

Abstract
Duplex Sequencing (DS) is a next-generation sequencing methodology capable of detecting a single mutation among >1 × 107 wild-type nucleotides, thereby enabling the study of heterogeneous populations and very-low-frequency genetic alterations. DS can be applied to any double-stranded DNA sample, but it is ideal for small genomic regions of <1 Mb in size. The method relies on the ligation of sequencing adapters harboring random yet complementary double-stranded nucleotide sequences to the sample DNA of interest. Individually labeled strands are then PCR-amplified, creating sequence ‘families’ that share a common tag sequence derived from the two original complementary strands. Mutations are scored only if the variant is present in the PCR families arising from both of the two DNA strands. Here we provide a detailed protocol for efficient DS adapter synthesis, library preparation and target enrichment, as well as an overview of the data analysis workflow. The protocol typically takes 1–3 d.
INTRODUCTION
Next-generation DNA sequencing (NGS) defines the modern genomic era. This powerful technology has revolutionized traditional genetics and has made feasible the emerging field of personalized medicine. It is now routine to sequence billions of nucleotides and to identify inherited clonal mutations. However, all NGS approaches have a relatively high error rate: on the order of one erroneous base call per 100–1,000 sequenced nucleotides (Table 1; ref. 1). Although this error rate is acceptable for studying inherited mutations, it greatly limits the analysis of subclonal mutations, which are defined as mutations that are present in only a fraction of cells within a population.
TABLE 1.
NGS platforms and their associated errors1.
| Platform | Primary error type | Background (%) |
|---|---|---|
| Pacific Biosciences | G/C deletions | 16 |
| Life Ion Torrent | Short deletions, homopolymers | 1 |
| ABI SOLiD | A-T bias | 0.2 |
| Illumina MiSeq | Single nucleotide | 0.1 |
| Illumina HiSeq | Single nucleotide | 0.1 |
There is growing need for technologies capable of resolving subclonal mutations. For example, genetic heterogeneity has long been proposed to be an intrinsic driver of cancer initiation and progression2. Recent tumor genome sequencing studies suggest that human cancers exhibit extreme levels of genetic heterogeneity3-6. Subclonal mutations are probably a major factor in cancer relapse and in rapid emergence of chemotherapy resistance7-9. However, the study of cancer subclones requires the confident detection of mutations that are present in <1% of cells—a level of resolution that cannot be obtained by conventional sequencing approaches. Similarly, the genetic diversity found within microbial populations underlies their ability to adapt to changing environments, including development of drug resistance10-13, but this genetic diversity is difficult to directly assess owing to the high background error rate of conventional NGS sequencing. Other fields with a similar need for robust low-frequency mutation detection include forensics14, paleogenomics15,16, evolution17 and toxicology18, as high-accuracy sequencing would allow one to assess the potential mutagenicity of new chemical compounds without the need for a genetic selection system to identify mutant genes.
The concept of DS
To overcome the high error rate of next-generation sequencing and thereby facilitate the study of subclonal and random mutations, we recently developed a highly sensitive sequencing methodology termed Duplex Sequencing (DS). DS yields unprecedented accuracy in sequencing of double-stranded DNA, with a >10,000-fold improvement compared with conventional NGS, and it has the unique ability to detect a single mutation among >107 sequenced bases19. DS takes advantage of the inherent complementarity of double-stranded DNA by using degenerate molecular tags20-26 to label each fragmented DNA molecule with its own unique DNA sequence. By tagging duplex DNA with adapters containing random yet complementary double-stranded nucleotide sequences, it becomes feasible to trace every sequence read back to one of the two strands of the original double-stranded DNA molecule (Fig. 1a). After adapter ligation, the individually labeled strands are PCR-amplified to create sequence families that share the same tag sequences derived from each of the two single parental strands (Fig. 1b). After sequencing, members of each tag family are grouped and a consensus sequence is established for each of the two strands to form ‘single-strand consensus sequences’ (SSCSs; Fig. 1c). The two complementary consensus sequences derived from the two strands of an individual DNA duplex are then compared with each other, and the base identity at each position is retained only if the two strands match perfectly at that position, yielding a ‘duplex consensus sequence’ (DCS; Fig. 1c). Mutations introduced during PCR by DNA polymerase misincorporations or arising from DNA damage will appear in only one of the two DNA strands and thus are not counted as real mutations.
Figure 1.

