

Abstract
Immune-mediated damage to medium-sized arteries results in wall remodeling with intimal hyperplasia, luminal stenosis and tissue ischemia. In the case of the aorta, vasculitis may result in dissection, aneurysm or rupture. The response-to-injury program of the blood vessel is a concerted action between the immune system and wall-resident cells, involving the release of growth and angiogenic factors from macrophages and giant cells and the migration and hyperproliferation of vascular smooth muscle cells. Innate immune cells, specifically, dendritic cells (DC) positioned in the vessel wall, have been implicated in the earliest steps of vasculitis. Pathogen-derived molecular patterns are capable of activating vascular DC and initiating adaptive immune responses. The pattern of the emerging vessel wall inflammation is ultimately determined by the initial insult. Ligands to toll-like receptor (TLR) 4, such as lipopolysaccharides, facilitate the recruitment of CD4 T cells that invade deep into the wall and distribute in a panarteritic pattern. Conversely, ligands for TLR5 condition vascular DC to support perivasculitic infiltrates. In essence, both innate and adaptive immune reactions collaborate to render the arterial wall susceptible to inflammatory damage. Unique features of the tissue microenvironment, including specialized DC, shape the course of the inflammatory response. Differences in vascular damage pattern encountered in different patients may relate to distinct instigators of vasculitis.
Arteries and the immune system—a two-way relationship
Arteries serve a vital function as the conduit for blood, delivering oxygen and nutrients to all tissues. They have a critical role in regulating blood pressure. Life is not sustainable without functional arteries and arterial disease, mostly in the form of atherosclerosis, is now the major cause of mortality in Western societies. However, recent data have emphasized that the contribution of arteries towards keeping the body functioning is even more essential than previously considered, and far exceeds blood transport and blood pressure regulation. In humans, medium-sized and large arteries are emerging as critical components of the body’s defense system [1], redefining our understanding of how the arterial network interacts with the immune system and how arteries become a target of immune-mediated disease.
Besides building a barrier between circulating immune cells and the tissue space, a function mainly achieved by endothelial cells (EC), macrovessels possess indigenous populations of dendritic cells (DC) [1–3], which are involved in immune surveillance and regulation of adaptive immune responses. Equipped with the ability to sense danger signals via toll-like receptors (TLR), these DC enable arteries to regulate localized and possibly systemic inflammatory responses [4]. This functional ability is a critical pathogenic element in large-vessel vasculitides, such as giant cell arteritis (GCA) and Takayasu arteritis, in which the site of inflammation is the arterial wall itself. In the emerging disease model for GCA, innate immune recognition events facilitated by wallembedded immune sentinels are considered to be crucially involved in the initiation of vasculitis [5] (Figure 1).
Figure 1.
Innate and adaptive immune responses lead to vascular damage in GCA. Tissue injury of the vascular wall in GCA is the cumulative effect of a cascade of immune events. Early steps relate to the functioning of the innate immune system with danger signals triggering artery-intrinsic DC. Induction of adaptive immune responses creates granulomatous infiltrates with tissue-injurious potential. The vascular wall responds with a remodeling program that is maladaptive and endangers the supply of blood and nutrients to dependent organs.
An indispensible contribution of the vessel wall provides a framework for the unique tropism for medium-sized and large vessels in GCA. Emerging data suggest that factors shaping the three-dimensional microenvironment in the vessel wall regulate pathogenic immune responses and may determine the outcome of inflammatory reactions placed within the layers of the vascular wall [6]. In GCA, the arterial cellular residents collaborate with recruited cellular components in the execution of diversified effector functions that are supported by the three-dimensional platform of the extracellular compartment. The ensuing tissue remodeling is thus the result of a two-pronged attack driven by specialized subsets of immune cells in combination with a destructive self-inflicted response to injury by the macrovessel (Figure 1).
Hierarchical studies have positioned the vascular DC at the helm of the immunological apparatus navigating the inflammatory response in diseased vessels. In normal arteries, the gatekeeper function of the vascular DC facilitates a commensal relationship with the vessel via the mitigation of potentially destructive immune responses. Under physiological conditions, the vessel wall is an immunoprivileged site, with local DC actively guarding this privilege. In experimental systems, and likely also in the patient, this privilege can be breached by innate stimuli that override the tolerogenic mechanisms of the DC [5]. This breakdown in self-tolerance leads to the propagation of a chronic inflammatory response distinguished by CD4 T-cell influx, formation of granulomatous infiltrates, neovascularization, intimal hyperplasia and luminal occlusion [7]. This review summarizes the known mechanisms underlying disease pathogenesis in GCA with particular emphasis on the role of the reciprocal interactions between the cellular components of the immune system and the structural elements that constitute the vessel wall (Figure 1).
TLRs in healthy and inflamed arteries
Indigenous DC integrated into the vessel wall are typically quiescent, which may be part of their tolerogenic capacity [3]. Losing the immature state is now considered an enabling step in the early stages of vessel wall inflammation, and once vasculitic infiltrates are established, DC continue to express a highly activated phenotype-releazing chemokines and cytokines [5]. In studies, where wall-residing DC are depleted the disease process is disrupted [5]. It seems that the integrity of the vessel and its protection from inflammation is ultimately regulated by the innate immune system, Utilizing TLR, vascular DC serve as sentinels and monitor events occurring in the walls of macroarteries. The family of TLRs confers dual functionality on DC by providing the thermostat that facilitates sensing of their surroundings and either allowing for the consequent maintenance of equilibrium under tolerizing conditions or initiating an inflammatory cascade.
