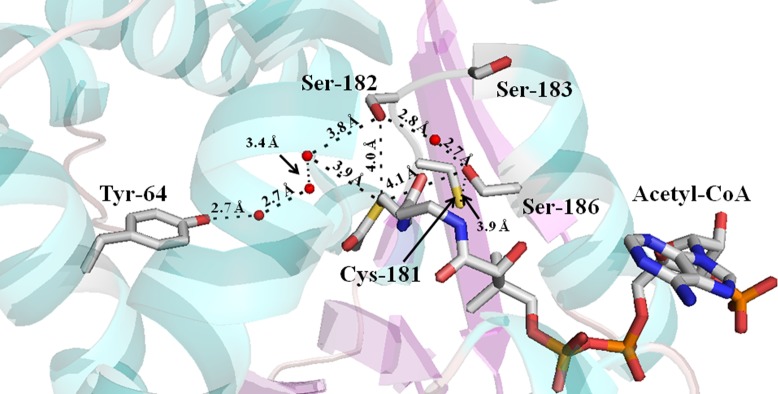
The general base in the AANATA catalytic cycle is likely Glu-47, Asp-142, or His-178, based on our pH–rate data, sequence alignment, and crystal structure (Figure 4). We mutated each of these residues to Ala to further define their respective role in catalysis. Mutation of Glu-47 has the greatest effect on catalysis, in that the kcat,app value decreases ∼15-fold and the (kcat/Km)app decreases 10–200-fold relative to wild-type values (Table 4). In addition, the E47A mutant exhibited the same Km,app-acetyl-CoA value as the wild type (within experimental error) but did show a 10–15-fold increase in Km,app-tyramine. The D142A and H178A mutants were catalytically competent, exhibiting kcat,app values that were 80–170% of the wild-type value. The small decreases observed in the (kcat/Km)app values for the D142A and H178A mutants relative to the wild-type value resulted largely from increases in the Km,app values for both acetyl-CoA and tyramine (Table 4).
Figure 4.

Crystal structure of D. melanogaster AANATA with bound acetyl-CoA identifying the possible general base residues. Stick model showing the proximity of Glu-47, His-178, and Asp-142 to acetyl-CoA. Red spheres represent water molecules.
Table 4. Steady-State Kinetic Constants for AANATA Site-Directed Mutants.
| Acetyl-CoA | ||||
|---|---|---|---|---|
| varianta,b | Km,app (μM) | kcat,app (s–1) | (kcat/Km)app (M–1 s–1) | (kcat/Km)app-mutant/(kcat/Km)app-wild-type × 100 (%) |
| wild type | 39 ± 12 | 16 ± 1 | (4.1 ± 1.3) × 105 | 100 |
| E47A | 32 ± 3 | 1.26 ± 0.04 | (3.9 ± 0.6) × 104 | 9.5 |
| P48A | 190 ± 10 | 1.4 ± 0.1 | (7.3 ± 0.5) × 103 | 1.8 |
| Y64A | 81 ± 5 | 8.6 ± 0.2 | (1.1 ± 0.1) × 105 | 27 |
| D142A | 80 ± 6 | 27.5 ± 0.6 | (3.4 ± 0.3) × 105 | 83 |
| R153A | 430 ± 60 | 106 ± 6 | (2.5 ± 0.4) × 105 | 61 |
| H178A | 120 ± 7 | 17.9 ± 0.4 | (1.5 ± 0.1) × 105 | 37 |
| C181A | 26 ± 1 | 19.1 ± 0.2 | (7.3 ± 0.3) × 105 | 178 |
| S186A | 29 ± 5 | 5.0 ± 0.3 | (1.7 ± 0.3) × 105 | 41 |
| S182A/S186A | 43 ± 4 | 8.9 ± 0.3 | (2.1 ± 0.2) × 105 | 51 |
| H220A | 160 ± 20 | 4.2 ± 0.2 | (2.6 ± 0.3) × 104 | 6.3 |
| Tyramine | ||||
|---|---|---|---|---|
| varianta,c | Km,app (μM) | kcat,app (s–1) | (kcat/Km)app (M–1 s–1) | (kcat/Km)app-mutant/(kcat/Km)app-wild-type × 100 (%) |
| wild type | 12 ± 1 | 19 ± 1 | (1.6 ± 0.2) × 106 | 100 |
| E47A | 160 ± 50 | 1.0 ± 0.1 | (6 ± 2) × 103 | 0.38 |
| P48A | 470 ± 50 | 0.86 ± 0.04 | (1.84 ± 0.2) × 103 | 0.12 |
| Y64A | 87 ± 17 | 8.8 ± 0.4 | (1.0 ± 0.2) × 105 | 6.3 |
| D142A | 17 ± 1 | 25 ± 1 | (1.5 ± 0.1) × 106 | 94 |
| R153A | 290 ± 20 | 98 ± 2 | (3.4 ± 0.3) × 105 | 21 |
| H178A | 25 ± 3 | 14.7 ± 0.5 | (5.9 ± 0.8) × 105 | 37 |
| C181A | 17 ± 2 | 22 ± 1 | (1.3 ± 0.1) × 106 | 81 |
| S186A | 73 ± 12 | 5.0 ± 0.1 | (6.9 ± 0.6) × 104 | 4.3 |
| S182A/S186A | 190 ± 20 | 8.3 ± 0.2 | (4.3 ± 0.3) × 104 | 2.7 |
| H220A | 82 ± 9 | 4.1 ± 0.2 | (4.9 ± 0.6) × 104 | 3.1 |
Kinetic constants are reported with the standard error (n = 3).
The reaction rate was measured at a fixed saturating concentration of tyramine while varying the concentration of acetyl-CoA.
The reaction rate was measured at a fixed saturating concentration of acetyl-CoA while varying the concentration of tyramine.
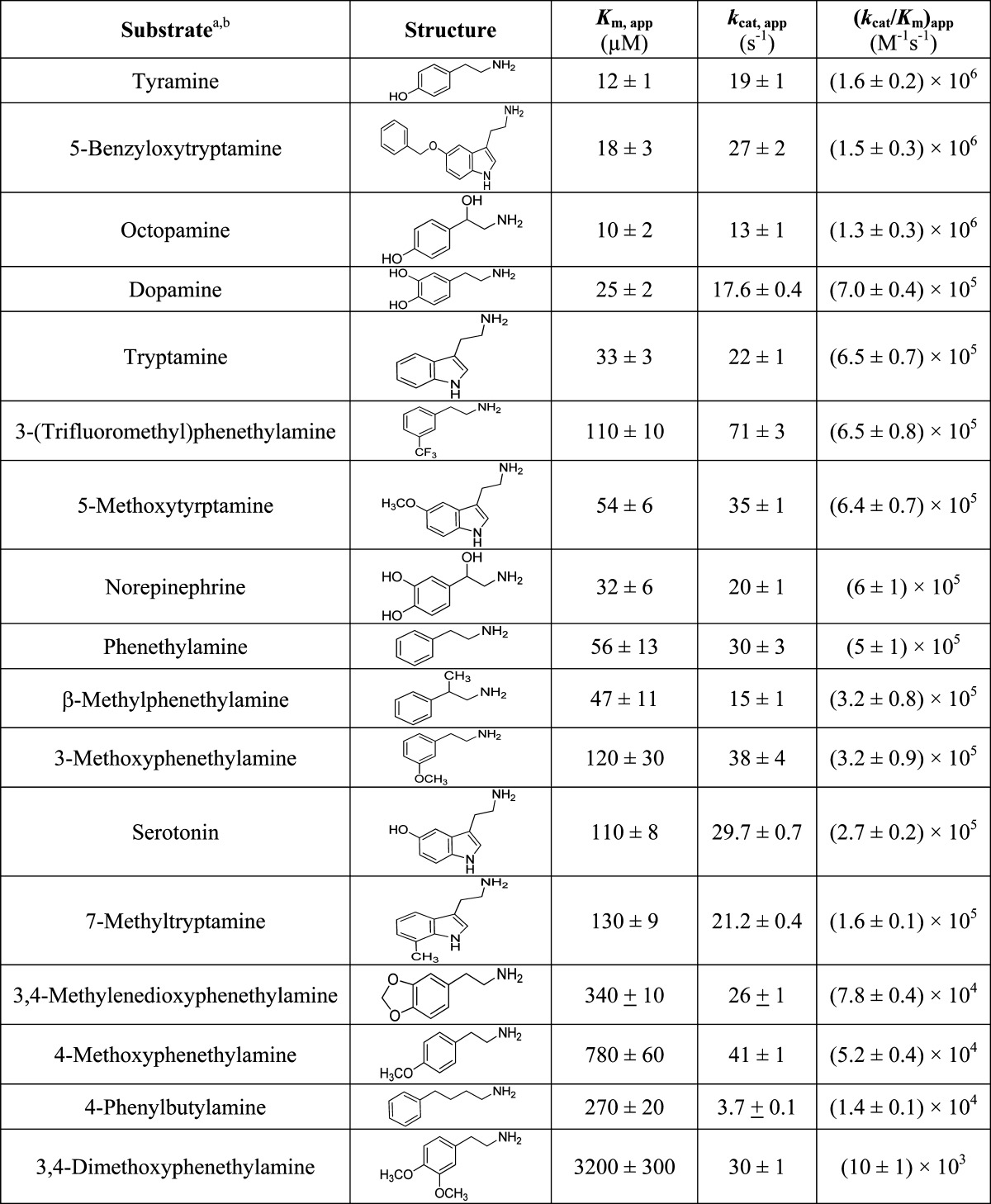
The AANATA crystal structure also points toward Tyr-64, Cys-181, Ser-182, or Ser-186 serving as the general acid (Figure 5) with a pKa,app of 9.8, visible in our pH–rate studies. Each of these residues was mutated to an Ala, and the kinetic parameters for each mutant were determined. The steady-state kinetic parameters for the C181A mutant are similar to the wild-type values. In contrast with the C181A mutant, the Y64A and S186A mutants were catalytically deficient, yielding (kcat/Km)app values that were 5–40% of the wild-type value, the decrease mostly came from increases in the Km,app values (Table 4). Also, we constructed and evaluated the S182A/S186A double mutant. The kinetic parameters for S182A/S186A were similar to those of the S186A single mutant, with the exception of an even higher Km,app-tyramine, such that the value increased from 12 μM for the wild type to 73 μM for the S186A mutant and to 190 μM for the S182A/S186A double mutant (Table 4).
