

Abstract
The mechanism by which cells undergo death determines whether dying cells trigger inflammatory responses or remain immunologically silent. Mitochondria play a central role in the induction of cell death, as well as in immune signaling pathways. Here, we identify of a mechanism by which mitochondria and downstream pro-apoptotic caspases regulate the activation of antiviral immunity. In the absence of active caspases, mitochondrial outer membrane permeabilization by Bax and Bak results in the expression of type I interferons (IFNs). This induction is mediated by mitochondrial DNA-dependent activation of the cGAS/STING pathway and results in the establishment of a potent state of viral resistance. Our results show that mitochondria have the capacity to simultaneously expose a cell-intrinsic inducer of the IFN response, and to inactivate this response in a caspase-dependent manner. This mechanism provides a dual control, which determines whether mitochondria initiate an immunologically silent or a pro-inflammatory type of cell death.
Introduction
Multicellular organisms are constantly exposed to the threat of viral infections. As a response, vertebrates have evolved several mechanisms of antiviral defense. These mechanisms include the production of type I interferons (IFNs) (Stetson and Medzhitov, 2006) and the suicide of infected cells (Upton and Chan, 2014).
Type I IFNs (IFNα and IFNβ) are cytokines of major importance for the innate antiviral response (Stetson and Medzhitov, 2006). They are produced after recognition of viral nucleic acids by toll-like receptors (TLRs) or by cytoplasmic proteins such as RIG-I like receptors (RLRs) or the cyclic GMP-AMP synthase (cGAS) (Cai et al., 2014; Kawai and Akira, 2011; Loo and Gale, 2011). After their secretion, type I IFNs bind to the type I IFN receptor (IFNAR) in an autocrine and paracrine manner. This signal induces the expression of hundreds of interferon-stimulated genes (ISGs) in the responding cell (Schneider et al., 2014). Overall, ISGs have the capacity to interfere with every step of viral replication and, as a consequence, the I FN response results in the establishment of a cellular state of viral resistance.
The programmed death, of infected cells limits the possibility for viruses to subvert the cellular machinery for their own replication (Best, 2008; Yatim and Albert, 2011). One of the best-described mechanisms of programmed cell death is apoptosis, which is mediated through the activation of members of the caspase family of proteases (Fuchs and Steller, 2011; Kumar, 2007; Taylor et al., 2008). The mitochondrial pathway of apoptosis is induced in response to cellular stress. It is regulated by the activities of pro- and anti-apoptotic members of the Bcl-2 family, which control the formation of the Bax/Bak channel that results in mitochondrial outer membrane permeabilization (MOMP) (Chipuk et al., 2010; Tait and Green, 2010; Youle and Strasser, 2008). Following MOMP, mitochondrial proteins, including cytochrome c are released in the cytosol. Together with Apaf-1 and caspase-9, cytosolic cytochrome c forms a protein complex called the apoptosome, which induces the activation of caspase-9 (Jiang and Wang, 2004; Riedl and Salvesen, 2007). The downstream effector caspases-3 and -7 are cleaved and activated by caspase-9, triggering a cascade of proteolytic events that culminates in the demise of the cell through apoptosis (Kroemer et al., 2009).
While caspases are key mediators of apoptotic cell death (Kumar, 2007), multiple mechanisms of caspase-independent cell death exist (Chipuk and Green, 2005; Tait et al., 2014; Vanden Berghe et al., 2014). The discovery of a broad diversity of non-apoptotic death pathways has led to a reevaluation of caspases as essential mediators of cell death. An appealing hypothesis to reconcile the evolutionary conservation of pro-apoptotic caspase signaling with the existence of multiple, and potentially redundant, death-inducing pathways is that caspase-dependent apoptosis is unique in its capacity to induce an immunologically silent form of cell death, while other types of cell death have pro-inflammatory or immunostimulatory properties (Martin et al., 2012; Tait et al., 2014). Indeed, necrotic cell death results in the release of molecules with pro-inflammatory properties, collectively termed damage-associated molecular patterns (DAMPs) or alarmins (Kroemer et al., 2013). Mounting evidence demonstrates that several DAMPs can be inactivated in a caspase-dependent manner during apoptosis, supporting the importance of caspases in maintaining cell death as immunologically silent. However, it is probable that a large spectrum of caspase-dependent mechanisms of immune regulation remain to be discovered (Martin et al., 2012).
In this study, we identify an unsuspected mechanism by which the mitochondrial events of apoptosis actively trigger the initiation of a cell intrinsic immune response, mediated by the expression of type I IFNs. Pro-apoptotic caspases, activated simultaneously by mitochondria, are required to inhibit that response and to maintain apoptosis immunologically silent. Therefore, mitochondria and caspases play a crucial role not only in the decision of the cell to live or to commit suicide, but also on the decision to die in a inflammatory or immunologically silent manner.
Results
Intrinsic apoptosis deficiency confers resistance to viral infection
As mice with genetic deficiencies in the intrinsic pathway of apoptosis die perinatlly (Hakem et al., 1998; Kuida et al., 1998; Lakhani et al., 2006; Yoshida et al., 1998), we generated mice with a floxed caspase-9 allele or a floxed caspase-3 allele (Figure S1A). With the initial objective of studying the role of the intrinsic pathway of apoptosis in immune cells, we crossed Casp9fl/fl mice and Casp3fl/fl Casp7-/- mice with Tie2-Cre(E+H) (Koni et al., 2001; Lakhani et al., 2006), to obtain mice with endothelial/hematopoietic tissue-specific deletion of the respective floxed alleles (Figure S1B-F). We observed that Casp9fl/fl Tie2-Cre+ and Casp3fl/fl Casp7-/- Tie2-Cre+ mice were highly resistant to viral infection in comparison to littermate controls. Indeed, lethality following intraperitoneal infection with encephalomyocarditis virus (EMCV, 2×103 TCID50), which was observed in control mice 6 days after infection, was delayed in Casp9fl/fl Tie2-Cre+ mice (Figure 1A). The prolonged survival and resistance to EMCV infection was associated with lower viral loads in the heart 2 days after infection, with undetectable expression of the EMCV genome in half of the Casp9fl/fl Tie2-Cre+ mice (Figure 1B). Furthermore, the deletion of caspase-9, or of both caspases-3 and -7, resulted in undetectable viral titers of vesicular stomatitis virus (VSV, 106 PFU) after intranasal infection (Figure 1C, D), highlighting a potent anti-viral state in vivo in pro-apoptotic caspase-deficient animals.
Figure 1. Loss of the intrinsic pathway of apoptosis enhances resistance to viral infection.
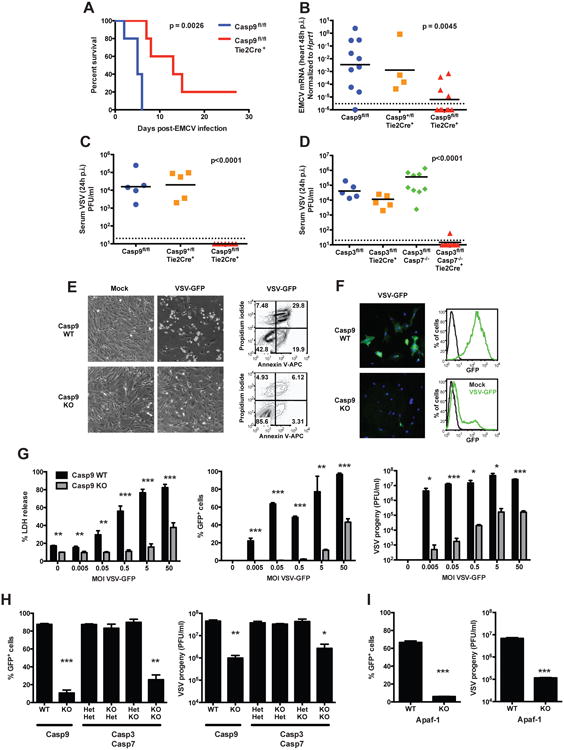
(A and B) Casp9fl/fl Tie2-Cre+ and control mice were infected intraperitoneally with EMCV (2×103 TCID50) and the survival was monitored (n=5 mice/group, p-value calculated by Mantel-Cox test) (A); or the mice were sacrificed 48h post-infection (p.i.) and viral loads in the heart were measured by real time RT-PCR (n=4-10 mice/group, combined from 3 independent experiments, p-value calculated by one-way ANOVA) (B). Each symbol represents an individual mouse and the black horizontal bars indicate geometric means. The dashed line indicates the limit of detection of the assay.
(C and D) Casp9fl/fl Tie2-Cre+ (C), Casp3fl/fl Casp7-/- Tie2-Cre+ (D) and control mice were infected intranasally with VSV (106 PFU) and sacrificed 24h later. Viral loads were measured in the plasma by plaque forming assay (n=5-7 mice/group, combined from at least 2 independent experiments, p-value calculated by one-way ANOVA).
(E and F) Casp9 WT and KO primary MEFs were infected in vitro with VSV-GFP (MOI = 0.5) and analyzed 24h later (E). The expression of virus-encoded GFP was analyzed by fluorescence microscopy (green, GFP; blue, counter-staining of nuclei with DAPI) or by flow cytometry (F).
(G) Casp9 WT and KO primary MEFs were infected with the indicated MOI of VSV-GFP, and assessed 24h later for cell death with LDH release assay (left panel), expression of GFP (middle) and viral progeny production by plaque assay (right). Results are presented as mean ± s.d. of triplicates, representative of at least 3 independent experiments.
(H and I) Casp3/7 double KO (H) or Apaf-1 KO (I) and respective control primary MEFs were infected with VSV-GFP (MOI = 0.5), and GFP expression and viral progeny were measured as in (G) (mean ± s.d. of duplicates, representative of 2 experiments).
*, p<0.05; **, p<0.01; ***, p<0.001 (two-tailed unpaired Student t-test, compared to respective WT or HetHet control).
See also Figure S1 and S2.