Among the major distinguishing features of the inflammatory vasculopathies is their distinct predilection for the colonization of unique vascular territories. GCA is no different in this regard and selectively targets extracranial branches of the aorta with manifestations in the head, neck and upper extremities [8]. These observations led to the recent investigation and characterization of six human vascular beds in order to gain insight into the tissue tropisms of these inflammatory diseases [1]. While TLRs are frequently expressed on DC, they are also found in monocytes, macrophages, and B cells.
The results of these profile studies, however, indicated that the functional TLR in the vascular tissues are essentially restricted to the CD11cþ wall-residing cells. All arteries examined abundantly expressed transcripts for most of the nine TLR tested. All arteries contained sequences specific for TLRs 2 and 4, and overall TLR-binding bacterial products were favored. The most surprizing finding was that the combination of TLR assigned a unique fingerprint to each of the six different vascular regions. The DC also showed differential localization, as an adventitial network was detected in all six arteries, while a second intimal DC network was identified only in the two largest arteries, the carotid and the aorta. The distinct TLR profiles not only conferred unique immunologic identities to each of the vascular beds, but also correlated with their functional heterogeneity as demonstrated by differential responses to stimulation with cognate ligands for TLRs 4 and 5 in arteries displaying disparate profiles for these two receptors.
The classic paradigm for DC maturation describes a population that resides in peripheral tissues as immature cells that have a characteristically high capacity for antigen capture via macropinocytosis, receptor-mediated endocytosis and phagocytosis. These cells express low levels of histocompatibility complex (MHC) and costimulatory molecules, and antigen-induced maturation drives the phenotypic and functional changes that endow the DC with the proficiency to bridge innate and adaptive immune responses. These events describe a typical antigenic response that is instigated by pathogen sensing and culminates in the initiation of an orchestrated inflammatory response. All available data indicate that this sequence of events is still maintained in GCA.
The localization of DC in peripheral tissues is widespread but not ubiquitous, and they are strategically located in certain tissues that include skin, the gut and the respiratory mucosa, all of which have immediate and frontline access to pathogens in the external environment. Their presence in artery walls, therefore, is novel. Early evidence that arteries may contain professional antigen-presenting cell (APC) came from histological studies identifying an S100þ cell population in human aor tic and carotid arteries [2].
Initially, these DC were associated with atherosclerotic disease, and it was not clear whether they were indigenous residents or responded to early tissue injury. Architectural analysis of the healthy temporal artery identified an indigenous resting population of DC localized at the media-adventitia border in the vessel wall. Further characterization studies showed that in temporal arteries of GCA patients this resting, immature population of DC is replaced by an activated, mature one that begins to infiltrate the media [5]. Triggering of TLR4 with its cognate ligand, lipopolysaccharide (LPS), is a well-known and potent maturation stimulus for DC and in vivo studies utilizing a human-severe combined immunodeficiency (SCID) mouse chimera have shown that TLR4 stimulation is sufficient to trigger a chain of events leading to recapitulation of the key features of vasculopathy observed in GCA. In situ triggering of TLR4, which is found on the resident population of DC in the vessel wall, induces DC differentiation, with the upregulation of MHC, CD83 and CD86, which in turn renders them capable of stimulating the T cells that mediate disease.
Vascular DCs as tolerance inducers
As medium-sized and large arteries are not exposed to the external world, the question arises about what benefit the host derives from sensing immune danger signals in the arterial wall. In essence, networks of DC in the vascular wall could potentially increase the risk of attracting tissue-damaging immune responses given their potency in the initiation of adaptive immune responses. The versatility of the DC, however, is likely the key feature that explains what appears to be a symbiotic relationship with the vessel wall, as it is equally competent in the mediation of peripheral tolerance. The execution of this latter function is essential for the regulation of self-reactivity outside of the thymus [9], and in healthy arteries the DC likely comprise a subset of steady state DC that patrol this peripheral checkpoint and defend the vessel against any potential attack by autoreactive T cells.
Given that the artery acts as a conduit for the blood, which serves as a major pipeline for the dissemination of both self- and foreign-antigens, the location of the DC in the proximal adventitia means that these resident cells are optimally positioned for sampling their environment, via the vasa vasorum, which are restricted to the adventitial layer of the vessel wall. These arterial wall sentinels may have an intrinsically high threshold for mounting a danger response within the confines of a microenvironment that has evolved to minimize collateral damage as a selective advantage to maintain the integrity of this non-regenerative and vital organ system.
Antigen-presenting functions in the vessel wall
Besides DC, monocytes and macrophages are known to have antigen-presenting and immunostimulatory functions. These cells are typically excluded from the healthy vessel wall, but are present in the inflammatory lesions of GCA [1,7]. It stands to reason then that these innate immune cells may also play key roles in driving T-cell inflammatory responses. The capacity of these cells to mediate development of T-cell effector functions was addressed utilizing bioengineered human arteries and was compared to that of DC by selective reconstitution of bioarteries with each of these three APC populations [6].
