

Abstract
Background
Triglycerides and their lipoprotein transport molecules are risk factors for heart disease. Observational studies have associated elevated levels of serum urate (SU) with triglycerides (Tg) and risk of heart disease. However, owing to unmeasured confounding, observational studies do not provide insight into the causal relationship between SU and Tg. The aim of this study was to test for a causal role of SU in increasing Tg using Mendelian randomisation that accounts for unmeasured confounding.
Methods and Results
Subjects were of European ancestry from the Atherosclerosis Risk in Communities (ARIC; n=5237) and Framingham Heart (FHS; n=2971) studies. Mendelian randomisation by the two-stage least squares regression method was done with SU as the exposure, a uric acid transporter genetic risk score as instrumental variable and Tg as the outcome. In ordinary linear regression SU was significantly associated with Tg levels (β=2.69 mmol/L change in Tg per mmol/L increase in SU). However, Mendelian randomisation-based estimation showed no evidence for a direct causal association of SU with Tg concentration - there was a non-significant 1.01 mmol/L decrease in Tg per mmol/L increase in SU attributable to the genetic risk score (P=0.21). The reverse analysis using a Tg genetic risk score provided evidence of a causal role for Tg in raising urate in men (PCorrected=0.018).
Conclusions
These data provide no evidence for a causal role for SU in raising Tg levels, consistent with a previous Mendelian randomisation report of no association between SU and ischaemic heart disease.
Keywords: urate, uric acid, triglycerides, Mendelian randomization, association study
Introduction
Elevated serum urate (SU) levels are a key risk factor for gout and nephrolithiasis. Hyperuricemia and gout are independently associated with all cause and cardiovascular disease (CVD) mortality and myocardial infarction in multivariate analyses (1-3). Clinical trials demonstrate cardiovascular benefits by lowering of elevated SU levels (reviewed in (4)). However an older meta-analysis did not fully support association of SU with coronary heart disease, with associative evidence decreased in studies that included adjustment for a wider range of possible confounders (5). More recently, application of Mendelian randomisation has provided no evidence for a causal role of urate in ischaemic heart disease (6). Therefore the potential causal role of hyperuricemia in the various categories of heart disease remains unresolved (7).
One risk factor for CVD is triglyceride (Tg) that, while not directly atherogenic, is an important biomarker of CVD risk owing to inclusion in atherogenic lipoproteins (8). Urate levels are positively associated with Tg levels independently of metabolic syndrome components (9), consistent with the possibility that urate could influence CVD risk through Tg levels. One approach to evaluate a possible cause-effect role for urate in a complex condition such as CVD is to evaluate relationships with sub-phenotypic, and presumably less complex, risk factors such as Tg levels.
A genome-wide association study has identified 28 loci that explain a small proportion (∼6%) of SU levels in European Caucasians (10). Half of this explained variance (∼3%) is attributed to renal and gut molecules (SLC2A9, SLC17A1, SLC22A11, SLC22A12, ABCG2) that regulate serum urate levels via regulation of excretion (10, 11). Mendelian randomisation, that exploits random assignment of alleles at conception, is a statistical genetics approach that can allow disentangling of cause and effect in the presence of potential confounding (12, 13). Given the relatively strong effect on urate levels, a genetic risk score comprised of the serum urate-associated variants at each locus or individual genetic variants, is useful as an ‘instrumental variable’ to test for a possible causal role for urate in related phenotypes. The Mendelian randomisation technique has been used to provide evidence against a causal role for urate in ischaemic heart disease, metabolic syndrome and reduced renal function (6, 14, 15). Conversely, use of a weight genetic instrumental variable demonstrates a causal role for increased body mass index (BMI) in raising urate levels (demonstrating BMI as an important confounding factor in observational studies of urate and metabolic conditions) (6, 16). Our aim, therefore, was to use Mendelian randomisation to test for a causal role for urate (the exposure) in raising Tg levels (the outcome), with the results expected to provide additional information to address the broader question of whether or not urate is causal in CVD.
Subjects and Methods
Subjects
Subjects of European ancestry were included from the Atherosclerosis Risk in Communities (ARIC; n=5237) and the Framingham Heart Study (FHS; n=2971). Demographic and clinical details of these study sets are described in Table S1. People taking antihypertensive medication and who self-reported physician-diagnosed kidney disease or gout were excluded from the analysis. The research procedures were in accordance with the ethical standards of the institutional review boards relevant to the ARIC and FHS data sets. Written informed consent was given by all participants.
Instrumental variable and statistical analysis
The Mendelian randomisation approach, two-stage least squares regression, was performed using a genetic risk score as an instrumental variable. The uric acid transporter instrumental variable was comprised of SNPs rs11942223 (SLC2A9), rs2231142 (ABCG2), rs1183201 (SLC17A1), rs2078267 (SLC22A11), and rs3825018 (SLC22A12) for both ARIC and FHS. A Tg instrumental variable was also used (17), comprised of rs10889353 (ANGPTL3), rs7557067 (APOB), rs2954029 (TRIB1), rs7819412 (XKR6-AMAC1L2), rs328 (LPL), rs3135506 (APOA5), rs662799 (APOA5), rs17216525 (NCAN-CLIP2-PBX4) and rs7679 (PLTP). An allele-counting genetic risk score consisted of each SNP coded 0–2 based on the number of alleles that associated with increased SU or Tg and scores were combined. Because the uric acid transporter genetic risk score was an adequate instrumental variable (Table 1) we did not use other SNPs with weaker effects on urate (10). We excluded from the Tg instrumental variable SNPs from the glycolytic locus GCKR and the MLXIPL locus (which includes the BAZ1B gene) because they are both also associated with SU (10) and there is evidence for pleoitropic effects of these loci on other SU- and Tg-related phenotypes (18,19). The individual genetic variants of the SU genetic risk score were also used as instrumental variables, as previously described (14).