Figure 5.
Crystal structure of D. melanogaster AANATA with bound acetyl-CoA identifying the possible general acid residues. Stick model showing the proximity of Tyr-64, Cys-181, Ser-182, Ser-183, and Ser-186 to acetyl-CoA. Red spheres represent water molecules.
Site-Directed Mutagenesis of Other Amino Acids Conserved between AANATA and Other AANAT-like Enzymes Found in Drosophila
Three residues, Pro-48, Arg-153, and His-220, were conserved among AANATA and the other Drosophila AANATL enzymes (Figure S3 of the Supporting Information) and seem necessary in maintaining the AANATA structure.37 Each of these residues was mutated to an Ala in an attempt to characterize their function (Figures S4 and S5 of the Supporting Information). The results are similar for both the P48A and H220A mutant enzymes, including (kcat/Km)app values that are 16–830-fold lower than the wild-type value resulting from decreases in the kcat,app values (16–870-fold lower) and increases in the Km,app values for the substrates (4–40-fold higher) (Table 4). Notably, the (kcat/Km)app-tyramine for the P48A mutant is 830-fold lower than that of the wild type. The pattern of results for the R153A mutant is different as the (kcat/Km)app value is decreased only 2–5-fold relative to that of the wild type. The outcome for the R153 mutant results from a 5–7-fold increase in kcat,app values that is balanced against 10–24-fold increases in the Km,app values for the acetyl-CoA and tyramine substrates (Table 4).
Salt Dependence of the Initial Rates
Arg-153 forms a salt bridge with Asp-46, and our evaluation of the R153A mutant indicates that the Arg-153–Asp-46 salt bridge is important in substrate binding and catalysis. The salt dependence of kinetic parameters, if any, could provide additional support for this suggestion. We found an increase in kcat,app and Km,app-acetyl-CoA as the concentration of NaCl added to the reaction buffer increased from 0 to 500 mM [0 mM NaCl, Table 1; 100 mM NaCl, (kcat,app)acetyl-CoA = 49 ± 1 and (Km,app)acetyl-CoA = 89 ± 10; 500 mM NaCl, (kcat,app)acetyl-CoA = 60 ± 1 and (Km,app)acetyl-CoA = 150 ± 10].
Coenzyme A (CoA-SH) Binding to Wild-Type AANATA and the R153A Mutant
The increase in kcat,app values for the R153A mutant was unexpected and may be related to a decrease in the binding affinity of the AANATA for CoA-SH, one product of the reaction. The equilibrium constant for the dissociation of CoA from the enzyme·CoA complex was obtained by measuring the quenching of intrinsic protein fluorescence at different CoA-SH concentrations for both wild-type AANATA and the R153A mutant. From these studies, we determined that the Kd,wild-type = 400 ± 50 μM and the Kd,R153A = 950 ± 110 μM (Figure 6).
Figure 6.

Determination of the coenzyme A dissociation constant by fluorescence titration for (A) the wild-type enzyme and (B) the R153A mutant.
Discussion
Comparison of AANATA and AANATB
AANATA differs from AANATB by an N-terminal truncation of 35 amino acids. Both variants are physiologically relevant in D. melanogaster, but differences in the expression patterns for each enzyme with respect to tissue distribution and stages of development have been identified.22 Although the differences in the expression patterns for the two enzymes suggest that AANATA and AANATB do not serve the same metabolic role in D. melanogaster, it is not known if truncation of AANATA to AANATB has any effect on the catalytic efficiency of the enzyme. A comparison of the kinetic constants for AANATA and AANATB shows that the two enzymes are catalytically equivalent, at least for the substrates included in our study. Thus, our data provide no clear insight regarding the differential expression of two AANAT variants in Drosophila. Most likely, the presence of AANATA and AANATB is important for differences in (a) subcellular localization for the two enzymes, (b) substrate specificities between the two enzymes that were not revealed by our work, (c) the modes regulation for the two enzymes, and/or (d) the post-translational modification of the two enzymes. Potential differences in post-translational modification patterns between AANATA and AANATB could be important because the activity of sheep serotonin N-acetyltransferase is regulated by the phosphorylation of Thr/Ser residues located in N-terminal or C-terminal regions of the protein.38−46
Substrate Specificity and the Potential Role of AANATA in Fatty Acid Amide Biosynthesis
We have had a long-standing interest in the biosynthesis of the fatty acid amides, a broad family of cell signaling lipids. One straightforward route to the fatty acid amides would be the reaction of a biogenic amine with an acyl-CoA, a possibility that has received relatively little attention. The identification of N-acyltransferases that catalyze the acyl-CoA-dependent formation of N-fatty acylglycines,47N-fatty acylserotonins,27 and N-fatty acyldopamines27 means that this chemistry should be examined in greater detail. Fatty acid amides are produced in D. melanogaster,48 but the enzymes responsible for their production in the fly are largely unknown. There is a single report of an enzyme, arylalkylamine N-acyltransferase like 2 (AANATL2), that catalyzes the formation of long-chain N-acylserotonins and N-acyldopamines.27 We undertook a detailed investigation of the substrate specificity of AANATA to address this question.
We first evaluated the substrate specificity for the acyl-CoA thioester substrates. The Km,app-acyl-CoA values were similar for acetyl-CoA through octanoyl-CoA, indicating that the AANATA active site can effectively bind the straight-chain acyl chain with lengths of eight or fewer carbon atoms. However, the kcat,app decreased ∼30-fold as the acyl change length increased from acetyl-CoA to octanoyl-CoA, with acetyl-CoA being the substrate with the highest (kcat/Km)app value. Lengthening the acyl chain to more than eight carbon atoms resulted in a dramatic effect on the kinetic parameters. Decanoyl-CoA is a poor substrate relative to the shorter-chain acyl-CoA substrates, with a Km,app value of 220 μM, >10-fold higher than that of octanoyl-CoA, and a kcat,app value of 0.04 s–1, 7-fold lower than that of octanoyl-CoA. Lauroyl-CoA and longer-chain acyl-CoA thioesters are not AANATA substrates but are inhibitors of the enzyme, with IC50 values of 0.4–1.0 μM. These data show that acetyl-CoA is best positioned for nucleophilic attack by the amino group of tyramine within the AANATA active site and that an increase in length alters the arrangement of the two substrates into suboptimal positions, resulting in a decrease in kcat,app. The AANATA active site must have sufficient flexibility to accommodate the lengthening acyl chain, up to eight carbon atoms, and still position the two substrates for chemistry with reasonable effectiveness as the (kcat/Km)app for octanoyl-CoA is 3% of the acetyl-CoA value. The limit of active site “tolerance” is an acyl chain of 10 carbon atoms because decanoyl-CoA is a poor substrate with a (kcat/Km)app value that is 0.04% of the acetyl-CoA value. Acyl-CoA thioesters with acyl chains longer than 10 carbon atoms bind to AANATA and are not substrates; the long-chain acyl-CoA thioesters either prevent tyramine binding in a position that allows chemistry to occur or actually prevent tyramine from binding. Thus, the long-chain acyl-CoA thioesters are inhibitors of AANATA.