Remarkably, TLR4 stimulation established a hierarchical response pattern in which DC emerged as the master stimulator, proving to be the most effective T-cell recruiter and showing the greatest upregulation of CD83, CD86 and CD40L. Conversely, while the macrophages were able to elicit T-cell recruitment, they failed to provide the necessary signals to promote T-cell activation. Monocytes neither recruited nor stimulated T cells. These studies conducted within the three-dimensional space of the bioengineered artery support earlier studies describing the absolute requirement for DC in the mediation of disease via the sensing of microbial patterns and reaffirmed the role of the DC as the major APC in vessel wall inflammation. It is currently unknown why only DCs are capable of serving as APC. This may be a direct reflection of the conditions encountered in the microspace of the adventitial, the medial and the intimal layers.
Another important component is that vessel walls are dynamic structures, undergoing rhythmic expansion and contraction. The migration pattern of cells entering the wall is highly directed, suggesting that such cells recognize wall motion and can orient their behavior accordingly. Underlying molecular mechanisms remain elusive, but bioengineered human arteries represent a promising experimental system to address these questions.
Adaptive immune responses in the unique environment of the arterial wall
While innate immune responses are instrumental in bringing immune cells to the artery [4], the tissue-damaging inflammation is ultimately maintained by adaptive immunity (Figure 1). Phenotypic studies have shown that Th1 cells are the dominant T-cell type in the vasculitic lesions of GCA [10,11]. Comparison of T-cell populations accumulated in the right and left temporal arteries of patients with GCA has demonstrated identical T-cell receptors [12], a finding that strongly supports the paradigm that antigen are the driving factor [13]. The identity of this antigen remains elusive. A recent immunophenotypic analysis has provided evidence supporting these earlier studies via a comparison of the T-cell receptor usage in peripheral blood lymphocytes with that of the lesion infiltrates in GCA patients at the time of diagnosis [14]. Disparities in the TCR V gene expansions in peripheral blood lymphocytes and tissue lymphocytes point to the possibility of a selective T-cell recruitment process or local expansion of T cells in situ. Non- randomness in the selection of T cells strongly supports a role for antigen.
The peripheral T-cell pool is assembled from a number of different subsets, e.g. naive and memory T cells. Memory T cells dominate the vasculitic infiltrates in GCA. However, multiple functional subpopulations contribute to the memory T-cell compartment. A recent study has analyzed whether vasculitic T cells originate from a particular subset or are representative of all memory T cells [15]. Experimental data support the concept that chemokine receptor 6 (CCR6)-expressing T cells are particularly effective at invading the wall layers and survive in the wall structure (Figure 2). CCR6 is expressed on several T-cell subsets and in particular has been shown to be an important functional marker for Th17 cells [16], a novel CD4 T-cell subset that has been implicated in the pathogenesis of autoimmune disease.
Figure 2.
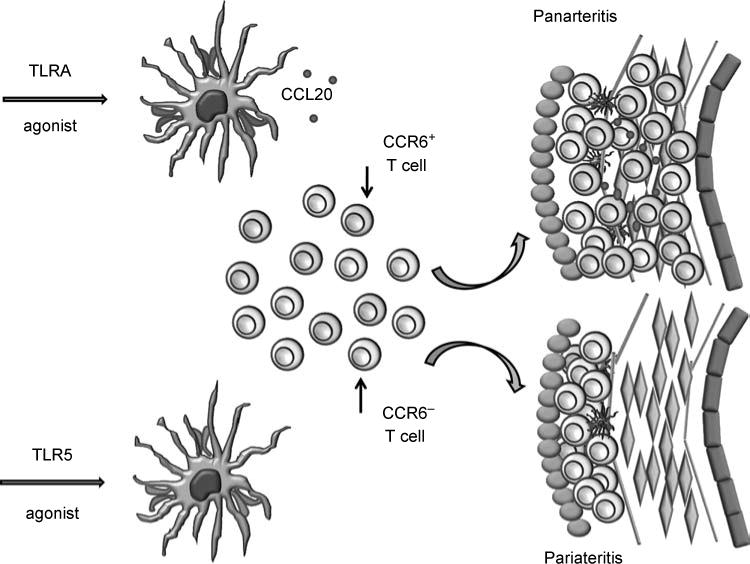
Vascular DC shape the arrangement and composition of T-cell responses in the arterial wall. Triggering of wall-embedded DC by different TLR ligands results in distinct architectures of vessel wall inflammation. TLR4 ligands induce the release of CCL20, the preferred recruitment of CCR6þ T cells and the establishment of wall-penetrating inflammation. TLR5 agonists facilitate T-cell recruitment with T cells clustering in the adventitia, assembling a perivascular infiltrate.
Indeed, CCR6-deficient T-cell subsets have been shown to diminish susceptibility to autoimmune diseases such as rheumatoid arthritis and multiple sclerosis [17], partially due to the demonstrated role of the receptor in directing the targeted migration of these subsets into inflammatory sites. To investigate the role of CCR6þ T cells in GCA, the authors made use of an experimental model in which normal human arteries are engrafted into SCID mice. Infusion of human T cells into such chimeras results in vessel wall inflammation if the arterial DCs are appropriately conditioned. In this study, two distinct conditioning regimes were compared. Wall-embedded DCs were either triggered with ligands for TLR4 (LPS) or with ligands for TLR5 (flagellin). Pretreatment with TLR4 ligands resulted in a typical transmural vasculitis with T cells clustering in the adventitia and infiltrating deep into the media.