Table 1. Association between uric acid transporter genetic risk score (instrumental variable) and serum urate, and between triglyceride genetic risk score and serum triglyceride.
| Uric acid transporter | Triglyceride | ||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| F-statistic | R | P | F-statistic | R | P | ||
| ARIC Europeans | All | 114.81 | 0.0215 | 1.72E-26 | 76.82 | 0.0145 | 2.60E-18 |
| Males | 63.05 | 0.0256 | 3.08E-15 | 39.71 | 0.0163 | 3.53E-10 | |
| Females | 99.25 | 0.0338 | 5.42E-23 | 36.56 | 0.0128 | 1.64E-09 | |
| FHS Europeans | All | 68.75 | 0.0226 | 1.69E-16 | 69.71 | 0.0228 | 1.32E-16 |
| Males | 29.68 | 0.0213 | 5.97E-08 | 36.28 | 0.0259 | 2.24E-09 | |
| Females | 90.91 | 0.0536 | 5.58E-21 | 45.43 | 0.0275 | 2.20E-11 | |
| Combined | All | 187.37 | 0.0223 | 3.38E-42 | 137.29 | 0.0165 | 1.79E-31 |
| Males | 93.33 | 0.0242 | 7.97E-22 | 74.77 | 0.0195 | 7.51E-18 | |
| Females | 183.43 | 0.0397 | 5.84E-41 | 65.5 | 0.0145 | 7.63E-16 | |
The F-statistic represents the strength and R2 the percent variance in SU explained by the instrumental variable
To test for a causal effect of SU on Tg levels, the change in SU resulting from the SU genetic risk score instrumental variable in the ordinary least squares regression (step 1) was regressed against Tg levels (outcome) (step 2). The estimates derived from the ordinary least squares regression between the explained variables (SU and Tg) and the two-stage least squares regression were then compared using the Durbin–Hausman test (20). The reverse Mendelian randomisation analysis with Tg as exposure and SU as outcome was done using the Tg genetic risk score described above.
All analyses were done using STATA version 8.0 (StataCorp, College Station, TX). The two-stage least squares analyses were conducted using the ivreg function on STATA 8.0, where the exposure represented the endogenous variable, outcome the dependent variable, and the appropriate genetic risk score the instrumental variable. Normal linear regression analyses were carried out using the regress function on STATA 8.0. A P<0.05 was regarded as significant. A correction factor of nine was applied for multiple testing (combined and separate data sets multiplied by sex analysis). All associations were adjusted for possible confounders (age, sex, BMI), the first two Eigen values of genome-wide single nucleotide polymorphism (SNP) principal component analysis (PCA) and by dataset when ARIC and FHS were combined, as previously described (14).
Power calculations
Power using the SU instrumental variable was calculated as described (21); implemented at http://glimmer.rstudio.com/kn3in/mRnd/), for uncorrected α=0.05 and using parameters from Tables 1,2 and S1, in the combined ARIC/FHS sample sets and in males and females separately (βOLS was the crude and βXY (an estimate of the unknown true causal relationship) the confounder adjusted regression estimate of urate on Tg). Power in the combined samples was 0.94, males only was 0.46 and females only (with a stronger instrumental variable and less variance in Tg levels) was 0.86.
Table 2. Mendelian randomisation analysis of SU against serum Tg using the uric acid transporter genetic risk score instrumental variable.
| Ordinary Least Square Regression | Two-stage Least Square | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Beta‡ | SE§ | P | Beta‡ | SE§ | P | DH P║ | |||
| ARIC Europeans | All | Crude* | 3.160 | 0.145 | 1.44E-100 | -1.235 | 1.075 | 0.250 | <0.0001 |
| Adjusted† | 2.474 | 0.181 | 6.13E-42 | -1.101 | 1.006 | 0.274 | 0.0002 | ||
| Males | Crude | 3.214 | 0.284 | 5.61E-29 | -1.191 | 1.864 | 0.523 | 0.012 | |
| Adjusted | 2.419 | 0.293 | 1.72E-16 | -1.340 | 1.826 | 0.463 | 0.031 | ||
| Females | Crude | 3.276 | 0.203 | 3.42E-56 | -1.332 | 1.198 | 0.266 | <0.0001 | |
| Adjusted | 2.369 | 0.218 | 5.44E-27 | -0.828 | 1.065 | 0.437 | 0.001 | ||
| FHS Europeans | All | Crude | 3.813 | 0.192 | 1.15E-82 | -1.272 | 1.418 | 0.370 | <0.0001 |
| Adjusted | 3.057 | 0.271 | 7.08E-29 | -0.924 | 1.311 | 0.481 | <0.0001 | ||
| Males | Crude | 4.525 | 0.452 | 8.26E-23 | -3.833 | 3.463 | 0.269 | <0.0001 | |
| Adjusted | 3.828 | 0.468 | 6.98E-16 | -1.980 | 2.816 | 0.482 | <0.0001 | ||
| Females | Crude | 3.261 | 0.241 | 1.80E-39 | -0.224 | 1.108 | 0.840 | <0.0001 | |
| Adjusted | 2.033 | 0.256 | 3.78E-15 | 0.032 | 0.999 | 0.974 | <0.0001 | ||
| Combined | All | Crude | 3.475 | 0.115 | 1.32E-191 | -1.093 | 0.838 | 0.192 | <0.0001 |
| Adjusted | 2.688 | 0.150 | 1.80E-70 | -1.007 | 0.797 | 0.206 | <0.0001 | ||
| Males | Crude | 3.701 | 0.244 | 1.27E-50 | -2.041 | 1.680 | 0.224 | 0.0002 | |
| Adjusted | 2.920 | 0.252 | 1.52E-30 | -1.520 | 1.547 | 0.326 | 0.0024 | ||
| Females | Crude | 3.483 | 0.151 | 6.13E-112 | -0.631 | 0.816 | 0.439 | <0.0001 | |
| Adjusted | 2.322 | 0.167 | 6.55E-43 | -0.564 | 0.767 | 0.463 | 0.0001 | ||
The center-side is the standard linear (ordinary least square) regression between the explained variables (SU and serum Tg) and the right-side is the two-stage least squares analysis.
Unadjusted
Adjusted by study data set (in combined), sex (in ‘All’), age, BMI and the first two Eigen values of genome-wide principal component analysis (in ARIC, FHS and combined).
Beta represents the change in serum Tg (mmol/L) attributed to a unit change in SU in the linear regression (on the center) and the change in serum Tg (mmol/L) caused by a unit change in SU attributed to the instrumental variable in the two-stage least squares analysis (on the right).
