

Abstract
Objective: AMP-activated protein kinase (AMPK) is an important regulator of multiple cellular pathways in the setting of energetic stress. Whether AMPK plays a critical role in hypoxic preconditioning (HPC), protecting cardiomyocytes against hypoxia reoxygenation (H/R) injury remains uncertain. Methods: H9c2 cells were preconditioned by exposing to 10 min of hypoxia and 30 min of reoxygenation. Then, the preconditioned and non-preconditioned cardiomyocytes were exposed to 90 min of hypoxia followed by 120 min of reoxygenation. Results: HPC protected H9c2 cells against H/R injury, the AMPK inhibitor or eNOS inhibitor abolished the effect of HPC. Compared with H/R group, HPC significantly increased the expression of p-AMPK (Thr172). HPC also markedly increased p-eNOS (Ser1177) expression, which was abolished by AMPK inhibition. HPC significantly increased PGC-1α expression, which were nullified by AMPK inhibition or eNOS inhibition. HPC attenuated the oxidative stress by increasing the SOD activity and decreasing the MDA and ROS level, which were abolished by AMPK inhibition or eNOS inhibition. Interestingly, the AMPK activator metformin mimicked the effects of HPC in part. Conclusions: These results indicated that HPC protects H9c2 cells against H/R injury by reducing oxidative stress partly via AMPK/eNOS/PGC-1α signaling pathway.
Keywords: Hypoxia reoxygenation injury, hypoxic preconditioning, oxidative stress, AMPK
Introduction
Several studies have clearly demonstrated that reperfusion after ischemia causes additional cell death and increases infarct size (IS), called myocardial ischemia/reperfusion (I/R) injury, that is the main cause of myocardial injury during the cardiac surgery [1,2], especially in the coronary artery bypass graft surgery. Reperfusion increases reactive oxygen species (ROS) production, and subsequently induces oxidative stress, which contributes to myocardial I/R injury [3]. It has been shown that reducing oxidative stress in cardiomyocytes can attenuate myocardial I/R injury [4], including inhibiting ROS production.
In recent years, cardiac ischemic preconditioning (IPC) has been extensively studied and found to be cardioprotective against I/R injury, its various roles in cardioprotection involve many factors, including reducing the inflammatory response, attenuating oxidative stress and an anti-apoptosis process [5,6]. However, the mechanisms of IPC for decreasing oxidative stress are still not very clear.
It is shown that hypoxia/reoxygenation (H/R) is used for simulating I/R in a cell culture model. As well as I/R injury, H/R injury is known to injure the cells through oxidant production and mitochondrial damage, which leads to the production of inflammatory cytokines and the activation of the inflammatory cell signaling pathways.
AMP-activated protein kinase (AMPK), a protein kinase that is emerging as an important regulator of multiple cellular pathways in the setting of energetic stress, is activated in response to alterations in cellular energy levels [7-9]. Its activation is mediated by increases in the intracellular AMP-to-ATP and creatine-to-phosphocreatine ratios through diverse mechanisms involving allosteric regulation of AMPK subunits, activation by an upstream AMPK kinase (AMPKK), and decreases in the activity of phosphatases [10]. Metabolic activators of AMPK include exercise, ischemia and glucose deprivation [11]. AMPK activation stimulates fatty acid oxidation, promotes glucose transport, accelerates glycolysis, and inhibits triglyceride and protein synthesis [12,13]. Additionally, it has been shown that AMPK activation also increases the phosphorylation and activity of endothelial nitric oxide synthase (eNOS) [14], and increases the expression of the transcriptional co-activator peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α).
PGC-1α, a well characterized positive regulator of mitochondrial function and oxidative metabolism [15,16], physiologically be induced or activated by conditions of shortage of, or increased demand for, energy, such as cold, physical activity and fasting, regulates the transcription of a group of genes involved in ROS detoxification [17]. Numbers studies have shown that PGC-1α regulates oxidative stress which are directly associated with myocardial I/R injury.
Considering the effects of AMPK and PGC-1α mainly focus on energy and oxidative stress, which were closely associated with H/R injury, AMPK and PGC-1α may play important roles in cardioprotection effect of HPC against H/R injury.
To our knowledge, there is no specific study about the role of AMPK in HPC and how HPC protects cardiomyocytes against I/R injury through AMPK- mediated signaling pathway. Herein, we hypothesize that hypoxic preconditioning (HPC) protects cardiomyocytes against hypoxia/reoxygenation injury by regulating oxidative stress, where AMPK-PGC-1α-mediated signaling was involved.
Materials and methods
Culture and experimental protocols for H9c2 cells
The rat embryonic-heart derived H9c2 cell line (the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences, Shanghai, China) were cultured before experimentation in Dulbecco’s modified Eagle’s medium low glucose medium (Life Technologies, Inc.) supplemented with 10% fetal bovine serum. Cells were routinely grown to 80% confluence in 75 cm2 flasks at 37°C in an atmosphere of 5% CO2 prior to passage and seeding for experiments. Twenty-four hours prior to experiments, the cells were made quiescent by serum starvation.
Hypoxic preconditioning was performed as described by Uchiyama et al [18] (Figure 1). Briefly, before hypoxic preconditioning, the medium was changed in Dulbecco’s modified Eagle’s medium containing no glucose. Hypoxic preconditioning was induced by incubating the cells in an airtight chamber in which O2 was replaced by N2. Hypoxic preconditioning was carried out by exposing cells to 10 min of hypoxia and 30 min of reoxygenation before second 90 min of hypoxia followed by 120 min of reoxygenation. The AMPK inhibitor Compound C (Sigma-Aldrich, Gillingham, UK) (5 μmol/L) and eNOS inhibitor L-NIO (Sigma-Aldrich, Gillingham, UK) (10 μmol/L) were treated at the beginning of hypoxic preconditioning. The AMPK activator Metformin (1,1-dimethylbiguanide hydrochloride) (Sigma-Aldrich, Gillingham, UK) (2 mmol/L) was treated at the beginning of hypoxia reoxygenation.