To determine whether this phenotype could be recapitulated in vitro, primary mouse embryonic fibroblasts (MEFs) isolated from Caspase-9 knockout (Kuida et al., 1998), from Caspase-3/-7 double knockout (Lakhani et al., 2006) and from Apaf-1 knockout mice (Yoshida et al., 1998) were infected with VSV. We observed that caspase-9 deficient cells (Casp9 KO) were only modestly affected by the infection with a recombinant strain of VSV expressing the green fluorescent protein (VSV-GFP), while wild type cells derived from littermate embryos (Casp9 WT) showed the typical phenotype of infected cells (cell rounding, detachment and death; Figure 1E). Fluorescence microscopy and flow cytometry analysis showed that only a small fraction of Casp9 KO cells expressed virus-encoded GFP (Figure 1F). To further substantiate this observation, Casp9 WT and KO MEFs were infected with VSV-GFP at various multiplicities of infection (MOI). By measuring cell death (percentage of LDH released) and viral infection and replication (GFP expression and plaque forming units), we observed that caspase-9 deficiency significantly reduced the susceptibility of cells to VSV infection at all MOIs tested (Figure 1G). Casp9 KO MEFs also displayed increased resistance to infection by EMCV (Figure S2A) or by herpes simplex virus type 2 (HSV-2) (Figure S2B). Similarly, Casp3/7 double KO and Apaf-1 KO MEFs showed resistance to VSV infection comparable to that observed in Casp9 KO (Figure 1H, I). These results demonstrate that deficiency in the intrinsic pathway of apoptosis, downstream of mitochondria, confers a strong broad-spectrum resistance to infection by RNA and DNA viruses, both in vivo and in vitro.
Constitutive activation of the type I IFN response
Type I IFNs are critical determinants of the cellular susceptibility to viral infections. They are constitutively expressed at low levels, and these steady state IFNs have profound physiological effects on homeostasis through tonic signaling in the absence of acute infection (Gough et al., 2012; Taniguchi and Takaoka, 2001). We measured the baseline expression levels of type I IFNs using a highly sensitive nested RT-PCR. We observed a modest increase in the basal levels of the mRNA encoding both IFNα and IFNβ in Casp9 KO MEFs compared to WT controls (Figure 2A), in the absence of any stimulation. We confirmed this result using a quantitative nested realtime PCR (Figure 2B) as well as a type I IFN bioactivity assay (Figure 2C). Increased steady state type I IFN mRNA was also induced in vitro and in vivo by Casp3/7 double deficiency (Figure 2D,E), as well as by the absence of Apaf-1 in MEFs (Figure 2F).
Figure 2. Inhibition of intrinsic apoptosis activates the IFN response.
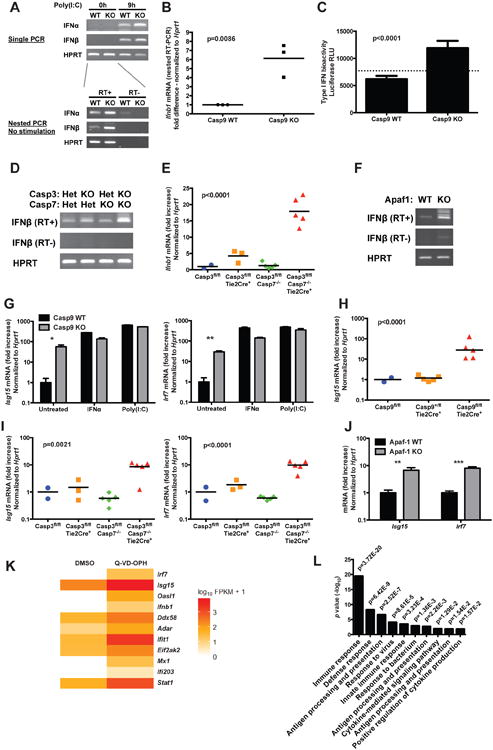
(A) The expression of IFNα and IFNβ mRNA was determined by RT-PCR in Casp9 WT and KO primary MEFs. Top: single RT-PCR on untreated cells and on cells transfected with poly(I:C) as a positive control. Bottom: nested RT-PCR on untreated cells (RT+, RNA reverse transcribed in cDNA; RT-, no reverse transcription).
(B) The steady-state expression of IFNβ mRNA expression in unstimulated primary MEFs was quantified by nested realtime RT-PCR. Each dot represents an independent experiment; p value: two-tailed unpaired Student t-test.
(C) Type I IFN bioactivity in the culture supernatant of unstimulated MEFs was measured using an ISRE-Luc reporter cell line (mean ± s.d. of 6 replicates, representative of 2 independent experiments; p value: two-tailed unpaired Student t-test; the dashed line indicates background from untreated reporter cells).
(D) Nested RT-PCR amplification of steady state IFNβ in Casp3/7 double deficient and control MEFs.
(E) IFNβ mRNA expression measured by realtime RT-PCR in Casp3/7 deficient and control spleen cells (n=2-5 mice/genotype; p value calculated by one-way ANOVA).
(F) Nested RT-PCR amplification of steady state IFNβ in Apaf-1 WT and KO MEFs.
(G) The expression of selected ISGs in Casp9 WT and KO primary MEFs was measured by realtime RT-PCR. IFNα and intracellular poly(I:C) were used as positive controls (mean ± s.d. of duplicates, representative of at least 5 independent experiments). *, p<0.05; **, p<0.01; ***, p<0.001; two-tailed unpaired Student t-test.
(H and I) ISG mRNA expression measured by realtime RT-PCR in Casp9 deficient and control white blood cells (H) or in Casp3/7 double deficient and control spleen cells (I) (n=2-5 mice/genotype; p value: one-way ANOVA).
(J) ISG mRNA expression measured by realtime RT-PCR in Apaf-1 WT and KO primary MEFs (mean ± s.d. of triplicates, representative of 3 independent experiments; p value calculated by two-tailed unpaired Student t-test).
(K) Heatmap of the expression of IFNβ and selected ISGs in WT primary MEFs stimulated for 48h with vehicle (DMSO) or with the caspase inhibitor Q-VD-OPH (10 μM).
(L) Gene Ontology analysis of the pathways overrepresented among genes differentially expressed between WT primary MEFs stimulated with vehicle or with Q-VD-OPH.
See also Figure S3 and S5, and Table S1.
In addition, we observed that interferon stimulated genes (ISGs) were constitutively expressed at elevated levels in vitro and in vivo in Casp9 KO (Figure 2G,H, S3A,B), in Casp3/7 double KO (Figure 2I) and in Apaf-1 KO cells (Figure 2J), in the absence of any stimulation. The maximal expression level of these ISGs, as induced by IFNα or intracellular poly(I:C), was comparable between Casp9 WT and KO cells.
To determine whether the pharmacological inhibition of caspases could recapitulate the phenotype caused by genetic deficiencies, we treated WT MEFs with broad-spectrum inhibitors of caspases (Z-VAD-fmk, Boc-D-fmk and Q-VD-OPH). These inhibitors induced an increased expression of ISGs (Figure 2K, S3C,D), similar to the effect of caspase or Apaf-1 deficiency. Surprisingly, a gene-ontology analysis of the genes differentially expressed between WT cells treated with DMSO or Q-VD-OPH revealed a highly significant overrepresentation of pathways related to immune responses (Figure 2L). While this transcriptional analysis does not take into account the direct proteolytic effects of caspases, it nevertheless reveals a profound effect of caspase inhibition on immune function.
Next, we compared the transcriptional changes induced by caspase inhibition in WT and IFNAR1 KO cells, which lack a critical subunit of the receptor for IFNα/β (Muller et al., 1994). We observed that the absence of the IFNAR receptor abrogated the transcriptional response of the cells to caspase inhibition by Q-VD-OPH (Figure 3A, Table S1), demonstrating the role of type I IFNs in this response.
Figure 3. Expression of ISGs and antiviral resistance are mediated by type I IFNs.
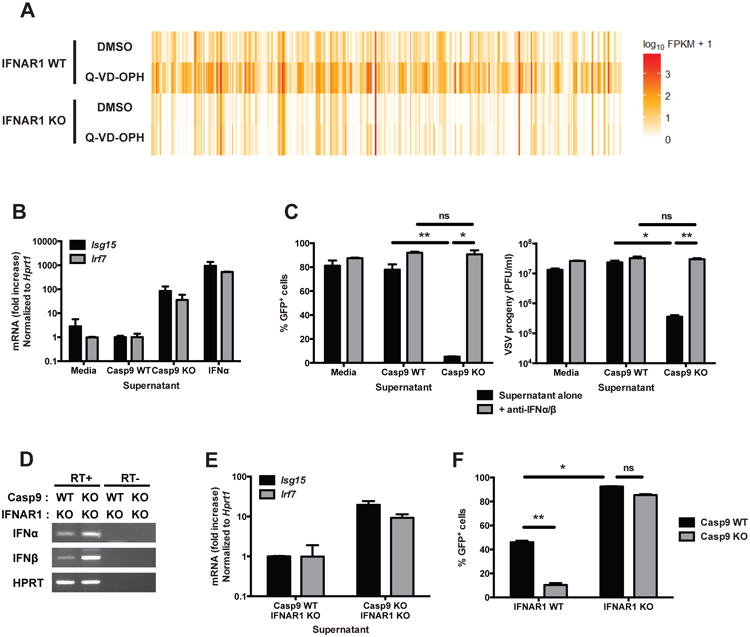
(A) Heatmap of the expression of genes differentially expressed (q<0.05, fold difference >= 5) in IFNAR1 WT and KO primary MEFs stimulated for 48h with vehicle (DMSO) or with the capase inhibitor Q-VD-OPH (10 μM), and determined by RNA sequencing on duplicate samples.
(B) Culture supernatants of confluent cultures of Casp9 WT and KO primary MEFs were collected. WT primary MEFs were then incubated for 16h in the presence of these supernatants or of recombinant IFNα (50 U/ml), and the expression level of selected ISGs was measured by realtime RT-PCR (mean ± s.d. of duplicates; representative of 2 independent experiments).