Overview of Duplex Sequencing. (a) Schematic of a Duplex Sequencing adapter, showing the random double-stranded tag and the invariant spacer sequence. (b) Ligation of the adapters to the sample DNA results in a unique 12-nt tag sequence on both ends of the molecule. PCR amplification of each strand of a DNA duplex results in two distinct, but related, PCR products. (c) Reads sharing unique α and β tag sequences are grouped together into tag families of form αβ or βα, and an SSCS is created for each tag family. Mutations are of three different types: sequencing mistakes (blue or purple dots); first-round PCR errors (brown dots); true mutations (green dots). Formation of the SSCS removes the first type of error, but not first-round PCR errors. Comparing SSCSs from the paired families with tags αβ and βα generates a DCS, which eliminates these first-round PCR errors. True mutations are scored if and only if they are present at the same position in both strands of the DNA. Figure is adapted from ref. 33, © 2013 Kennedy et al.
Alternatives to DS
As the need for accurate sequencing methods has increased, three main strategies besides DS have emerged: (i) single-cell sequencing4,27-29, (ii) single-stranded molecular barcoding20,21,23 and (iii) circle sequencing (CirSeq)30,31. Although each approach has unique strengths, all three of these methods involve sequencing DNA derived from a single strand of a double-stranded molecule. Misincorporation events by DNA polymerase occurring during the first round of amplification will inherently be propagated to the daughter molecules, and they are likely to be erroneously scored as mutations. In the case of single-cell sequencing, the use of random primer sequences in conjunction with a strand-displacing DNA polymerase results in random priming from the newly synthesized DNA and the generation of ‘copies of copies’, thus propagating any initial misincorporation events to all of the reads. Because all the reads are derived from a single cell, the propagated error would be incorrectly called a genetic variant. A similar process can occur in CirSeq and singlestranded barcoding, whereby a misinsertion event occurring during the first round of synthesis can be propagated to subsequently synthesized daughter molecules. When sequenced, these subpopulations of related molecules will all contain a copy of the original misincorporation event, and they will be erroneously scored as a mutation. However, because the same mutation is unlikely to occur in unrelated molecules, these artifactual variants would give the appearance of a subclonal mutation. DNA polymerases typically used in library construction make misinsertions at a frequency between 10−4 and 10−6, which can lead to thousands of false positives on a typical sequencing run. Damaged or degraded DNA is particularly sensitive to this form of error because of the prevalence of DNA adducts that cause erroneous base pairings during polymerization. Removal of DNA damage with the addition of glycosylases or in vitro repair kits has been shown to reduce the number of false mutations in these samples31,32. However, not all mutagenic lesions are recognized by these enzymes, nor is the fidelity of repair perfect, thus limiting their utility in error correction.
Advantages of DS
The primary advantage of DS over other approaches is its superior accuracy. In our original publication on DS19, we demonstrated the direct measurement of the mutation frequency for the M13 bacteriophage as 2.5 × 10−6 mutations per base pair. We have subsequently measured mutation frequencies as low as 5 × 10−8 in human nuclear DNA (E.J.F. and L.A.L., unpublished results). In addition, we recently applied DS to measuring the mutation frequency of human mtDNA in brain tissue from young and old individuals33. We showed that the frequency of mtDNA point mutations is 10–100-fold lower than that previously reported, and the frequency increases approximately fivefold over an 80-year lifespan. Surprisingly, we observed no significant age-associated increase in the mutations most commonly associated with oxidative damage to mtDNA. This finding is inconsistent with free-radical theories of aging and suggests that previous studies indicating that reactive oxygen species induced mutations increase with age may be the result of PCR artifacts.
Similarly to our results with mtDNA, we have found that DS can help mitigate artifactual mutations derived from chemically damaged DNA. Numerous studies using NGS to sequence DNA from formalin-fixed tissues have reported a high number of false mutations34-36. Because complementary artifacts from damage events are unlikely to occur at the same corresponding position on paired DNA strands, the use of sequencing information from both strands to correct these errors makes DS extremely resistant to damage-induced misincorporations. By using DS, we have sequenced paired formalin-fixed and unfixed tissue samples, and we found only a twofold change in the mutation frequency, indicating that DS is useful for removing mutational artifacts even in extremely damaged and degraded DNA (M.J.P., E.J.F. and L.A.L., unpublished results).
Limitations of DS
DS is a uniquely sensitive method that is capable of detecting and quantifying mutations that occur at extremely low frequencies; however, this ability comes at a cost. Owing to the method’s reliance on sequencing multiple PCR duplicates of both DNA strands, DS requires much larger sequencing capacity than conventional NGS to produce a given depth of sequencing. At present, the use of current NGS technology with DS to sequence large genomes or genome targets >1–2 Mbp in size to a high depth of coverage is prohibitively expensive; however, as the cost of sequencing continues to fall, we anticipate that this will become increasingly practical.
Overview of the procedure
Sequencing library construction for DS is similar to the standard Illumina library preparation protocol. The protocol follows the basic standard steps of DNA shearing by sonication, size selection, end repair, 3′ dA-tailing, adapter ligation, PCR amplification and, optionally, targeted DNA capture. We have made several important updates and optimizations to the protocol since its initial publication, which have substantially increased its reliability and reproducibility. Specifically, we have re-designed the sequencing adapters to allow for more efficient dA-tailing of the sample DNA. As part of this new design, the adapters require a different synthesis method that we have included in Boxes 1 and 2. An additional change in the protocol involves the amount of DNA used during the PCR step. In the original publication, we specified that ~40 attomoles of DNA was optimal. However, we have since determined that the PCR input amount depends on a number of factors, including the number of reads devoted to a sample, the use of targeted DNA capture, genome and target size, and gene and target copy number. We formally outline the relationship between these parameters and provide the optimal DNA input amounts to maximize the final amount of data. Finally, we have included the option to use targeted DNA capture, which greatly expands the utility of DS beyond small, highly pure genomes, such as mtDNA and other small plasmids that were presented in our original publication.
Box 1. Synthesis of DS adapters  TIMING 2 d.
TIMING 2 d.
The adapter synthesis protocol generates a sufficient amount of adapters for several hundred samples.
-
Anneal the two oligonucleotides (MWS51 and MWS55 in Table 3) by combining 100 μl of each 100 μM oligonucleotide in a 0.2-ml PCR tube; heat the tube to 95 °C for 5 min in a thermocycler with a heated lid. Turn off the machine and leave it for 1 h. Remove and save 1 μl for quality control; label it as ‘annealed adapters’.
 CRITICAL STEP It is extremely important that the oligonucleotides be allowed to cool slowly. Make sure that the thermocycler does not automatically cool after the heating cycle.
CRITICAL STEP It is extremely important that the oligonucleotides be allowed to cool slowly. Make sure that the thermocycler does not automatically cool after the heating cycle. - Extend the annealed adapters by mixing the components in the table below by gently pipetting up and down, and then splitting into two 0.2-ml PCR tubes and incubating for 1 h at 37 °C.
Reagent Volume (μl) Final concentration DNA from step 1 199 Variable 10× NEB buffer #2 27.9 1× 10 mM dNTP mix 27.9 1 mM ddH2O 11.6 N/A Klenow exo− (5 U/μl) 11.6 0.5 U/μl -
Ethanol-precipitate the DNA and resuspend it with 200 μl of ddH2O. We recommend saving 1 μl of the resuspended adapters for quality control purposes; label as ‘extended adapters’.
 CRITICAL STEP It is essential that the two strands never melt. If they do, the complementarity of the tags will be lost, and DS will not work. The strands could melt if the DNA is overdried after precipitation.
CRITICAL STEP It is essential that the two strands never melt. If they do, the complementarity of the tags will be lost, and DS will not work. The strands could melt if the DNA is overdried after precipitation.? TROUBLESHOOTING
- Cleave the extended adapters by mixing the components in the table below by carefully pipetting up and down and then dividing into four 0.2-ml PCR tubes. Incubate the tubes for 16 h at 37 °C in a thermocycler with a heated lid. After 16 h, remove and save 1 μl for quality control purposes; label as ‘cut adapters’.
Reagent Volume (μl) Final concentration DNA from step 3 200 Variable 10× NEB CutSmart buffer 50 1× ddH2O 235 N/A HpyCH4III (5 U/μl) 15 0.5 U/μl Add 50 μl of 3 M sodium acetate (pH 5.2) and mix. The final volume should be 550 μl.
-
Divide the solution into aliquots in 1.5-ml tubes, 275 μl per tube (~2 tubes total), and add 675 μl (i.e., 2.5 volumes) of room-temperature 100% ethanol to each tube; mix the tubes by inversion.
? TROUBLESHOOTING
Centrifuge the mixture for 30 min at 4 °C at >10,000g.
Remove the supernatant, add 1 ml of 75% (vol/vol) ethanol to each tube and mix by inversion. Immediately centrifuge the tubes for 30 min at 4 °C at >10,000g.
Remove the supernatant. Dry the tube for 10 min inverted on a paper towel, and then for 5 min upright.
 CRITICAL STEP Dry the DNA pellet for the exact times specified. Overdrying the DNA could lead to strand melting.
CRITICAL STEP Dry the DNA pellet for the exact times specified. Overdrying the DNA could lead to strand melting.Resuspend the pellet in each tube with 100 μl TElow. After resuspension, pool all the tubes together for a final volume of 200 μl. Remove and save 1 μl for quality control; label as ‘final adapters’.
Divide the adapters into 50-μl aliquots, and place them at −80 °C for long-term storage. The final concentration will be ~50 μM.
(Optional) Radiolabel the sample from each step and run it on an 8 M urea, 14% (wt/vol) acrylamide gel (Fig. 2a). Although this step is optional, it is strongly recommended. See Box 2 for a detailed protocol.! CAUTION γ-32P-ATP is radioactive. Take appropriate steps to avoid exposure to radioactive compounds.? TROUBLESHOOTING
Box 2. Optional quality control steps for adapter synthesis using radiolabeled adapters and PAGE  TIMING 1 d.
TIMING 1 d.
The following steps provide a method by which to evaluate the synthesis of the adapters. We have found that the use of radiolabeling with polyacrylamide electrophoresis provides the most consistent and sensitive way to evaluate the success of the adapter synthesis steps. Agarose electrophoresis could also be used; however, the fluorescent dyes used require double-stranded DNA. Therefore, the evaluation of the extension step (step 3) will be limited. Technical solutions, such as the Agilent TapeStation 2200 or Bioanalyzer 2100, do not have enough resolution to detect the 9-bp size change that occurs during enzymatic restriction (step 4) and should be avoided.
Dilute each DNA sample from steps 1, 3, 4 and 5 from Box 1 in 9 μl of ddH2O.
- For each DNA sample, mix the following components and incubate the mixture at 37 °C for 30 min.! CAUTION γ-32P-ATP is radioactive. Take appropriate steps to avoid exposure.
Reagent Volume (μl) Final concentration DNA from step 1 1 Variable 10× NEB polynucleotide kinase (PNK) buffer 1 1× γ-32P-ATP (6,000 Ci/mmol) 0.25 0.4 μM ddH2O 16.25 N/A T4 PNK (10 U/μl) 0.5 0.5 U/μl Add 20 μl of the denaturing gel-loading buffer containing sequencing gel dye mix for a total volume of 40 μl.
Boil the DNA:buffer mixture for 1 min.
Mix 100 ml of the 8 M urea, 14% (wt/vol) polyacrylamide gel mixture with 130 μl of ammonium persulfate and 75 μl of tetramethylethylenediamine (TEMED). Pour it rapidly into the sequencing gel plate setup, as described by the manufacturer.
-
Run 5 μl of the final DNA:buffer mixture on the 8 M urea, 14% (wt/vol) polyacrylamide gel at 60 W for 1.5–2 h or until the bromophenol blue band reaches three-fourths of the way to the bottom of the gel.
 CRITICAL STEP The small fragment that is removed by ethanol precipitation migrates near the bottom of the bromophenol blue marker. Do not run the gel for too long to avoid loss of the fragment. See Figure 3 for an example gel.
CRITICAL STEP The small fragment that is removed by ethanol precipitation migrates near the bottom of the bromophenol blue marker. Do not run the gel for too long to avoid loss of the fragment. See Figure 3 for an example gel.
In addition to the updated protocol, we also include a comprehensive computational pipeline that we use to process and analyze our data (Fig. 2). The computational workflow for DS uses a number of standard software packages to process the sequencing data; these include the Burrows-Wheeler Aligner (BWA)37, SAMtools38, Picard and the Genome Analysis Toolkit (GATK)39,40, as well as several custom Python scripts. The computational workflow is broken down into three major steps: (i) Tag parsing and initial alignment; (ii) SSCS assembly; and (iii) DCS assembly. The latest version of the DS software package can be downloaded from https://github.com/loeblab/Duplex-Sequencing.
Figure 2.

Schematic of the basic computational workflow for Duplex Sequencing. The blue numbers correspond to the steps in the PROCEDURE.
Each read obtained from a DS run consists of a 12-nt tag sequence, followed by an invariant 5-bp sequence corresponding to the ligation site. First, the invariant 5-bp sequence is computationally removed from each read, and the 12-nt tag present on each of the two paired-end reads is combined to a single 24-nt tag that is stored in the read header. Sequences with ambiguous nucleotides or homopolymers greater than nine bases within the tag are discarded. These steps are all performed by the custom python script called ‘tag_to_header.py’ (Supplementary Fig. 1). The reads are then aligned to the reference genome using BWA37. After alignment, reads sharing the same tag sequence and genomic coordinates are identified and grouped to form ‘tag families’ with a python script called ‘ConsensusMaker.py’. By default, the script requires three members to result in a tag family. The family members are then compared at each sequence position, and the identity of a position is kept only when at least 70% of the members have the same sequence at that position. Positions that cannot form a consensus are replaced by an ‘N’ and are considered undefined. The resulting data are referred to as SSCSs (Supplementary Fig. 2). We have experimented with requiring more than three members per family or >70% sequence agreement, but we have found that this reduces data yield without any appreciable change in the method’s accuracy (Supplementary Fig. 3).
Next, the two related SSCS reads corresponding to the two initial DNA strands are grouped together and compared by a script called ‘DuplexMaker.py’. The 24-nt tag associated with each sequence read consists of two 12-nt sequences, and the tags corresponding to a pair of SSCS reads can be grouped by virtue of being transposed relative to one another. Specifically, if the two 12-nt sub-tag sequences are designated α and β, then a sequence with a tag αβ in read 1 is compared with the sequence having the tag βα in read 2. The paired strand SSCS reads are then compared at each position, with only matching bases being kept to produce a DCS. Non-matching bases are considered undefined, and they are replaced by ‘N’. Reads containing a high proportion of Ns (>30%) are filtered out during this step (Supplementary Fig. 4). The processed DCS data are then re-aligned to the reference genome.
Experimental design
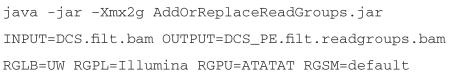
In the following sections, we will highlight important considerations that should be taken into account for each stage in the protocol, as well as provide details about the changes and improvements we have made since the initial publication19.
Sample requirements
The amount of fragmented DNA actually used in the PCR amplification step is typically in the low-attomole range, so the theoretical amount of starting DNA needed for DS is extremely low. However, when feasible, it is most convenient to start with excess amounts of DNA to allow for easy quantification of the DNA throughout the initial sample preparation steps and to account for expected sample loss at the enzymatic and purification steps. Typically, 1–3 μg of DNA is required for targeted DNA capture and 100–300 ng of DNA for smaller genomes, such as purified plasmid or mtDNA, that do not require targeted capture. When performing DNA isolation, it is imperative to avoid any manipulations that could result in melting of the two DNA strands, such as heating above 80 °C, the use of chaotropic agents such as urea or overdrying the DNA during ethanol precipitations.
DNA fragmentation
We physically shear the DNA by sonication using a Covaris acoustic ultrasonicator. The shearing time is varied depending on the size of the genome, with shorter times for small genomes (such as mtDNA or viral DNA) and longer times for nuclear DNA. Specific shearing parameters are given in Equipment Setup below. Other methods such as nebulization or enzymatic digestion should both be compatible with DS, but would probably require optimization. ‘Tagmentation’ methods, which involve simultaneous fragmentation of the sample DNA and ligation of sequencing adapters by a transposase (e.g., Illumina’s Nextera DNA sample prep kit), are currently incompatible with DS because they make use of an invariant transposon sequence that is incompatible with molecular barcodes. Because the DNA strands are not marked with molecular barcodes, it is impossible to identify unique DNA molecules and perform the comparison between complementary strands.
Size selection
Many protocols for next-generation sequencing library preparation use PAGE, with excision and purification of the desired fragment range from the gel. However, gel-based size selection can result in melting of the two DNA strands, precluding its use with DS. In addition, the use of gels increases the introduction of DNA damage during UV transillumination. To mitigate these problems, we perform size selection with Ampure XP beads, which has the additional benefits of higher recovery and greater speed.
End repair and dA-tailing
Similar to the standard Illumina library construction protocol, the sheared DNA is subjected to end repair and 3′-end dA-tailing. Notably, in our initial description of DS19, we used A-tailed adapters and enzymatically added a 3′ dT overhang on the fragmented DNA sample. However, since then, we have determined enzymatic T-tailing to be considerably less efficient than A-tailing (Supplementary Fig. 5), which results in a substantially reduced number of final reads. We have re-engineered the adapters to have a 3′ dT overhang (Fig. 1a, see ‘Adapter synthesis’ section and Boxes 1 and 2), which simultaneously increases the final data yield (Supplementary Table 1) and makes the protocol more consistent with conventional NGS library preparations.
Adapter synthesis
DS adapters are constructed by annealing two oligonucleotides, one of which contains a 12-nt singlestranded randomized tag sequence. A DNA polymerase is used to copy the degenerate tag sequence, thereby converting it to a double-stranded form (Fig. 3). The extended product contains an HpyCH4III restriction site downstream of the tag sequence. Cleavage of this site results in a 3′ dT overhang on the end of the final adapter (Figs. 1a and 3b), which can be ligated to the 3′ dA overhang on the DNA fragment library to be sequenced. Twelve random nucleotides per adapter (24 nt per final ligated molecule) significantly exceed the degeneracy needed to ensure unique labeling of every molecule in a library. However, a tag length of <12 is incompatible with the Illumina sequencer because of technical limitations of the platform’s ‘phasing’ requirement and should be avoided.
Figure 3.