Conversely, pretreatment with a TLR5 agonist led to a perivasculitic pattern of inflammation in which T cells accumulated in the adventitia but failed to invade into the media. To answer the question of whether different T-cell populations could contribute to vasculitis, markers of T-cell recruitment and activation were compared. T-cell activation as quantified by IFN-g was similarly induced in arteries with trans- mural and perivascular infiltrates. However, T cells migrating deep into the vessel wall were markedly enriched for CCR6þ T cells. Subsequent experiments showed preferential recruitment of CCR6þ T cells into the media in LPS-treated human artery-SCID chimeras, an invasive property that was abrogated following depletion of the CCR6þ cells or upon blockade with an antibody specific for the CCR6 receptor. This skewing of the T-cell responsive phenotype was shown to be driven by a differential and enhanced production of CCL20, the unique ligand for the CCR6 receptor [18], by TLR4- triggered DC, a response which was not mirrored by flagellin-stimulated DC.
This work, for the first time, demonstrates selectivity for functional T-cell subsets driving large- vessel vasculitis. Phenotyping of panarteritic T cells in temporal artery specimens from GCA patients confirmed the dominance of CCR6þ T cells. Most importantly, the recruitment of CCR6þ T cells could be linked to the stimulatory conditions of wall-residing DC, assigning to them a true checkpoint function in attracting T cells and assembling the population of T cells mediating vasculitis. The distinct features of TLR4- and TLR5-induced vasculitis, which correspond to two distinct histomorphological patterns of disease in patients with GCA [19], re-emphasize that the nature of the original trigger is crucial in directing pathogenic immune responses in large-vessel vasculitis (Figure 2).
These observations should reinvigorate the discussion about how pathogen-derived motifs render the artery susceptible to inflammation. It is now clear that TLR ligands can modulate the function of DC profoundly, both by stimulating and by inducing inhibitory receptors [20]. Dissecting GCA cases based on the nature of the innate and adaptive immune responses in the artery could provide valuable clues towards disease instigators.
Damaging the arterial wall in GCA—a multiplicity of effector pathways
Multiple T-cell effector functions including IFN-g production, cytotoxicity and tissue invasiveness are relevant in arterial injury [21,22]. The ultimate mediators of tissue injury are often macrophages and wall-resident cells that are guided by T cells (Table I). Two components of the disease process participate in pathologic consequences: a systemic inflammatory syndrome and the tissue lesions in the vascular wall [23]. The systemic inflammatory component is of high relevance in the diagnostic process and is used by most physicians to monitor therapy. Studies have detected increased serum levels of cytokines such as IL-6 in patients with active disease and corresponding decreases in patients demonstrating remission of clinical symptoms following treatment [24–26], where these cytokines originate is not entirely understood. They may be released in part by the inflamed vessel walls, or may partially derive from highly activated monocytes/macrophages in the circulation [27].
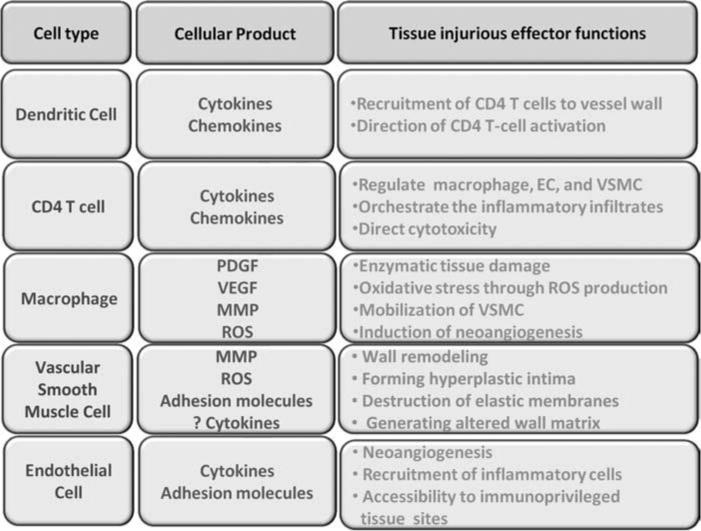
Table I.
The spectrum of cells and mechanisms involved in vessel wall damage in large vessel vasculitis
| Cell type | Cellular product | Tissue injurious effect functions |
|---|---|---|
| Dendritic cell | Cytokines Chemokines |
Recruitment of CD4 T cells to vessel wall Direction of CD4 T-cell activation |
| CD4 T cell | Cytokines Chemokines |
Regulate macrophage, EC and VSMC Orchestrate the inflammatory infiltrates Direct cytotoxicity |
| Macrophage | PDGF VEGF MMP ROS |
Enzymatic tissue damage Oxidative stress through ROS production Mobilization of VMSC Induction of neoangiogenesis |
| VSMC | MMP ROS Adhesion |
Wall remodelling Forming hyperplastic intima |
| Endothelial cell | Cytokines Adhesion molecules |
Neoangiogenesis Recruitment of inflammatory cells Accessibility to immunoprivileged tissue sites |
Whether lymphoid tissues, such as the bone marrow and lymph nodes, are in a state of activation resulting in excessive production of pro-inflammatory cytokines is currently not known. In patients with polymyalgia rheumatica, a forme fruste of GCA, inflammatory lesions around joints best described as bursitis and tendonitis have been described that could be a potential source of circulating cytokines [28]. It is cytokine studies of the arterial wall, however, from which the most insight has been gained thus far regarding the evolution of vascular damage. That CD4 T cells mediate inflammatory responses in GCA is undisputed, as the adoptive transfer of tissue-derived T cells into human temporal artery-SCID mouse chimeras accelerates disease and T-cell depletion leads to elimination of the vascular lesions and loss of characteristic inflammatory cytokines [5,13,29].