(C) WT primary MEFs were incubated for 16 hours with conditioned supernatants from Casp9 WT or KO MEFs, in the presence or absence of anti-IFNα and anti-IFNβ neutralizing antibodies (300 NU/ml each). The cells were then washed, infected with VSV-GFP (MOI = 0.5, 24h) and the expression of GFP (left panel) and viral progeny production (right panel) were measured (mean ± s.d. of triplicates, representative of 3 independent experiments).
(D) The expression of IFNα and IFNβ mRNA was detected by nested RT-PCR in unstimulated Casp9 WT/IFNAR1 KO and Casp9 KO/IFNAR1 KO primary MEFs (RT+, RNA reverse transcribed in cDNA; RT-, no reverse transcription).
(E) WT primary MEFs were incubated for 16h with conditioned media from Casp9/IFNAR1 WT/KO or KO/KO MEFs, and the expression levels of ISGs were measured by real-time RT-PCR.
(F) Casp9/IFNAR1 double KO and control primary MEFs were infected with VSV-GFP (MOI = 0.5, 24h) and the expression of GFP was measured by flow cytometry (mean ± s.d. of duplicates, representative of 3 experiments).
*, p<0.05; **, p<0.01; ns, not significant; pairwise comparisons following two-way ANOVA.
See also Figure S4 and Tables S1 and S2.
To demonstrate that ISG expression and viral resistance in Casp9 KO cells was also due to type I IFNs, supernatants from confluent unstimulated Casp9 WT and KO MEFs were transferred to cultures of WT MEFs for 24h. The conditioned supernatant from Casp9 KO cells induced an increase in the expression of ISGs by WT MEFs (Figure 3B), to levels similar to those measured in Casp9 KO cells (compare with Figure 2G). Next, we pre-treated WT cells with conditioned supernatants collected from Casp9 WT and KO cells in the absence or presence of neutralizing anti-IFNα/β antibodies. The cells were then washed and infected with VSV-GFP. The supernatants from Casp9 KO cell cultures conferred resistance to VSV infection in WT cells, and this effect was completely abolished by the presence of anti-IFNα/β neutralizing antibodies in the conditioned media during the pre-treatment of the cells (Figure 3C). Similarly, conditioned supernatants from Casp9 KO cells failed to confer resistance to viral infection when used to pre-treat IFNAR1 KO MEFs (Figure S4). These results demonstrate that the ISG-inducing activity and the resistance to VSV infection are mediated by the elevated concentrations of type I IFNs in the supernatant of Casp9 KO cells.
To further confirm this result, we generated Casp9/IFNAR1 double KO MEFs. Like Casp9 KO cells, Casp9/IFNAR1 double KO cells expressed increased steady-state levels of IFNα/β (Figure 3D), and their supernatant contained ISG-inducing activity (Figure 3E). However, in the absence of IFNAR1, Casp9 WT and KO cells were equally susceptible to VSV infection (Figure 3F). As Casp9/IFNAR1 double KO cells are deficient in apoptosis but are nevertheless susceptible to VSV, this result shows that viral resistance is not a direct consequence of defective cell death.
Caspase-9 deficient mice die during embryonic development or shortly after birth, and this phenotype has been attributed to apoptosis defects in the developing brain (Hakem et al., 1998; Kuida et al., 1998). However, we wanted to determine whether the constitutive activation of the IFN response and high expression of ISGs could contribute to this lethality. To this end, we compared the viability of caspase-9 KO mice in pre- and post-natal life, in the presence or absence of IFNAR1 (Table S2). The absence of IFNAR1 did not rescue the embryonic lethality, showing that constitutive type I IFNs/ISGs expression is not responsible.
Aberrant expression of type I IFNs is the cause of several autoimmune disorders (Stetson, 2009). Surprisingly however, despite constitutive expression of type I IFNs and ISGs, conditional Casp9 KO or Casp3/7 double KO mice did not show any increase in total serum immunoglobulin or in anti-nuclear antibodies, two diagnostic characteristics of autoimmune diseases (Figure S5). We speculate that this absence of autoimmunity despite constitutive IFN response is due to pleiotropic functions of caspases, and probable functional deficiencies in other mechanisms involved in the development of (auto)immunity.
Taken together, these observations demonstrate that in the absence of a functional pathway of intrinsic apoptosis, an increased expression of steady-state type I IFNs is sufficient to induce ISG expression and viral resistance is established. Such unexpected findings raise the intriguing questions as to what ligands and mechanisms govern type I IFN response in dying cells, how healthy cells contain unwanted IFN production and finally, by what means do pro-apoptotic caspases affect these processes.
Bax/Bak-dependent induction of type I IFN
The intrinsic pathway of apoptosis is activated upon mitochondrial outer membrane permeabilization (MOMP) by the Bax/Bak channel (Jiang and Wang, 2004). We thus wanted to determine whether mitochondria and Bax/Bak dependent permeabilization were also involved in regulating the IFN response. Unlike deficiency in Apaf-1 or caspases, the absence of Bax and Bak (Figure 4A, 4B) or the overexpression of the antagonist protein Bcl-2 (Figure S6A), although preventing cell death induced by viral infection (Figure 4A, 4B, S6B, S6C), did not confer any resistance to viral infection (Figure 4B and S6C). This observation could suggest that Apaf-1 and caspases regulate IFN expression independently of mitochondrial events. However, further investigations revealed a more subtle role of Bax and Bak in this process. Indeed, unlike WT cells, Bax/Bak deficient cells treated with the caspase inhibitor Q-VD-OPH failed to induce the expression of ISGs (Figure 4C). This observation suggests that Bax/Bak are actually required for the induction of the IFN response in the absence of active caspases. As Bax/Bak-dependent permeabilization of mitochondrial outer membrane occurs, in cultures of unstimulated cells, only in a small percentage of dying cells or in cells undergoing incomplete MOMP, we hypothesized that the pharmacological inhibition of Bcl-2 could favor Bax/Bak-dependent MOMP and amplify the IFN response caused by caspase deficiency or inhibition. Consistently, we observed an induction of the expression of IFNβ in Casp9 KO cells treated with the Bcl-2 inhibitor ABT-737 (Oltersdorf et al., 2005) (Figure 4D). In contrast, Casp9 WT cells did not express IFNβ in response to Bcl-2 inhibition, showing the role of caspases in regulating this response. As the constitutive expression of ISGs in Casp9 KO (Figure 2, S3) could contribute to the response, we next treated WT cells with a combination of the Bcl-2 inhibitor ABT-737 and the caspase inhibitor Q-VD-OPH. The combination of Bcl-2/caspase inhibition (ABT-737 + Q-VD-OPH) resulted in a robust expression of IFNβ after 3-6h of stimulation (Figure 4E). Other cytokines, such as IL-6 and TNFα, were only moderately induced (Figure S6D). The induction of IFNβ mRNA by Bcl-2/caspase co-inhibition was entirely dependent on the presence of Bax/Bak (Figure 4F). The treatment of human PBMCs with ABT-737 and Q-VD-OPH also induced high expression of IFNβ (Figure 4G), showing that the mechanism of Bax/Bak-dependent caspase-regulated induction of type I IFN is conserved between species. Importantly, inhibiting caspases in the context of cell-extrinsic, caspase-8-dependent apotosis induction did not induce the expression of IFNβ, showing the specificity of this process for mitochondria-dependent apoptosis (Figure S6E).
Figure 4. Bax/Bak-dependent induction of the IFN response in the absence of active caspases.
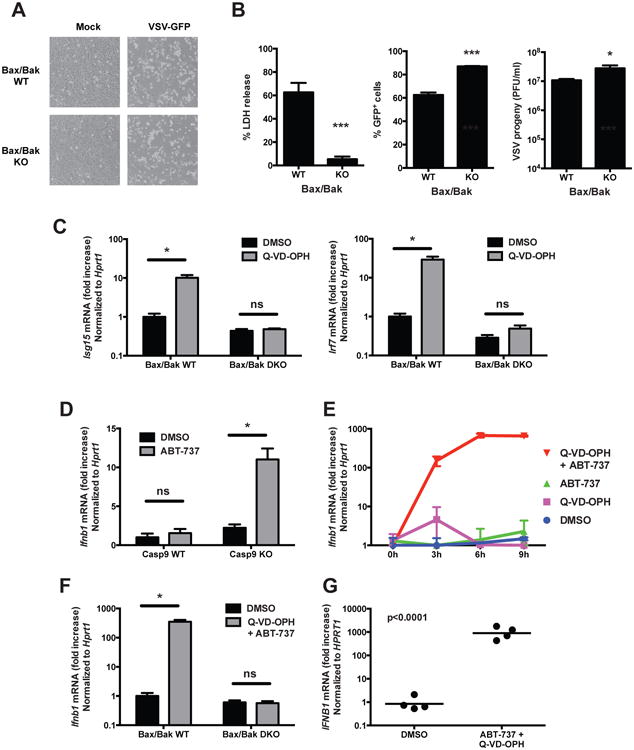
(A and B) Bax/Bak double KO and control immortalized MEFs were infected with VSV-GFP (MOI = 0.5) and their morphology was observed by microscopy (A) and cell death, GFP expression and viral progeny production were determined (B) (mean ± s.d. of triplicates, representative of 3 experiments).
*, p < 0.05; ***, p < 0.001 (two-tailed unpaired t-test).
(C) Bax/Bak WT and double KO MEFs were treated with vehicle (DMSO) or with the caspase inhibitor Q-VD-OPH (10 μM) and the expression of ISGs was measured 48h later by RT-PCR (mean ± s.d. of triplicates, representative of 3 experiments). *, p < 0.05; ns, not significant; pairwise comparisons following two-way ANOVA.
(D) Expression of IFNβ mRNA by Casp9 WT and KO immortalized MEFs after 6h of treatment with vehicle (DMSO) or with the Bcl-2 inhibitor ABT-737 (10 μM) (mean ± s.d. of triplicates, representative of 3 independent experiments).