Quality control of the sequencing adapters at each step of synthesis. (a) Representative 14% (wt/vol) polyacrylamide gel for each step of the adapter synthesis. Lane numbers correspond to the following steps in the synthesis protocol described in Box 1. Lane 1: step 1 (annealed adapters); lane 2: step 3 (extended adapters); lane 3: step 4 (cut adapters); and lane 4: step 10 (final adapters). Band sizes are as follows: a = 58 nt; b = 56 nt; c = 83 nt; d = 75 nt; e = 48 nt; and f = 9 nt. (b) Schematic of the adapters at each step on the synthesis. Lane and gel band designations correspond to the designations in a.
Adapter ligation
The DNA:adapter ratio is a crucial variable, as too few adapters lead to inefficient ligation, and the use of excess adapters can result in adapter dimers, which, owing to their small size, preferentially amplify during PCR. The presence of these adapter dimers interferes with DNA sequencing. To minimize the presence of adapter dimers, we determine the molar concentration of DNA molecules in the preligated DNA library on an Agilent TapeStation 2200 or Bioanalyzer 2100 and then use 20-fold molar excess of adapter relative to DNA. Unligated adapters and adapter dimers are then removed by purification with Ampure XP beads (Supplementary Fig. 6).
Setting tag family size by PCR amplification
The sequencing library is PCR-amplified for the purpose of creating multiple copies of each strand of a double-stranded DNA molecule. The number of DNA fragments used in the PCR, along with the fraction of a sequencing lane dedicated to a particular sample, are the primary adjustable variables that dictate the number of sequencing reads that share the same tag sequence (i.e., tag family size), which strongly influences the final number of DCSs formed. If there are too few reads sharing the same tag sequence (i.e., small tag family size), a consensus sequence cannot be calculated; conversely, too many reads having the same tag sequence (i.e., large tag family size) wastes sequencing capacity without appreciably improving data yield. Because the number of reads varies between different tag families and occur in a distribution (Fig. 4), we use ‘peak family size’ as our preferred metric to refer to tag family sizes generated under a given set of conditions. This distribution occurs during PCR amplification, and it is the result of different amplification efficiencies of the DNA molecules present in the library. By plotting the proportion of reads belonging to tag families of the same size (e.g., tag families can have the same number of reads as different tags) as a function of tag family size (the ‘PE_reads. tagstats’ file generated in Step 60 of the PROCEDURE section provides these data), we typically observe a distribution of tag family sizes with a solitary peak at a tag family size of one, which is probably the result of sequencing errors in the tag region, and a broader distribution centered at the peak family size (see Fig. 4a as an example). We formally define peak family size as the tag family size >1 containing the highest proportion of reads. On the basis of the analysis of samples with different values for the peak family size, we have determined that a peak family size of six maximizes the efficiency of DS: that is, requiring the smallest number of raw reads to produce a single DCS (Fig. 5a). Although a peak family size of six is optimal, it should be noted that data yield continues to increase, albeit at a decreasing overall efficiency, for peak family sizes ranging between six and 16 members (Fig. 5b). As the peak family size approaches 16 reads per tag family, nearly all additional reads are redundant and consume sequencing capacity without yielding further DCS data. Therefore, when deciding on DNA input amounts for PCR, we aim for a peak family size ranging between six and 12 members. This range allows for some tolerance in peak family size because of pipetting or measurement error, while maximizing the final number of DCSs that we obtain.
Figure 4.

Representative tag family size distributions. (a) Optimal family size distribution. (b) Tag family size distribution that is too small because of too much PCR input. (c) Tag family size distribution that is too large because of too little PCR input. We have found that a family size that is centered around approximately six members maximizes the final number of DCS reads. Samples that exhibit a small peak family size can be sequenced again and the raw sequencing data from the two sequencing runs can be combined and analyzed. Importantly, further sequencing of a sample with a large peak family size will not increase the final depth of coverage.
Figure 5.