Furthermore, temporal artery biopsy studies have provided a tissue cytokine profile which suggests that GCA is a Th1-driven disease in which local IFN-g production drives disease progression [11,22]. This proinflammatory cytokine regulates the differentiation of the tissue-infiltrating macrophages that contribute to tissue injury via the production of proinflammatory cytokines, growth factors, reactive oxygen intermediates (ROI) and matrix metalloproteinases (MMP) [27,29,30]. In essence, much of the vascular injury could be described as unintentional destruction caused by a displaced adaptive immune response.
Vascular smooth muscle cells (VSMC)—a cell population prone to respond to injury
Smooth muscle cells are a major component of the vessel wall defining its contractile functions as well as providing precursor cells to heal and repair wall damage. In that process, migratory ability, proliferative expansion and matrix production all contribute to the repair response. In GCA, T cells and macrophages typically accumulate between the medial smooth muscle cell fibers and VSMC are a direct target of inflammatory attack. Specifically, VSMC have been shown to be subjected to oxidative stress in GCA [30,31], and conversely may also be a source of reactive oxygen species.
VSMC are not only injured by the inflammatory milieu, but they also actively participate in its generation and maintenance. The anatomical separation of the media from the intima is demarcated by the internal elastic lamina (IEL), whose fragmentation represents one of the pathological hallmarks of this disease [32]. Apart from its contractile functions, VSMC have known synthetic functions; and MMP, one of its secretory products, has been purported as the mediator of this IEL degradation. MMP are involved in numerous pathologies including autoimmune and cardiovascular diseases [33], resulting mainly from their ability to degrade components of the extracellular matrix such as collagen and elastin.
Specifically, in a clinical study employing histomorphological analyses of GCA patient temporal artery biopsies, a significant association was found between MMP-9 and IEL destruction in diseased vessels [34]. While both MMP-2 and MMP-9 were detected in the inflammatory cells and the medial and intimal layers as previously reported, only MMP-9 showed this specific association in these patients. Evidence collected through colocalization studies have identified VSMC as a major source of MMP-9 in an experimental model of a systemic vasculitic disease characterized by vessel wall remodeling and aneurysm formation [35].
Disease progression was marked by increased enzymatic activity of this endopeptidase, whose expression is regulated by proinflammatory cytokines such as TNFa. The demonstrated ability of VSMC to secrete MMP positions these cells as potential key contributors to the vascular remodeling that characterizes GCA.
Neoangiogenesis and concentric intimal hyperplasia are additional features of the maladaptive arterial response in GCA that give rise to luminal occlusion and consequently to tissue ischemia that accounts for the clinical symptoms that plague these patients [36]. The vasa vasorum network, conventionally confined to the adventitia in healthy arteries, is found to be extended to the medial and intimal layers of the inflamed artery, a phenomenon that correlates closely with IEL fragmentation as well as with the presence of the multinucleated giant cells from which the disease derives its name. The giant cells and macrophages located at the media-intima junction contribute to the program of tissue injury via the production of proinflammatory cytokines, ROI and MMP [8]. The extensive arsenal of these cells includes growth factors such as platelet-derived growth factor and vascular endothelial growth factors (VEGF), both of which drive neovascularization and intimal thickening via their stimulatory effects on VSMC [37].
The signals thus provided promote VSMC proliferation and may accelerate their migration into the intima. While the source of and precise mechanisms by which VSMC cells attain access to the intima remain unclear, the detection of MMP-9 in the VSMC layer suggests its active involvement, which is supported by its known degradative functions and its demonstrated role in smooth muscle cell-matrix attachment. What remains undisputed, however, is the cumulative effect of a maladaptive repair response that leads to the emergence of new vasa vasorum, intimal lesions and luminal stenosis?
ECs—unintended helpers of wall remodeling and wall injury
Just as the role of the artery extends beyond its function in hemodynamic regulation, so too does the role of the arterial endothelium extend beyond merely serving as a physical interface between the blood and extravascular tissues. Among their physiological functions, vascular EC regulate inflammatory and immune reactions, as well as vascular remodeling that define the vascular lesions of GCA [38]. The mediumsized and large arteries affected by GCA essentially have two populations of EC, and both are likely to have a pathogenic role. However, the EC lining the artery’s macrolumen may be less relevant in the events driving GCA than the micro-EC defining the vasa vasorum and outgrowing microvessels supporting the wall remodeling process. ECs are major targets of cytokines produced by the inflammatory cells forming the vasculitic lesions including DC, macrophages and CD4 T cells. Notably IL-6, whose systemic levels correlate closely with disease status [25,26], has potent effects on EC proliferation and tubular formation, and in vivo and ex vivo models have identified this cytokine as a stimulus for the induction of angiogenesis in large vessels.