Figure 1.
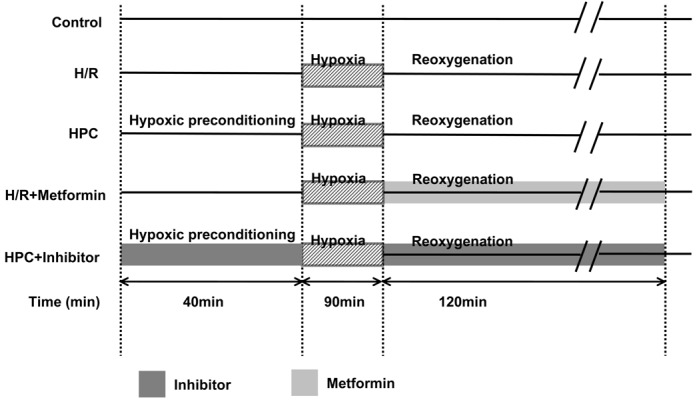
Protocols for studies of hypoxic preconditioning (HPC) in H9c2 cells. HPC = hypoxic preconditioning containing 10 min of hypoxia and 30 min of reoxygenation before second 90 min of hypoxia followed by 120 min of reoxygenation, Control = time-matched normoxia culture of cardiomyocytes, H/R = unprotected cardiomyocytes exposed to 90 min of hypoxia followed by 120 min of reoxygenation, inhibitors were administered at the beginning of hypoxic preconditioning. Metformin was treated at the beginning of hypoxia reoxygenation. Three individual experiments in each group were performed.
Cell viability assessment
The cell viability was evaluated by CCK-8 assay (Dojindo Molecular Technologies, Inc.). H9c2 cells were plated in the 96-well plates (2.0x104 cell per well) and incubated for 24 h before experiments. The cells were washed with DHanks buffer solution. Two hundred microlitres of CCK-8 solution was added to each well and incubated for an additional 1 h at 37°C. The optical density (OD) of each well at 450 nm was recorded on a Microplate Reader (Thermo, Varioskan Flash). The cell viability (% of control) is expressed as the percentage of (ODtest-ODblank)/(ODcontrol-ODblank), where ODcontrol is the optical density of the control sample and ODblank is the optical density of the wells without H9c2 cells.
Apoptosis assay
Cell apoptosis was measured by Annexin V-FITC Apoptosis Detection Kit (Bipec Biopharma, MA, USA) according to the manufacturer’s protocol. The cells were analyzed by FACScanTM flow cytometer (BD Biosciences, CA, USA). The percentages of total apoptotic cell were calculated by summing the percentages of cells in early apoptosis (Annexin V-positive but PI-negative) and late apoptosis (Annexin V-positive and PI-positive).
Maleic dialdehyde (MDA) and superoxide dismutase (SOD) activity
MDA level and superoxide dismutase (SOD) activity in H9c2 cells were measured as markers of oxidative stress using MDA kit and SOD kit (Nanjing Jiancheng) according to the manufacturer’s protocol. The concentration of MDA was calculated by the absorbance coefficient and one unit of SOD was defined as the enzyme amount causing 50% inhibition of the pyrogallic acid reduction rate. The MDA level and SOD activity were expressed as % control.
Intracellular ROS detection
H9c2 cells were loaded with 4 μmol H2DCF-DA in PBS at 37°C for 15 min after being exposed to 120 min of reoxygenation. Images were acquired by a fluorescence microscope at 488 nm excitation and 525 nm emission wavelengths at room temperature, and fluorescence intensity was measured using Image-Pro® Plus software.
Western blot
H9c2 cells collected from each group were lysed in the lysis buffer RIPA (Beyotime biotechnology, China) containing one tablet of a protease inhibitor cocktail (Beyotime biotechnology, China), and then incubated for 20 min on ice, and centrifuged at 12000 rpm for 15 min at 4°C. The supernatant was collected as total cellular protein. Protein concentration was quantified by bicinchoninic acid assay (BCA, Beyotime biotechnology, China), and 40 μg of the protein was loaded for SDS-polyacrylamide gelelectrophoresis. Proteins were transferred onto PVDF membranes with an electroblotting apparatus. Membranes were blocked for 2 h in Tris-buffered saline (TBS) containing 5% BSA, incubated with primary antibodies for phosphor-eNOS (Ser1177, Thr495), eNOS, phosphor-AMPK (Thr172), AMPK and PGC-1α (Santa Cruz, CA, USA) overnight and washed for 3 times (10 min each) with TBS containing 0.1% Tween-20. The membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies for 2 h each and then washed for 3 times (10 min each) with TBS containing 0.1% Tween-20. Immunoreactive bands were detected by enhanced chemiluminescence (ECL) (Pierce, IL, USA) and quantitated by Kodak Molecular Imaging Software
Real-time quantitative RT-PCR
Total RNAs were prepared using TRIzol® reagent (Invitrogen) according to the manufacturer’s instructions and used for the detection of peroxisome proliferator activated receptor gamma coactivator-1 alpha (PGC-1α) mRNAs. β-actin was used as internal control. Briefly, 3 μg of total RNA was retrotranscripted in the presence of random primers and Moloney murine leukemia virus according to the manufacturer’s instructions. PCRs of PGC-1α and β-actin cDNA (30 cycles of 15 s melting at 95°C, 30 s annealing at 56°C, and 30 s of extension at 72°C) were performed with Platinum TaqDNA polymerase (Invitrogen) using the primers listed below:
Rat PGC-1α forward, 5’-GCACACATCGCAATTCTCCC-3’, PGC-1α reverse, 5’-CTCTCTGCGGTATTCGTCCC-3’; Rat β-actin forward, 5’-CTATCGGCAATGAGCGGTTCC-3’, β-actin reverse, 5’-TGTGTTGGCATAGAGGTCTTTACG-3’.