(E) Expression of IFNβ mRNA by WT primary MEFs at the indicated time points after stimulation with vehicle (DMSO), Bcl-2 inhibitor (ABT-737, 10 μM), caspase inhibitor (Q-VD-OPH, 10 μM) or both inhibitors (mean ± s.d. of duplicates, representative of 3 independent experiments).
(F) Expression of IFNβ mRNA by BaxBak WT and double KO immortalized MEFs after 6h of treatment with vehicle or ABT-737 + Q-VD-OPH (mean ± s.d. of triplicates, representative of 3 independent experiments).
(G) Expression of IFNβ mRNA by human PBMCs after 6h of treatment with vehicle or ABT-737 + Q-VD-OPH (n=4 healthy donors, results combined from 2 independent experiments; p value calculated by two-tailed unpaired Student t-test).
See also Figure S6.
Taken together, these results uncover a novel immunomodulatory role for the mitochondria in innate immunity, a process tightly regulated by proapototic caspases in which Bax/Bak induced MOMP facilitates the release of a mitochondrial factor with the capacity to stimulate type I IFN expression and promote viral resistance.
Activation of the cGAS/STING pathway
To identify the putative mitochondrial factor that induces type I IFN expression in response to Bcl-2/caspases co-inhibition, we first determined which interferon-inducing pathway is involved. We used two criteria to determine the involvement of a candidate sensor or signaling molecule in this process: (i) the activation of this candidate factor after ABT-737 + Q-VD-OPH treatment, and (ii) the absence of IFNβ expression, in response to the same treatment, in cells lacking the candidate factor.
Interferon regulatory factors (IRFs), and in particular IRF-3 and IRF-7, are major transcription factors required for the expression of type I IFNs (Tamura et al., 2008) and they are activated by the upstream kinase TBK1. Consistent with the expression of IFNβ, the inhibition of Bcl-2 and caspases (ABT-737 + Q-VD-OPH) induced the phosphorylation of TBK1 and IRF-3 in Bax/Bak sufficient cells, but not in Bax/Bak KO cells (Figure 5A). The phosphorylation of TBK1 and IRF-3 after transfection of HT-DNA (herring testes DNA, a stimulator of the interferon response that serves as a positive control), was not affected by the absence of Bax/Bak. The expression of IFNβ in response to ABT-737 + Q-VD-OPH was completely abrogated in IRF-3/7 double-deficient cells (Figure 5B). These results demonstrate the critical involvement of TBK1 and IRFs in Bax/Bak-dependent caspase-regulated type I IFN induction. Two intracellular pathways converge on the TBK1/IRFs-dependent transcription of type I IFNs: the RLR/MAVS-dependent pathway activated by intracellular viral RNA (Loo and Gale, 2011), and the cGAS/STING pathway of cytosolic DNA recognition (Cai et al., 2014). MAVS deficiency did not affect the response to ABT-737 + Q-VD-OPH (Figure 5C), excluding a role of the cytosolic RNA recognition pathway. In contrast, the Bax/Bak-dependent, caspase-regulated IFN production was entirely dependent on the cGAS/STING pathway of cytosolic DNA recognition. The cGAS/STING pathways is induced upon recognition of double stranded DNA by cGAS (Cai et al., 2014). cGAS then acquires its enzymatic activity and synthesizes cGAMP, a dinucleotide that binds to and activates STING. The treatment with ABT-737+Q-VD-OPH resulted in detectable amounts of cGAMP in cell extracts, indicative of cGAS activity (Figure 5D). Furthermore, cGAS deficiency or STING deficiency completely prevented the IFNβ response to ABT-737 + Q-VD-OPH (Figure 5E,F). In contrast, TLR signaling was dispensable for the response to ABT-737 + Q-VD-OPH (Figure S7). These results unequivocally identify cGAS/STING as the pathway through which type I IFNs are induced by Bcl-2/caspase co-inhibition.
Figure 5. Activation of the cGAS/STING pathway of IFN induction.
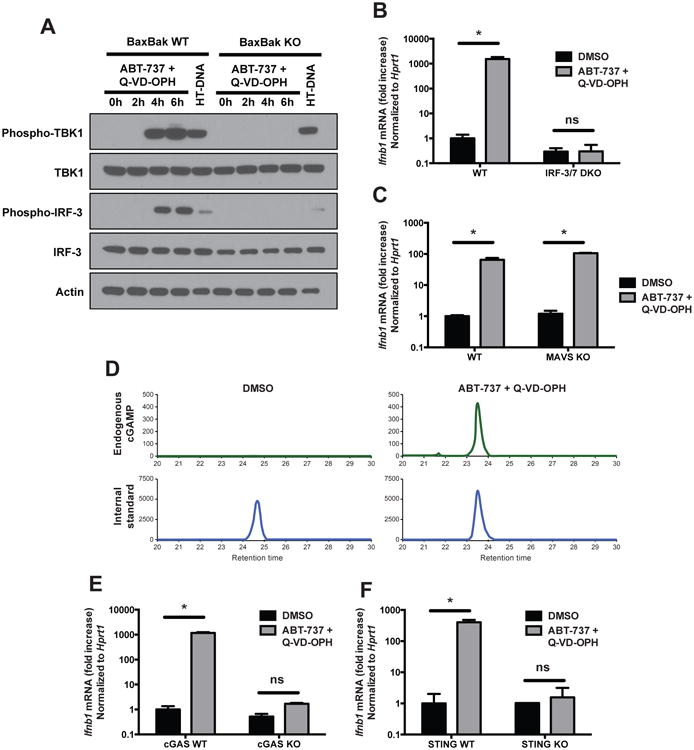
(A) Western blot analysis of the phosphorylation of TBK1 and IRF-3 in BaxBak WT and KO cells treated with combined Bcl-2/caspase inhibitors (ABT-737 + Q-VD-OPH, 10 μM each), or transfected with HT-DNA as a positive control (3 μg/ml, 3h). Result representative of 3 independent experiments.
(B and C) Expression of IFNβ mRNA by IRF-3/7 double KO (B), MAVS KO (C) and control WT primary MEFs after 6h of treatment with vehicle (DMSO) or with ABT-737 + Q-VD-OPH (mean ± s.d. of triplicates, representative of 2 experiments). *, p<0.05; ns, not significant; pairwise comparisons following two-way ANOVA.
(D) cGAMP measurement in cell extracts of WT primary MEFs stimulated for 4h with vehicle (DMSO) or ABT-737 + Q-VD-OPH. Result representative of 2 independent experiments.
(E and F) Expression of IFNβ mRNA by cGAS WT and KO bone marrow-derived macrophages (E) and by STING WT and KO primary MEFs (F) after 6h of treatment with vehicle or ABT-737 + Q-VD-OPH (mean ± s.d. of 3 or 2 replicates, respectively).
See also Figure S7.
Next, we determined whether the cGAS/STING/TBK1/IRFs pathway was also responsible for the increased steady state IFN response in caspase-deficient cells. Interestingly, we observed constitutive phosphorylation of TBK1 in Casp9 KO cells and this phosphorylation could be further induced by treatment with ABT-737 or transfected HT-DNA (Figure 6A). That observation prompted us to test whether other components of the pathway are constitutively active in the absence of functional caspases. Upon stimulation, IRF-3 is ubiquitinylated and degraded through a process of negative feedback loop (Saitoh et al., 2006). We observed lower levels of total IRF-3 protein in Casp9 KO cells, suggestive of constitutive activation followed by degradation of IRF-3. Similarly, activated STING is phosphorylated and marked for lysosomal degradation (Konno et al., 2013). Again suggestive of constitutive activation, STING protein abundance was reduced in Casp9 KO cells, and the level of STING could be restored to WT levels after treatment with chloroquine, a potent inhibitor of lysosomal acidification (Figure 6B). These observations suggest that the STING pathway is constitutively active in Casp9 KO cells, and likely contributes to the IFN-dependent induction of ISG expression. Furthermore, the constitutive activation of STING suggests that the inhibitory activity of caspases acts upstream of STING activation.
Figure 6. cGAS/STING-dependent constitutive ISG expression in the absence of active caspases.
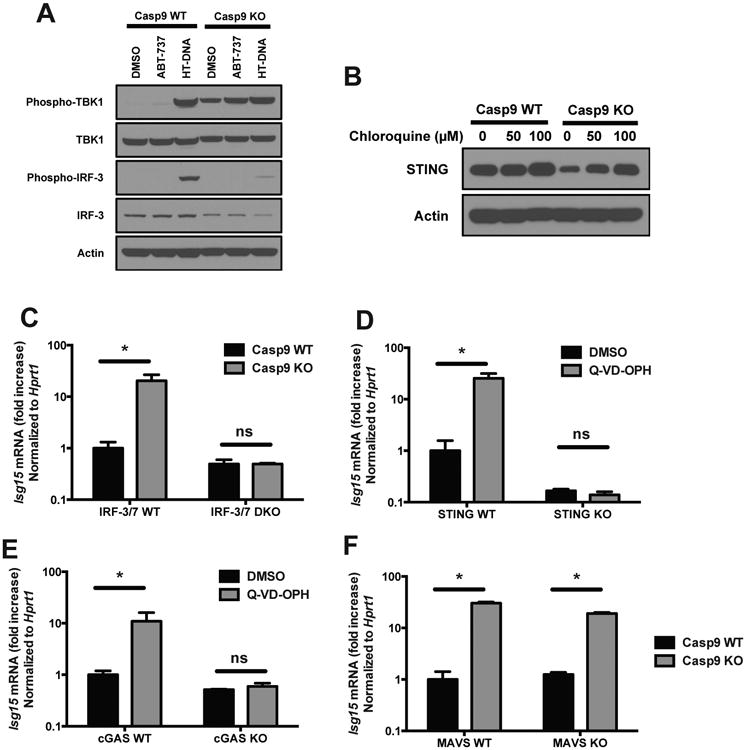
(A) Western blot analysis of the phosphorylation of TBK1 and IRF-3 in Casp9 WT and KO cells treated for 6h with vehicle (DMSO), with the Bcl-2 inhibitor ABT-737 (10 μM), or transfected with HT-DNA as a positive control (3 μg/ml, 3h). Result representative of 3 independent experiments.