Macrophages and giant cells in the lesion are major producers of IL-6 [27], and these tissue-infiltrating cells further contribute to the growth of microcapillaries via the production of VEGF, a stimulant of EC proliferation. Neoangiogenesis serves to perpetuate the inflammatory response given that the vasa vasorum provides the physiologic port of entry for the invading lymphocytes, thereby increasing the number of portals by which the inflammatory reinforcements can access and maintain the ongoing inflammation. ECs, however, are not passive targets in the inflammatory response and are themselves responsible for cytokine production in response to paracrine signals.
These signals modulate the EC synthesis of proinflammatory cytokines and adhesion molecules. The outcome of endothelial activation then is a cycle of cytokine production in response to cytokine signaling by vascular infiltrates. The contributory role of the endothelium in the evolution of vascular disease is further supported by studies in bioengineered vessels. In order to address the role of EC on immunomodulatory functions of APC, tubular constructs with or lacking an endothelial monolayer were seeded with APC and compared for their T-cell stimulatory capacity [6]. As indicated by enhanced DC activation markers as well as increased T-cell recruitment into the tubular constructs, the results showed an amplification of the immune response mediated by DC and macrophages that correlated with the presence of the endothelial layer. These observations confirm the active involvement of the endothelial layer in pathogenesis and the bi-directionality of the interplay between the vessel wall and the immune cells that maintain chronic inflammatory responses leading to the progressive building of the vascular lesions of GCA.
Conclusion
In essence, much of the vascular injury in GCA could be described as a result of inadvertent damage caused by a displaced adaptive immune response (Table I and Figure 1). Emerging data support the model that T cells involved in vasculitic responses are selected and that CCR6þ T cells are uniquely capable of invading all layers of the vessel wall (Figure 2). The blood vessel is not only a passive bystander in the disease process but determines two checkpoints (Figure 3). Indigenous DC sense danger signals, and depending on the nature of these signals, coordinates distinct adaptive immune reactions. Derivatives from gram-negative bacteria, e.g. LPSs, induce a panarteritic distribution of T cells. Motifs derived from flagellated bacteria, e.g. flagellin, promote perivasculitic architecture of the evolving inflammation. As the disease process progresses the vessel wall has another opportunity to direct pathologic consequences. Many, but not all, patients develop lumen-occlusive intimal hyperplasia. This response-to-injury program is driven by VSMC, supported by growth and angiogenic factors released by invading immune cells. The formidable role of the blood vessel in dictating early and late stages of vasculitis needs to be considered in a novel approach towards treating GCA (Figure 3). So far, all therapeutic efforts are focused on suppressing the immune system. Resetting functions of the blood vessel may be a more productive way of avoiding the life-threatening consequences of this large vessel vasculitis.
Figure 3.
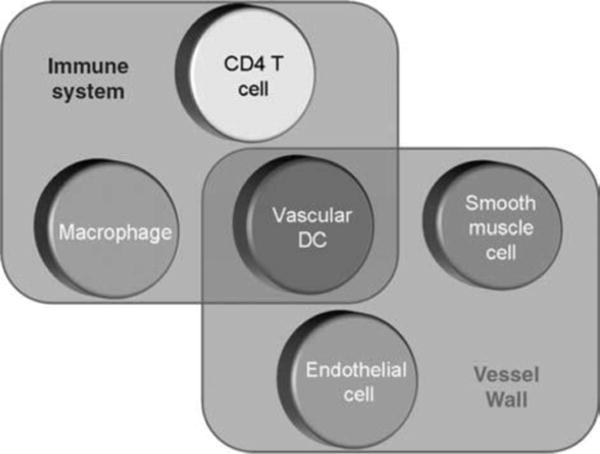
Immune cells and vessel wall cells collaborate in mediating vascular damage in GCA. Vascular DC are an indigenous cell population in the artery’s wall and respond to danger signals by recruiting T cells and macrophages. Macrophages secrete ROI, enzymes, and growth factors. ECs participate in cell recruitment and neoangiogenesis, supporting the remodeling of the wall structure. Vascular smooth muscle cells proliferate and migrate to form the hyperplastic and lumen-obstructive intima but also contribute to wall damage by secreting tissue-injurious enzymes and imposing oxidative stress.