Statistics
Data are expressed as mean ± SEM. Statistical significance was assessed by Student’s t test or ANOVA with subsequent post hoc Tukey test where appropriate. All statistics was calculated by GraphPad 5.0. An error probability of P < 0.05 was regarded as significant.
Results
AMPK and eNOS were involved in the protection of HPC against H/R injury in H9c2 cells
First, we detected the cell viability by CCK-8 assay to estimate whether HPC protects H9c2 cells against H/R injury (Figure 2), in which the formation of formazan dye depends on the mitochondria activity. Compared with Control group, H/R markedly decreased the cell viability, which was reversed by HPC. However, the AMPK inhibitor Compound C and eNOS inhibitor L-NIO abolished the protection effect of HPC. Interestingly, the AMPK activator metformin mimicked the effect of HPC in protecting against H/R injury. The similar results were shown in the cell apoptosis measured by Annexin V-FITC Apoptosis Detection Kit (Figure 3). These data suggested that HPC protects H9c2 cells against H/R injury partly through AMPK and eNOS-related pathway.
Figure 2.
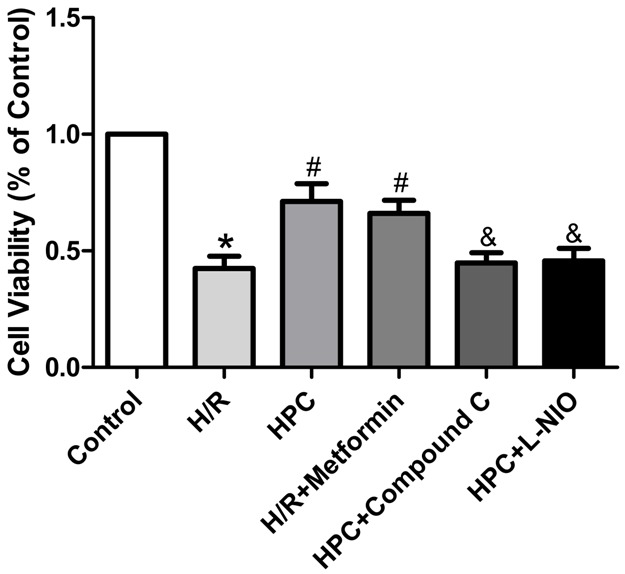
AMPK and eNOS were involved in protection of HPC against H/R injury in H9c2 cells. CCK-8 cell viability assay showing H9c2 cells proliferation. HPC increased the cells proliferation against H/R injury, which was abolished by AMPK inhibitor Compound C and eNOS inhibitor L-NIO, and mimicked by AMPK activator metformin in part. *P < 0.05 vs. Control; #P < 0.05 vs. H/R; &P < 0.05 vs. HPC.
Figure 3.
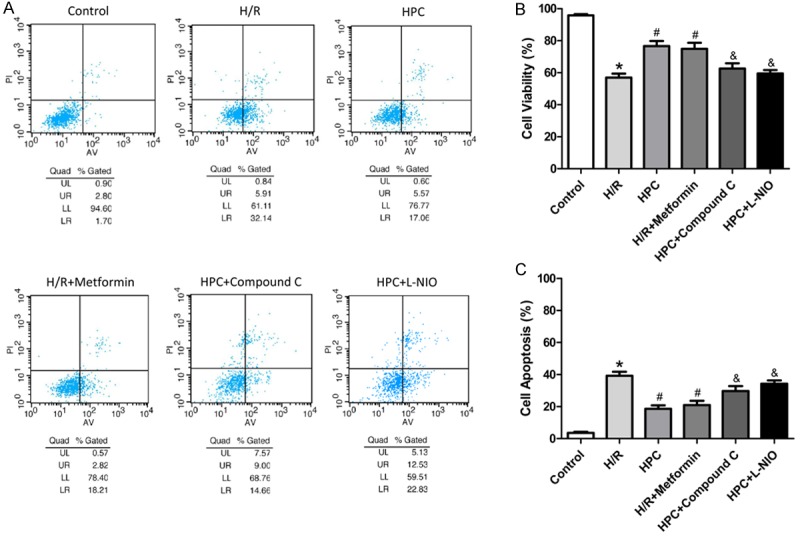
AMPK and eNOS were involved in cardioprotection of HPC against H/R injury. Cell apoptosis assay measured by flow cytometry method (FCM) showing H9c2 cells apoptosis. HPC increased cell survival and decreased cell apoptosis against H/R injury, which was abolished by the AMPK inhibitor Compound C and eNOS inhibitor L-NIO, and mimicked by the AMPK activator metformin. *P < 0.05 vs. Control; #P < 0.05 vs. H/R; &P < 0.05 vs. HPC.
Cardioprotection of HPC was mediated through the activation of AMPK
We next investigated whether HPC could activate AMPK in H/R injured H9c2 cells. The expression of p-AMPK (Thr172) and AMPK were measured (Figure 4A, 4B). Compared with H/R group, HPC significantly increased the phosphorylation of AMPK in H9c2 cells, which was similar to the effect of the AMPK activator metformin. These results showed that AMPK plays an essential role in the protection of HPC against H/R injury.
Figure 4.