Optimal peak family size. Replicates of the same sample at different lane fractions. (a) Plot compares peak family size to the number of final DCSs that are formed for every read originally dedicated to a sample. The maximum efficiency of DCS formations occurs at a peak family size of six and corresponds to ~40 raw reads being required to form one DCS read. (b) The total number of DCSs increases until a peak family size of ~16 is reached. A peak family size >16 does not result in an increase in the final number of DCSs.
To achieve greater sequencing depth for a given target, or to achieve the same depth for a larger genome or genome target, more DCSs are needed. To increase the number of reads obtained in DS, it is necessary to proportionally increase both the input amounts of DNA for PCR and the dedicated lane fraction to maintain a peak family size of six (Table 2). Nonproportional changes to PCR input and lane fraction lead to suboptimal peak family sizes and substantial decreases in DS efficiency (Figs. 4b,c and 5a). The choice of lane fraction (i.e., the number of reads devoted to a particular sample) will be influenced by the size of the targeted region and the desired depth of coverage. In particular, for a given lane fraction, the final sequencing depth of a sample will decrease as the size of the genome target increases. The following formula can be used to estimate the number of reads to devote to a sample:
Where N is the number of paired-end reads devoted to a sample (i.e., lane fraction), D is the desired average depth of coverage, G is the genome or genome target size in base pairs and R is the postanalysis read length in bases (76 in our analysis). The value of 40 is an empirically determined pseudo-constant that approximately corresponds to the average number of unprocessed read-pairs needed to form a single DCS. This value roughly corresponds to the product of the optimal peak family size (i.e., six) and the number of SSCSs typically needed to form a single DCS (we typically obtain SSCS:DCS ratios between four and ten, with an average of around six). However, because not all raw reads are of high quality, several extra reads are typically present that fail to form a DCS, and this tends to raise the number of raw reads needed to form a single DCS to ~40. Notably, this formula provides a rough estimate of the depth that will be obtained provided that the peak family size and SSCS:DCS ratio are optimal. After determining the per-sample lane fraction, this value can be referred to in Table 2 to determine the amount of target DNA that should be used in the PCR to obtain the appropriate peak family size of six.
TABLE 2.
DNA input amounts for PCR.
| Reads per sample | Attomoles of DNA (with capture) |
attomoles of DNA (with no capture) |
|---|---|---|
| 8 × 106 | 4 | 40 |
| 16 × 106 | 8 | 80 |
| 24 × 106 | 12 | 120 |
| 32 × 106 | 16 | 160 |
| 40 × 106 | 20 | 200 |
| 48 × 106 | 24 | 240 |
| 56 × 106 | 28 | 280 |
| 64 × 106 | 32 | 320 |
| 72 × 106 | 36 | 360 |
| 80 × 106 | 40 | 400 |
For samples that do not make use of targeted capture, such as purified mtDNA or plasmid DNA, we have found that a PCR input of 40 amol of DNA produces an optimal peak family size of six when devoting eight million paired-end reads to a sample (i.e., ~5% sequencing capacity of a HiSeq2500 lane). For the 16.5-kb human mtDNA, this typically results in a depth of 500–1,000. Given the linear relationship between PCR input and lane fraction needed to keep the peak family size constant, 200 attomoles of input mtDNA for 25% of a HiSeq lane results in a depth of 2,500–5,000.
When performing targeted capture, a PCR input amount of 4 amol of target DNA is used for ~8 million paired-end reads (i.e., ~5% sequencing capacity of a HiSeq2500 lane). However, in contrast to a noncapture sample, this value refers specifically to the amount of target DNA being captured in the sample, which is generally much less than the amount of total DNA. The amount of total DNA used in the PCR is dependent on its relative copy number and the size of the genomic target, and it can be estimated by the following formula:
Where m is the desired amount of target DNA in attomoles of fragments (Table 2), G is the size of total genome in base pairs, T is the size of the target DNA in base pairs and N is the number of copies of the target per copy of the nontarget portion of the genome. For single-copy genes, N will have a value of 1 and can be ignored. However, in the case of multicopy targets such as mtDNA or ribosomal genes, N can be on the order of hundreds or thousands of copies per nontarget genome and can markedly influence the amount of input DNA. The value of N can be reasonably estimated by performing copy number analysis with quantitative PCR (qPCR) using primers that are specific to the target and nontarget DNA. Similar to the nontarget DNA inputs, the target DNA input amounts should also be proportionally increased with increasing lane fraction (Table 2).
The number of PCR cycles is an important variable. Additional cycles beyond the exponential phase can result in the appearance of nonspecific, higher-molecular-weight products (Fig. 6a,b). To avoid this complication, we carefully monitor the number of PCR cycles. For samples requiring <10 fmol of input DNA, such as for mtDNA, we monitor the reaction by qPCR and remove the reactions one or two cycles before the SYBR Green signal plateaus, which typically occurs between cycles 15 and 20. Notably, the specific plateau point will be dependent on the amount of DNA used in the PCR and should be carefully monitored.
Figure 6.
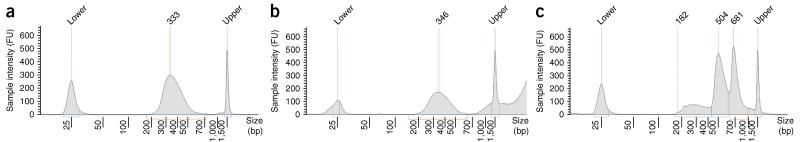
Example Agilent TapeStation 2200 electropherograms. (a) Electropherogram of an optimal post-PCR sample at Step 47. (b) Electropherogram from Step 47 showing higher molecular species resulting from too many PCR cycles. See Experimental design for details on determining the number of PCR cycles. (c) Electropherogram of the postligation at Step 36; note that the double peaks are normal. The peaks can vary in size and intensity without affecting the final results.
If a sample requires >10 fmol of DNA, such as when performing targeted capture of nuclear DNA, we have found that the SYBR Green signal can be unreliable. As a benchmark for determining the number of cycles that should be used during PCR, we have found that samples requiring ~600 fmol of total input DNA should be stopped between cycles six and eight. Importantly, for every twofold increase or decrease in the amount of DNA used in the PCR, the reactions should be stopped one cycle earlier or later, respectively. When first setting up DS for a new experimental system or genomic target, we recommend testing the number of cycles needed to maximize yield while avoiding higher-molecular-weight products by pausing the thermocycler every two cycles and carefully removing a small aliquot from the reaction. The aliquots can then be quantified on an Agilent TapeStation 2200 or Bioanalyzer 2100 to determine the optimal cycle number (Fig. 6a,b).
Targeted DNA capture
Targeted capture of the sequences of interest is often desirable. We have used the Agilent SureSelectXT target enrichment system. We have designed and used several custom-designed targeted capture sets against both nuclear and mtDNA from a number of organisms, including humans, mice and flies, with good success. Hybridization-based enrichment methods offered by other manufacturers would probably work as well. However, we have not evaluated these technologies, and they are likely to require some optimization. Of note, PCR-based enrichment methods are not compatible with DS because the targeted duplex DNA is melted into its single-stranded components during PCR, and the complementarity of the original DNA is lost.
Capture is readily performed on large targets, or on high-copy-number targets such as mtDNA. However, capture is inefficient when targeting small, low-copy-number regions. The reason is that capture typically results in a 10,000-fold enrichment of the target DNA. For example, the mitochondrial genome, owing to its high copy number, composes ~0.2% of the total mass of DNA in a human cell. Therefore, a 10,000-fold enrichment will result in nearly 100% purity of the targeted DNA. However, a 20-kb locus in genomic DNA, which is present in a single copy per haploid genome, composes only 0.0007% of the total DNA. Thus, a 10,000-fold enrichment will result in only 7% of the final DNA being on-target. However, we have found that repeating the targeted capture steps typically results in >85% of reads mapping to the target sequence.
Quality metrics
The overall efficiency of DS is calculated by dividing the number of DCS reads by the number of raw reads, and in our hands it can range from 1 to 10%. We have found that two primary factors determine overall data yield: the peak family size and the SSCS:DCS ratio. The peak family size is determined by the amount of DNA used for the PCR and the fraction of a lane dedicated to a sample, and it should optimally be adjusted to six, as discussed previously (see the ‘Setting tag family size by PCR amplification’ section; Fig. 5a). Should a sample exhibit a peak family size that is smaller than what is desired, then an optimal peak family size can be obtained by resequencing the same sample using the information in Supplementary Table 2 and combining the data with the previous run(s). Importantly, the resequenced sample must be a technical replicate; performing the protocol again on the same biological sample will not work. The SSCS:DCS ratio is the other key factor. A ratio of two is the theoretically ideal and would indicate that every SSCS can find its partner and form a DCS. We typically obtain SSCS:DCS ratios between four and ten, with an average of ~6. An excessively high ratio (i.e., >14–15) can occur for several reasons. If peak family sizes are too small, then families with at least three members might not form from both DNA strands. This issue is readily overcome by increasing the family size, as noted above, although it does not appreciably improve above peak sizes of 5–6 (Fig. 5b). If a poor ratio is seen despite adequate family sizes, this suggests that only one of the two DNA strands was successfully ligated and amplified during PCR. In such cases, the sample library will need to be remade in order to obtain more data, with attention to optimizing ligation conditions and library tailing steps.
Finally, mutations should theoretically be distributed throughout all possible read positions; however, we observe an increased frequency of mutations at the 5′ and 3′ends of reads. These artifacts are likely due to errors that occur during the end-repair process and alignment. These artifacts can be mitigated by performing local re-alignment with GATK, followed by clipping the first and last five bases of each read39.
MATERIALS
REAGENTS
DNA of interest
 CRITICAL It must be purified under non-strand-denaturing conditions.
CRITICAL It must be purified under non-strand-denaturing conditions.Agencourt AMPure XP beads (Beckman Coulter, cat. no. A63880) Alternatively, Agencourt RNAclean XP (Beckman Coulter, cat. no. A63987) may be used
Klenow fragment (3′®5′exo−; New England Biolabs (NEB) cat. no. M0212L, includes NEB buffer #2)
10× TBE electrophoresis buffer (Sigma-Aldrich, cat. no. T4415)
TElow (Affymetrix, cat. no. 75793)
Nuclease-free water (Life Technologies, cat. no. AM9937)
T4 polynucleotide kinase (NEB, cat. no. M0201; includes reaction buffer)
HpyCH4III (NEB, cat. no. R0618; includes CutSmart buffer)
dNTPs, 2.5 mM each (Promega, cat. no. U1511)
Ethanol (100%, 200 proof; Sigma-Aldrich, cat. no. E7023)! CAUTION Ethanol is flammable. Keep it away from flames.
Sodium acetate (Sigma-Aldrich, cat. no. S2889)
γ-32P-ATP (6,000 Ci/mmol; MP Biomedicals, cat. no. 0135001U01)! CAUTION γ-32P-ATP is radioactive. Take appropriate care when handling it.
Urea (Sigma-Aldrich, cat. no. U6504)
Acrylamide (Supplied as a 19:1 30% (wt/vol) acrylamide/bis-acrylamide solution; Bio-Rad, cat. no. 161-0154)! CAUTION Acrylamide is a known neurotoxin; handle it with care.
Denaturing gel loading buffer (Life Technologies, cat. no. AM8546G)
DS adapter oligonucleotides (IDT; see Table 3 for sequences)
 CRITICAL Oligonucleotides must be PAGE purified.
CRITICAL Oligonucleotides must be PAGE purified.Sequencing adapter PCR oligonucleotide primers (IDT; see Table 3 for sequences
NEBNext end-repair module (NEB, cat. no. E6050L; includes reaction buffer)
NEBNext dA-tailing module (NEB, cat. no. E6053L; includes reaction buffer)
Ultrapure T4 ligase (600,000 U/ml; Enzymatics, cat. no. L6030-HC-L; supplied with 10× Ultrapure ligation buffer)
KAPA HiFi HotStart DNA polymerase (1 U/ml; KAPA Biosystems, cat. no. KK2502; supplied with 5× fidelity buffer and 10 mM dNTP mix)
SYBR Green (10,000×; Life Technologies, cat. no. S-7563)
Agilent DNA high-sensitivity D1K TapeStation kit (Agilent, cat. no. 5067-5363 and 5067-5364). Alternatively, an Agilent high-sensitivity DNA for the Bioanalyzer 2100 may be used (Agilent, cat. no. 5067-4626)
Agilent SureSelectXT target enrichment Set (protocol version 1.6; Agilent)