(B) Western blot analysis of STING in Casp9 WT and KO cells treated for 16h with the indicated concentrations of chloroquine.
(C) Caspase-9 KO mice were crossed with IRF-3/7 DKO and the expression of ISGs in embryo heads was measured by RT-PCR. Results shown are mean ± s.d. of 3 embryos for each genotype.
(D and E) STING WT and KO primary MEFs (D) or cGAS WT and KO bone marrow-derived macrophages (E) were treated with vehicle (DMSO) or with the caspase inhibitor Q-VD-OPH (10 μM) and ISG expression was measured 48h later by RT-PCR (mean ± s.d. of triplicates, representative of 2 independent experiments).
(F) Caspase-9 KO mice were crossed with MAVS KO and the expression of ISGs in embryo heads was measured by RT-PCR. Results shown are mean ± s.d. of 2 embryos for each genotype.
*, p<0.05; ns, not significant; pairwise comparisons following two-way ANOVA.
We next confirmed the role of the STING pathway at the genetic level. Similarly to the response to ABT-737+Q-VD-OPH, the constitutive expression of ISGs in Casp9 KO cells or in cells treated for 48h with the caspase inhibitor Q-VD-OPH was entirely abrogated in cells deficient for IRF-3/7, STING or cGAS (Figure 6C-E). In contrast, the RNA recognition pathway was not involved, as the expression of ISGs was not affected by MAVS deficiency (Figure 6F).
Taken together, these results demonstrate that the putative mitochondrial factor released after MOMP and that induces type I IFN in the absence of caspases is a ligand for the cytosolic DNA sensor cGAS. These observations demonstrate the existence of a regulated mechanism of activation of the cGAS pathway by an endogenous ligand. This ligand is sequestered in mitochondria in healthy cells, it is released in a Bax/Bak-dependent manner in dying cells and its function is intrinsically regulated in a caspase-dependent manner.
mtDNA-dependent expression of type I IFNs
We hypothesized that the mitochondrial ligand recognized by the DNA sensor cGAS could be mitochondrial DNA (mtDNA). To test this possibility, we used a well-established protocol of ethidium bromide (EtdBr)-mediated depletion of mtDNA (Hashiguchi and Zhang-Akiyama, 2009). The addition of low concentrations of EtdBr (150-450 ng/ml) to the culture medium results in the intercalation of EtdBr into mtDNA and prevents its replication, but it does not affect the replication of genomic DNA. This treatment induced an approximately 10-fold reduction in mtDNA (Figure 7A). When we treated mtDNA depleted cells with both Bcl-2 and caspase inhibtiors, the expression of IFNβ was strongly inhibited compared to control cells (Figure 7B,C). The phosphorylation of TBK1 and IRF-3 in response to the combined inhibitors was also abolished in mtDNA-depleted cells (Figure 7D). These results implicate mtDNA as the major inducer of type I IFN in this system. In contrast, the response to transfected HT-DNA was not affected by the EtdBr treatment (Figure 7B,C), showing that the cGAS/STING pathway remained functional. The response to other stimuli, such as transfected poly(I:C) and LPS, was also maintained after EtdBr treatment (Figure 7C) showing that other platforms of innate immune activation are unaffected by EtdBr treatment and mtDNA depletion. The expression of IFNβ by Casp9 KO cells in response to Bcl-2 inhibition, and the constitutive phosphorylation of TBK1 in unstimulated Casp9 KO cells were also abrogated after depletion of mtDNA with EtdBr (Figure 7E,F).
Figure 7. mtDNA mediates the induction of type I IFN expression.
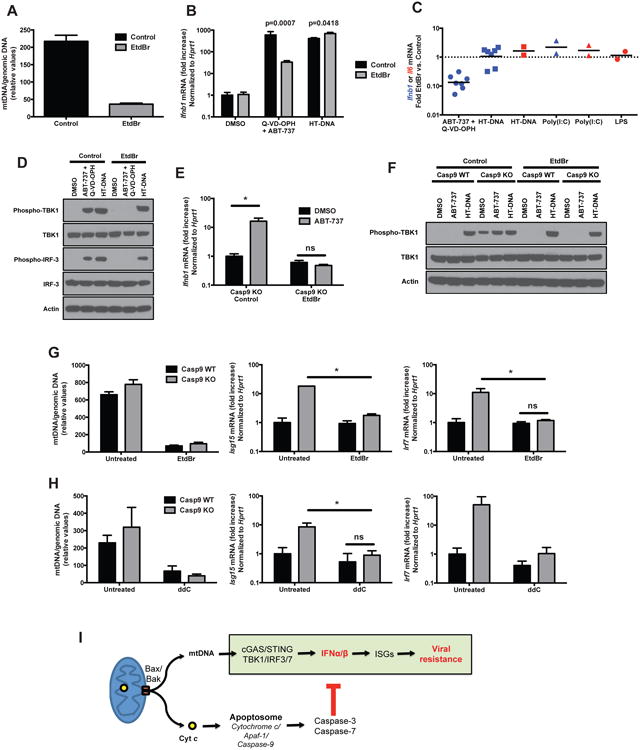
(A) Ratio of mitochondrial DNA (dloop) to genomic DNA (Tert) measured by RT-PCR on total extracts of WT immortalized MEFs treated for 4 days in EtdBr (150 ng/ml) and then maintained in culture for 16h without treatment (mean ± s.d. of duplicates, representative of at least 5 independent experiments).
(B) WT immortalized MEFs treated with EtdBr (150 ng/ml) as in (A) were stimulated with combined Bcl-2/caspase inhibitors (ABT-737 + Q-VD-OPH, 10 μM each) or transfected HT-DNA (3 μg/ml) for 6h and the expression of IFNβ mRNA was measured by realtime RT-PCR (mean ± s.d. of duplicates). p-values calculated by two-tailed unpaired Student t test.
(C) Fold inhibition by EtdBr pre-treament, of the induction of IFNβ (blue symbols) or IL-6 (red symbols) mRNA in cells stimulated with ABT-737 + Q-VD-OPH, transfected with HT-DNA, transfected with poly(I:C) or stimulated with LPS. Each dot represents an individual experiment.
(D) Western blot analysis of the phosphorylation of TBK1 and IRF-3 induced by ABT-737 + Q-VD-OPH (10 μM each, 6h), or by transfection of HT-DNA (3 μg/ml, 3h), in control WT immortalized MEFs or in the same cells pre-treated as in (A) with EtdBr (450 ng/ml). Result representative of 3 independent experiments.
(E) Casp9 KO immortalized MEFs treated or not with EtdBr (450 ng/ml) as in (A) were stimulated with vehicle (DMSO) or the Bcl-2 inhibitor ABT-737 (10 μM) for 6h and the expression of IFNβ mRNA was measured by realtime RT-PCR (mean ± s.d. of duplicates, representative of 2 independent experiments).
(F) Western blot analysis of the phosphorylation of TBK1 after treatment with vehicle (DMSO) or the Bcl-2 inhibitor ABT-737 (10 μM, 6h), or after transfection of HT-DNA (3 μg/ml, 3h), in Casp9 WT and KO immortalized MEFs, pre-treated or not with EtdBr (450 ng/ml) as in (A). Results representative of 3 independent experiments.
(G and H) Casp9 WT and KO primary MEFs were treated for 4 days with ethidium bromide (150 ng/ml) (G) or immortalized MEFs were treated for 6 days with dideoxycytidine (ddC, 40 μg/ml) (H). The ratio of mitochodrial to genomic DNA was measured by realtime PCR on total extracts (left panel) and the expression of ISGs was determined by realtime RT-PCR. Results are shown as mean ± s.d. of triplicates, representative of 3 and 2 independent experiments, respectively.
(I) Schematic model representation of Bax/Bak-dependent, caspase-regulated activation by mtDNA of the cGAS/STING pathway of type I IFN induction.
*, p<0.05; ns, not significant; pairwise comparisons following two-way ANOVA.
Similarly to the response to ABT-737+Q-VD-OPH, the constitutive expression of ISGs in Casp9 KO cells was reversed by the treatment with EtdBr (Figure 7G), again implicating a role for mtDNA. We confirmed this result with an independent protocol of mtDNA depletion: ddC is an inhibitor of mitochondrial DNA polymerase γ, and does not affect the function of nuclear DNA polymerases (Kaguni, 2004). ddC efficiently depleted mtDNA and reduced the expression of ISGs (Figure 7H), similarly to the EtdBr treatment.
Together, these results show that mtDNA is an endogenous ligand that is released from mitochondria via Bax/Bak, and that induces type I IFN expression through the cGAS/STING pathway. Caspases play a crucial role in preventing this cell-intrinsic immune response, thus maintaining the immunologically silent nature of mitochondria-dependent apoptotic cell death (Figure 7I).
Discussion
The physiological role of regulated cell death is to maintain homeostasis, and dysregulated cell death can result in cancer, autoimmune and inflammatory disorders, immunodeficiency or neurodegeneration. The highly regulated process of caspase-dependent apoptosis is unique in its capacity to induce a non-inflammatory type of cell death (Martin et al., 2012; Tait et al., 2014). In contrast, caspase-independent cell death generally induces an inflammatory response through release of molecules, termed DAMPs, into the extracellular environment. These DAMPs contribute to the recruitment and activation of inflammatory cells of the immune system such as granulocytes and monocytes/macrophages. Here, we identify a mechanism by which dying cells expose an intracellular DAMP that activates a cell-intrinsic innate immune response. This type of cell-intrinsic immune activation in dying cells could occur while the physical integrity of the plasma membrane is still intact, or even in cells that will eventually recover and will not undergo death.