Standard error
Durbin-Hausman P value
Results
Mean SU and serum Tg levels according to uric acid transporter genetic risk score are shown in Table S2. There was, as expected, a clear relationship between increased genetic risk score and SU, with the genetic risk score explaining 2.15%, 2.26% and 2.23% of variance in serum urate in the ARIC, FHS and combined cohorts, respectively (Table 1). However, as the genetic risk score increases, no clear change was observed in serum triglycerides in any of the study groups (Table S2).
Mendelian randomisation was performed using the two-stage least squares approach. The ordinary least squares regression analysis showed that an increase of 1 mmol/L in SU was associated with a significant increase in Tg levels (β=2.69 mmol/L, P=1.80×10-70; Figure 1, Table 2). However, use of the two-stage least squares approach as a quantitative measure for the exposure (SU) on outcome (serum Tg) showed no evidence for a causal role of SU in raising serum Tg levels in any sample set, as no significant change was observed in Tg values from each unit increase in SU attributable to the genetic risk score using two-stage least squares (P > 0.05; Figure 1, Table 2). Notably, each genetically attributed unit increase in SU was consistently associated with a decrease in serum Tg, with significant Durbin-Hausman P-values (all <0.002) (Table 2) indicating possible reverse causality.
Figure 1.
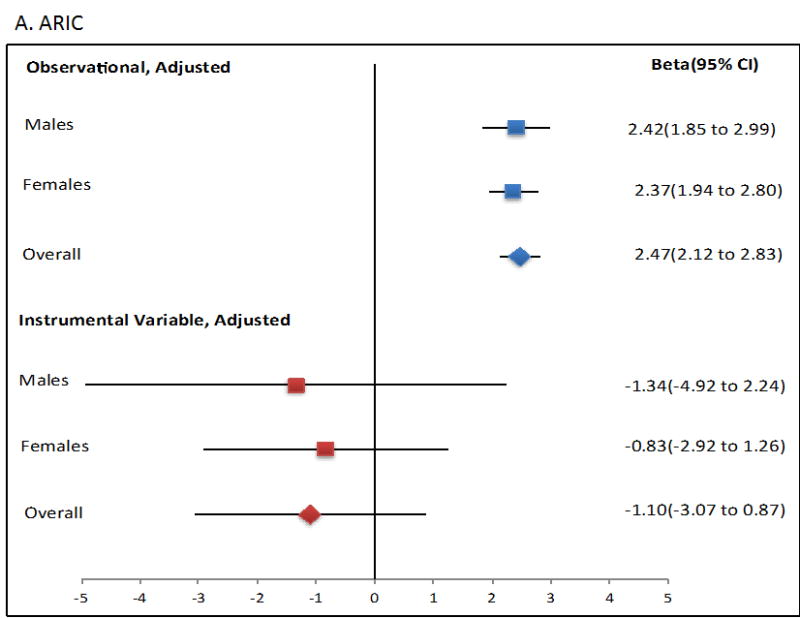
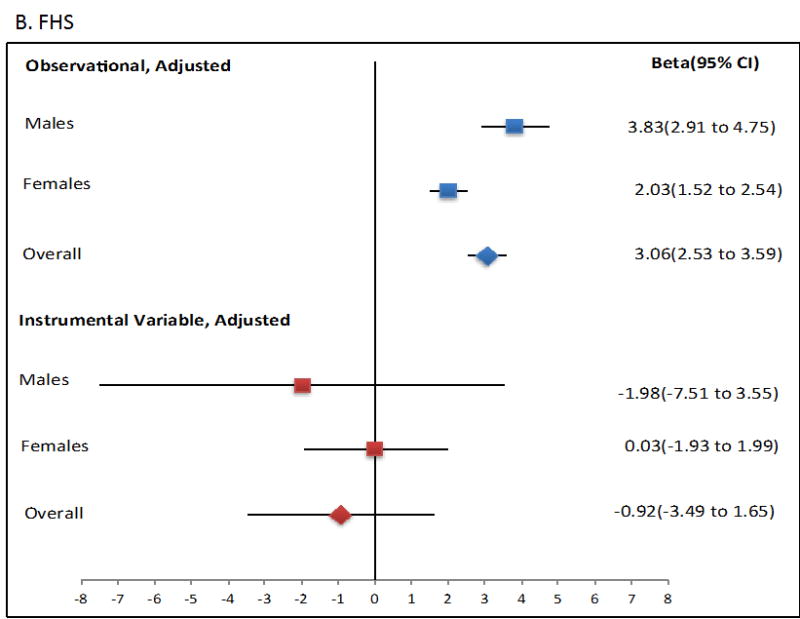
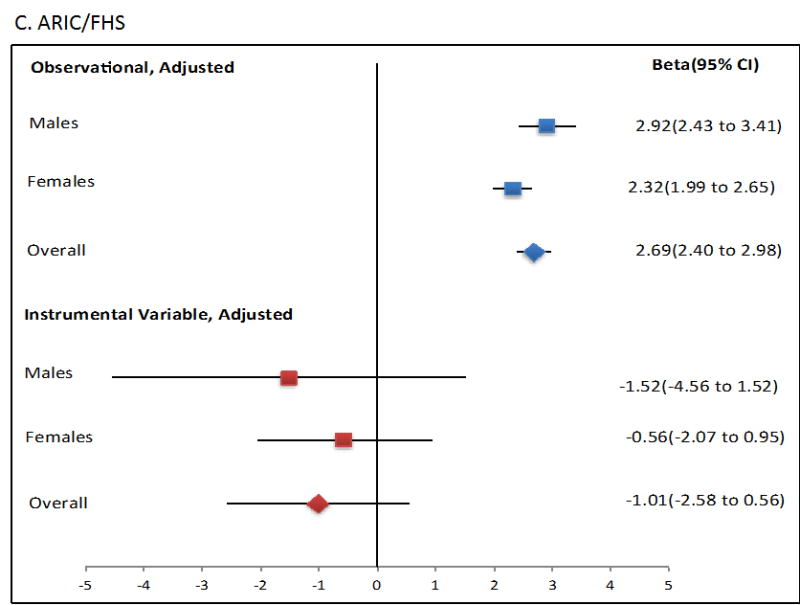
Forest plot showing observational and instrumental variable estimates of the effect of Tg (mmol/L) on SU (mmol/L) in ARIC (A), FHS (B) and combined ARIC/FHS (C). The instrumental variable estimate represents the increase in Tg per mmol/L increase in SU attributable to the genetic risk score.