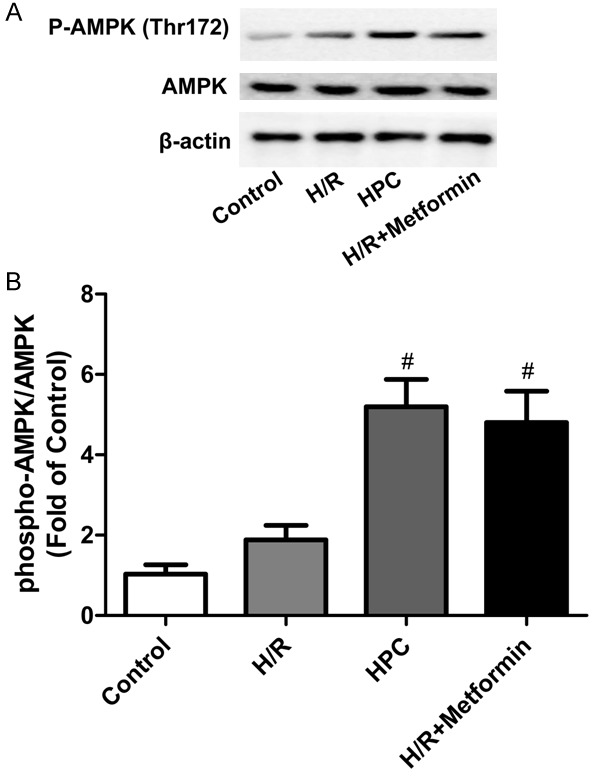
Expression of p-AMPK and AMPK in H9c2 cells. HPC and the AMPK activator metformin significantly increased the expression of p-AMPK compared with H/R group. A: Representative immunoblots of phosphorylated AMPK at residue threonine 172 (p-AMPKThr172) and total AMPK at 120 min of reoxygenation. B: Densitometric analysis of the phosphorylated state of AMPKThr172. Bars represent the ratio of phosphorylated AMPK to total AMPK. #P < 0.05 vs. H/R.
Cardioprotection of HPC was also mediated through eNOS
It has been reported that the phosphorylation of eNOS at serine residue 1177 (eNOSSer1177) close to the carboxyterminal is a critical requirement for eNOS activation and it is mediated by AMPK during myocardial ischemia [19]. Phosphorylation at threonine residue 495 (eNOSThr495) also influences the activity of eNOS. Phosphorylation at this site inhibits NO synthesis, whereas dephosphorylation can promote NO synthesis [20]. Therefore, we determined whether the cardioprotection of HPC was mediated through eNOS. The expression of p-eNOS Ser1177, p-eNOSThr495 and eNOS were measured. As shown in Figure 5A-C, HPC significantly increased eNOSSer1177 phosphorylation compared with H/R group and did not alter eNOSThr495 phosphorylation. The effects of HPC were abolished by AMPK inhibition and mimicked by AMPK activation. Considering that eNOS inhibition abolished the protection effect of HPC shown in Figures 2 and 3, these results suggested that eNOS also plays an important role in cardioprotection of HPC, and HPC regulates eNOS phosphorylation through AMPK-mediated pathway.
Figure 5.
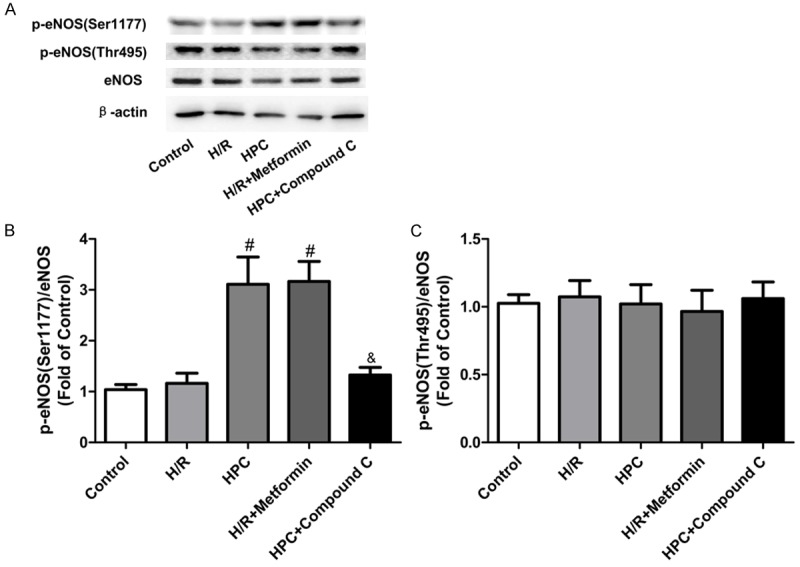
Expression of eNOS and p-eNOS in H9c2 cells. HPC increased eNOSSer1177 phosphorylation compared with H/R group in H9c2 cells, which was abolished by the AMPK inhibitor Compound C and mimicked by the AMPK activator metformin. HPC did not alter eNOSThr495 phosphorylation. A: Representative immunoblots of phosphorylated eNOS at serine residue 1177 (p-eNOSSer1177), theronine residue 495 (p-eNOSThr495), and total eNOS at 120 min of reoxygenation. B: Densitometric analysis of the phosphorylated state of eNOSSer1177. C: Densitometric analysis of the phosphorylated state of eNOSThr495. #P < 0.05 vs. H/R; &P < 0.05 vs. HPC.
PGC-1α acted as the downstream target of AMPK-eNOS-mediated signaling in the cardioprotection of HPC
To further investigate the mechanism of HPC protection, we next sought to find the downstream target protein regulated by AMPK/eNOS. As shown in Figure 6, we measured the expression of PGC-1α mRNA and protein. Compared with the control group, H/R decreased PGC-1α mRNA level in H9c2 cells, which was markedly reversed by HPC. However, the AMPK inhibitor compound C and eNOS inhibitor L-NIO abolished the effects of HPC. The AMPK activator metformin mimicked the effects of HPC. The same trends were shown in PGC-1α protein level. These results showed that AMPK/eNOS/PCG-1α axis may make an important role in the cardioprotection of HPC.