Another singular aspect of this process is the dual role played by mitochondria. Mitochondrial membrane permeabilization is a point of no return in the decision to initiate cell suicide. Bax/Bak-dependent apoptosis is generally considered as a noninflammatory type of cell death. However, our results show that Bax and Bak contribute actively to the induction of the IFN response. The concomitant activation of caspases is required to maintain this type of cell death immunologically silent. This mechanism likely provides the cell with an additional level of control over the decision of whether to die with or without alerting the immune system. One physiological situation in which Bax/Bak-dependent induction of type I IFNs could occur is in the context of infection by viruses that express caspase inhibitors (Best, 2008; Callus and Vaux, 2007; Tait et al., 2014). Those virally encoded caspase inhibitors represent an evolutionary response of viruses to host antiviral defenses (i.e. the suicide of infected cells) (Best, 2008; Callus and Vaux, 2007). Our model suggests that sensing caspase inhibition could be a mechanism by which host cells trigger an antiviral response, independently of the physical sensing of viral nucleic acids.
Three important questions remain to be elucidated:
What is the nature of the mtDNA involved and how does it come into contact with cGAS? Given the size of the Bax/Bak pore, it is likely that small fragments of mtDNA, rather than entire copies of mitochondrial genome are released. Since such fragments are experimentally difficult to detect and to quantify reliably, further studies are needed to investigate this possibility. Studying the process of mtDNA release is complicated by the technical challenges of isolating cytosolic and mitochondrial fractions while maintaining the absolute integrity of mitochondria (without release of mtDNA) and of nucleic acids. Furthermore, our knowledge of the physiological mechanisms of mtDNA turnover and degradation is still incomplete (Clay Montier et al., 2009).
How do caspases prevent mtDNA-dependent activation of the cGAS pathway? Our results suggest that caspases act upstream of STING activation. cGAS or a regulator of cGAS could be targets for caspase-dependent cleavage. Another possibility is that caspase-dependent nucleases (Nagata, 2005) could degrade mtDNA, thus preventing its binding to cGAS. Finally, caspases could affect the release of mtDNA from mitochondria.
In which cells does this process occur in vivo? Dying cells, or cells that undergo incomplete MOMP and do not die, are the most probable source of Bax/Bak-dependent caspase-regulated type I IFNs. Alternatively, all cells could produce type I IFN at low levels and/or transiently, due to leakiness of the Bax/Bak pore in healthy cells.
Answering those questions experimentally will require the development of novel experimental protocols that would allow the simultaneous monitoring of mtDNA release, of cGAS activity and of caspase activation at the single-cell level.
A surprising observation is that caspase-deficient animals do not develop any symptoms of autoimmune disease, despite the constitutive activation of the type I IFN response. This observation suggests that caspase deficiency could affect the function of other aspects of the immune response. Future studies will determine how caspases contribute to the regulation of adaptive immunity.
Finally, numerous pharmacological inhibitors of caspases have been developed and are being tested in clinical trials, with the aim of preventing tissue damage caused by pathological cell death (Callus and Vaux, 2007). Our results suggest that the consequences of a chronic IFN response induced by these inhibitors should be evaluated. Conversely, we propose that caspase inhibitors could be used to induce an IFN response (i.e. the expression of ISGs) while minimizing the adverse effects caused by interferon therapies (Vilcek, 2006), as caspase inhibition induces only very low levels of type I IFNs.
It has long been known that apoptosis is an immunologically silent form of cell death but the molecular basis of this is unknown. We demonstrate a new function of mitochondria in the induction of a type I IFN-mediated cell intrinsic immune response in dying cells. Caspases, activated by mitochondria, are required to silence that immune process in apoptotic cells.
Experimental procedures
Mice
Conditional knockout mice with a floxed caspase-9 allele or a floxed caspase-3 allele were generated as described in the extended experimental procedures and illustrated in Figure S1. Caspase-9, Caspase-3, Caspase-7, Apaf-1, IFNAR1, IRF-3, IRF-7, MAVS and cGAS deficient mice have been reported previously (Honda et al., 2005; Kuida et al., 1998; Kuida et al., 1996; Lakhani et al., 2006; Li et al., 2013; Muller et al., 1994; Sato et al., 2000; Sun et al., 2006; Yoshida et al., 1998). All animal experimentations were performed in compliance with Yale Institutional Animal Care and Use Committee protocols.
Cell cultures
Primary MEFs were generated from caspase-9 KO, IFNAR1 KO, caspase-9/IFNAR1 double KO, caspases-3/-7 double KO, Apaf-1 KO, caspase-9/IRF-3/IRF-7 triple KO, and caspase-9/MAVS double KO and respective littermate control embryos (E16.5-18.5). All primary MEFs used for experiments were from passage 4 or less. Bax/Bak double KO and control immortalized MEFs were provided by Dr. C. Thompson (University of Pennsylvania) (Wei et al., 2001), and primary STING KO MEFs by Dr. G. Barber (University of Miami) (Ishikawa et al., 2009). SV40-immortalized Casp9 WT and KO MEFs were reported previously (Masud et al., 2007).
Herring testis DNA (HT-DNA, Sigma-Aldrich, 3 μg/ml) and poly(I:C) (Invivogen, 1 μg/ml) were transfected using Lipofectamine 2000 (Invitrogen, 3 μl/ml). IFNα (used at 50 U/ml) and anti IFNα/β antibodies (used at 300 neutralizing U/ml each) were obtained from Hycult biotech and PBL Assay Science, respectively. Z-VAD-fmk, Boc-D-fmk (EMD Millipore), Q-VD-OPH (MP Biomedicals) and ABT-737 (SantaCruz Biotechnology) were used at 10 μg/ml. Staurosporine and Etoposide were obtained from Sigma-Aldrich and used at 0.01 μM or 10 μM, respectively.
Viral infections
Mice were infected with EMCV by intraperitoneal injection of 2×103 TCID50 of the virus diluted in 100 μl PBS. Mice were sacrificed and heart harvested 48h later, or survival was monitored for 27 days.
For VSV infection, mice were anesthetized with methoxyflurane (Anafane) and 106 PFU of VSV in 50 μl PBS were administered intranasally. The mice were sacrificed 24h later. Blood was collected by cardiac puncture and transferred in heparinized tubes. The samples were then centrifuged (2 minutes at 3000 rpm) and dilutions of the plasma were used for viral titration.
For in vitro infections, cells were plated at a density of 105 cells/ml in 6-well plates. The next day, the cells were infected with VSV-GFP, VSV-DsRed, HSV-2 or EMCV (diluted in 500 μl of DMEM without FBS) for 1h with gentle shaking every 15 minutes. The cells were then washed and incubated in 1 ml of media containing 10% FBS. The dose of virus used for infection and the duration of the incubation are indicated in figure legends. Cell death was measured by flow cytometry after staining with Annexin V-APC and propidium iodide (BD Biosciences) or by LDH release assay (CytoTox 96 assay, Promega).
Type I IFN bioassay
To measure type I IFN bioactivity, undiluted culture supernatants were transferred on cultures of the L929-pISRE-Luc reporter cell line (Jiang et al., 2005). The luciferase activity in cell lysates was measured 24h later (Dual-Luciferase Reporter Assay, Promega)
Mitochondrial DNA depletion
Cells were treated with ethidium bromide (150 ng/ml or 450 ng/ml for 4 days; Sigma-Aldrich) or dideoxycytidine (40 μg/ml for 6 days; Sigma-Aldrich), RNA was extracted and the expression of ISGs was measured by realtime RT-PCR. For induction of IFNβ expression by mtDNA-depleted cells, the cells were cultivated for 4 days in the presence of EtdBr, replated, cultivated overnight in the absence of EtdBr and then stimulated as indicated. To measure the efficiency of mtDNA depletion, total extracts were prepared by resuspending the cells in NaOH 50 mM, incubation at 95°C for 1h, and neutralization by adding 10% volume Tris 1M pH 7.5. The ratio of mtDNA (DLoop) vs. genomic DNA (TERT) was measured by SybrGreen realtime PCR using the following primer pairs:
DLoop Forward: AATCTACCATCCTCCGTGAAACC
DLoop Reverse: TCAGTTTAGCTACCCCCAAGTTTAA
TERT Forward: CTAGCTCATGTGTCAAGACCCTCTT
TERT Reverse: GCCAGCACGTTTCTCTCGTT
Statistical analysis
The means of two groups were compared using two-tailed unpaired Student t test. When three groups were compared, we used a one way ANOVA test. When there were two variables, we used a two way ANOVA test, followed by Games-Howell or Tukey post hoc test to compare pairs of means. Survival curves were compared using Mantel-Cox test.
Supplementary Material
Figure S1: Generation and validation of caspase-9 and caspase-3 conditional KO mice (related to Figure 1).
(A) Schematic representation of the targeting strategy used to flank exon VI of the Casp9 gene with LoxP sites and to excise it, or exon II of the Casp3 gene. The arrows A, B and C indicate the position of the primers used to determine the genotype of the mice by PCR.
(B) PCR identification, using primers B and C, of the different caspase-9 alleles in genomic DNA of ear (in which the Tie2-Cre transgene is not expressed and the floxed allele is not deleted) and in white blood cells (WBC, in which the floxed allele is deleted due to Tie2-Cre expression) of mice of the indicated genotypes.
(C) Western blot analysis of the caspase-9 protein in thymocytes of mice of the indicated genotypes.
(D) Thymocytes from the indicated mice were treated with the death-inducing agents etoposide (10 μM) or staurosporine (0.01 μM) and cell survival was measured by Annexin V and propidium iodide staining.
(E) Western blot analysis of the caspase-3 protein in thymocytes of mice of the indicated genotypes.
(F) Thymocytes from the indicated mice were treated with the death-inducing agents etoposide (10 μM) or staurosporine (0.01 μM) and cell survival was measured by Annexin V and propidium iodide staining.