The use of an instrumental variable in Mendelian randomisation requires that it fulfill three assumptions (13). The instrument used should be (i) adequately correlated with the exposure (SU), (ii) independent from confounders (eg age, sex, BMI), and (iii) would influence the outcome via the exposure and not via any pleiotropic effects. The urate instrument satisfies assumption one in Europeans (Table 1) where the F-statistic is ∼30 or greater in all analyses. Regarding assumption two, we tested by linear regression for association between the uric acid transporter instrumental variable and major confounders age, sex and BMI, in addition to eGFR, SBP, high and low density cholesterol and ancestry using Eigen values from principal component analysis (PCA)– there was no evidence for association with any of these variables excepting PCA1 (Table S4). This association was observed in the FHS dataset, presumably caused by the familial correlation within the FHS. However, there was no evidence for association of Tg with PCA1 in the FHS sample set (age, sex, BMI adjusted P=0.30), arguing against violation of assumption 2 caused by the familial structure. For assumption three, the possibility of pleiotropic effects (ie, effects on Tg levels aside from or in addition to a direct effect of the urate exposure) of the uric acid transporter instrumental variable on Tg levels is very difficult to eliminate (14). To mitigate possible violations of assumption three, the two-stage least squares Mendelian randomisation was adjusted by age, sex, BMI, and Eigen vectors. Previously, using the same genetic risk score as an instrumental variable in two-stage least squares Mendelian randomisation we provided evidence for a protective role of urate on renal function (14). However, the effect size of the individual component SNPs of the genetic risk score on urate did not correlate with the two-stage least squares effect estimate on renal function (14). Therefore, here, the individual variants were also tested (Table 3). At rs11942223 (SLC2A9) and rs2078267 (SLC22A11) negative ß-values and significant Durbin-Hausman P values were observed, suggesting that these variants may causally influence Tg levels, with lifetime exposure to the urate-increasing allele correlating with reduced serum Tg (Table 3; ß= -0.408 mmol/L, PDurbin-Hausman<0.00001 and ß= -5.262 mmol/L, PDurbin-Hausman=0.002, respectively).
Table 3. Mendelian randomisation analysis of SU versus Tg using uric acid transporter single genetic variants as instrumental variables in the combined ARIC and FHS sample set.
| F-statistics | R2 | Beta* | SE† | P | DHP‡ | |||
|---|---|---|---|---|---|---|---|---|
| All | ||||||||
| rs11942223 (SLC2A9) | 228.71 | 0.0271 | -0.408 | 0.704 | 0.562 | <0.00001 | ||
| rs2231142 (ABCG2) | 61.14 | 0.0074 | 1.211 | 1.401 | 0.387 | 0.286 | ||
| rs2078267 (SLC22A11) | 19.13 | 0.0023 | -5.262 | 3.037 | 0.083 | 0.002 | ||
| rs1183201 (SLC17A1) | 15.11 | 0.0018 | 0.103 | 2.347 | 0.965 | 0.261 | ||
| rs3825018 (SLC22A12) | 11.93 | 0.0015 | -3.156 | 3.535 | 0.372 | 0.072 | ||
| Males | ||||||||
| rs11942223 (SLC2A9) | 75.64 | 0.0197 | -0.924 | 1.775 | 0.599 | 0.022 | ||
| rs2231142 (ABCG2) | 53.48 | 0.014 | 0.027 | 2.098 | 0.990 | 0.157 | ||
| rs2078267 (SLC22A11) | 8.24 | 0.0022 | -7.633 | 5.832 | 0.191 | 0.028 | ||
| rs1183201 (SLC17A1) | 11.64 | 0.0031 | 1.115 | 4.122 | 0.787 | 0.659 | ||
| rs3825018 (SLC22A12) | 5.63 | 0.0015 | -2.426 | 5.446 | 0.656 | 0.299 | ||
| Females | ||||||||
| rs11942223 (SLC2A9) | 316.16 | 0.0665 | -0.200 | 0.591 | 0.735 | <0.00001 | ||
| rs2231142 (ABCG2) | 37.24 | 0.0083 | 2.657 | 1.841 | 0.149 | 0.855 | ||
| rs2078267 (SLC22A11) | 15.64 | 0.0035 | -3.320 | 2.936 | 0.258 | 0.031 | ||
| rs1183201 (SLC17A1) | 17.25 | 0.0039 | -0.428 | 2.412 | 0.859 | 0.239 | ||
| rs3825018 (SLC22A12) | 4.75 | 0.0011 | -2.940 | 3.804 | 0.440 | 0.125 | ||
Adjusted by study data set, sex (in ‘All’), age, BMI and the first two Eigen values of genome-wide principal component analysis
Beta represents the change in serum Tg (mmol/L) attributed to a unit change in SU attributed to the instrumental variable in the two-stage least squares analysis.
Standard error
Durbin-Hausman P value
Finally, we performed the reverse Mendelian randomisation, testing for a causal role for Tg in altering SU levels (Table 4; Figure 2; Table S3), using a genetic risk score of adequate strength (Table 1; R2=0.0165; F-statistic is ∼35 or greater in all analyses). This revealed evidence for a causal role of increased Tg in raising SU levels in men (β=0.021; P=0.002; PCorrected=0.018), with consistent evidence in each of the ARIC and FHS cohorts (β=0.020; P=0.037 and β=0.022; P=0.016, respectively). There was no evidence for a causal role for Tg in raising SU in women, with a Durbin-Hausman P of 0.002 providing evidence for possible reverse causality. The genetic risk score instrumental variable was not associated with confounders age, sex and BMI (Table S5), although there was evidence for association with PCA1 in the FHS data set. Weak association between PCA1 and SU in the FHS sample set (age, sex, BMI adjusted P=0.023) does not suggest serious violation of assumption 2 caused by the familial structure.