Amino donors are defined herein as compounds that contain a free amino moiety. The substrate specificity of AANATA was evaluated using a variety of arylalkylamine amino donors, and these data show that tyramine is the amino donor with the highest (kcat/Km)app value. The kinetic constants for tyramine, with both AANATA and AANATB, were very similar (for AANATB, Km,app-tyramine = 20 ± 3 μM and kcat,app-tyramine = 16 ± 1 s–1 compared to the data for AANATA in Table 3).
We evaluated a number of biologically important arylalkylamines as AANATA substrates, including tyramine, octopamine, dopamine, tryptamine, norepinephrine, and serotonin. These arylalkylamines are found in vivo and potentially could be found as the amine partner in the fatty acid amide family. All are respectable AANATA substrates with tyramine exhibiting the highest (kcat/Km)app value (at saturating acetyl-CoA) and serotonin the worst, with a (kcat/Km)app,tyramine/(kcat/Km)app,serotonin ratio of 6 (Table 3). The differences in the kinetic parameters among this set of biologically relevant arylalkylamines are reflected in the Km,app values, whereas the kcat,app values are similar for these amino donor substrates. Our results for the substrate specificity of AANATA with respect to both acyl-CoA and biologically relevant arylalkylamines provide evidence that the fatty acid amides can be produced by the conjugation of biogenic amines to an acyl-CoA. However, AANATA is probably not involved in the in vivo biosynthesis of long-chain fatty acid amides because the long-chain fatty acyl-CoA thioesters are not substrates. The major function of AANATA in the cell is likely amine acetylation, a reaction important in neurotransmitter inactivation, melatonin biosynthesis, and cuticle sclerotization.
Our discovery of the AANATA-catalyzed acetylation of 5-methoxytryptamine opens up an alternative route for melatonin biosynthesis. Melatonin is produced from l-tryptophan in a series of four steps: hydroxylation to 5-hydroxytryptophan, decarboxylation to serotonin, acetylation to N-acetylserotonin, and methylation to N-acetyl-5-methoxytryptamine (melatonin).49,50 Our data for the AANATA-catalyzed acetylation of serotonin and 5-methoxytryptamine [(kcat/Km)app,5-methoxytryptamine/(kcat/Km)app,serotonin ∼ 2] show that both of these amines are effective AANATA substrates, similar to that reported by Falcon et al.51 It seems plausible that melatonin biosynthesis could involve either serotonin acetylation followed by methylation or serotonin methylation followed by acetylation. One question is whether 5-methoxytryptamine is biosynthesized in vivo to allow direct acetylation to melatonin. The level of 5-methoxytryptamine was shown to increase in a time-dependent manner in hamster skin cells upon incubation with serotonin.52 Further studies are required to address this possible route to melatonin in D. melanogaster.
In addition to the biologically relevant amines, a larger group of arylalkylamine substrates were analyzed to understand the general structural features of the amino donor substrates that affect AANATA binding and/or catalysis. These include indole and phenyl ring modifications, the length of the spacer group between the amino group and the phenyl ring, modification of the α- and β-positions on the ethylamine spacer group, and other non-arylalkylamines that may function as amine substrates.
The ring-substituted analogues of tyramine included herein are dopamine, phenethylamine, 3-(trifluoromethyl)phenethylamine, 3-methoxyphenethylamine, 4-methoxyphenethylamine, 3,4-methylenedioxyphenethylamine, 3,4-methoxyphenethylamine, and 7-methyltryptamine. All are respectable AANATA substrates with kcat,app values that are approximately the same as that for tyramine (19 s–1) or higher. The tyramine analogue with the highest kcat,app value of 71 s–1 is 3-(trifluoromethyl)phenethylamine. However, there is considerable variation in the Km,app values among the tyramine series, ranging from 12 μM for tyramine to 3200 μM for 3,4-dimethoxyphenethylamine (Table 3). Similar trends were observed in comparing the kinetic parameters between tryptamine and the ring-substituted tryptamine analogue: little variation in the kcat,app values with greater differences being found for the Km,app values.
Tyramine and tryptamine exhibit relatively high (kcat/Km)app values, allowing us also to examine how changes in the spacer group between the amine moiety and the phenyl or indole ring affect AANAT catalysis. First, we determined that the cognate amino acids, tyrosine and tryptophan, were neither substrates nor inhibitors of AANATA, as were the results for tyrosine methyl ester. These data demonstrate that the presence of an α-carboxylate or α-carboxylate ester dramatically decreases AANATA binding affinity, mainly because of steric interference in the active site. In contrast with our data about the effect of α-substitution of tyramine, we find that modification at the β-position has an only small effect on binding and catalysis as octopamine, norepinephrine, and β-methylphenethylamine are all AANATA substrates with (kcat/Km)app values that are 20–80% of the value for tyramine. The relatively minimal effect of β-substitution is on binding affinity as all of the β-substituted tyramines have approximately the same kcat,app values. This pattern holds true for the comparison of dopamine to norepinephrine (β-hydroxyldopamine), which both have approximately the same Km,app, kcat,app, and (kcat/Km)app values. Also, we interrogated the importance of the spacer length between the amino group and the phenyl ring using another set of tyramine analogues. Decreasing the spacer length from two methylene groups to one methylene group yields benzylamine, C6H5-CH2-NH2. Benzylamine is not a substrate. Increasing the spacer length from two methylene groups to four methylenes yields 4-phenylbutylamine, C6H5-(CH2)4-NH2. 4-Phenylbutylamine is a poor AANATA substrate, exhibiting a (kcat/Km)app that is 120-fold decreased relative to that of tyramine, with the largest extent of the decrease reflected in an ∼20-fold increase in Km,app [12 μM for tyramine vs 270 μM for 4-phenylbutylamine (Table 3)]. Our specificity studies of the amino donor substrates again demonstrate that the AANATA active site is flexible, able to accommodate a variety of structures particularly analogues of tyramine or tryptamine, with either a β-substitution of the ethylamine moiety or the addition of hydrophobic bulk to the phenyl or indole rings. AANATA is less tolerant of substitution at the α-substitution of the ethylamine moiety or changing the length of the methylene spacer between the amino group and the ring. Substitution at the α-position seems to eliminate binding to AANATA, while alternation in the length of space hinders the optimal positioning of the amino donor for nucleophilic attack of acetyl-CoA.
The availability of recombinant AANATA has afforded us the opportunity to explore the specificity of this enzyme for both its acyl-CoA and amine substrates. The structure–activity information obtained from this work will be valuable for the future development of inhibitors targeted against the N-acyltransferases, particularly those involved in fatty acid amide biosynthesis. Errors in fatty acid amide metabolism are correlated to human disease,53−56 and considerable effort has been devoted to the development of inhibitors for the major enzyme responsible for fatty acid amide degradation, fatty acid amide hydrolase (FAAH).57
Kinetic Mechanism and Inhibitor Analysis
An intersecting initial velocity double-reciprocal plot indicates that a ternary complex is formed prior to catalysis for AANATA. The dead-end inhibitor patterns for oleoyl-CoA and tyrosol point toward an ordered sequential mechanism, with acetyl-CoA binding first, followed by tyramine binding to generate the AANATA·acetyl-CoA·tyramine ternary complex before catalysis occurs. An ordered sequential mechanism with acetyl-CoA binding first suggests that acetyl-CoA binding drives a conformational change to convert the amine binding pocket from a low-affinity state to a high-affinity state. Oleoyl-CoA is a known inhibitor of human and sheep serotonin N-acetyltransferase, and the inhibition is not a detergent effect of the C18 tail because the IC50 for oleic acid is 500-fold higher than that for oleoyl-CoA.58,59 Tyrosol is an analogue of tyramine, with the amine being replaced with a hydroxyl group. A rate was not observed for tyrosol acetylation at 100 mM, which shows that AANATA will not catalyze the O-acylation of at least this substrate. Tryptophol, a similar analogue of tryptamine, is an inhibitor of sheep serotonin N-acetyltransferase and was useful in studies to define the kinetic mechanism of this enzyme.60 An ordered sequential mechanism for D. melanogaster AANATA is consistent with isothermal calorimetry data demonstrating little binding of dopamine to AANATA in the absence of acetyl-CoA,37 as well as with the kinetic mechanism proposed for sheep serotonin N-acetyltransferase.60
Site-Directed Mutagenesis To Match Catalytic Amino Acids to the Measured pKa,app Values
We employed protein sequence alignments, pH–rate studies, site-directed mutagenesis of targeted amino acids, and information from the AANATA crystal structure37 to propose a chemical mechanism for the AANATA-catalyzed formation of N-acylarylalkylamides. Primary sequence alignment of AANATA with other D. melanogaster AANATL enzymes identified a number of conserved residues that might function in catalysis. A primary sequence alignment of D. melanogaster AANATA with sheep serotonin N-acetyltransferase produced a low sequence alignment score (<12%), and the catalytic core of the mammalian ortholog61−66 is not conserved in the fly enzyme. Therefore, AANATA could catalyze the formation of the same N-acetylarylalkylamide products as the mammalian ortholog with either a completely different chemical mechanism or, at minimum, different catalytic amino acid residues.