 CRITICAL The use of target enrichment is optional for this protocol. The target enrichment section uses Agilent SureSelectXT. The use of other targeted capture products will probably work; however, they have not been evaluated and their compatibility is unknown.
CRITICAL The use of target enrichment is optional for this protocol. The target enrichment section uses Agilent SureSelectXT. The use of other targeted capture products will probably work; however, they have not been evaluated and their compatibility is unknown.SureSelectXT reagent kit, HSQ, 16 (Agilent, cat. no. G9611A) This item is optional and is needed only if targeted capture is performed
TABLE 3.
Sequences of the adapter and PCR oligonucleotides.
| Name | Sequence | Purpose |
|---|---|---|
| MWS51 | 5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′ | Duplex sequencing adapter oligos. |
| MWS55 | 5′-TCTTCTACAGTCANNNNNNNNNNNNAGATCGGAAGAGCACACGTCTGAACTCCAGTCAC-3′ | Annealed adapters prepared as described in Box 1 and used in PROCEDURE Step 30 |
| MWS13 | 5′-AATGATACGGCGACCACCGAG-3′ | PCR primers used at PROCEDURE Steps 38 |
| MWS20 | 5′-GTGACTGGAGTTCAGACGTGTGC-3′ | and 51 to generate amplified tag families, |
| MWS21 | 5′-CAAGCAGAAGACGGCATACGAGATXXXXXXGTGACTGGAGTTCAGACGTGTGC-3′ | with (Steps 38B and 51) or without (Step 38A) targeted capture |
Xs represent the sample indexing sequence. We recommend using the Illumina TruSeq index sequences available on the Illumina website.
EQUIPMENT
Eight-well PCR strip tubes (0.2 ml; BioExpress, cat. no. T-3035-1)
DynaMag-96 side magnetic plate separator (Life Technologies, cat. no. 12331D)
Vacuum centrifuge
Microcentrifuge tubes (1.7 ml, Eppendorf)
qPCR thermocycler (Bio-Rad)
qPCR-compatible eight-well PCR strip tubes (0.2 ml; Bio-Rad, cat. no. TLS0851)
qPCR-compatible eight-well PCR strip caps (Bio-Rad, cat. no. TCS0803)
Microcentrifuge (Eppendorf, cat. no. 5424 000.410)
Heating block (37 °C)
Heating stir plate
Covaris Sonicator S220 (Covaris)
Sonication tubes (Covaris, cat. no. 520045)
Agilent TapeStation 2200 (Agilent, cat. no. G2964AA) or Agilent Bioanalyzer 2100 (Agilent, cat. no. G2938C)
Illumina HiSeq2500 and associated equipment (Illumina): NextSeq500, HiSeq1000 and HiSeq2000 are all compatible with this protocol. The MiSeq uses a substantially reduced number of clusters relative to the other Illumina platforms and, although not incompatible with this protocol, is not recommended. Non-Illumina sequencing platforms have not been evaluated and would require re-designing of the adapters
Pipette tips
BWA software package (http://bio-bwa.sourceforge.net/): the protocol is known to work with versions ≤0.6.2. The software distributed with this protocol has been designed for use with BWA; different aligners may not be compatible
SAMtools software package (http://samtools.sourceforge.net/): the protocol is known to work with versions ≤0.1.18. The software distributed with this protocol has been designed for use with SAMtools; the use of other software to manipulate sam/bam files may not be compatible
Python software package (http://python.org/): the scripts used in this protocol have been tested with v2.7.x. They are not currently compatible with Python 3.x
BioPython software package (http://biopython.org/): the scripts used in this protocol are compatible with v1.62.
Pysam software package (http://code.google.com/p/pysam/): the scripts used in this protocol are compatible with v0.7.5
Genome Analysis Toolkit (GATK) software package (http://www.broadinstitute.org/gatk/): the scripts used in this protocol are known to be compatible with v2.4-9
Picard software package (http://picard.sourceforge.net/): the scripts used in this protocol are known to be compatible with v1.107
Distance software package: the scripts used in this protocol are compatible with v0.1.3.
REAGENT SETUP
Urea 8 M, polyacrylamide gel mixture 14% (wt/vol)
Add 480 g of urea to 466 ml of acrylamide and 100 ml of 10× TBE. Allow the urea to dissolve by slowly stirring with mild heat using a stir plate. After the urea is dissolved, bring the volume up to 1 liter with ddH2O. Store it at 4 °C for up to 1–2 months.
Sodium acetate solution, 3 M
Add 24.6 g of sodium acetate and dissolve it in 90 ml of ddH2O. Adjust the pH to 5.2 with 1 M HCl, and then add ddH2O to adjust the final volume to 100 ml. Sterilize the solution by autoclaving. This solution can be stored indefinitely at room temperature (20–25 °C).
Ethanol solution, 75% (vol/vol)
Add 25 ml of ddH2O to 75 ml of 100% ethanol. This solution can be stored at room temperature for up to 1 month.
SYBR Green, 12.5×
Dilute 1 μl of 10,000× SYBR Green in 799 μl of ddH2 and divide the solution into 50-μl aliquots; store the aliquots at −20 °C for up to 6 months.
Sequencing adapter oligonucleotides
Dissolve the oligos in TE or ddH2O in separate microcentrifuge tubes to a final concentration of 100 μM each. Immediately freeze the oligonucleotides at −20 °C until further use. Oligonucleotides can be stored at this temperature for 6–12 months.
Sequencing adapter PCR primers
Dissolve each respective PCR primer in TE or ddH2O in separate microcentrifuge tubes to a final concentration of 20 μM each. Immediately freeze the oligonucleotides at −20 °C until further use. Primers can be stored at this temperature for 6–12 months.
AMPure XP magnetic beads
The beads are normally stored at 4 °C. Allow the beads to warm to room temperature before use.  CRITICAL The beads will not function properly if they are not at room temperature.
CRITICAL The beads will not function properly if they are not at room temperature.
EQUIPMENT SETUP
Covaris S220 Sonicator
The following settings are used to shear nuclear DNA to ~300 bp (range 100–500 bp): duty cycle, 10%; intensity, 5; cycles/burst, 200; and time, 20 s × 6. For small genomes, such as mtDNA, viral or plasmid DNA, the following settings should be used: duty cycle, 10%; intensity, 5; cycles/burst, 100; and time, 20 s × 3.  CRITICAL The water-bath temperature should be 4 °C, and the water bath should be degassed for 30 min before shearing.
CRITICAL The water-bath temperature should be 4 °C, and the water bath should be degassed for 30 min before shearing.
PROCEDURE
Sonication of DNA  TIMING 1 h
TIMING 1 h
 CRITICAL The following procedure is presented for a single sample; however, the protocol can be scaled up for an arbitrary number of DNA samples.
CRITICAL The following procedure is presented for a single sample; however, the protocol can be scaled up for an arbitrary number of DNA samples.
For each library, dilute the DNA into a final volume of 130 μl of TElow. For samples that do not use targeted capture, we typically start with 200–500 ng of DNA. For targeted capture, we typically start with 1–3 μg of DNA.
Transfer the DNA to a Covaris sonication tube and shear DNA using the settings outlined in Equipment Setup.
 CRITICAL STEP Ensure that there are no air bubbles in the bottom of the tube after loading the sample. In the case of air bubbles, gently tap or shake to bring the solution to the bottom of the well. In addition, the water bath temperature should be 4 °C, and the water bath should be degassed for 30 min before shearing.
CRITICAL STEP Ensure that there are no air bubbles in the bottom of the tube after loading the sample. In the case of air bubbles, gently tap or shake to bring the solution to the bottom of the well. In addition, the water bath temperature should be 4 °C, and the water bath should be degassed for 30 min before shearing.Vortex the room-temperature AMPure XP bead mixture to resuspend any magnetic particles that may have settled.
 CRITICAL STEP The beads must be at room temperature before use. A cold bead mixture significantly reduces yield.
CRITICAL STEP The beads must be at room temperature before use. A cold bead mixture significantly reduces yield.Split the 130 μl of sonicated DNA into 2 × 65-μl aliquots in 0.2-ml PCR tubes.
Add 130 μl of AMPure XP beads to each PCR tube (2:1 bead:sample ratio) and mix well by vortexing or by pipetting up and down. The 2:1 ratio is used to ensure the maximal retention of all DNA.
Incubate the mixture for 5 min at room temperature.
Place PCR tubes onto the DynaMag-96 side magnetic plate separator (or equivalent) for at least 2 min or until all the beads are out of solution. Visually confirm that the beads have moved to the side of the tubes and that the solution is clear.
Aspirate the supernatant from each tube and discard it. Keep the tubes on the magnetic plate separator.
Carefully dispense 200 μl of room-temperature 75% (vol/vol) ethanol to each PCR tube and incubate the tubes for 30 s at room temperature.
Aspirate out the ethanol and discard it. Repeat Steps 9 and 10 once more for a total of two washes.
Allow the beads to dry for 5 min at room temperature until there are no visible traces of ethanol.
 CRITICAL STEP Overdrying or underdrying the beads reduces yield.
CRITICAL STEP Overdrying or underdrying the beads reduces yield.Remove the PCR tubes from the magnetic plate separator and add 40 μl of ddH2O to only one of the two tubes of each sample and resuspend.
Carefully transfer the resuspended bead mixture to the second sample PCR tube and resuspend the beads. Let the bead mixture sit for at least 2 min.
Place the tube containing the combined sample back on the magnetic plate separator for 2 min or until the supernatant is clear. Transfer the supernatant to a new 0.2-ml PCR tube. Discard the beads.
 PAUSE POINT Samples can be stored at −20 °C for several weeks.
PAUSE POINT Samples can be stored at −20 °C for several weeks.
End repair of sonicated DNA and size selection  TIMING 1 h
TIMING 1 h
-
15Combine the components in the table below in a 0.2-ml PCR tube and mix them carefully by pipetting up and down. Incubate the mixture in a thermocycler for 30 min at 20 °C.
Reagent Volume (μl) Final concentration DNA from Step 14 40 Variable 10× NEBNext end-repair buffer 5 1× NEBNext end-repair enzyme Mix 5 N/A -
16
Add 35 μl of AMPure XP beads (0.7:1 bead:sample ratio) and incubate the tube at room temperature for 5 min. This selects against large (>500 bp), poorly sheared DNA fragments.
-
17
Place the tube containing the end-repaired DNA onto the magnetic plate separator for at least 2 min or until all the beads are out of solution, and then transfer the supernatant (should be 85 μl) to a new 0.2-ml PCR tube. Discard the beads.
-
18
Add 55 μl of AMPure XP beads to the sample and incubate the tube at room temperature for 5 min. Place the PCR tube onto the magnetic plate separator for at least 2 min or until all the beads are out of solution, and discard the 140-μl supernatant. Keep the beads.
-
19
Repeat the double 75% (vol/vol) ethanol wash outlined in Steps 9–11.
-
20
Remove the PCR tube from the magnet and add 42 μl of ddH2O to each sample and gently pipette up and down until the beads are resuspended. Let the bead mixture sit for at least 2 min.
-
21
Place the PCR tube back onto the magnetic plate separator for at least 2 min or until all the beads are out of solution, and then transfer the supernatant to a new 0.2-ml PCR tube.
 PAUSE POINT Samples can be stored at −20 °C for up to several weeks.
PAUSE POINT Samples can be stored at −20 °C for up to several weeks.
3′ A-tailing of blunt-ended DNA library  TIMING 1 h
TIMING 1 h
-
22Combine the components in the table below in a 0.2-ml PCR tube and mix them carefully by pipetting up and down. Incubate the mixture in a thermocycler for 30 min at 37 °C.
Reagent Volume (μl) Final concentration DNA from Step 21 42 Variable 10× NEBNext dA-tailing buffer 5 1× NEBNext Klenowexo- 3 -
23
Add 60 μl of AMPure XP beads (1.2:1 bead:sample ratio) and mix. The bead ratio selects against DNA fragments <150 bp in size. Incubate the tube for 5 min at room temperature.
-
24
Place A-tailed DNA sample onto the magnetic plate separator for at least 2 min or until all the beads are out of solution, and then remove and discard the supernatant.
-
25
Repeat the double 75% (vol/vol) ethanol wash outlined in Steps 9–11.
-
26
Remove the tube from the magnetic plate separator and resuspend the beads in 37 μl of ddH2O. Let the bead mixture sit for at least 2 min.
-
27
Place the sample back onto the magnetic plate separator for at least 2 min or until all the beads are out of solution, and then transfer the 37 μl of eluent to a new 0.2-ml PCR tube.
 PAUSE POINT Samples can be stored at −20 °C for several weeks.
PAUSE POINT Samples can be stored at −20 °C for several weeks. -
28
Remove 2 μl from the A-tailed library and quantify the amount of DNA using the high-sensitivity kits for either the Agilent TapeStation 2200 or Agilent Bioanalyzer 2100, according to the manufacturer’s instructions.? TROUBLESHOOTING
Ligation of DS adapters to sample DNA  TIMING 1 h
TIMING 1 h
-
29
By using the amount of sample DNA determined in Step 28, calculate the amount of DS adapters (synthesized as described in Boxes 1 and 2) that will be needed for a 20:1 molar excess relative to the total DNA concentration in the final ligation reaction. If necessary, dilute the adapters in ddH2O to an easily pipetteable volume.
-
30Mix the following components, in order, in a 0.2-ml PCR tube, and mix them carefully by pipetting up and down. As noted in Step 29, the volumes of adapters and water that are used will need to be adjusted on the basis of the DNA concentration of your sample, but the total volume should be 60 μl.