Table 4. Mendelian randomisation analysis of serum Tg against serum urate using the triglyceride genetic risk score instrumental variable.
| Ordinary Least Square Regression | Two-stage Least Square | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Beta‡ | SE§ | P | Beta‡ | SE§ | P | DH P║ | |||
| ARIC Europeans | All | Crude* | 0.026 | 0.001 | 1.44E-100 | 0.010 | 0.010 | 0.304 | 0.113 |
| Adjusted† | 0.014 | 0.001 | 6.13E-42 | 0.008 | 0.008 | 0.336 | 0.436 | ||
| Males | Crude | 0.016 | 0.001 | 5.61E-29 | 0.015 | 0.011 | 0.165 | 0.955 | |
| Adjusted | 0.012 | 0.001 | 1.72E-16 | 0.020 | 0.010 | 0.037 | 0.356 | ||
| Females | Crude | 0.026 | 0.002 | 3.42E-56 | -0.010 | 0.015 | 0.520 | 0.011 | |
| Adjusted | 0.017 | 0.002 | 5.44E-27 | -0.012 | 0.014 | 0.387 | 0.029 | ||
| FHS Europeans | All | Crude | 0.031 | 0.0016 | 1.15E-82 | 0.007 | 0.011 | 0.505 | 0.020 |
| Adjusted | 0.014 | 0.0012 | 7.08E-29 | 0.012 | 0.007 | 0.104 | 0.802 | ||
| Males | Crude | 0.015 | 0.0015 | 8.26E-23 | 0.020 | 0.009 | 0.035 | 0.607 | |
| Adjusted | 0.012 | 0.0015 | 6.98E-16 | 0.022 | 0.009 | 0.016 | 0.279 | ||
| Females | Crude | 0.032 | 0.0023 | 1.80E-39 | -0.016 | 0.016 | 0.286 | 0.0005 | |
| Adjusted | 0.019 | 0.0023 | 3.78E-15 | -0.005 | 0.013 | 0.709 | 0.050 | ||
| Combined | All | Crude | 0.029 | 0.001 | 1.32E-191 | 0.005 | 0.008 | 0.491 | 0.001 |
| Adjusted | 0.014 | 0.001 | 1.80E-70 | 0.009 | 0.006 | 0.095 | 0.306 | ||
| Males | Crude | 0.016 | 0.001 | 1.27E-50 | 0.017 | 0.007 | 0.025 | 0.860 | |
| Adjusted | 0.012 | 0.001 | 1.52E-30 | 0.021 | 0.007 | 0.002 | 0.166 | ||
| Females | Crude | 0.031 | 0.001 | 6.13E-112 | -0.023 | 0.013 | 0.076 | <0.00001 | |
| Adjusted | 0.018 | 0.001 | 6.55E-43 | -0.008 | 0.010 | 0.403 | 0.002 | ||
The center-side is the standard linear (ordinary least square) regression between the explained variables (SU and serum Tg) and the right-side is the two-stage least squares analysis.
Unadjusted
Adjusted by study data set (in combined), sex (in ‘All’), age, BMI, first two Eigen values of genome-wide SNP (in ARIC, FHS and combined).
Beta represents the change in SU (mmol/L) attributed to a unit change in Tg in the linear regression (on the center) and the change in SU (mmol/L) caused by a unit change in serum Tg attributed to the instrumental variable in the two-stage least squares analysis (on the right).
Standard error
Durbin-Hausman P value
Figure 2.
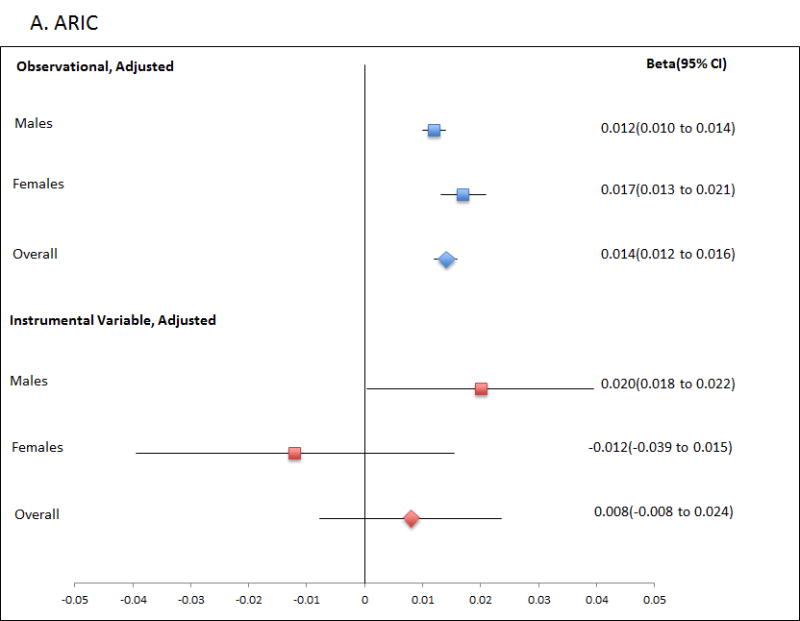
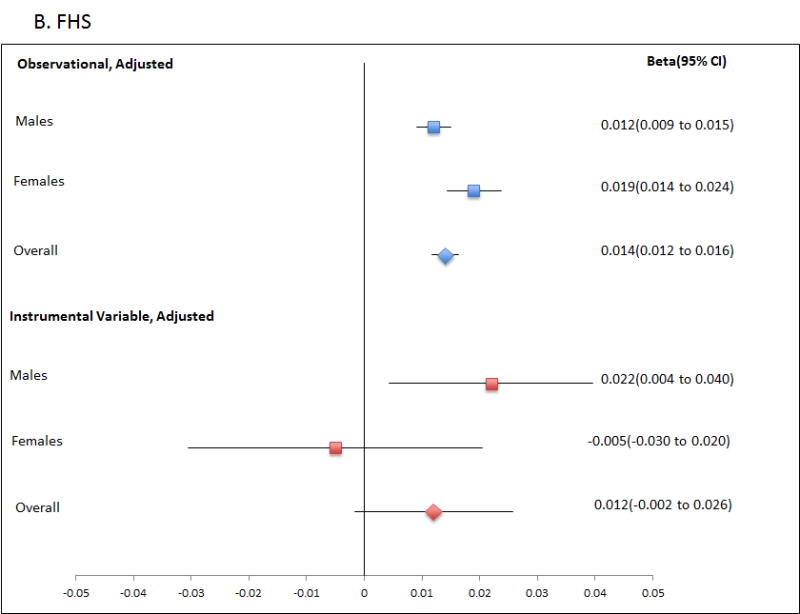
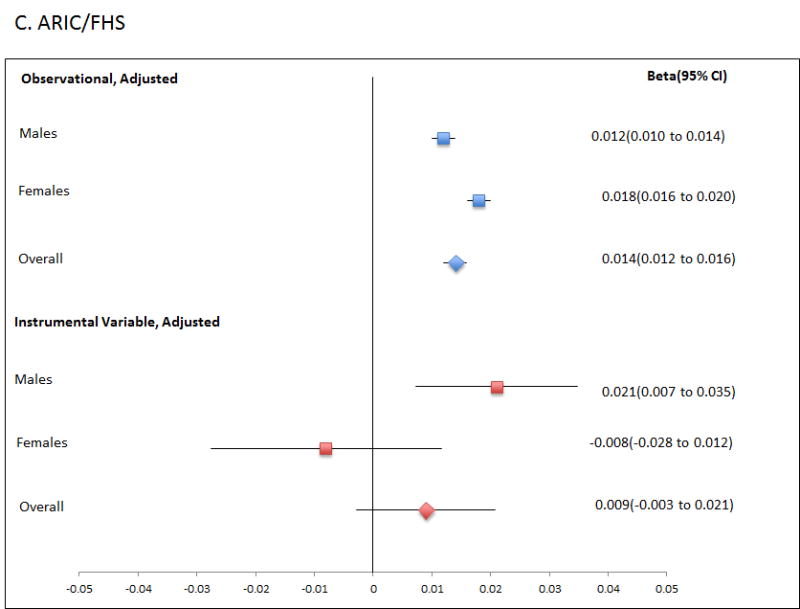
Forest plot showing observational and instrumental variable estimates of the effect of Tg (mmol/L) on SU (mmol/L) in ARIC (A), FHS (B) and combined ARIC/FHS (C). The instrumental variable estimate represents the increase in SU per mmol/L increase in Tg attributable to the genetic risk score.
Discussion
Use of the uric acid transporter genetic risk score in the two-stage least squares Mendelian randomization procedure showed that each standard unit increase in SU due to the genetic risk score was not directly associated with increased serum Tg levels. However the significant Durbin-Hausman P values did provide some evidence for reverse causality, with an increased uric acid transporter genetic risk score associated with reduced Tg levels, in a direction opposite to the relationship between SU and serum Tg. The analysis of the individual genetic variants, rs11942223 (SLC2A9) and rs2078267 (SLC22A11) in particular, also provided evidence by the Durbin-Hausman