Figure 6.
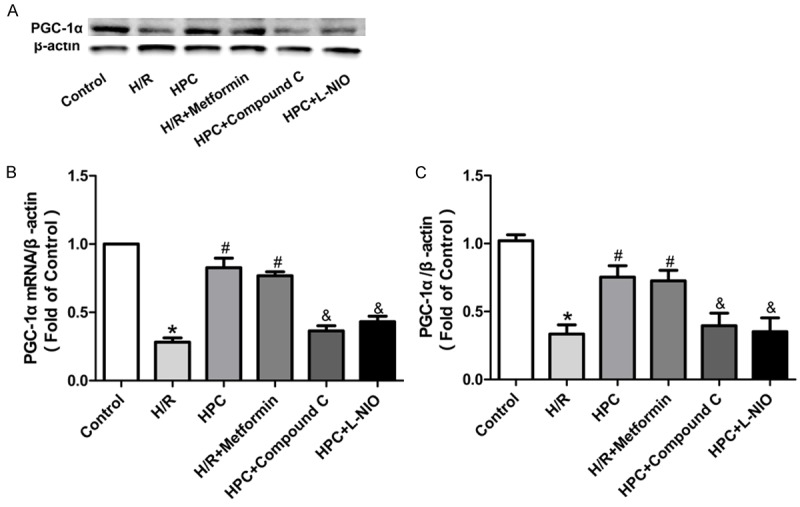
Protein and mRNA level of PGC-1α in H9c2 cells. HPC reversed the expression of PGC-1α protein and mRNA, which was mimicked by the AMPK activator metformin. The AMPK inhibitor Compound C and the eNOS inhibitor L-NIO inhibited the effect of HPC. A: Representative immunoblots of PGC-1α at 120 min of reoxygenation. B: mRNA levels of PGC-1α. C: Densitometric analysis of PGC-1α. *P < 0.05 vs. Control; #P < 0.05 vs. H/R; &P < 0.05 vs. HPC.
HPC attenuated oxidative stress via AMPK/eNOS/PGC-1α pathway
Considering that PGC-1α regulates level of oxidative stress in skeletal muscle [21], we sought to determine whether HPC regulates oxidative stress through PGC-1α signaling pathway to protect H9c2 cells. Compared with the Control group, H/R significantly increased the ROS level and MDA level, which were decreased by HPC. However, AMPK inhibitor and the eNOS inhibitor both abolished the effects of HPC against oxidative stress (Figure 7A, 7C). The AMPK activator metformin mimicked the effects of HPC. The similar results were shown in SOD activity, HPC reversed the reduction of SOD activity by H/R, which was mimicked by AMPK activation, and abolished by AMPK inhibition or eNOS inhibition (Figure 7B).
Figure 7.
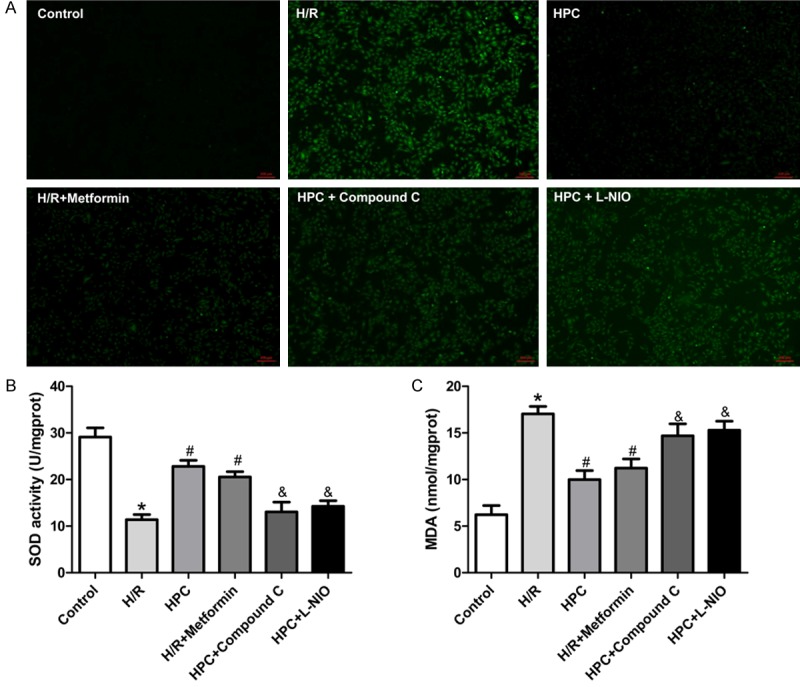
HPC regulated the SOD activity and production of ROS and MDA in H9c2 cells. H/R decreased the SOD activity and increased the ROS and MDA level, which was reversed by HPC. The AMPK activator metformin mimicked the effects of HPC. The AMPK inhibitor Compound C and eNOS inhibitor L-NIO inhibited the effects of HPC. A: Intracellular ROS level in H9c2 cells. B: MDA activity in H9c2 cells. C: SOD activity in H9c2 cells. *P < 0.05 vs. Control; #P < 0.05 vs. I/R; &P < 0.05 vs. HPC.
These data suggested that hypoxic preconditioning protects cardiomyocytes against hypoxia/reoxygenation injury by regulating oxidative stress, where AMPK/eNOS/PGC-1α axis was involved.
Discussion
This study was designed to explore the mechanism of HPC-induced cardioprotection against H/R injury in H9c2 cells. The major findings of this work are: (1) AMPK and eNOS inhibition abolished the cardioprotection of HPC in H9c2 cells subjected to H/R. (2) AMPK activation mimicked the cardioprotection of HPC. (3) HPC protects cardiomyocytes against H/R injury by reducing oxidative stress in which AMPK/eNOS/PGC-1α axis plays an important role. Taken together, these findings demonstrate for the first time that HPC protects cardiomyocytes against H/R injury partly through AMPK/eNOS/PGC-1α signaling pathway.