Our pH–rate studies implicate two chemical species with pKa,app values of 7.0 and 9.8 in AANATA catalysis (Figure 3). The pKa,app value of 7.0 was observed in the kcat,app and (kcat/Km)app profiles and, most likely, reflects deprotonation by Glu-47. Evidence consistent with the proposal that Glu-47 serves as a general base in the AANAT catalytic cycle includes the conservation of Glu-47 among the D. melanogaster family of AANATL enzymes, the catalytic deficiency of the E47A mutant, and the disappearance of the pKa,app of 7.0 in the pH–rate profile of the E47A mutant. Cheng et al.36 reported that the log(kcat/Km)app versus pH profile for the E47A mutant is linear, with a slope of 0.3, probably resulting from hydroxide chemically rescuing Glu-47 as the base in AANATA catalysis. Furthermore, a Glu residue has been proposed as the general base in other GNAT enzymes such as members of the Gcn5/PCAF family of histone N-acetyltransferases67,68 and human spermidine/spermine N1-acetyltransferase.99−70
A pKa,app of 9.8 was observed in the pH–rate profiles for only kcat,app, indicating that this chemical species contributes to catalysis only after the first irreversible step (Figure 3A,B). We must point out that Ellman’s reagent is unstable and the acyl-CoA substrates are subject to rapid base-catalyzed hydrolysis at pH >9.5. Because we measure AANAT activity by assaying CoA release using Ellman’s reagent, our pH–rate studies are confined to pH ≤9.5. Thus, the lack of a pKa,app of ≥9.8 in our (kcat/Km)app profiles may reflect our inability to conduct measurements at pH >9.5, and the presence of a pKa,app of 9.8 for in the kcat,app profiles is based on a fit of our data, all generated at pH ≤9.5, to eq 9. Cheng et al.37 extended their studies to pH 10 and found a clearly defined pKa,app of 9.0 ± 0.2 in their pH–rate profiles for (kcat/Km)app. Experimental differences between our work and that of Cheng et al.37 render direct comparisons of the respective data sets difficult, but it seems likely that a chemical species with a pKa of 9–10 does participate in AANATA catalysis.
With this in mind, we sought to identify the chemical species that would serve as a general acid in AANATA; possibilities include Tyr-64, Cys-181, Ser-182, Tyr-185, and Ser-186 (Figure 5). Cys-181 and Tyr-185 were eliminated because the corresponding alanine substitution mutants (C181A and Y185A) showed little to no effect on the kcat,app relative to the wild-type value (Table 4 and ref (36)). Tyr-64 is located within a nonpolar binding pocket for the amino donor substrate,37 and comparison of the Y64A mutant to the wild type reveals increases in the Km,app values for both substrates (2-fold for acetyl-CoA and 6-fold for tyramine) and an only ∼2-fold decrease in the kcat,app values (Table 4). Most likely, Tyr-64 is involved in substrate binding and is not the general acid in AANATA catalysis. Ser-182 and Ser-186 remain as potential general acids. The mutation of both to Ala decreases kcat,app relative to that of the wild type, 12-fold for the S182A mutant and 3–5-fold for the S186A mutant (Table 4 and ref (36)). The pH–rate profiles for the S182A and S186A mutants are pH-independent37 at pH ≥7.0, meaning that the pKa,app of 9.0 is not observed for these two mutant enzymes. On the basis of these data and the proximity of both Ser-182 and Ser-186 to the acetyl-CoA thiol group, 4.0 and 4.1 Å, respectively (Figure 7), it is conceivable that either Ser-182 or Ser-186 is the general acid in AANATA catalysis. In fact, Cheng et al.37 argue that Ser-186 is the general acid, functioning to protonate the departing thiolate anion of CoA to complete the catalytic cycle. An argument could be made that Ser-182 and Ser-186 serve redundant roles in catalysis where one of these serines is the general acid, whereas the other serine serves to depress the pKa of the catalytic serine. In the absence of the catalytic serine, the other serine could chemically “rescue” the function of the other serine.
Figure 7.

Crystal structure of D. melanogaster AANATA with bound acetyl-CoA showing a possible “proton wire” involving the general base, Glu-47.
We do not favor either Ser-182 or Ser-186 being the general acid in AANATA catalysis. This would require a significant depression of 3–5 pH units in the pKa for Ser-182 or Ser-186, and we would expect the S182A/S186A double mutant to be dramatically impaired as a catalyst relative to either the S182A or S186A single mutant. Instead, we find that the S182A/S186A double mutant exhibits a kcat,app similar to that of the S186A mutant; the largest difference for S182A/S186A is an increased Km,app for tyramine. A tyrosine serves as the general acid for other GNAT enzymes, but the mutation studies eliminate the two active tyrosine residues, Tyr-64 and Tyr-185, as possibilities. There are no other obvious candidate amino acids to serve as a general acid within the acetyl-CoA binding pocket.37
We propose that the pKa,app of 9.8 in our pH–rate profiles (and the pKa,app of 9.0 from ref (37)) represents the pKa of the thiol group of CoA, which has been reported to range from 9.6 to 10.4.71,72 Formation of the protonated CoA-SH product results from the collapse of the tetrahedral intermediate (Scheme 1), as proposed for other N-acyltransferases.73−75 The deprotonation of CoA-SH to CoA-S–, in the AANATA active site, inhibits product release, accounting for the decrease in kcat,app at pH >9. This was likely not observed in the (kcat/Km)app profiles because of either the difficulty in measuring the kinetic constants at high pH (instability of DTNB and/or the rapid hydrolysis of acetyl-CoA) or the fact that the protonation of CoA-S– occurs after the first irreversible step in catalysis. Our suggestion obviates the need for an active site residue that serves as the general acid and is consistent with the data available for AANATA.
Scheme 1. Proposed Chemical Mechanism for D. melanogaster AANATA.

R has different acyl-chain lengths for acyl-CoA. The dashed arrows represent ambiguities in the position of the irreversible step caused by differences between the pH–rate profiles reported herein and those reported by Cheng et al.37
Site-Directed Mutagenesis of Amino Acids That Are Important in Substrate Binding and AANAT Structure
As detailed previously, we mutated a number of amino acids in AANATA in an attempt to match an active site amino acid to the pKa,app values identified in the pH–rate profiles. The set of AANATA mutants that we produced also provide information regarding the amino acids involved in substrate binding and point toward structural changes in the protein. In silico docking of dopamine into the AANATA structure with acetyl-CoA bound suggests that the amino donor substrate binds into a hydrophobic binding pocket containing Phe-43, Leu-61, Tyr-64, Ile-116, and Ile-145.37 Our results are consistent with the docking results as the Km,app-tyramine values for the E47A, P48A, and Y64A mutants are >6-fold higher than the wild-type value (Table 4). The Km,app-tyramine values for H220A and S186A single mutants also are ≥6-fold higher than the wild-type value, indicating that these amino acids contribute to the binding of the amino donor substrate. Of the mutants with a 6-fold increase in the Km,app-tyramine value relative to the wild-type value, only the E47A and S186A mutants showed no difference in Km,app-acetyl-CoA, indicating that Glu-47 and Ser-186 contribute little to acetyl-CoA binding. The Km,app-tyramine value for the S182A/S186A double mutant (190 μM) is significantly higher than the wild-type value (12 μM), suggesting that Ser-182 and Ser-186 function synergistically in maintaining the proper active site architecture for optimal binding to the amino donor substrate. The binding pocket for acetyl-CoA is more extensive than the pocket for the amino donor substrate and includes the amino acids involved in catalysis.