Reagent Volume (μl) Final concentration DNA from Step 27 35 Variable 10× Ultrapure ligation buffer 6 1× Duplex Sequencing adapters (50 μM) Variable Variable ddH2O Variable N/A Ultrapure T4 ligase (600 U/μl) 6 60 U/μl Total volume 60 -
31
Incubate the tube for 15 min at 25 °C in a thermocycler (no heated lid).
-
32
Add 60 μl of AMPure beads (1.2× bead:sample ratio) and mix. This ratio selects against DNA fragments <150 bp in size. Incubate the tube for 5 min at room temperature.
-
33
Place the PCR tube containing the ligated sample onto the magnetic plate separator for at least 2 min or until all the beads are out of solution. Remove and discard the supernatant.
-
34
Repeat the double 75% (vol/vol) ethanol washes outlined in Steps 9–11, with the exception that the beads be fully resuspended in the ethanol by vortexing before being placed on the magnetic separator. Dry the beads for 5 min at 37 °C in a heat block with the tube cap opened.
 CRITICAL STEP Resuspending the magnetic beads during the ethanol washes is essential for removing adapter dimers that form during ligation. Failure to do so will adversely affect the downstream PCR steps. Removal of any residual ethanol by drying at 37 °C is also important, as ethanol inhibits PCR.
CRITICAL STEP Resuspending the magnetic beads during the ethanol washes is essential for removing adapter dimers that form during ligation. Failure to do so will adversely affect the downstream PCR steps. Removal of any residual ethanol by drying at 37 °C is also important, as ethanol inhibits PCR. -
35
Remove samples from the magnetic separator and add 30 μl of TElow to each sample. After 2 min, place the samples back on the magnetic separator for 2 min or until the supernatant is clear. Transfer the 30 μl of supernatant to a new 0.2-ml PCR tube.
 PAUSE POINT Samples can be stored at −20 °C for several weeks.
PAUSE POINT Samples can be stored at −20 °C for several weeks.
PCR amplification  TIMING 1 h
TIMING 1 h
-
36
Quantify the molar amount of adapter-ligated DNA library from Step 35 on an Agilent TapeStation 2200 or Bioanalyzer 2100. The ligation reaction can result in multiple peaks; we quantify all DNA seen between 200 and 900 bp (Fig. 6c).? TROUBLESHOOTING
-
37Determine the number of attomoles of input DNA required for PCR on the basis of target size and lane fraction. For noncapture experiments, we use 40 amol of input for every 8 million paired-end reads. For capture of mtDNA, we use ~2 fmol of total DNA for the same number of reads. For nuclear DNA capture (20 kb target size), we use ~600 fmol of total DNA per 8 million reads. The formula for estimating the total DNA input is
where m is the desired amount of target DNA in attomoles of fragments, G is the size of total genome in base pairs, T is the size of the target DNA in base pairs and N is the number of copies of the target per copy of the nontarget portion of the genome. (Refer to the INTRODUCTION and Table 2 for further details.) -
38Generate amplified tag families by qPCR. To generate tag families for small homogeneous genomes such as mtDNA or plasmids that do not require targeted capture, follow option A. To generate duplicate families of genomic targets that require targeted capture, follow option B.
- Generation of duplicate families without targeted capture
- Mix the components in the table below in a qPCR-compatible PCR tube, and mix carefully by pipetting up and down. The volumes of DNA and water that are used will need to be adjusted on the basis of the DNA concentration and the desired input amount of the sample, but the total volume should be 50 μl. See Table 3 for MWS13 and MWS21 primer sequences.
Reagent Volume (μl) Final concentration ddH2O Variable N/A 5× KAPA fidelity buffer 10 1× 10 mM dNTP mix 1.5 0.3 mM 20 μM Primer MWS13 1 0.4 μM 20 μM Primer MWS21 1 0.4 μM 12.5× SYBR Green 1 0.25× DNA (From Step 37) Variable Variable KAPA HiFi DNA polymerase (1 U/μl) 1 0.02 U/μl Total volume 50 - Incubate the mixture in a qPCR thermocycler as follows:
Cycle Denature Anneal Extend 1 95 °C for 4 min — — 2-variable 98 °C for 20 s 60 °C for 20 s 72 °C for 15 s  CRITICAL STEP The number of PCR cycles is a crucial variable; a difference of a single cycle can result in nonspecific, higher-molecular-weight products appearing (Fig. 6b). See ‘PCR Amplification’ in the INTRODUCTION for details on determining the number of PCR cycles.
CRITICAL STEP The number of PCR cycles is a crucial variable; a difference of a single cycle can result in nonspecific, higher-molecular-weight products appearing (Fig. 6b). See ‘PCR Amplification’ in the INTRODUCTION for details on determining the number of PCR cycles.
- Generation of duplicate families for targeted DNA capture
- Dilute the DNA from Step 37 to a final volume of 30 μl with ddH2O (see ‘PCR Amplification’ in the INTRODUCTION and Table 2). We have found that splitting the input DNA into three equal aliquots and performing a separate PCR on each sample aliquot provides enough PCR product for targeted capture (each PCR will contain one-third of the total input DNA).
- For each PCR, mix the components in the tables below in a qPCR-compatible PCR tube and mix carefully by pipetting up and down (see Table 3 for MWS13 and MWS20 primer sequences):
Reagent Volume (μl) Final concentration ddH2O 24.5 N/A 5× KAPA fidelity buffer 10 1× 10 mM dNTP mix 1.5 0.3 mM 20 μM primer MWS13 1 0.4 μM 20 μM primer MWS20 1 0.4 μM 12.5× SYBR Green 1 0.25× DNA (from Step 38B(i)) 10 Variable KAPA HiFi DNA polymerase (1 U/μl) 1 0.02 U/μl  CRITICAL STEP It is important that the nonindexed primer MWS20 be used and not MWS21.
CRITICAL STEP It is important that the nonindexed primer MWS20 be used and not MWS21. - Incubate the mixture in a qPCR thermocycler as follows:
Cycle Denature Anneal Extend 1 95 °C for 4 min — — 2-Variable 98 °C for 20 s 60 °C for 20 s 72 °C for 15 s  CRITICAL STEP The number of PCR cycles is a crucial variable; a difference of a single cycle can result in nonspecific, higher-molecular-weight products appearing (Fig. 6b). See ‘PCR Amplification’ in the INTRODUCTION for details on determining the number of PCR cycles.
CRITICAL STEP The number of PCR cycles is a crucial variable; a difference of a single cycle can result in nonspecific, higher-molecular-weight products appearing (Fig. 6b). See ‘PCR Amplification’ in the INTRODUCTION for details on determining the number of PCR cycles.
-
39
Transfer and pool all the PCRs from a single sample into a single 1.7-ml microcentrifuge tube.
-
40
Add 1.0 volumes of AMPure XP beads to the pooled PCRs (typically 50 μl of beads for every 50 μl of PCR). The bead ratio selects against DNA fragments <200 bp in size. Incubate the mixture for 5 min at room temperature.
-
41
Place the PCR sample onto a magnetic separator for at least 2 min or until all the beads are out of solution, and then remove and discard the supernatant.
-
42
Remove the beads from the magnetic plate and resuspend them in 200 μl of room-temperature 75% (vol/vol) ethanol by vortexing.
 CRITICAL STEP Failure to completely resuspend the beads during this step can lead to the retention of nonspecific PCR products (Supplementary Fig. 6).
CRITICAL STEP Failure to completely resuspend the beads during this step can lead to the retention of nonspecific PCR products (Supplementary Fig. 6). -
43
Place the microcentrifuge tube onto a magnetic separator for at least 2 min or until all the beads are out of solution, and then remove and discard the supernatant.
-
44
Repeat Steps 42 and 43. Let the beads dry for 5 min.
-
45
Remove the microcentrifuge tube from the magnetic plate separator and resuspend the beads in 20 μl of ddH2O. Let the bead mixture sit for at least 2 min.
-
46
Place the sample onto the magnetic separator for at least 2 min or until all the beads are out of solution, and then transfer the 20 μl of eluent to a new 0.2-ml PCR tube.
 PAUSE POINT Samples can be stored at −20 °C for several weeks.
PAUSE POINT Samples can be stored at −20 °C for several weeks. -
47
Quantify the final bead-purified PCR products between 200 and 900 bp using an Agilent TapeStation 2200 or Bioanalyzer 2100. If targeted capture is being performed, continue to Step 48; otherwise, the sample can be submitted for sequencing at this point.? TROUBLESHOOTING
(Optional) Targeted DNA capture  TIMING 24 h
TIMING 24 h
-
48
Remove 200 ng of purified PCR product from Step 47 and lyophilize it completely using a vacuum centrifuge. Resuspend it in 1.7 μl of ddH2O. ? TROUBLESHOOTING
-
49
Perform targeted DNA capture according to the Agilent SureSelectXT instructions (version 1.6). Alternatively, we routinely perform the targeted capture at 0.25× volume reduction, relative to those recommended by the manufacturer. The lower volume capture greatly reduces cost, with no adverse effects noted. The 0.25× capture follows the standard Agilent SureSelectXT protocol with all reagent volumes reduced by three-quarters. The final capture DNA should be eluted in 30 μl of ddH2O.
-
50PCR-amplify the 30 μl of captured sample by mixing the components in the table below in a qPCR-compatible PCR tube and mix carefully by pipetting up and down (see Table 3 for MWS13 and MWS21 primer sequences):
Reagent Volume (μl) Final concentration ddH2O 5 N/A 5× KAPA fidelity buffer 10 1× 10 mM dNTP mix 1 0.2 mM 20 μM primer MWS13 1 0.4 μM 20 μM primer MWS21 1 0.4 μM 12.5× SYBR Green 1 0.25× Captured DNA (from Step 49) 30 Variable KAPA HiFi DNA polymerase (1 U/μl) 1 0.02 U/μl  CRITICAL STEP For DNA prepared using target capture, it is essential to use all the DNA from the targeted capture to maintain sample diversity.
CRITICAL STEP For DNA prepared using target capture, it is essential to use all the DNA from the targeted capture to maintain sample diversity. -
51Incubate the mixture in a qPCR thermocycler as follows:
Cycle Denature Anneal Extend 1 95 °C for 4 min — — 2-30 98 °C for 20 s 60 °C for 20 s 72 °C for 15 s  CRITICAL STEP The number of PCR cycles is a crucial variable; a difference of a single cycle can result in nonspecific, higher-molecular-weight products appearing. We monitor the reaction by the SYBR Green signal, and we remove the tube one or two cycles before the signal plateaus. The SYBR Green signal typically begins to plateau between cycles 15 and 22.
CRITICAL STEP The number of PCR cycles is a crucial variable; a difference of a single cycle can result in nonspecific, higher-molecular-weight products appearing. We monitor the reaction by the SYBR Green signal, and we remove the tube one or two cycles before the signal plateaus. The SYBR Green signal typically begins to plateau between cycles 15 and 22. -
52
Purify each completed PCR by repeating Steps 40–44.
 PAUSE POINT Samples can be stored at −20 °C for several weeks.
PAUSE POINT Samples can be stored at −20 °C for several weeks. -
53
Quantify the final bead-purified PCR products between 200 and 900 bp using an Agilent TapeStation 2200 or Bioanalyzer 2100 and submit for sequencing.? TROUBLESHOOTING
Bioinformatic processing: prepare a reference genome for use with BWA and SAMtools  TIMING variable
TIMING variable
-
54
First, download and decompress the genome of interest. For the human nuclear and mitochondrial genomes, we use v37 of the human reference genome from the 1,000 Genomes Project. This reference genome can be obtained at ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/technical/reference/human_g1k_v37.fasta.gz. Alternatively, the hg19 reference genome from the University of California, Santa Cruz (UCSC) Genome Browser can be used.
-
55
The reference genome must be indexed before being used with BWA. Follow the instructions on the website (http://bio-bwa.sourceforge.net/bwa.shtml) using the recommended options relevant to the genome of interest.
 CRITICAL STEP The bioinformatics steps outlined in this protocol are specific to BWA. We have not used other sequencing aligners, and their compatibility with the downstream bioinformatics steps is not known.
CRITICAL STEP The bioinformatics steps outlined in this protocol are specific to BWA. We have not used other sequencing aligners, and their compatibility with the downstream bioinformatics steps is not known.
Bioinformatic processing: parsing and filtering duplex tags  TIMING variable
TIMING variable
-
56
Concatenate the 12-nt tag sequences from the paired reads and evaluate for tag quality using ‘tag_to_header.py’ using the following command:
 This command results in two fastq files (one for each read) that are ready to be aligned. A flowchart schematic can be found in Supplementary Figure 1.
This command results in two fastq files (one for each read) that are ready to be aligned. A flowchart schematic can be found in Supplementary Figure 1. CRITICAL STEP If samples are multiplexed within the same sequencing lane, then these samples should be sorted into separate files based on their index sequence before tag parsing.
CRITICAL STEP If samples are multiplexed within the same sequencing lane, then these samples should be sorted into separate files based on their index sequence before tag parsing. -
57
Align each read to the reference genome:

-
58
Make a single paired-end .sam file:

-
59
Convert to .bam format and sort by position:
Bioinformatics processing: making SSCSs  TIMING variable
TIMING variable
-
60
Run the Python program ‘ConsensusMaker.py’ to collapse PCR duplicates into SSCS:

A detailed flowchart schematic of the program can be found in Supplementary Figure 2.
-
61
Sort the SSCS reads:
Bioinformatics processing: making DCSs  TIMING variable
TIMING variable
-
62
Construct DCSs from SSCSs using ‘DuplexMaker.py’.

A detailed flowchart schematic of the program can be found in Supplementary Figure 3.
-
63
Align each DCS .fastq to the reference genome:

-
64
Make a paired-end .sam file for the DCS data:

-
65
Convert to .bam format and sort by position:

Bioinformatics processing: prepare files for analysis  TIMING variable
TIMING variable
-
66
Index the final sorted DCS .bam file:
-
67
Filter out unmapped reads from the final DCS .bam file:
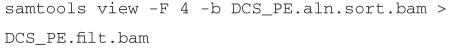
-
68
Add readgroups field to the header of the final DCS .bam file with Picard to allow for compatibility with the GATK using Picard tools. An example command is provided below:
-
69
Index the final sorted DCS .bam file:
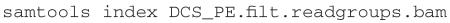
-
70
Perform local re-alignment of the reads using GATK. First identify the genome targets for local re-alignment. An example command is as follows:

This command is followed by the actual local re-alignment. An example command is as follows:

 CRITICAL STEP Alignment increases the occurrence of false mutations that occur at the 3′ and 5′ ends of the reads, so this step is highly recommended. In addition, local re-alignment must be done after the last alignment step.
CRITICAL STEP Alignment increases the occurrence of false mutations that occur at the 3′ and 5′ ends of the reads, so this step is highly recommended. In addition, local re-alignment must be done after the last alignment step. -
71
Perform end-trimming of DCS reads. An example command that trims five bases from both the 3′ and 5′ ends of each read is provided:

 CRITICAL STEP Failure to clip the ends of reads increases the occurrence of false positives that occur during the enzymatic steps used during sequencing library preparation.
CRITICAL STEP Failure to clip the ends of reads increases the occurrence of false positives that occur during the enzymatic steps used during sequencing library preparation.
Bioinformatics analysis: quality metrics and basic mutation analysis  TIMING variable
TIMING variable
 CRITICAL The processed DCS .bam files can be used in any downstream analysis that is desired. We present the basic data analysis protocol that we currently use.
CRITICAL The processed DCS .bam files can be used in any downstream analysis that is desired. We present the basic data analysis protocol that we currently use.
-
72
Plot the tag family size distribution (see Fig. 4 as an example). The information for this plot is found in the ‘PE_reads. tagstats’ file created in Step 60. Plot the family size (column 1) on the x axis and the fraction of total reads (column 3) on the y axis.? TROUBLESHOOTING
-
73
Make a pileup file from the final DCS reads using the following example command:

-
74
Count the number of unique mutations present in the final DCS sequences and calculate their frequencies:

The optional argument -u invokes unique mutation counting; if it is left out, all mutations are counted, instead of only counting each type of mutation at each position once. The argument -d specifies that a minimum depth of 100 is required to score a position, and -C indicates that only mutations present at a clonality of <1% are counted. The output file lists the number and frequency of mutations with Wilson confidence intervals for each type of mutation, as well as total mutation frequency.
-
75
Locate the genomic position of each mutation:

This program outputs a tab-delimited file containing the following information for each genomic position in the reference genome: chromosome, position, wild-type base identity, depth, total number of mutations observed, number of nonreference Ts, number of nonreference Cs, number of nonreference Gs, number of nonreference As, number of nonreference insertions, and number of nonreference deletions.
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 4.
TABLE 4.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
|
Box 1, steps 3 and 6 |
Sample becomes cloudy upon addition of ethanol |
Precipitation of enzymes and buffer salts |
Precipitate does not affect downstream steps or library preparation. Continue with the protocol |
|
Box 1, Step 12 |
Adapters are not properly synthesized (bands in lane 4 of Fig. 3a do not match) |
Adapters are not fully extended (Fig. 3a, lane 2 does not match) |
Replace reagents, re-anneal oligonucleotides and perform the extension reaction again |
| Adapters are incompletely cut (Fig. 2a, lane 3 does not match) |
Add an additional 2 μl of HpyCH4III and incubate overnight at 37 °C. If necessary, replace enzyme and start the synthesis over again |
||
| Restriction fragment is still present after final ethanol precipitation (Fig. 3a, band f still present in lane 4) |
Perform ethanol precipitation again using the protocol outlined in step 5 of Box 1 |
||
| 28, 36 | No DNA is detectable during quantification |
Ethanol concentration used during the washes is too low |
Remake the 75% (vol/vol) ethanol and start over |
| Not enough DNA 1s used during library preparation |
Repeat the library preparation with a larger amount of input DNA |
||
| 47, 53 | Dimerized adapter or nonspecific PCR products are present after cleaning with AMPure XP beads |
Adapter dimers are not efficiently removed |
Bring the volume up to 50 μl with ddH2O and repeat the bead clean-up (Steps 40-44; supplementary Fig. 6b) |
| No DNA is detectable during quantitation or large-molecular-weight products are present |
DNA concatemerization owing to too many PCR cycles |
Run a titration of the PCR cycle number. Quantify the unpurified PCR on a TapeStation or Bioanalyzer to confirm that the expected product is present (see Fig. 6a for an example). Rerun the PCR steps as outlined in the PROCEDURE and remove the samples at the determined cycle |
|
| Ethanol concentration used during the washes is too low |
Remake the 75% (vol/vol) ethanol and repeat PCR | ||
| 48 | Not enough DNA is present to perform targeted capture |
Not enough DNA is produced during PCR amplification |
Perform additional PCRs under the same conditions to obtain additional DNA, pool all the reactions, and purify the DNA (Steps 38B-47) |
| 72 | Not enough depth is obtained and the peak family size is too small |
Too much DNA is used during the initial PCR or too small a lane fraction was used |
Sequence the sample again (do not re-do the PCR) and pool the raw sequencing reads prior to data analysis. See Supplementary Table 2 to determine the lane fraction needed to obtain a peak family size of six |
| Not enough depth is obtained and the peak family size is too large |
Too little DNA is used during the initial PCR |
Depth is directly proportional to the number of DCS reads, which is directly proportional to the number of DNA molecules used as input in the PCR. If the number of molecules was too low, the depth will be too low. Further resequencing will only increase the family size, not the depth. Divide your peak family size by six, which is the optimal peak family size. Then increase the DNA amount in the PCR proportionally. For example, if the obtained peak family size is 24, 24/6 = 4, 4× more DNA is needed in the PCR |
 TIMING
TIMING
Steps 1–14, sonication of DNA: 1 h
Steps 15–21, end repair of sonicated DNA and size selection: 1 h
Steps 22–28, 3′ dA-tailing of end-repaired DNA: 1 h
Steps 29–35, ligation of DS adapters to sample DNA: 1 h
Steps 36–47, PCR amplification: 1 h
Steps 48–53 (optional), targeted DNA capture: 24 h
Steps 54–75, bioinformatic processing: variable and dependent on computational resources
Box 1, synthesis of DS adapters: 2 d
Box 2, optional quality control steps for adapter synthesis using radiolabeled adapters and PAGE: 1 d
ANTICIPATED RESULTS
We have applied DS to a number of biological systems, including the M13mp2 phagemid19, human mtDNA from frozen brain tissue33 and human nuclear DNA from both frozen and formalin-fixed samples (M.J.P., E.J.F. and L.A.L.; unpublished results). We consistently observe a >50,000-fold reduction in the apparent mutation frequency in these samples, relative to conventional NGS methods. We have found that SSCS formation is capable of removing almost all sequencer-derived false positives, which account for >99% of all artifacts. However, this step is unable to remove artifacts arising from first-round PCR errors that have been propagated to all tag family members. The mutational spectrum of the SSCS typically shows a high frequency of G®T mutations, which is the result of fixation of oxidative damage occurring during the DNA purification and processing steps19. Formation of the DCS reads consistently removes these artifacts, as well as those from other damage events, and it results in a further 90–99+% reduction in mutational artifacts.
Supplementary Material
ACKNOWLEDGMENTS
We thank D. Crispin and A. Lawrence for testing the protocol as written and for helping to clarify several important steps of the protocol. We also thank A. Herr for helpful comments and discussion. Support for this research was provided by the following US National Institutes of Health grants from the National Institute on Aging (NIA) and the National Cancer Institute (NCI): NIA P01-AG001751, NCI P01-CA77852, NCI R01-CA160674 and NCI R01-CA102029 to L.A.L. S.R.K. was further supported by the Genetic Approaches to Aging Training grant (NIA T32-AG000057).
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS S.R.K., M.W.S., J.J.S. and E.J.F. developed the original protocol. S.R.K., M.W.S. and B.F.K. developed the data analysis software. S.R.K., M.W.S., E.J.F., M.J.P., E.H.A., J.-C.S., K.J.K. and R.-A.R. optimized the protocol. S.R.K., M.W.S., J.J.S., B.F.K. and L.A.L. wrote the paper.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Glenn TC. Field guide to next-generation DNA sequencers. Mol. Ecol. Resour. 2011;11:759–769. doi: 10.1111/j.1755-0998.2011.03024.x. [DOI] [PubMed] [Google Scholar]
- 2.Loeb LA, Springgate CF, Battula N. Errors in DNA replication as basis of malignant change. Cancer Res. 1974;34:2311–2321. [PubMed] [Google Scholar]
- 3.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bolli N, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014;5:2997. doi: 10.1038/ncomms3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lawrence MS, et al. Mutational heterogeneity in cancer and the for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kreso A, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science. 2013;339:543–548. doi: 10.1126/science.1227670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson BE, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343:189–193. doi: 10.1126/science.1239947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmitt MW, Prindle MJ, Loeb LA. Implications of genetic heterogeneity in cancer. Ann. NY Acad. Sci. 2012;1267:110–116. doi: 10.1111/j.1749-6632.2012.06590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Z, et al. Patterns of diversifying selection in the phytotoxin-like scr74 gene family of Phytophthora infestans. Mol. Biol. Evol. 2005;22:659–672. doi: 10.1093/molbev/msi049. [DOI] [PubMed] [Google Scholar]
- 11.Srivatsan A, et al. High-precision, whole-genome sequencing of laboratory strains facilitates genetic studies. PLoS Genet. 2008;4:e1000139. doi: 10.1371/journal.pgen.1000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holt KE, et al. High-throughput sequencing provides insights into genome variation and evolution in Salmonella typhi. Nat. Genet. 2008;40:987–993. doi: 10.1038/ng.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loh E, Salk JJ, Loeb LA. Optimization of DNA polymerase mutation rates during bacterial evolution. Proc. Natl. Acad. Sci. USA. 2010;107:1154–1159. doi: 10.1073/pnas.0912451107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Budowle B, van Daal A. Extracting evidence from forensic DNA analyses: future molecular biology directions. Biotechniques. 2009;46:339–340. 342–350. doi: 10.2144/000113136. [DOI] [PubMed] [Google Scholar]
- 15.Hofreiter M, Jaenicke V, Serre D, von Haeseler A, Paabo S. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. 2001;29:4793–4799. doi: 10.1093/nar/29.23.4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knapp M, Hofreiter M. Next-generation sequencing of ancient DNA: requirements, strategies and perspectives. Genes. 2010;1:227–243. doi: 10.3390/genes1020227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carlson CA, et al. Decoding cell lineage from acquired mutations using arbitrary deep sequencing. Nat. Methods. 2012;9:78–80. doi: 10.1038/nmeth.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Besaratinia A, et al. A high-throughput next-generation sequencing-based method for detecting the mutational fingerprint of carcinogens. Nucleic Acids Res. 2012;40:e116. doi: 10.1093/nar/gks610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmitt MW, et al. Detection of ultra-rare mutations by next-generation sequencing. Proc Natl. Acad. Sci. USA. 2012;109:14508–14513. doi: 10.1073/pnas.1208715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA. 2011;108:9530–9535. doi: 10.1073/pnas.1105422108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hiatt JB, Patwardhan RP, Turner EH, Lee C, Shendure J. Parallel, tag-directed assembly of locally derived short sequence reads. Nat. Methods. 2010;7:119–122. doi: 10.1038/nmeth.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCloskey ML, Stoger R, Hansen RS, Laird CD. Encoding PCR products with batch-stamps and barcodes. Biochem. Genet. 2007;45:761–767. doi: 10.1007/s10528-007-9114-x. [DOI] [PubMed] [Google Scholar]
- 23.Jabara CB, Jones CD, Roach J, Anderson JA, Swanstrom R. Accurate sampling and deep sequencing of the HIV-1 protease gene using a primer ID. Proc. Natl. Acad. Sci. USA. 2011;108:20166–20171. doi: 10.1073/pnas.1110064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kivioja T, et al. Counting absolute numbers of molecules using unique molecular identifiers. Nat. Methods. 2012;9:72–74. doi: 10.1038/nmeth.1778. [DOI] [PubMed] [Google Scholar]
- 25.Casbon JA, Osborne RJ, Brenner S, Lichtenstein CP. A method for counting PCR template molecules with application to next-generation sequencing. Nucleic Acids Res. 2011;39:e81. doi: 10.1093/nar/gkr217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shiroguchi K, Jia TZ, Sims PA, Xie XS. Digital RNA sequencing minimizes sequence-dependent bias and amplification noise with optimized single-molecule barcodes. Proc. Natl. Acad. Sci. USA. 2012;109:1347–1352. doi: 10.1073/pnas.1118018109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zong C, Lu S, Chapman AR, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science. 2012;338:1622–1626. doi: 10.1126/science.1229164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Fan HC, Behr B, Quake SR. Genome-wide single-cell analysis of recombination activity and de novo mutation rates in human sperm. Cell. 2012;150:402–412. doi: 10.1016/j.cell.2012.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, et al. Clonal evolution in breast cancer revealed by single-nucleus genome sequencing. Nature. 2014;512:155–160. doi: 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Acevedo A, Brodsky L, Andino R. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature. 2014;505:686–690. doi: 10.1038/nature12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lou DI, et al. High-throughput DNA sequencing errors are reduced by orders of magnitude using circle sequencing. Proc. Natl. Acad. Sci. USA. 2013;110:19872–19877. doi: 10.1073/pnas.1319590110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diegoli TM, Farr M, Cromartie C, Coble MD, Bille TW. An optimized protocol for forensic application of the PreCR repair mix to multiplex STR amplification of UV-damaged DNA. Forensic Sci. Int. Genet. 2012;6:498–503. doi: 10.1016/j.fsigen.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 33.Kennedy SR, Salk JJ, Schmitt MW, Loeb LA. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013;9:e1003794. doi: 10.1371/journal.pgen.1003794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yost SE, et al. Identification of high-confidence somatic mutations in whole genome sequence of formalin-fixed breast cancer specimens. Nucleic Acids Res. 2012;40:e107. doi: 10.1093/nar/gks299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kerick M, et al. Targeted high throughput sequencing in clinical cancer settings: formaldehyde fixed-paraffin embedded (FFPE) tumor tissues, input amount and tumor heterogeneity. BMC Med. Genomics. 2011;4:68. doi: 10.1186/1755-8794-4-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spencer DH, et al. Comparison of clinical targeted next-generation sequence data from formalin-fixed and fresh-frozen tissue specimens. J. Mol. Diagn. 2013;15:623–633. doi: 10.1016/j.jmoldx.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Durbin R. Fast and accurate short-read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKenna A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.