Figure S2 : Reduced susceptibility of Casp9 KO cells to infection by EMCV and HSV-2 (related to Figure 1).
(A) Casp9 WT and KO MEFs were infected with EMCV and viral progeny production was determined by TCID50 assay 12 hours after infection with the indicated MOI of EMCV (mean ± s.d. of duplicates).
(B) Casp9 WT and KO MEFs were infected with HSV-2 (MOI = 0.1). The expression of the HSV envelope gp. was determined by flow cytometry (left panel) and viral progeny production was measured by plaque assay (right panel) at the indicated time points after infection (mean ± s.d. of triplicates).
Results are representative of two independent experiments.
Figure S3 : Increased expression of ISGs, but not inflammatory cytokines, in the absence of active caspases (related to Figure 2).
(A) The expression level of selected ISGs and pro-inflammatory cytokines was measured by real-time PCR in Casp9 WT and KO primary MEFs. Treatment with IFNα (50 U/ml) or intracellular poly(I:C) were used as positive controls to induce the activation of the type I IFN response (mean ± s.d. of duplicates; N.D. : not detectable). *, p<0.05; **, p<0.01; ***, p<0.001; ns, not significant (two-tailed unpaired Student t-test)
(B) Embryonic tissues (liver and head) were isolated from embryos at day E16.5 and the expression of ISG15 and IRF-7 were determined by realtime RT-PCR (mean ± s.d.).
(C) WT primary MEFs were treated with vehicle (DMSO), with caspase inhibitors (Z-VAD-fmk or Boc-D-fmk, 10 μM), or a positive control (intracellular poly(I:C)) and the expression levels of ISG15 and IRF-7 were measured by realtime RT-PCR (mean ± s.d. of duplicates, representative of 3 independent experiments).
(D) The expression level of IRF-7 was measured in IFNAR1 WT and KO MEFs treated with vehicle or Q-VD-OPH (10 μM) (mean ± s.d. of triplicates, representative of 2 independent experiments). *, p<0.05; ns, not significant; pairwise comparisons following two-way ANOVA.
Figure S4 : The supernatant of Casp9 KO cells requires type I IFN signaling in targer cells to confer resistance to VSV infection (related to Figure 3).
IFNAR1 WT or KO primary MEFs were incubated for 16 hours in the presence of conditioned supernatants from Casp9 WT and KO MEFs. The cells were washed and infected with VSV-GFP (MOI = 0.5, 24h). The percentage of GFP-positive cells was determined by flow cytometry (left) and viral progeny production was measured by plaque assay (right). Results represent mean ± s.d. of triplicates.
Figure S5 : Caspase deficiency does not induce autoimmunity (related to Figure 2).
(A and B) Total IgG (A) and total IgM (B) concentrations were measured by ELISA in the serum of mice of the indicated genotype. Each dot represents a mouse and the horizontal bars indicate mean values. Black dots, 7-10 months old mice; red dots, 15-17 months old mice.
(C) The presence of antinuclear antibodies in the serum of the same mice was determined by indirect fluorescence antibody technique.
Figure S6 : Bcl-2 and Bax/Bak-dependent regulation of the IFN response (related to Figure 4).
(A) Wildtype immortalized MEFs were transduced with MSCV-Bcl2-ires-GFP or control retroviruses, and transduced cells were selected by flow cytometry sorting based on GFP expression. The cells were treated with staurosporine and cell survival was measured by Annexin V and propidium iodide staining to confirm the functional efficacy of Bcl-2 overexpression.
(B) The cells were infected with VSV-DsRed (MOI = 0.5) and their morphology was observed by microscopy 24h later.
(C) Infected cells were analyzed for cell death (LDH release assay, left panel), DsRed expression (middle panel) and viral progeny production (right panel). Results are presented as mean ± s.d. of triplicates.
(D) Expression of TNFα and IL-6 mRNA by WT primary MEFs at the indicated time points after stimulation with vehicle (DMSO), Bcl-2 inhibitor (ABT-737, 10 μM), caspase inhibitor (Q-VD-OPH, 10 μM) or both inhibitors (mean ± s.d. of duplicates).
(E) WT immortalized MEFs were treated for 6h with ABT-737 + cycloheximide (CHX, 10 μM each) (to induce mitochondria-dependent, caspase-9 dependent apoptosis) or with TNFα (10 ng/ml) + CHX (to induce mitochondria-independent, caspase-8 dependent apoptosis), in the presence of vehicle (DMSO) or the caspase inhibitor Q-VD-OPH (10 μM), and the expression of IFNβ mRNA was measured by realtime RT-PCR (mean ± s.d. of duplicates). CHX is required for the induction of apoptosis by TNFα; it was added to the ABT-737 stimulation in this experiment in order to control for the potential effect of translation inhibition on the IFN response.
Figure S7 : Type I IFN expression independent of TLR signaling (related to Figure 5).
Expression of IFNβ mRNA by WT, TRIF/MyD88 double KO and TLR9 KO bone marrow-derived macrophages after 6h of treatment with vehicle (DMSO) or ABT-737 + Q-VD-OPH (10 μM each; mean ± s.d. duplicates).
Table S1 : RNA sequencing analysis of cells treated with caspase inhibitor (related to Figures 2 and 3).
RNA sequencing data of genes differentially expressed (q<0.05, fold difference >= 5) in IFNAR1 WT and KO primary MEFs stimulated for 48h with vehicle (DMSO) or with the capase inhibitor Q-VD-OPH (10 μM), and determined by RNA sequencing on duplicate samples.
Table S2 : Survival of Casp9 KO mice (related to Figure 3).
Numbers of frequencies of Casp9+/+, Casp9+/- and Casp9-/- mice obtained from the intercross caspase-9 heterozygous (with or without additional deficiency in IFNAR1) at the indicated embryonic (E) and postnatal (P) days.
Acknowledgments
We thank C. Thompson, G. Barber, T. Mak, R. Medzhitov, B. Beutler and R. Verma for providing reagents; A. Ferrandino, L. Evangelisti, J. Stein and C. Hughes for ES cell work; F. Sutterwala and N. Palm for comments on the manuscript; J. Alderman for managerial support; and C. Lieber for manuscript submission. This work was funded by NIAID AI082030, DOD W81XWH and Blavatnik Family Foundation M157176 (to R.A.F.); by NIH R01-AI093967 and Cancer Prevention and Research Institute of Texas RP120718-P3 (to Z.J.C); NIH R01-AG047632 and the United Mitochondrial Disease Foundation (to G.S.S.); by a Grant-In-Aid for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to T.T.); and by a postdoctoral fellowship PF-13-035-01-DMC from the American Cancer Society (to A.P.W.). Christian C D Harman is a Howard Hughes Medical Institute International Student Research fellow. Z.J.C., A.I. and R.A.F. are Investigators of the Howard Hughes Medical Institute.
Footnotes
Author contributions: A.R., R.J., T.L., M.d.Z. and B.Y. performed experiments. A.R., R.J., C.H. and R.A.F. analyzed results. C.Y.K., S.A.L. and A.R. generated conditional knockout mice. A.P.W., Y.W., T.T., G.S.S., Z.J.C. and A.I. provided scientific advice and reagents. A.R. and R.A.F. conceived the project and wrote the manuscript. R.A.F. supervised research.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Best SM. Viral subversion of apoptotic enzymes: escape from death row. Annual review of microbiology. 2008;62:171–192. doi: 10.1146/annurev.micro.62.081307.163009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Chiu YH, Chen ZJ. The cGAS-cGAMP-STING Pathway of Cytosolic DNA Sensing and Signaling. Molecular cell. 2014;54:289–296. doi: 10.1016/j.molcel.2014.03.040. [DOI] [PubMed] [Google Scholar]
- Callus BA, Vaux DL. Caspase inhibitors: viral, cellular and chemical. Cell death and differentiation. 2007;14:73–78. doi: 10.1038/sj.cdd.4402034. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Green DR. Do inducers of apoptosis trigger caspase-independent cell death? Nature reviews Molecular cell biology. 2005;6:268–275. doi: 10.1038/nrm1573. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Molecular cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay Montier LL, Deng JJ, Bai Y. Number matters: control of mammalian mitochondrial DNA copy number. Journal of genetics and genomics = Yi chuan xue bao. 2009;36:125–131. doi: 10.1016/S1673-8527(08)60099-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742–758. doi: 10.1016/j.cell.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough DJ, Messina NL, Clarke CJ, Johnstone RW, Levy DE. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity. 2012;36:166–174. doi: 10.1016/j.immuni.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W, et al. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–352. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- Hashiguchi K, Zhang-Akiyama QM. Establishment of human cell lines lacking mitochondrial DNA. Methods in molecular biology (Clifton, NJ) 2009;554:383–391. doi: 10.1007/978-1-59745-521-3_23. [DOI] [PubMed] [Google Scholar]
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annual review of biochemistry. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Huber M, Kalis C, Keck S, Galanos C, et al. CD14 is required for MyD88-independent LPS signaling. Nature immunology. 2005;6:565–570. doi: 10.1038/ni1207. [DOI] [PubMed] [Google Scholar]
- Kaguni LS. DNA polymerase gamma, the mitochondrial replicase. Annual review of biochemistry. 2004;73:293–320. doi: 10.1146/annurev.biochem.72.121801.161455. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA. Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. The Journal of experimental medicine. 2001;193:741–754. doi: 10.1084/jem.193.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno H, Konno K, Barber GN. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell. 2013;155:688–698. doi: 10.1016/j.cell.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annual review of immunology. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell death and differentiation. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- Kumar S. Caspase function in programmed cell death. Cell death and differentiation. 2007;14:32–43. doi: 10.1038/sj.cdd.4402060. [DOI] [PubMed] [Google Scholar]
- Lakhani SA, Masud A, Kuida K, Porter GA, Jr, Booth CJ, Mehal WZ, Inayat I, Flavell RA. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science (New York, NY) 2006;311:847–851. doi: 10.1126/science.1115035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science (New York, NY) 2013;341:1390–1394. doi: 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo YM, Gale M., Jr Immune signaling by RIG-I-like receptors. Immunity. 2011;34:680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SJ, Henry CM, Cullen SP. A perspective on mammalian caspases as positive and negative regulators of inflammation. Molecular cell. 2012;46:387–397. doi: 10.1016/j.molcel.2012.04.026. [DOI] [PubMed] [Google Scholar]