There are amino acids uniquely involved in acetyl-CoA binding, for which there is a change of only the value of Km,app-acetyl-CoA upon mutation. This was observed for the D142A and H178A mutants: Km,app-tyramine values that are approximately the same as that of the wild type and Km,app-acetyl-CoA values that are 2–3-fold higher than that of the wild type. Both Km,app-tyramine and Km,app-acetyl-CoA values for the P48A, Y64A, R153A, and H220A mutants are all different from and greater than the wild-type values (Table 4). These amino acids either are involved in domains within AANATA that overlap between the acetyl-CoA and amino donor binding pockets or are key to maintaining the AANATA structure in a conformation that is optimal for substrate binding. Overlap between the binding sites for the two substrates is expected because the binding pocket for acetyl-CoA is more extensive than the pocket for the amino donor substrate36 and should include the amino acids that are required to position the amino group of the amino donor for nucleophilic attack at the carbonyl of the acetyl-CoA thioester moiety. In addition, the synergistic effect of acetyl-CoA binding to convert the amine binding pocket from a low-affinity state to a high-affinity state means that specific amino acids important for acetyl-CoA binding can also have a dramatic effect on amine binding.
In comparing the kinetic constants of our mutants to those of wild-type AANATA (Table 4), we find that three of our mutants stand out: E47A, P48A, and R153A. We argue that Glu-47 is the catalytic base and find, as anticipated, that E47A is catalytically deficient with relatively low kcat,app values. The AANATA structure containing bound acetyl-CoA37 reveals a set of ordered water molecules between the carboxylate of Glu-47 and the carbonyl of the acetyl-CoA thioester. The distance from the carboxyl of Glu-47 to the carbonyl of the acetyl-CoA thioester is 6.8 Å. We suggest that Glu-47 functions as a general base to allow the water-assisted deprotonation of the positively charged amino group of the donor substrate (a “proton wire”) and to properly position the neutral amine for attack at the carbonyl of the acetyl-CoA thioester (Scheme 1). Elimination of the anionic carboxylate of Glu-47 by mutation to Ala would alter the positions of the ordered water molecules (perhaps, even resulting in their loss) and would decrease the affinity for only the amino donor substrate, as found for the E47A mutant.
Pro-48 is found at the beginning of a flexible loop and is positioned within the predicted binding pocket for the amine donor substrate (Figure S6 of the Supporting Information). We superimposed the AANATA crystal structure [Protein Data Bank (PDB) entry 3TE4]37 with that of sheep serotonin N-acetyltransferase (PDB entry 1CJW)61 using Protein Binding Site Tools (ProBiS Tools) and found that Pro-48 from D. melanogaster AANATA is equivalent to Pro-64 from sheep serotonin N-acetyltransferase. In sheep serotonin N-acetyltransferase, Pro-64 is also positioned on a flexible loop that undergoes a major structural change to alter the amino donor substrate binding pocket from a low- to high-affinity state.76,77 In the low-affinity state, Pro-64 occupies the acetyl-CoA binding pocket, whereas in the high-affinity state, Pro-64 moves ∼8 Å to form a base-staking interaction with the tryptamine moiety of the tryptamine–acetyl-CoA bisubstrate inhibitor cocrystallized with the sheep enzyme. Our data for the P48A mutant, low kcat,app and high Km,app-tyramine and Km,app-acetyl-CoA values, suggest that Pro-48 in AANATA functions like Pro-64 in sheep serotonin N-acetyltransferase and further hints that protein dynamics could regulate catalysis for AANATA. His-220 is in van der Waals contact with Pro-48 in AANATA (Figure 8). Replacement of His-220 with Ala will alter this interaction and, as seen in the data for the P48A mutant, would lead to the observed increases in the Km,app-tyramine and Km,app-acetyl-CoA values for the H220A mutant.
Figure 8.
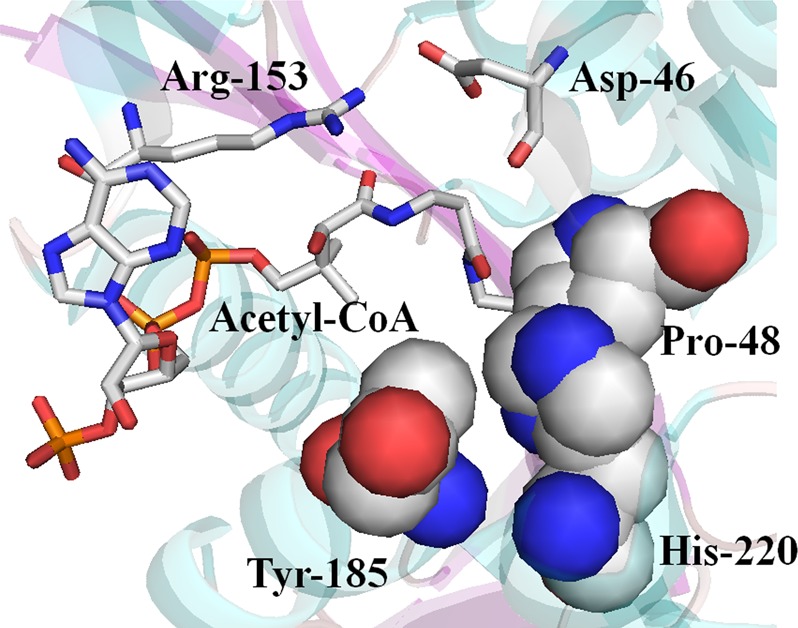
Crystal structure of D. melanogaster AANATA depicting the structural importance of His-220. CPK schematic of His-220 found in AANATA along with secondary structure highlighting van der Waals interactions between His-220 of Tyr-185 and His-220 of Pro-48.
The R153A mutant is intriguing, with values higher than wild-type values for kcat,app, Km,app-tyramine, and Km,app-acetyl-CoA. The ∼6-fold increase in kcat,app is balanced against 10–20-fold increases in the Km,app values, such that the (kcat/Km)app value for the R153A mutant is lower than that of the wild type by 40–80%. As revealed from the AANATA structure with acetyl-CoA bound,37 Arg-153 participates in a water-mediated hydrogen bond with the carbonyl amide of the pantothenate moiety (atom O9P) of acetyl-CoA and in a salt bridge to Asp-46 (Figure 9). The increase in the Km,app values for both substrates suggests that Arg-153 is critical not only in the acetyl-CoA binding pocket but also in forming the binding pocket for the amine substrate. The increase in the kcat,app value suggests that Arg-153 contributes to a rate-determining step in catalysis, related to the salt bridge between Arg-153 and Asp-46. We suspect that the Arg-153–Asp-46 salt bridge is critical to an AANATA conformation that decreases the rate of CoA-SH release, such that product release is, at least, partially rate-limiting. Partially rate-determining CoA-SH release has been identified in many other N-acetyltransferases.20,66,78,79 Elimination of the Arg-153–Asp-46 salt bridge in the R153A increases the rate of CoA-SH release, reflected in the increase in kcat,app, but decreases the affinity of AANATA for both substrates, reflected in the increases in the Km,app values. Another approach to decrease the influence of the Arg-153–Asp-46 salt bridge on the rate of CoA-SH release would be to increase the salt concentration in the reaction buffer. Our prediction is that a greater salt concentration would mimic the pattern of results we obtained for the R153A mutant, which is exactly what we found. The values of kcat,app and Km,app-acetyl-CoA increased as the concentration of added NaCl increased from 0 to 500 mM. Similar data were obtained for sheep serotonin N-acetyltransferase,25 which was argued to increase the rate of CoA-SH release by shielding charge–charge interactions of the enzyme with the anionic phosphates of acetyl-CoA. Also, we determined the Kd for binding of CoA-SH to both wild-type AANATA and the R153A mutant by measuring the quenching of intrinsic protein fluorescence as a function of CoA-SH concentration [Kd,wild-type = 400 ± 50 μM, and Kd,R153A = 950 ± 110 μM (Figure 6)]. The 2.4-fold decrease in affinity for the binding of CoA-SH to the R153A mutant is in reasonable agreement with the (kcat,app)wild-type/(kcat,app)R153A ratio of 5–6 (Table 4). The salt-dependent increase in both kcat,app and Km,app-acetyl-CoA and the increase in Kd for CoA-SH for the R153A mutant provide evidence that the salt bridge between Arg-153 and Asp-46 is important for decreasing the rate of release of CoA-SH from wild-type AANATA. Similar to our data for the P48A mutant, our data for the R153A mutant point toward protein dynamics regulating catalysis for AANATA.