Ischemic preconditioning as a potent cardioprotective method against I/R injury was first reported by Murry et al. in 1986, which is the induction of a brief episode of ischemia and reperfusion in myocardium to markedly reduce tissue damage induced by prolonged ischemia [22,23]. In this study we corroborated previous observations that H9c2 undergo significant stress and cell death under H/R conditions, and HPC significantly improves the viability of H9c2 cells against H/R injury [24,25].
It has been reported that oxidative stress contributes to cardiac dysfunction and myocardial damage under a variety of conditions, such as ischemia-reperfusion [26,27]. Increased oxidative stress is an imbalance between pro-oxidants and anti-oxidants, which can lead to deleterious effects on cell biology and survival [28]. In order to corroborate that HPC protects H9c2 cells against H/R injury mainly by reducing oxidative stress and to explore the mechanism of attenuating oxidative stress by HPC, we measured the SOD activity and production of ROS and MDA in H9c2 cells. Our results showed that H/R markedly increased the oxidative stress level in H9c2 cells compared with the Control group, which was reduced by HPC. Interestingly, AMPK activation mimicked the effects of HPC, and AMPK inhibition and eNOS inhibition both abolished the cardioprotection of HPC. These data indicated that HPC decreases oxidative stress against H/R injury, in which AMPK and eNOS are needed.
AMPK, which is expressed in a number of tissues, including the heart, brain, and skeletal muscle, activated by a rise in the AMP: ATP ratio (ie in a low ATP or energy depleted state) [29,30], is an important regulator of diverse cellular pathways and is considered to be a “fuel gauge” or “master switch” for cellular energy levels [31]. Previous studies showing that the transduction of dominant-negative AMPK impairs ischemia induced glucose transport and fatty acid metabolism, increases left ventricular dysfunction and susceptibility to cellular damage [32,33]. Increasing evidence suggest that AMPK might also function as a sensor by responding to oxidative stress [34,35]. Most importantly, AMPK modulates endogenous antioxidant gene expression and/or suppress the production of oxidants. AMPK promotes cardiovascular homeostasis by ensuring an optimum redox balance on the heart and vascular tissues [36]. Indeed, we found that HPC enhanced the activity of AMPK by increasing the expression of p-AMPK (Thr172) compared with H/R group, AMPK inhibition abolished the anti-oxidative effects of HPC, and AMPK activation mimicked the effects of HPC. These data suggested that HPC reduces oxidative stress partly through AMPK-mediated signaling pathway. In agreement with macrophage migration inhibitory factor provides cardioprotection during ischemia/reperfusion by reducing oxidative stress through AMPK signaling pathway [37].
The phosphorylation and activity of AMPK are increased within minutes of the onset of myocardial ischemia and remain elevated for at least 48 h following reperfusion [38,39]. Our data showed that HPC has the ability to augment the I/R-induced increase in AMPK activity at the time of reperfusion. Since AMPK promotes ATP generation [40] and attenuates cardiomyocyte apoptosis [41], that HPC amplify signaling through AMPK during reperfusion is beneficial to the H/R injured cells.
eNOS, as a source of NO, plays important physiological and pathological roles in the cardiovascular system. Studies have shown that phosphorylation of eNOS could be increased in an AMPK-dependent manner [42,43]. Our data showed that HPC significantly increased the expression of p-eNOSSer1177 in H/R injured cells, which was mimicked by AMPK activation and abolished by AMPK inhibition, and eNOS inhibition abolished the protection of HPC. These data suggested that AMPK-eNOS-mediated signaling plays an important role in the protection effect of HPC, which may act the role through increasing the production of NO.
It is shown that downstream targets of PGC-1α include both genomic and mitochondrial genes involved in oxidative metabolism such as cytochrome c and SOD activity [44,45]. To determine whether PGC-1α was involved in the AMPK-eNOS-mediated signaling pathway, we measured the expression of PGC-1α and found that HPC reversed H/R induced reduction of PGC-1α, which was mimicked by AMPK activation and abolished by AMPK inhibition or eNOS inhibition. Additionally, AMPK inhibition or eNOS inhibition also abolished the reduction of oxidative stress by HPC. These results indicated that HPC protects H9c2 cells against H/R injury through PGC-1α, the downstream target protein of AMPK-eNOS-mediated signaling.
In conclusion, it is the first time reporting that hypoxic preconditioning protects H9c2 cells against hypoxia/reoxygenation injury partly via AMPK/eNOS/PGC-1α signaling pathway. These findings complete the mechanism of HPC in protecting against H/R injury.
Acknowledgements
This study was supported by Natural Science Foundation of China (No. 81173052, 30973534).
Disclosure of conflict of interest
None.