test for a causal effect of the urate-increasing alleles of the variants in lowering Tg levels. It is not feasible to eliminate the possibility of residual confounding contributing to this observation. Given a possibly weak causal relationship which would reduce power it will be important to provide more evidence for the potential reverse causation by testing for association of increased uric acid transporter genetic risk score with reduced Tg levels by two stage least squares analysis in larger cohorts than those studied here. However, we were adequately powered to detect a causal relationship equivalent to the confounder adjusted population ordinary least squares regression of urate on Tg (Table 2).
Whether or not the effect of rs11942223 and rs2078267 owes to urate per se or to other pleiotropic effects that can be ascribed to the physiological (functional) effect tagged by these SNPs (which would violate the third requirement of a Mendelian randomisation instrumental variable) is unclear. Similar two-stage least squares analysis that provided evidence for a protective role in renal function of the same urate-increasing genetic variants used here concluded that this effect was consistent with the possibility that the physiological action of the genetic variants (rs2078267 in SLC22A11 encoding OAT4, in particular) in raising SU is responsible for the improved renal function (14). The strongest evidence here by the Durbin-Hausman test of a causal role for increased urate to reduce triglyceride levels came from rs11942223 (SLC2A9) (P<0.0001), with support also from rs2078267 in SLC22A11 (Table 3). Arguing against a role for SU per se causing the Tg-lowering effect are the data from ABCG2 rs2231142 – this instrumental variable was strong (F=61.14, r2=0.0074) yet the β in the two-stage least squares analysis was +1.211 mmol/L (indicating the SU-raising genetic effect of rs2231142 could also raise Tg levels) with a non-significant Durbin-Hausman P of 0.29. The possibility of the activity of ABCG2 concomitantly raising SU and serum Tg requires testing in a larger sample set. With respect to the SLC2A9 effect, one environmental exposure relevant to both urate and Tg levels is sugar-sweetened beverage consumption. The SLC2A9 transporter exchanges sugars (glucose and fructose) for uric acid, with uric acid transport modified by fructose and glucose (22, 23). Controlled feeding studies show that consumption of sugar-sweetened beverage increases both SU and serum Tg (24,25), and consumption of sugar-sweetened beverage is associated with increased SU and serum Tg (26-28). Collectively these observations make it conceivable that the activity of SLC2A9 in raising urate lowers Tg levels, for example by influencing the availability of sugar for Tg synthesis via glycolysis. Together with our previous Mendelian randomisation study testing for a causal role for urate in renal function (14), these results emphasise the difficulty of identifying a urate instrumental variable for studying the causal relationship of urate with various metabolic phenotypes that has no pleiotropic effects. Whilst this could be regarded as a deficiency, studies such as this do, however, allow new biological insights into physiological aspects of urate metabolism and generation of testable experimental hypotheses. For example, what are the urate and Tg-related phenotypes of rodent models humanized for the individual instrumental variables that indicate pleiotropic effects.