Figure 9.

Crystal structure of D. melanogaster AANATA with bound acetyl-CoA depicting the Asp-46–Arg-153 salt bridge. Stick model showing the proximity of Asp-46 and Arg-153 to acetyl-CoA. Red spheres represent water molecules.
Chemical Mechanism for AANATA catalysis
A chemical mechanism for the catalytic cycle for AANATA, consistent with our new data and previously published data,21,37 is illustrated in Scheme 1. The mechanism features (a) chemistry occurring after the formation of the AANATA·acetyl-CoA·tyramine complex, (b) Glu-47 serving as a general base to facilitate water-assisted deprotonation of tyramine, (c) nucleophilic attack of deprotonated tyramine at the carbonyl of the acetyl-CoA thioester to form a zwitterionic tetrahedral intermediate, (d) collapse of the tetrahedral intermediate with the concomitant protonation of CoA, and (e) ordered product release with CoA-SH departing last to regenerate AANATA for the next round of catalysis. Our only evidence of the ordered product release is the data showing that CoA-SH binds to AANATA in the absence of N-acetyltyramine (Figure 7) and is consistent with the ordered binding of substrates, acetyl-CoA binding first. The release of CoA-SH is partially rate-determining because of the AANATA conformation that has a relatively high affinity for CoA-SH. The Arg-153–Asp-46 salt bridge is critical to maintaining AANATA in this conformation, and the affinity for CoA-SH increases as the CoA-S– thiolate forms at higher pH.
One issue remaining with this mechanism is the roles played by Ser-182 and Ser-186 in catalysis, given that Ser-186 was proposed to serve as the general acid in AANATA catalysis.37 A preponderance of the data for the S182A, S186A, and S182A/S186A mutants seem to rule out a direct catalytic role for both Ser-182 and Ser-186, and the transfer of a proton from a serine hydroxyl to a thiol is thermodynamically unfavorable. Instead, we suggest that Ser-182 and Ser-186 are involved in an elaborate network of hydrogen bonds that function in both substrate binding and catalysis (Figure 4). Mutation of Ser-182 and/or Ser-186 to alanine alters the hydrogen bonding network, changing the active site architecture. This alteration of the active site is reflected in the changes observed in the kcat,app and Km,app values for the S182A, S186A, and S182A/S186A mutants.
Conclusions
We determined that D. melanogaster AANATA will catalyze the formation of N-acylarylalkylamides from a broad array of corresponding acyl-CoA and arylalkylamine substrates. These data validate the potential role of N-acyltransferases in fatty acid amide biosynthesis and provide structure–activity data for the future development of inhibitors to this class of enzymes. A chemical mechanism for AANATA, consistent with all the data available for this enzyme,21,37 is illustrated in Scheme 1. Substrate binding is sequential ordered with acetyl-CoA binding first, meaning that catalysis occurs only after the formation of an AANAT·acetyl-CoA·arylalkylamine ternary complex. We identified Glu-47 as the general base involved in the water-assisted deprotonation of the positively charged amino group of the donor substrate via an active site “proton wire”. The deprotonation of a species with a pKa,app of 9.8 was attributed to the formation of the tightly bound CoA thiolate. Finally, our mutagenesis data suggest that protein dynamics regulate substrate binding and catalysis in AANATA. The active site amino acids, Asp-46, Pro-48, Arg-153, and His-220, seem to be critical to the regulatory dynamics of the enzyme.
Acknowledgments
Support for this work has been provided by the Mass Spectrometry and Peptide Facility, Department of Chemistry, University of South Florida. We thank Elisabeth Nevins Caswell for a thorough reading and revision of the manuscript.
Glossary
Abbreviations
- GNAT
GCN5-related N-acetyltransferase
- AANATL
arylalkylamine N-acetyltransferase like enzyme
- LC/QTOF-MS
liquid chromatography/quadrupole time-of-flight mass spectroscopy
- MES
2-(N-morpholino)ethanesulfonic acid
- AMeP
2-amino-2-methyl-1-propanol hydrochloride
- Tris
tris(hydroxymethyl)aminomethane
- IC50
half-maximal inhibitory constant.
Supporting Information Available
Mutant PCR primers (Table S1), LC/QTOF-MS data for product characterization (Table S2), and a list of arginines found in other GNAT enzymes that are conserved with Arg-153 (Table S3), and figures showing cloning and purification data, mutant SDS–PAGE gels, a D. melanogaster AANATL primary sequence alignment, and the AANATA active site depicting the location of Pro-48 with respect to bound acetyl-CoA. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
⊥ A.-M.C.: University of Florida College of Medicine, M-108 Health Science Center, P.O. Box 100216, Gainesville, FL 32610-0216.
This work has been supported, in part, by grants from the Florida Center for Excellence for Biomolecular and Targeted Therapeutics (FCoE-BITT Grant GALS020), the University of South Florida (a Research Scholarship grant from the College of Arts and Sciences), the Shirley W. and William L. Griffin Foundation, the National Institute of Drug Abuse of the National Institutes of Health (Grant R03-DA034323 to D.J.M.), and a Dissertation Completion Fellowship from the Office of Graduate Studies at the University of South Florida to D.R.D.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Blenau W.; Baumann A. (2001) Molecular and pharmacological properties of insect biogenic amine receptors: Lessons from Drosophila melanogaster and Apis mellifera. Arch. Insect Biochem. Physiol. 48, 13–38. [DOI] [PubMed] [Google Scholar]
- Marecos C.; Ng J.; Kurian M. A. (2014) What is new for monoamine neurotransmitter disorders?. J. Inherited Metab. Dis. 4, 619–626. [DOI] [PubMed] [Google Scholar]
- Coleman C. M.; Neckameyer W. S. (2004) Substrate regulation of serotonin and dopamine synthesis in Drosophila. Invertebr. Neurosci. 5, 85–96. [DOI] [PubMed] [Google Scholar]
- Coleman C. M.; Neckameyer W. S. (2005) Serotonin synthesis by two distinct enzymes in Drosophila melanogaster. Arch. Insect Biochem. Physiol. 59, 12–31. [DOI] [PubMed] [Google Scholar]
- Han Q.; Ding H.; Robinson H.; Christensen B. M.; Li J. (2010) Crystal structure and substrate specificity of Drosophila 3,4-dihydroxyphenylalanine decarboxylase. PLoS One 5, e8826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole S. H.; Carney G. E.; McClung C. A.; Willard S. S.; Taylor B. J.; Hirsh J. (2005) Two functional but noncomplementing Drosophila tyrosine decarboxylase genes: Distinct roles for neural tyramine and octopamine in female fertility. J. Biol. Chem. 280, 14948–14955. [DOI] [PubMed] [Google Scholar]
- Blumenthal E. M. (2009) Isoform- and cell-specific function of tyrosine decarboxylase in the Drosophila Malpighian tubule. J. Exp. Biol. 212, 3802–3809. [DOI] [PubMed] [Google Scholar]
- Monastirioti M.; Linn C. E. Jr.; White K. (1996) Characterization of Drosophila tyramine β-hydroxylase gene and isolation of mutant flies lacking octopamine. J. Neurosci. 16, 3900–3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray E. E.; Small S. N.; McGuirl M. A. (2006) Expression and characterization of recombinant tyramine β-monooxygenase from Drosophila: A monomeric copper-containing hydroxylase. Protein Expression Purif. 47, 162–170. [DOI] [PubMed] [Google Scholar]
- Hess C. R.; McGuirl M. M.; Klinman J. P. (2008) Mechanism of the insect enzyme, tyramine β-monooxygenase, reveals differences from the mammalian enzyme, dopamine β-monooxygenase. J. Biol. Chem. 283, 3042–3049. [DOI] [PubMed] [Google Scholar]