References
- 1.Arslan F, Smeets MB, O’Neill LA, Keogh B, McGuirk P, Timmers L, Tersteeg C, Hoefer IE, Doevendans PA, Pasterkamp G. Myocardial ischemia/reperfusion injury is mediated by leukocytic Toll-like receptor-2 and reduced by systemic administration of a novel anti-Toll-like receptor-2 antibody. Circulation. 2010;121:80–90. doi: 10.1161/CIRCULATIONAHA.109.880187. [DOI] [PubMed] [Google Scholar]
- 2.Gong D, Zhang Y, Zhang H, Gu H, Jiang Q, Hu S. Aldehyde Dehydrogenase-2 Activation during Cardioplegic Arrest Enhances the Cardioprotection against Myocardial Ischemia-Reperfusion Injury. Cardiovas Toxicol. 2012;12:350–358. doi: 10.1007/s12012-012-9179-6. [DOI] [PubMed] [Google Scholar]
- 3.Lundberg JO, Carlström M, Larsen FJ, Weitzberg E. Roles of dietary inorganic nitrate in cardiovascular health and disease. Cardiovas Res. 2011;89:525–532. doi: 10.1093/cvr/cvq325. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Zhang ZZ, Wu Y, Zhan J, He XH, Wang YL. Honokiol protects rat hearts against myocardial ischemia reperfusion injury by reducing oxidative stress and inflammation. Exp Ther Med. 2013;5:315–319. doi: 10.3892/etm.2012.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoole SP, Heck PM, Sharples L, Khan SN, Duehmke R, Densem CG, Clarke SC, Shapiro LM, Schofield PM, O’Sullivan M. Cardiac remote ischemic preconditioning in coronary stenting (CRISP Stent) study a prospective, randomized control trial. Circulation. 2009;119:820–827. doi: 10.1161/CIRCULATIONAHA.108.809723. [DOI] [PubMed] [Google Scholar]
- 6.Turer AT, Hill JA. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. Am J Cardiol. 2010;106:360–368. doi: 10.1016/j.amjcard.2010.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tian R, Musi N, D’Agostino J, Hirshman MF, Goodyear LJ. Increased adenosine monophosphate-activated protein kinase activity in rat hearts with pressure-overload hypertrophy. Circulation. 2001;104:1664–1669. doi: 10.1161/hc4001.097183. [DOI] [PubMed] [Google Scholar]
- 8.Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003;546:113–120. doi: 10.1016/s0014-5793(03)00560-x. [DOI] [PubMed] [Google Scholar]
- 9.Kemp BE, Stapleton D, Campbell DJ, Chen ZP, Murthy S, Walter M, Gupta A, Adams JJ, Katsis F, van Denderen B, Jennings IG, Iseli T, Michell BJ, Witters LA. AMP-activated protein kinase, super metabolic regulator. Biochem Soc Trans. 2003;31:162–168. doi: 10.1042/bst0310162. [DOI] [PubMed] [Google Scholar]
- 10.Iseli TJ, Turner N, Zeng XY, Cooney GJ, Kraegen EW, Yao S, Ye Y, James DE, Ye JM. Activation of AMPK by bitter melon triterpenoids involves CaMKKbeta. PLoS One. 2013;8:e62309. doi: 10.1371/journal.pone.0062309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hardie DG. The AMP-activated protein kinase pathway-new players upstream and downstream. J Cell Sci. 2004;117:5479–5487. doi: 10.1242/jcs.01540. [DOI] [PubMed] [Google Scholar]
- 12.Russell RR 3rd, Bergeron R, Shulman GI, Young LH. Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am J Physiol. 1999;277:H643–H649. doi: 10.1152/ajpheart.1999.277.2.H643. [DOI] [PubMed] [Google Scholar]
- 13.Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 14.Zhang W, Wang Q, Wu Y, Moriasi C, Liu Z, Dai X, Wang Q, Liu W, Yuan ZY, Zou MH. Endothelial cell-specific liver kinase B1 deletion causes endothelial dysfunction and hypertension in mice in vivo. Circulation. 2014;129:1428–1439. doi: 10.1161/CIRCULATIONAHA.113.004146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sahin E, DePinho RA. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature. 2010;464:520–528. doi: 10.1038/nature08982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H, Jia H, Liu J, Ao N, Yan B, Shen W, Wang X, Li X, Luo C, Liu J. Combined R-α-lipoic acid and acetyl-L-carnitine exerts efficient preventative effects in a cellular model of Parkinson’s disease. J Mol Cell Cardiol. 2010;14:215–225. doi: 10.1111/j.1582-4934.2008.00390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Austin S, Klimcakova E, St-Pierre J. Impact of PGC-1α on the topology and rate of superoxide production by the mitochondrial electron transport chain. Free Radical Bio Med. 2011;51:2243–2248. doi: 10.1016/j.freeradbiomed.2011.08.036. [DOI] [PubMed] [Google Scholar]
- 18.Uchiyama T, Engelman RM, Maulik N, Das DK. Role of Akt signaling in mitochondrial survival pathway triggered by hypoxic preconditioning. Circulation. 2004;109:3042–3049. doi: 10.1161/01.CIR.0000130647.29030.90. [DOI] [PubMed] [Google Scholar]
- 19.Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power DA, de Montellano PRO, Kemp BE. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999;443:285–289. doi: 10.1016/s0014-5793(98)01705-0. [DOI] [PubMed] [Google Scholar]
- 20.Mount PF, Kemp BE, Power DA. Regulation of endothelial and myocardial NO synthesis by multi-site eNOS phosphorylation. J Mol Cell Cardiol. 2007;42:271–279. doi: 10.1016/j.yjmcc.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 21.Yuzefovych L, Wilson G, Rachek L. Different effects of oleate vs. palmitate on mitochondrial function, apoptosis, and insulin signaling in L6 skeletal muscle cells: role of oxidative stress. Am J Physiol Endoc M. 2010;299:E1096–E1105. doi: 10.1152/ajpendo.00238.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohen MV, Baines CP, Downey JM. Ischemic preconditioning: from adenosine receptor to KATP channel. Annu Rev Physiol. 2000;62:79–109. doi: 10.1146/annurev.physiol.62.1.79. [DOI] [PubMed] [Google Scholar]