The reverse Mendelian randomisation analysis by two-stage least squares provided direct evidence for a role of Tg in increasing urate levels in males only (Figure 2; Table 4) with consistent and significant effect sizes attributable to the Tg genetic risk score observed in each of the ARIC and FHS cohorts. The effect is in a direction consistent with the observational data, as evidenced by positive two stage least squares beta values and non-significant Durbin-Hausman P values. A very similar Tg instrumental variable to that used by us (ours excluded MLXIPL) was previously used as an instrumental variable in a Mendelian randomisation study that reported evidence of a protective role for raised serum Tg in type 2 diabetes but no evidence for a causal role in glucose levels or insulin resistance (17). These findings are ostensibly incongruous. Furthermore, given the established observational positive relationship between urate and these traits, our evidence supporting a causal role for Tg in raising urate is not obviously consistent with no causal role for Tg in glucose levels or insulin resistance. The findings using this Tg genetic instrumental variable, and the individual components, need to be extended in a larger Mendelian randomisation study that provides adequate power for evaluating the individual components, in order to disentangle the complex cause-effect relationship between Tg and other metabolic phenotypes.
One possible deficiency in our study design requires noting. The inherent property of Mendelian randomisation should ensure that confounding owing to sugar-sweetened beverage exposure, for example, is ruled out by equivalent lifetime exposure between the randomized genotype groups. However this is based on the assumption of no effect modification by such confounders on the instrumental variable. In the case of SLC2A9 rs11942223 and sugar-sweetened beverage exposure there is evidence for non-additive interaction on the control of SU – with sugar-sweetened beverage exposure the main urate-lowering effect of the C-allele is reversed (26). This possibility was not included in our two-stage least squares procedure.
Our results provided no evidence for a causal role for urate per se in raising Tg levels, a risk factor for CVD (8). These results are in agreement with those of Palmer et al.(6) who, using SLC2A9 rs7442295 (in complete linkage disequilibrium with rs11942223) as an instrumental variable for urate exposure, provided no evidence in a study of 7,172 cases and 61,502 controls for a causal association between urate and ischaemic heart disease and between urate and blood pressure. The observational studies that show independent association of SU with CVD and risk factors may be confounded by exposure to environmental factors that both raise urate levels and increase risk of CVD, with sugar exposure being a good example (26, 29). These exposures are very difficult to measure and account for in observational studies, and highlight the power of Mendelian randomisation, with different genotype groups having equivalent lifetime exposures to such confounders, in studying causal relationships. Our data illuminate a small aspect of the overall urate-CVD relationship, using an instrumental variable that explains a fraction of urate (2.2%) controlled by genetic variation in uric acid transporters. Further research should use Mendelian randomisation in larger well phenotyped cohorts to replicate and extend our findings and to examine the consequence of urate exposure on specific CVD-related phenotypes such as endothelial dysfunction and inflammation.
Supplementary Material
Acknowledgments
ARIC and FHS analyses (project #834) were approved by the relevant Database of Genotype and Phenotype (www.ncbi.nim.nih/gov/gap) Data Access Committees. The authors thank the staff and participants of the ARIC study for their important contributions. The FHS and the Framingham SHARe project are conducted and supported by the National Heart, Lung, and Blood Institute in collaboration with Boston University. The Framingham SHARe data used for the analyses described in this manuscript were obtained through dbGaP. This manuscript was not prepared in collaboration with investigators of the FHS and does not necessarily reflect the opinions or views of the FHS, Boston University, or the National Heart, Lung and Blood Institute.
Funding Sources: This work was supported by the Health Research Council of New Zealand, Arthritis New Zealand, New Zealand Lottery Health, and the University of Otago. The ARIC study is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute contracts N01-HC-55015, N01-HC-55016, N01-HC-55018, N01-HC-55019, N01-HC-55020, N01-HC-55021, N01-HC-55022, R01HL087641, R01HL59367, and R01HL086694; National Human Genome Research Institute contract U01HG004402; and National Institutes of Health contract HHSN268200625226C. The ARIC infrastructure was partly supported by Grant Number UL1RR025005, a component of the National Institutes of Health and NIH Roadmap for Medical Research.
Footnotes
Conflict of Interest Disclosures: None
References
- 1.Chen JH, Chuang SY, Chen HJ, Yeh WT, Pan WH. Serum uric acid level as an independent risk factor for all-cause, cardiovascular, and ischemic stroke mortality: A chinese cohort study. Arthritis Care Res. 2009;61:225–232. doi: 10.1002/art.24164. [DOI] [PubMed] [Google Scholar]
- 2.Krishnan E, Baker JF, Furst DE, Schumacher HR. Gout and the risk of acute myocardial infarction. Arthritis Rheum. 2006;54:2688–2696. doi: 10.1002/art.22014. [DOI] [PubMed] [Google Scholar]
- 3.Kuo CF, See LC, Luo SF, Ko YS, Lin YS, Hwang JS, et al. Gout: an independent risk factor for all-cause and cardiovascular mortality. Rheumatology. 2010;49:141–146. doi: 10.1093/rheumatology/kep364. [DOI] [PubMed] [Google Scholar]
- 4.Kanbay M, Segal M, Afsar B, Kang DH, Rodriguez-Iturbe B, Johnson RJ. The role of uric acid in the pathogenesis of human cardiovascular disease. Heart. 2013;99:759–766. doi: 10.1136/heartjnl-2012-302535. [DOI] [PubMed] [Google Scholar]