- Dewhurst S. A.; Croker S. G.; Ikeda K.; McCaman R. E. (1972) Metabolism of biogenic amines in Drosophila nervous tissue. Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol. 43, 975–981. [DOI] [PubMed] [Google Scholar]
- Wright T. R. (1987) The genetics of biogenic amine metabolism, sclerotization, and melanization in Drosophila melanogaster. Adv. Genet. 24, 127–222. [PubMed] [Google Scholar]
- Sekeris C. E.; Karlson P. (1966) Biosynthesis of catecholamines in insects. Pharmacol. Rev. 18, 89–94. [PubMed] [Google Scholar]
- Karlson P.; Sekeris C. E. (1962) N-Acetyl-dopamine as sclerotizing agent of insect cuticle. Nature 195, 183–184. [Google Scholar]
- Brunet P. C. J. (1980) The metabolism of the aromatic amino-acids concerned in the cross-linking of insect cuticle. Insect Biochem. Mol. Biol. 10, 467–500. [Google Scholar]
- Andersen S. O. (2010) Insect cuticular sclerotization: A review. Insect Biochem. Mol. Biol. 40, 166–178. [DOI] [PubMed] [Google Scholar]
- Finocchiaro L.; Callebert J.; Launay J. M.; Jallon J. M. (1988) Melatonin biosynthesis in Drosophila: Its nature and its effects. J. Neurochem. 50, 382–387. [DOI] [PubMed] [Google Scholar]
- Anisimov V. N. (2003) Effects of exogenous melatonin: A review. Toxicol. Pathol. 31, 589–603. [DOI] [PubMed] [Google Scholar]
- Dyda F.; Klein D. C.; Hickman A. B. (2000) GCN5-related N-acetyltransferases: A structural overview. Annu. Rev. Biophys. Biomol. Struct. 29, 81–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetting M. W.; S de Carvalho L. P.; Yu M.; Hegde S. S.; Magnet S.; Roderick S. L.; Blanchard J. S. (2005) Structure and functions of the GNAT superfamily of acetyltransferases. Arch. Biochem. Biophys. 433, 212–226. [DOI] [PubMed] [Google Scholar]
- Maranda B.; Hodgetts R. (1977) Characterization of dopamine acetyltransferase in Drosophila melanogaster. Insect Biochem. Mol. Biol. 7, 33–43. [Google Scholar]
- Brodbeck D.; Amherd R.; Callaerts P.; Hintermann E.; Meyer U. A.; Affolter M. (1998) Molecular and biochemical characterization of the aaNAT1 (Dat) locus in Drosophila melanogaster: Differential expression of two gene products. DNA Cell Biol. 17, 621–633. [DOI] [PubMed] [Google Scholar]
- Klein D. C.; Weller J. L. (1970) Indole metabolism in the pineal gland: A circadian rhythm in N-acetyltransferase. Science 169, 1093–1095. [DOI] [PubMed] [Google Scholar]
- Klein D. C.; Roseboom P. H.; Coon S. L. (1996) New light is shining on the melatonin rhythm enzyme: The first postcloning view. Trends Endocrinol. Metab. 7, 106–112. [DOI] [PubMed] [Google Scholar]
- Zheng W. P.; Cole P. A. (2002) Serotonin N-acetyltransferase: Mechanism and inhibition. Curr. Med. Chem. 9, 1187–1199. [DOI] [PubMed] [Google Scholar]
- Amherd R.; Hintermann E.; Walz D.; Affolter M.; Meyer U. A. (2000) Purification, cloning, and characterization of a second arylalkylamine N-acetyltransferase from Drosophila melanogaster. DNA Cell Biol. 19, 697–705. [DOI] [PubMed] [Google Scholar]
- Dempsey D. R.; Jeffries K. A.; Anderson R. L.; Carpenter A. M.; Rodriquez Opsina S.; Merkler D. J. (2014) Identification of an arylalkylamine N-acyltransferase from Drosophila melanogaster that catalyzes the formation of long-chain N-acylserotonins. FEBS Lett. 588, 594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell E. K.; Merkler D. J. (2008) Biosynthesis, degradation and pharmacological importance of the fatty acid amides. Drug Discovery Today 13, 558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S. N.; Hunt H. D.; Horton R. M.; Pullen J. K.; Pease L. R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77, 51–59. [DOI] [PubMed] [Google Scholar]
- Ellman G. L. (1959) Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82, 70–77. [DOI] [PubMed] [Google Scholar]
- Morrison J. F.; Uhr M. L. (1966) The function of bivalent metal ions in the reaction catalysed by ATP:creatine phosphotransferase. Biochim. Biophys. Acta 122, 57–74. [DOI] [PubMed] [Google Scholar]
- Schachter D.; Taggart J. V. (1954) Glycine N-acylase: Purification and properties. J. Biol. Chem. 208, 263–275. [PubMed] [Google Scholar]
- Dempsey D. R.; Bond J. D.; Carpenter A. M.; Rodriguez Ospina S.; Merkler D. J. (2014) Expression, purification, and characterization of mouse glycine N-acyltransferase in Escherichia coli. Protein Expression Purif. 97, 23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K. C.; Yew W. W.; Tam C. M.; Leung C. C. (2013) WHO group 5 drugs and difficult multidrug-resistant tuberculosis: A systematic review with cohort analysis and meta-analysis. Antimicrob. Agents Chemother. 57, 4097–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernon A. (2013) Treatment of latent tuberculosis infection. Seminars in Respiratory and Critical Care Medicine 34, 67–86. [DOI] [PubMed] [Google Scholar]
- Zumla A.; Raviglione M.; Hafner R.; von Reyn C. F. (2013) Tuberculosis. N. Engl. J. Med. 368, 745–755. [DOI] [PubMed] [Google Scholar]
- Cheng K. C.; Liao J. N.; Lyu P. C. (2012) Crystal structure of the dopamine N-acetyltransferase-acetyl-CoA complex provides insights into the catalytic mechanism. Biochem. J. 446, 395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters K. E.; Morrissey J. J.; Loos P. J.; Lovenberg W. (1977) Pineal protein phosphorylation during serotonin N-acetyltransferase induction. Proc. Natl. Acad. Sci. U.S.A. 74, 1928–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obsil T.; Ghirlando R.; Klein D. C.; Ganguly S.; Dyda F. (2001) Crystal structure of the 14-3-3ζ:serotonin N-acetyltransferase complex. A role for scaffolding in enzyme regulation. Cell 105, 257–267. [DOI] [PubMed] [Google Scholar]
- Klein D. C. (1985) Photoneural regulation of the mammalian pineal gland. Ciba Found. Symp. 117, 38–56. [DOI] [PubMed] [Google Scholar]
- Roseboom P. H.; Coon S. L.; Baler R.; McCune S. K.; Weller J. L.; Klein D. C. (1996) Melatonin synthesis: Analysis of the more than 150-fold nocturnal increase in serotonin N-acetyltransferase messenger ribonucleic acid in the rat pineal gland. Endocrinology 137, 3033–3045. [DOI] [PubMed] [Google Scholar]
- Zhan-Poe X.; Craft C. M. (1999) Biochemical characterization of recombinant serotonin N-acetyltransferase. J. Pineal Res. 27, 49–58. [DOI] [PubMed] [Google Scholar]
- Zheng W.; Zhang Z.; Ganguly S.; Weller J. L.; Klein D. C.; Cole P. A. (2003) Cellular stabilization of the melatonin rhythm enzyme induced by nonhydrolyzable phosphonate incorporation. Nat. Struct. Biol. 10, 1054–1057. [DOI] [PubMed] [Google Scholar]
- Zheng W.; Schwarzer D.; Lebeau A.; Weller J. L.; Klein D. C.; Cole P. A. (2005) Cellular stability of serotonin N-acetyltransferase conferred by phosphonodifluoromethylene alanine (Pfa) substitution for Ser-205. J. Biol. Chem. 280, 10462–10467. [DOI] [PubMed] [Google Scholar]
- Ganguly S.; Weller J. L.; Ho A.; Chemineau P.; Malpaux B.; Klein D. C. (2005) Melatonin synthesis: 14-3-3-dependent activation and inhibition of arylalkylamine N-acetyltransferase mediated by phosphoserine-205. Proc. Natl. Acad. Sci. U.S.A. 102, 1222–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szewczuk L. M.; Tarrant M. K.; Sample V.; Drury W. J. III; Zhang J.; Cole P. A. (2008) Analysis of serotonin N-acetyltransferase regulation in vitro and in live cells using protein semisynthesis. Biochemistry 47, 10407–10419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waluk D. P.; Schultz N.; Hunt M. C. (2010) Identification of glycine N-acyltransferase-like 2 (GLYATL2) as a transferase that produces N-acyl glycines in humans. FASEB J. 24, 2795–2803. [DOI] [PubMed] [Google Scholar]