- 23.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Ge X, Liu X. The cardioprotective effect of postconditioning is mediated by ARC through inhibiting mitochondrial apoptotic pathway. Apoptosis. 2009;14:164–172. doi: 10.1007/s10495-008-0296-4. [DOI] [PubMed] [Google Scholar]
- 25.Park M, Youn B, Zheng Xl, Wu D, Xu A, Sweeney G. Globular adiponectin, acting via AdipoR1/APPL1, protects H9c2 cells from hypoxia/reoxygenation-induced apoptosis. PLoS One. 2011;6:e19143. doi: 10.1371/journal.pone.0019143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Negoro S, Kunisada K, Fujio Y, Funamoto M, Darville MI, Eizirik DL, Osugi T, Izumi M, Oshima Y, Nakaoka Y. Activation of signal transducer and activator of transcription 3 protects cardiomyocytes from hypoxia/reoxygenation-induced oxidative stress through the upregulation of manganese superoxide dismutase. Circulation. 2001;104:979–981. doi: 10.1161/hc3401.095947. [DOI] [PubMed] [Google Scholar]
- 27.Fukuda KI, Asoh S, Ishikawa M, Yamamoto Y, Ohsawa I, Ohta S. Inhalation of hydrogen gas suppresses hepatic injury caused by ischemia/reperfusion through reducing oxidative stress. Biochem Bioph Res Co. 2007;361:670–674. doi: 10.1016/j.bbrc.2007.07.088. [DOI] [PubMed] [Google Scholar]
- 28.Al Ahmad A, Gassmann M, Ogunshola OO. Involvement of oxidative stress in hypoxia-induced blood-brain barrier breakdown. Microvasc Res. 2012;84:222–225. doi: 10.1016/j.mvr.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 29.Zhao Y, Tan Y, Dai J, Wang B, Li B, Guo L, Cui J, Wang G, Li W, Cai L. Zinc deficiency exacerbates diabetic down-regulation of Akt expression and function in the testis: essential roles of PTEN, PTP1B and TRB3. J Nutr Biochem. 2012;23:1018–1026. doi: 10.1016/j.jnutbio.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 30.Takikawa M, Kumagai A, Hirata H, Soga M, Yamashita Y, Ueda M, Ashida H, Tsuda T. 10-Hydroxy-2-decenoic acid, a unique medium-chain fatty acid, activates 5’-AMP-activated protein kinase in L6 myotubes and mice. Mol Nutr Food Res. 2013;57:1794–1802. doi: 10.1002/mnfr.201300041. [DOI] [PubMed] [Google Scholar]
- 31.Hardie DG. Minireview: The AMP-activated protein kinase cascade: The key sensor of cellular energy status. Endocrinology. 2003;144:5179–5183. doi: 10.1210/en.2003-0982. [DOI] [PubMed] [Google Scholar]
- 32.Jones SP, Greer JJ, Kakkar AK, Ware PD, Turnage RH, Hicks M, van Haperen R, de Crom R, Kawashima S, Yokoyama M, Lefer DJ. Endothelial nitric oxide synthase overexpression attenuates myocardial reperfusion injury. Am J Physiol Heart Circ Physiol. 2004;286:H276–H282. doi: 10.1152/ajpheart.00129.2003. [DOI] [PubMed] [Google Scholar]
- 33.Russell RR, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ, Young LH. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Onken B, Driscoll M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans healthspan via AMPK, LKB1, and SKN-1. PLoS One. 2010;5:e8758. doi: 10.1371/journal.pone.0008758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu Y, Viana M, Thirumangalathu S, Loeken M. AMP-activated protein kinase mediates effects of oxidative stress on embryo gene expression in a mouse model of diabetic embryopathy. Diabetologia. 2012;55:245–254. doi: 10.1007/s00125-011-2326-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shirwany NA, Zou MH. AMPK in cardiovascular health and disease. Acta Pharmacol Sin. 2010;31:1075–1084. doi: 10.1038/aps.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koga K, Kenessey A, Powell SR, Sison CP, Miller EJ, Ojamaa K. Macrophage migration inhibitory factor provides cardioprotection during ischemia/reperfusion by reducing oxidative stress. Antioxid Redox Sign. 2011;14:1191–1202. doi: 10.1089/ars.2010.3163. [DOI] [PubMed] [Google Scholar]
- 38.Russell RR 3rd, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ, Young LH. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baron SJ, Li J, Russell RR 3rd, Neumann D, Miller EJ, Tuerk R, Wallimann T, Hurley RL, Witters LA, Young LH. Dual mechanisms regulating AMPK kinase action in the ischemic heart. Circ Res. 2005;96:337–345. doi: 10.1161/01.RES.0000155723.53868.d2. [DOI] [PubMed] [Google Scholar]
- 40.Winder WW, Hardie DG. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am J Physiol. 1996;270:E299–304. doi: 10.1152/ajpendo.1996.270.2.E299. [DOI] [PubMed] [Google Scholar]
- 41.He C, Zhu H, Li H, Zou MH, Xie Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes. 2013;62:1270–1281. doi: 10.2337/db12-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thors B, Halldorsson H, Thorgeirsson G. Thrombin and histamine stimulate endothelial nitric-oxide synthase phosphorylation at Ser1177 via an AMPK mediated pathway independent of PI3K-Akt. FEBS Lett. 2004;573:175–180. doi: 10.1016/j.febslet.2004.07.078. [DOI] [PubMed] [Google Scholar]
- 43.Thors B, Halldorsson H, Thorgeirsson G. eNOS activation mediated by AMPK after stimulation of endothelial cells with histamine or thrombin is dependent on LKB1. BBA Mol Cell Res. 2011;1813:322–331. doi: 10.1016/j.bbamcr.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 44.Hood DA. Invited Review: contractile activity-induced mitochondrial biogenesis in skeletal muscle. J Appl Physiol. 2001;90:1137–1157. doi: 10.1152/jappl.2001.90.3.1137. [DOI] [PubMed] [Google Scholar]
- 45.Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RAV, Lazarowski ER, Damian VA, Masliah E, La Spada AR. PGC-1α rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med. 2012;4:142ra197. doi: 10.1126/scitranslmed.3003799. [DOI] [PMC free article] [PubMed] [Google Scholar]