Acknowledgments
This work was funded in part by grants from the National Institutes of Health (RO1 AR42527, RO1 AR41974, R01 AI44142, R01 AI57266, RO1 EY11916 and R01 AG15043), by core grant P30-EY06360 (Department of Ophthalmology) also from the National Institutes of Health, and by a departmental grant (Department of Ophthalmology) from Research to Prevent Blindness. Dr Newman is a recipient of a Research to Prevent Blindness Lew R. Wasserman Merit Award. The authors thank Tamela Yeargin for editing the manuscript.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.Pryshchep O, Ma-Krupa W, Younge BR, Goronzy JJ, Weyand CM. Vessel-specific toll-like receptor profiles in human medium and large arteries. Circulation. 2008;118(12):1276–1284. doi: 10.1161/CIRCULATIONAHA.108.789172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bobryshev YV. Dendritic cells and their involvement in atherosclerosis. Curr Opin Lipidol. 2000;11(5):511–517. doi: 10.1097/00041433-200010000-00009. [DOI] [PubMed] [Google Scholar]
- 3.Krupa WM, Dewan M, Jeon MS, Kurtin PJ, Younge BR, Goronzy JJ, Weyand CM. Trapping of misdirected dendritic cells in the granulomatous lesions of giant cell arteritis. Am J Pathol. 2002;161(5):1815–1823. doi: 10.1016/S0002-9440(10)64458-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma-Krupa W, Kwan M, Goronzy JJ, Weyand CM. Toll-like receptors in giant cell arteritis. Clin Immunol. 2005;115(1):38–46. doi: 10.1016/j.clim.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 5.Ma-Krupa W, Jeon MS, Spoerl S, Tedder TF, Goronzy JJ, Weyand CM. Activation of arterial wall dendritic cells and breakdown of self-tolerance in giant cell arteritis. J Exp Med. 2004;199(2):173–183. doi: 10.1084/jem.20030850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han JW, Shimada K, Ma-Krupa W, Johnson TL, Nerem RM, Goronzy JJ, Weyand CM. Vessel wall-embedded dendritic cells induce T-cell autoreactivity and initiate vascular inflammation. Circ Res. 2008;102(5):546–553. doi: 10.1161/CIRCRESAHA.107.161653. [DOI] [PubMed] [Google Scholar]
- 7.Weyand CM, Goronzy JJ. Medium- and large-vessel vasculitis. N Engl J Med. 2003;349(2):160–169. doi: 10.1056/NEJMra022694. [DOI] [PubMed] [Google Scholar]
- 8.Weyand CM, Ma-Krupa W, Goronzy JJ. Immunopathways in giant cell arteritis and polymyalgia rheumatica. Autoimmun Rev. 2004;3(1):46–53. doi: 10.1016/S1568-9972(03)00064-8. [DOI] [PubMed] [Google Scholar]
- 9.Villadangos JA, Schnorrer P. Intrinsic and cooperative antigen- presenting functions of dendritic-cell subsets in vivo. Nat Rev Immunol. 2007;7(7):543–555. doi: 10.1038/nri2103. [DOI] [PubMed] [Google Scholar]
- 10.Weyand CM, Tetzlaff N, Bjornsson J, Brack A, Younge B, Goronzy JJ. Disease patterns and tissue cytokine profiles in giant cell arteritis. Arthritis Rheum. 1997;40(1):19–26. doi: 10.1002/art.1780400105. [DOI] [PubMed] [Google Scholar]
- 11.Weyand CM, Hicok KC, Hunder GG, Goronzy JJ. Tissue cytokine patterns in patients with polymyalgia rheumatica and giant cell arteritis. Ann Intern Med. 1994;121(7):484–491. doi: 10.7326/0003-4819-121-7-199410010-00003. [DOI] [PubMed] [Google Scholar]
- 12.Weyand CM, Schonberger J, Oppitz U, Hunder NN, Hicok KC, Goronzy JJ. Distinct vascular lesions in giant cell arteritis share identical T cell clonotypes. J Exp Med. 1994;179(3):951–960. doi: 10.1084/jem.179.3.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brack A, Geisler A, Martinez-Taboada VM, Younge BR, Goronzy JJ, Weyand CM. Giant cell vasculitis is a T cell- dependent disease. Mol Med. 1997;3(8):530–543. [PMC free article] [PubMed] [Google Scholar]
- 14.Schaufelberger C, Andersson R, Nordborg E, Hansson GK, Nordborg C, Wahlstrom J. An uneven expression of T cell receptor V genes in the arterial wall and peripheral blood in giant cell arteritis. Inflammation. 2008;31(6):372–383. doi: 10.1007/s10753-008-9088-9. [DOI] [PubMed] [Google Scholar]
- 15.Deng J, Ma-Krupa W, Gewirtz AT, Younge BR, Goronzy JJ, Weyand CM. Toll-like receptors 4 and 5 induce distinct types of vasculitis. Circ Res. 2009 doi: 10.1161/CIRCRESAHA.108.185777. forthcoming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, Parente E, Fili L, Ferri S, Frosali F, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204(8):1849–1861. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, Yamaguchi T, Nomura T, Ito H, Nakamura T, et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med. 2007;204(12):2803–2812. doi: 10.1084/jem.20071397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schutyser E, Struyf S, Van Damme J. The CC chemokine CCL20 and its receptor CCR6. Cytokine Growth Factor Rev. 2003;14(5):409–426. doi: 10.1016/s1359-6101(03)00049-2. [DOI] [PubMed] [Google Scholar]
- 19.Chatelain D, Duhaut P, Loire R, Bosshard S, Pellet H, Piette JC, Sevestre H, Ducroix JP. Small-vessel vasculitis surrounding an uninflamed temporal artery: A new diagnostic criterion for polymyalgia rheumatica? Arthritis Rheum. 2008;58(8):2565–2573. doi: 10.1002/art.23700. [DOI] [PubMed] [Google Scholar]