- Jeffries K. A.; Dempsey D. R.; Behari A. L.; Anderson R. L.; Merkler D. J. (2014) Drosophila melanogaster as a model system to study long-chain fatty acid amide metabolism. FEBS Lett. 588, 1596–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilvarg C. (1961) Metabolism of amino acids. Annu. Rev. Biochem. 30, 239–268. [Google Scholar]
- Finocchiaro L.; Callebert J.; Launay J. M.; Jallon J. M. (1988) Melatonin biosynthesis in Drosophila: Its nature and its effects. J. Neurochem. 50, 382–387. [DOI] [PubMed] [Google Scholar]
- Falcon J.; Coon S. L.; Besseau L.; Cazamea-Catalan D.; Fuentes M.; Magnanou E.; Paulin C. H.; Boeuf G.; Sauzet S.; Jorgensen E. H.; Mazan S.; Wolf Y. I.; Koonin E. V.; Steinbach P. J.; Hyodo S.; Klein D. C. (2014) Drastic neofunctionalization associated with evolution of the timezyme AANAT 500 Mya. Proc. Natl. Acad. Sci. U.S.A. 111, 314–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slominski A.; Baker J.; Rosano T. G.; Guisti L. W.; Ermak G.; Grande M.; Gaudet S. J. (1996) Metabolism of serotonin to N-acetylserotonin, melatonin, and 5-methoxytryptamine in hamster skin culture. J. Biol. Chem. 271, 12281–12286. [DOI] [PubMed] [Google Scholar]
- Hermanson D. J.; Marnett L. J. (2011) Cannabinoid, endocannabinoids, and cancer. Cancer Metastasis Rev. 30, 599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroof N.; Pardon M. C.; Kendall D. A. (2013) Endocannabinoid signalling in Alzheimer’s disease. Biochem. Soc. Trans. 41, 1583–1587. [DOI] [PubMed] [Google Scholar]
- Skaper S. D.; Facci L.; Fusco M.; della Valle M. F.; Zusso M.; Costa B.; Guisti P. (2014) Palmitoylethanolamide, a naturally occurring disease-modifying agent in neuropathic pain. Inflammopharmacology 22, 79–94. [DOI] [PubMed] [Google Scholar]
- Masoodi M.; Lee E.; Eiden M.; Bahlo A.; Shi Y.; Ceddia R. B.; Baccei C.; Prasit P.; Spaner D. E. (2014) A role for oleoylethanolamine in chronic lymphocytic leukemia. Leukemia 28, 1381–1387. [DOI] [PubMed] [Google Scholar]
- Bisogno T.; Maccarrone M. (2013) Latest advances in the discovery of fatty acid amide hydrolase inhibitors. Expert Opin. Drug Discovery 8, 509–522. [DOI] [PubMed] [Google Scholar]
- Ferry G.; Loynel A.; Kucharczyk N.; Bertin S.; Rodriguez M.; Delagrange P.; Galizzi J. P.; Jacoby E.; Volland J. P.; Lesieur D.; Renard P.; Canet E.; Fauchere J. L.; Boutin J. A. (2000) Substrate specificity and inhibition studies of human serotonin N-acetyltransferase. J. Biol. Chem. 275, 8794–8805. [DOI] [PubMed] [Google Scholar]
- Khalil E. M.; Cole P. A. (1998) A potent inhibitor of the melatonin rhythm enzyme. J. Am. Chem. Soc. 120, 6195–6196. [Google Scholar]
- De Angelis J.; Gastel J.; Klein D. C.; Cole P. A. (1998) Kinetic analysis of the catalytic mechanism of serotonin N-acetyltransferase (EC 2.3.1.87). J. Biol. Chem. 273, 3045–3050. [DOI] [PubMed] [Google Scholar]
- Hickman A. B.; Namboodiri M. A.; Klein D. C.; Dyda F. (1999) The structural basis of ordered substrate binding by serotonin N-acetyltransferase: Enzyme complex at 1.8 Å resolution with a bisubstrate analog. Cell 97, 361–369. [DOI] [PubMed] [Google Scholar]
- Zheng W.; Scheibner K. A.; Ho A. K.; Cole P. A. (2001) Mechanistic studies on the alkyltransferase activity of serotonin N-acetyltransferase. Chem. Biol. 8, 379–389. [DOI] [PubMed] [Google Scholar]
- Scheibner K. A.; De Angelis J.; Burley S. K.; Cole P. A. (2002) Investigation of the roles of catalytic residues in serotonin N-acetyltransferase. J. Biol. Chem. 277, 18118–18126. [DOI] [PubMed] [Google Scholar]
- Wolf E.; De Angelis J.; Khalil E. M.; Cole P. A.; Burley S. K. (2002) X-ray crystallographic studies of serotonin N-acetyltransferase catalysis and inhibition. J. Mol. Biol. 317, 215–224. [DOI] [PubMed] [Google Scholar]
- Klein D. C. (2007) Arylalkylamine N-acetyltransferase: “The timezyme”. J. Biol. Chem. 282, 4233–4237. [DOI] [PubMed] [Google Scholar]
- Khalil E. M.; De Angelis J.; Cole P. A. (1998) Indoleamine analogs as probes of the substrate selectivity and catalytic mechanism of serotonin N-acetyltransferase. J. Biol. Chem. 273, 30321–30327. [DOI] [PubMed] [Google Scholar]
- Tanner K. G.; Trievel R. C.; Kuo M. H.; Howard R. M.; Berger S. L.; Allis C. D.; Marmorstein R.; Denu J. M. (1999) Catalytic mechanism and function of invariant glutamic acid 173 from the histone acetyltransferase GCN5 transcriptional coactivator. J. Biol. Chem. 274, 18157–18160. [DOI] [PubMed] [Google Scholar]
- Yuan H.; Marmorstein R. (2013) Histone acetyltransferases: Rising ancient counterparts to protein kinases. Biopolymers 99, 98–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman C. S.; Huang H.; Pegg A. E. (1995) Role of the carboxyl terminal MATEE sequence of spermidine/spermine N1-acetyltransferase in the activity and stabilization by the polyamine analog N1,N12-bis(ethyl)spermine. Biochemistry 34, 13423–13430. [DOI] [PubMed] [Google Scholar]
- Bewley M. C.; Graziano V.; Jiang J.; Matz E.; Studier F. W.; Pegg A. E.; Coleman C. S.; Flanagan J. M. (2006) Structures of wild-type and mutant human spermidine/spermine N1-acetyltransferase, a potential therapeutic drug target. Proc. Natl. Acad. Sci. U.S.A. 103, 2063–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde S. S.; Chandler J.; Vetting M. W.; Yu M.; Blanchard J. S. (2007) Mechanistic and structural analysis of human spermidine/spermine N1-acetyltransferase. Biochemistry 46, 7187–7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Merck Index, 12th ed. (1996) pp 417–418, Merck and Co., White House Station, NJ. [Google Scholar]
- Pitman I. H.; Morris I. J. (1980) Coenzyme A: pKa and γ values. Aust. J. Chem. 33, 1625–1630. [Google Scholar]
- Berndsen C. E.; Denu J. M. (2008) Catalysis and substrate selection by histone/protein lysine acetyltransferases. Curr. Opin. Struct. Biol. 18, 682–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoden J. B.; Cook P. D.; Schaffer C.; Messner P.; Holden H. M. (2009) Structural and functional studies of QdtC: An N-acetyltransferase required for the biosynthesis of dTDP-3-acetamido-3,6-dideoxy-α-d-glucose. Biochemistry 48, 2699–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoden J. B.; Holden H. M. (2010) Molecular structure of WlbB, a bacterial N-acetyltransferase involved in the biosynthesis of 2,3-diacetamido-2,3-dideoxy-d-mannuronic acid. Biochemistry 49, 4644–4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman A. B.; Klein D. C.; Dyda F. (1999) Melatonin biosynthesis: The structure of serotonin N-acetyltransferase at 2.5 Å resolution suggests a catalytic mechanism. Mol. Cell 3, 23–32. [DOI] [PubMed] [Google Scholar]
- Pavlicek J.; Coon S. L.; Ganguly S.; Weller J. L.; Hassan S. A.; Sackett D. L.; Klein D. C. (2008) Evidence that proline focuses movement of the floppy loop of arylalkylamine N-acetyltransferase (EC 2.3.1.87). J. Biol. Chem. 283, 14552–14558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draker K. A.; Northrop D. B.; Wright G. D. (2003) Kinetic mechanism of the GCN5-related chromosomal aminoglycoside acetyltransferase AAC(6′)-Ii from Enterococcus faecium: Evidence of dimer subunit cooperativity. Biochemistry 42, 6565–6574. [DOI] [PubMed] [Google Scholar]
- Farazi T. A.; Manchester J. K.; Gordon J. I. (2000) Transient-state kinetic analysis of Saccharomyces cerevisiae myristoylCoA:protein N-myristoyltransferase reveals that a step after chemical transformation is rate limiting. Biochemistry 39, 15807–15816. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.