- 5.Wheeler JG, Juzwishin KD, Eiriksdottir G, Gudnason V, Danesh J. Serum uric acid and coronary heart disease in 9,458 incident cases and 155,084 controls: prospective study and meta-analysis. PLoS Med. 2005;2:e76. doi: 10.1371/journal.pmed.0020076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palmer TM, Nordestgaard BG, Benn M, Tybjærg-Hansen A, Smith GD, Lawlor DA, et al. Association of plasma uric acid with ischaemic heart disease and blood pressure: mendelian randomisation analysis of two large cohorts. Br Med J. 2013;347:f4262. doi: 10.1136/bmj.f4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shah A, Keenan RT. Gout, hyperuricemia, and the risk of cardiovascular disease: cause and effect? Curr Rheumatol Rep. 2010;12:118–124. doi: 10.1007/s11926-010-0084-3. [DOI] [PubMed] [Google Scholar]
- 8.Miller M, Stone NJ, Ballantyne C, Bittner V, Criqui MH, Ginsberg HN, et al. Triglycerides and cardiovascular disease a scientific statement from the American Heart Association. Circulation. 2011;123:2292–2333. doi: 10.1161/CIR.0b013e3182160726. [DOI] [PubMed] [Google Scholar]
- 9.Keenan T, Blaha MJ, Nasir K, Silverman MG, Tota-Maharaj R, Carvalho JA, et al. Relation of uric acid to serum levels of high-sensitivity C-reactive protein, triglycerides, and high-density lipoprotein cholesterol and to hepatic steatosis. Am J Cardiol. 2012;110:1787–1792. doi: 10.1016/j.amjcard.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kottgen A, Albrecht E, Teumer A, Vitart V, Krumsiek J, Hundertmark C, et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat Genet. 2013;45:145–154. doi: 10.1038/ng.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Merriman TR, Choi HK, Dalbeth N. The genetic basis of gout. Rheum Dis Clin North Am. 2014;40:279–290. doi: 10.1016/j.rdc.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 12.Didelez V, Sheehan N. Mendelian randomization as an instrumental variable approach to causal inference. Stat Meth Med Res. 2007;16:309–330. doi: 10.1177/0962280206077743. [DOI] [PubMed] [Google Scholar]
- 13.Lawlor DA, H R, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27:1133–1163. doi: 10.1002/sim.3034. [DOI] [PubMed] [Google Scholar]
- 14.Hughes K, Flynn T, de Zoysa J, Dalbeth N, Merriman TR. Mendelian randomization analysis associates increased serum urate, due to genetic variation in uric acid transporters, with improved renal function. Kidney Int. 2014;85:344–351. doi: 10.1038/ki.2013.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKeigue PM, C H, Wild S, Vitart V, Hayward C, Rudan I, Wright AF, Wilson JF. Bayesian methods for instrumental variable analysis with genetic instruments (‘Mendelian randomization’): example with urate transporter SLC2A9 as an instrumental variable for effect of urate levels on metabolic syndrome. Int J Epidemiol. 2010;39:907–918. doi: 10.1093/ije/dyp397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lyngdoh T, V P, Marques-Vidal P, Rousson V, Waeber G, Vollenweider P, Bochud M. Serum uric acid and adiposity: deciphering causality using a bidirectional Mendelian randomization approach. PLoS One. 2012;7:e39321. doi: 10.1371/journal.pone.0039321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Silva NM, Freathy RM, Palmer TM, Donnelly LA, Luan J, Gaunt T, et al. Mendelian randomization studies do not support a role for raised circulating triglyceride levels influencing type 2 diabetes, glucose levels, or insulin resistance. Diabetes. 2011;60:1008–1018. doi: 10.2337/db10-1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Böger CA, Gorski M, Li M, Hoffmann MM, Huang C, Yang Q, et al. Association of eGFR-Related Loci Identified by GWAS with Incident CKD and ESRD. PLoS Genet. 2011;7:e1002292. doi: 10.1371/journal.pgen.1002292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li N, van der Sijde MR, Lifelines Cohort Study Group. Bakker SJ, Dullaart RP, van der Harst P, et al. Pleiotropic effects of lipid genes on plasma glucose, HbA1c and HOMA-IR levels. Diabetes. 2014;63:3149–3158. doi: 10.2337/db13-1800. [DOI] [PubMed] [Google Scholar]
- 20.Hausman JA. Specification Tests in Econometrics. Econometrica. 1978;46:1251–1271. [Google Scholar]
- 21.Brion MJA, Shakhbazov K, Visscher PM. Calculating statistical power in Mendelian randomization studies. J Epidemiol. 2013;42:1497–1501. doi: 10.1093/ije/dyt179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caulfield MJ, Munroe PB, O'Neill D, Witkowska K, Charchar FJ, Doblado M, et al. SLC2A9 is a high-capacity urate transporter in humans. PLoS Med. 2008;7:e197. doi: 10.1371/journal.pmed.0050197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Witkowska K, Smith KM, Yao SY, Ng AM, O'Neill D, Karpinski E, et al. Human SLC2A9a and SLC2A9b isoforms mediate electrogenic transport of urate with different characteristics in the presence of hexoses. Am J Physiol Renal Physiol. 2012;15:F527–F539. doi: 10.1152/ajprenal.00134.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le MT, Frye RF, Rivard CJ, Cheng J, McFann KK, Segal MS, et al. Effects of high-fructose corn syrup and sucrose on the pharmacokinetics of fructose and acute metabolic and hemodynamic responses in healthy subjects. Metabolism. 2012;61:641–651. doi: 10.1016/j.metabol.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stanhope KL, Schwarz JM, Keim NL, Griffen SC, Bremer AA, Graham JL, et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest. 2009;119:1322–1334. doi: 10.1172/JCI37385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Batt C, Phipps-Green A, Black MA, Cadzow M, Merriman ME, Topless R, et al. Sugar-sweetened beverage consumption: A risk factor for prevalent gout with SLC2A9 genotype-specific effects on serum urate and risk of gout. Ann Rheum Dis. 2013 Sep 11; doi: 10.1136/annrheumdis-2013-203600. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi JW, Ford ES, Gao X, Choi HK. Sugar-sweetened soft drinks, diet soft drinks, and serum uric acid level: the Third National Health and Nutrition Examination Survey. Arthritis Rheum. 2008;59:109–116. doi: 10.1002/art.23245. [DOI] [PubMed] [Google Scholar]
- 28.Dhingra R, Sullivan L, Jacques PF, Wang TJ, Fox CS, Meigs JB, et al. Soft drink consumption and risk of developing cardiometabolic risk factors and the metabolic syndrome in middle-aged adults in the community. Circulation. 2007;116:480–488. doi: 10.1161/CIRCULATIONAHA.107.689935. [DOI] [PubMed] [Google Scholar]
- 29.Fung TT, Malik V, Rexrode KM, Manson JE, Willett WC, Hu FB. Sweetened beverage consumption and risk of coronary heart disease in women. Am J Clin Nutr. 2009;89:1037–1042. doi: 10.3945/ajcn.2008.27140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.