- 20.Groschel S, Piggott KD, Vaglio A, Ma-Krupa W, Singh K, Goronzy JJ, Weyand CM. TLR-mediated induction of negative regulatory ligands on dendritic cells. J Mol Med. 2008;86(4):443–455. doi: 10.1007/s00109-008-0310-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weyand CM, Younge BR, Goronzy JJ. T cells in arteritis and atherosclerosis. Curr Opin Lipidol. 2008;19(5):469–477. doi: 10.1097/mol.0b013e32830bfdc2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagner AD, Bjornsson J, Bartley GB, Goronzy JJ, Weyand CM. Interferon-gamma-producing T cells in giant cell vasculitis represent a minority of tissue-infiltrating cells and are located distant from the site of pathology. Am J Pathol. 1996;148(6):1925–1933. [PMC free article] [PubMed] [Google Scholar]
- 23.Weyand CM, Goronzy JJ. Giant-cell arteritis and polymyalgia rheumatica. Ann Intern Med. 2003;139(6):505–515. doi: 10.7326/0003-4819-139-6-200309160-00015. [DOI] [PubMed] [Google Scholar]
- 24.Martinez-Taboada VM, Alvarez L, RuizSoto M, Marin-Vidalled MJ, Lopez-Hoyos M. Giant cell arteritis and polymyalgia rheumatica: Role of cytokines in the pathogenesis and implications for treatment. Cytokine. 2008;44(2):207–220. doi: 10.1016/j.cyto.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 25.Roche NE, Fulbright JW, Wagner AD, Hunder GG, Goronzy JJ, Weyand CM. Correlation of interleukin-6 production and disease activity in polymyalgia rheumatica and giant cell arteritis. Arthritis Rheum. 1993;36(9):1286–1294. doi: 10.1002/art.1780360913. [DOI] [PubMed] [Google Scholar]
- 26.Weyand CM, Fulbright JW, Hunder GG, Evans JM, Goronzy JJ. Treatment of giant cell arteritis: Interleukin-6 as a biologic marker of disease activity. Arthritis Rheum. 2000;43(5):1041–1048. doi: 10.1002/1529-0131(200005)43:5<1041::AID-ANR12>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 27.Wagner AD, Goronzy JJ, Weyand CM. Functional profile of tissue-infiltrating and circulating CD68 þ cells in giant cell arteritis. Evidence for two components of the disease. J Clin Invest. 1994;94(3):1134–1140. doi: 10.1172/JCI117428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salvarani C, Cantini F, Olivieri I, Barozzi L, Macchioni L, Niccoli L, Padula A, De Matteis M, Pavlica P. Proximal bursitis in active polymyalgia rheumatica. Ann Intern Med. 1997;127(1):27–31. doi: 10.7326/0003-4819-127-1-199707010-00005. [DOI] [PubMed] [Google Scholar]
- 29.Brack A, Rittner HL, Younge BR, Kaltschmidt C, Weyand CM, Goronzy JJ. Glucocorticoid-mediated repression of cytokine gene transcription in human arteritis-SCID chimeras. J Clin Invest. 1997;99(12):2842–2850. doi: 10.1172/JCI119477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rittner HL, Hafner V, Klimiuk PA, Szweda LI, Goronzy JJ, Weyand CM. Aldose reductase functions as a detoxification system for lipid peroxidation products in vasculitis. J Clin Invest. 1999;103(7):1007–1013. doi: 10.1172/JCI4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rittner HL, Kaiser M, Brack A, Szweda LI, Goronzy JJ, Weyand CM. Tissue-destructive macrophages in giant cell arteritis. Circ Res. 1999;84(9):1050–1058. doi: 10.1161/01.res.84.9.1050. [DOI] [PubMed] [Google Scholar]
- 32.Weyand CM, Wagner AD, Bjornsson J, Goronzy JJ. Correlation of the topographical arrangement and the functional pattern of tissue-infiltrating macrophages in giant cell arteritis. J Clin Invest. 1996;98(7):1642–1649. doi: 10.1172/JCI118959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: The good, the bad, and the ugly. Circ Res. 2002;90(3):251–262. [PubMed] [Google Scholar]
- 34.Rodriguez-Pla A, Bosch-Gil JA, Rossello-Urgell J, Huguet-Redecilla P, Stone JH, Vilardell-Tarres M. Metalloproteinase-2 and -9 in giant cell arteritis: Involvement in vascular remodeling. Circulation. 2005;112(2):264–269. doi: 10.1161/CIRCULATIONAHA.104.520114. [DOI] [PubMed] [Google Scholar]
- 35.Lau AC, Duong TT, Ito S, Yeung RS. Matrix metalloproteinase 9 activity leads to elastin breakdown in an animal model of Kawasaki disease. Arthritis Rheum. 2008;58(3):854–863. doi: 10.1002/art.23225. [DOI] [PubMed] [Google Scholar]
- 36.Kaiser M, Younge B, Bjornsson J, Goronzy JJ, Weyand CM. Formation of new vasa vasorum in vasculitis. Production of angiogenic cytokines by multinucleated giant cells. Am J Pathol. 1999;155(3):765–774. doi: 10.1016/S0002-9440(10)65175-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaiser M, Weyand CM, Bjornsson J, Goronzy JJ. Plateletderived growth factor, intimal hyperplasia, and ischemic complications in giant cell ar teritis. Arthritis Rheum. 1998;41(4):623–633. doi: 10.1002/1529-0131(199804)41:4<623::AID-ART9>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 38.Kofler S, Nickel T, Weis M. Role of cytokines in cardiovascular diseases: A focus on endothelial responses to inflammation. Clin Sci (Lond) 2005;108(3):205–213. doi: 10.1042/CS20040174. [DOI] [PubMed] [Google Scholar]