- Masud A, Mohapatra A, Lakhani SA, Ferrandino A, Hakem R, Flavell RA. Endoplasmic reticulum stress-induced death of mouse embryonic fibroblasts requires the intrinsic pathway of apoptosis. The Journal of biological chemistry. 2007;282:14132–14139. doi: 10.1074/jbc.M700077200. [DOI] [PubMed] [Google Scholar]
- Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science (New York, NY) 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Nagata S. DNA degradation in development and programmed cell death. Annual review of immunology. 2005;23:853–875. doi: 10.1146/annurev.immunol.23.021704.115811. [DOI] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nature reviews Molecular cell biology. 2007;8:405–413. doi: 10.1038/nrm2153. [DOI] [PubMed] [Google Scholar]
- Saitoh T, Tun-Kyi A, Ryo A, Yamamoto M, Finn G, Fujita T, Akira S, Yamamoto N, Lu KP, Yamaoka S. Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nature immunology. 2006;7:598–605. doi: 10.1038/ni1347. [DOI] [PubMed] [Google Scholar]
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annual review of immunology. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson DB. Connections between antiviral defense and autoimmunity. Current opinion in immunology. 2009;21:244–250. doi: 10.1016/j.coi.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity. 2006;25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Sun Q, Sun L, Liu HH, Chen X, Seth RB, Forman J, Chen ZJ. The specific and essential role of MAVS in antiviral innate immune responses. Immunity. 2006;24:633–642. doi: 10.1016/j.immuni.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nature reviews Molecular cell biology. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- Tait SW, Ichim G, Green DR. Die another way - non-apoptotic mechanisms of cell death. Journal of cell science. 2014;127:2135–2144. doi: 10.1242/jcs.093575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annual review of immunology. 2008;26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Takaoka A. A weak signal for strong responses: interferon-alpha/beta revisited. Nature reviews Molecular cell biology. 2001;2:378–386. doi: 10.1038/35073080. [DOI] [PubMed] [Google Scholar]
- Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nature reviews Molecular cell biology. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- Upton JW, Chan FK. Staying Alive: Cell Death in Antiviral Immunity. Molecular cell. 2014;54:273–280. doi: 10.1016/j.molcel.2014.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nature reviews Molecular cell biology. 2014;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- Vilcek J. Fifty years of interferon research: aiming at a moving target. Immunity. 2006;25:343–348. doi: 10.1016/j.immuni.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science (New York, NY) 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatim N, Albert ML. Dying to replicate: the orchestration of the viral life cycle, cell death pathways, and immunity. Immunity. 2011;35:478–490. doi: 10.1016/j.immuni.2011.10.010. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nature reviews Molecular cell biology. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Generation and validation of caspase-9 and caspase-3 conditional KO mice (related to Figure 1).
(A) Schematic representation of the targeting strategy used to flank exon VI of the Casp9 gene with LoxP sites and to excise it, or exon II of the Casp3 gene. The arrows A, B and C indicate the position of the primers used to determine the genotype of the mice by PCR.
(B) PCR identification, using primers B and C, of the different caspase-9 alleles in genomic DNA of ear (in which the Tie2-Cre transgene is not expressed and the floxed allele is not deleted) and in white blood cells (WBC, in which the floxed allele is deleted due to Tie2-Cre expression) of mice of the indicated genotypes.
(C) Western blot analysis of the caspase-9 protein in thymocytes of mice of the indicated genotypes.
(D) Thymocytes from the indicated mice were treated with the death-inducing agents etoposide (10 μM) or staurosporine (0.01 μM) and cell survival was measured by Annexin V and propidium iodide staining.
(E) Western blot analysis of the caspase-3 protein in thymocytes of mice of the indicated genotypes.
(F) Thymocytes from the indicated mice were treated with the death-inducing agents etoposide (10 μM) or staurosporine (0.01 μM) and cell survival was measured by Annexin V and propidium iodide staining.
Figure S2 : Reduced susceptibility of Casp9 KO cells to infection by EMCV and HSV-2 (related to Figure 1).
(A) Casp9 WT and KO MEFs were infected with EMCV and viral progeny production was determined by TCID50 assay 12 hours after infection with the indicated MOI of EMCV (mean ± s.d. of duplicates).
(B) Casp9 WT and KO MEFs were infected with HSV-2 (MOI = 0.1). The expression of the HSV envelope gp. was determined by flow cytometry (left panel) and viral progeny production was measured by plaque assay (right panel) at the indicated time points after infection (mean ± s.d. of triplicates).
Results are representative of two independent experiments.
Figure S3 : Increased expression of ISGs, but not inflammatory cytokines, in the absence of active caspases (related to Figure 2).
(A) The expression level of selected ISGs and pro-inflammatory cytokines was measured by real-time PCR in Casp9 WT and KO primary MEFs. Treatment with IFNα (50 U/ml) or intracellular poly(I:C) were used as positive controls to induce the activation of the type I IFN response (mean ± s.d. of duplicates; N.D. : not detectable). *, p<0.05; **, p<0.01; ***, p<0.001; ns, not significant (two-tailed unpaired Student t-test)
(B) Embryonic tissues (liver and head) were isolated from embryos at day E16.5 and the expression of ISG15 and IRF-7 were determined by realtime RT-PCR (mean ± s.d.).
(C) WT primary MEFs were treated with vehicle (DMSO), with caspase inhibitors (Z-VAD-fmk or Boc-D-fmk, 10 μM), or a positive control (intracellular poly(I:C)) and the expression levels of ISG15 and IRF-7 were measured by realtime RT-PCR (mean ± s.d. of duplicates, representative of 3 independent experiments).
(D) The expression level of IRF-7 was measured in IFNAR1 WT and KO MEFs treated with vehicle or Q-VD-OPH (10 μM) (mean ± s.d. of triplicates, representative of 2 independent experiments). *, p<0.05; ns, not significant; pairwise comparisons following two-way ANOVA.
Figure S4 : The supernatant of Casp9 KO cells requires type I IFN signaling in targer cells to confer resistance to VSV infection (related to Figure 3).
IFNAR1 WT or KO primary MEFs were incubated for 16 hours in the presence of conditioned supernatants from Casp9 WT and KO MEFs. The cells were washed and infected with VSV-GFP (MOI = 0.5, 24h). The percentage of GFP-positive cells was determined by flow cytometry (left) and viral progeny production was measured by plaque assay (right). Results represent mean ± s.d. of triplicates.
Figure S5 : Caspase deficiency does not induce autoimmunity (related to Figure 2).
(A and B) Total IgG (A) and total IgM (B) concentrations were measured by ELISA in the serum of mice of the indicated genotype. Each dot represents a mouse and the horizontal bars indicate mean values. Black dots, 7-10 months old mice; red dots, 15-17 months old mice.
(C) The presence of antinuclear antibodies in the serum of the same mice was determined by indirect fluorescence antibody technique.
Figure S6 : Bcl-2 and Bax/Bak-dependent regulation of the IFN response (related to Figure 4).
(A) Wildtype immortalized MEFs were transduced with MSCV-Bcl2-ires-GFP or control retroviruses, and transduced cells were selected by flow cytometry sorting based on GFP expression. The cells were treated with staurosporine and cell survival was measured by Annexin V and propidium iodide staining to confirm the functional efficacy of Bcl-2 overexpression.
(B) The cells were infected with VSV-DsRed (MOI = 0.5) and their morphology was observed by microscopy 24h later.
(C) Infected cells were analyzed for cell death (LDH release assay, left panel), DsRed expression (middle panel) and viral progeny production (right panel). Results are presented as mean ± s.d. of triplicates.
(D) Expression of TNFα and IL-6 mRNA by WT primary MEFs at the indicated time points after stimulation with vehicle (DMSO), Bcl-2 inhibitor (ABT-737, 10 μM), caspase inhibitor (Q-VD-OPH, 10 μM) or both inhibitors (mean ± s.d. of duplicates).
(E) WT immortalized MEFs were treated for 6h with ABT-737 + cycloheximide (CHX, 10 μM each) (to induce mitochondria-dependent, caspase-9 dependent apoptosis) or with TNFα (10 ng/ml) + CHX (to induce mitochondria-independent, caspase-8 dependent apoptosis), in the presence of vehicle (DMSO) or the caspase inhibitor Q-VD-OPH (10 μM), and the expression of IFNβ mRNA was measured by realtime RT-PCR (mean ± s.d. of duplicates). CHX is required for the induction of apoptosis by TNFα; it was added to the ABT-737 stimulation in this experiment in order to control for the potential effect of translation inhibition on the IFN response.
Figure S7 : Type I IFN expression independent of TLR signaling (related to Figure 5).
Expression of IFNβ mRNA by WT, TRIF/MyD88 double KO and TLR9 KO bone marrow-derived macrophages after 6h of treatment with vehicle (DMSO) or ABT-737 + Q-VD-OPH (10 μM each; mean ± s.d. duplicates).
Table S1 : RNA sequencing analysis of cells treated with caspase inhibitor (related to Figures 2 and 3).
RNA sequencing data of genes differentially expressed (q<0.05, fold difference >= 5) in IFNAR1 WT and KO primary MEFs stimulated for 48h with vehicle (DMSO) or with the capase inhibitor Q-VD-OPH (10 μM), and determined by RNA sequencing on duplicate samples.
Table S2 : Survival of Casp9 KO mice (related to Figure 3).
Numbers of frequencies of Casp9+/+, Casp9+/- and Casp9-/- mice obtained from the intercross caspase-9 heterozygous (with or without additional deficiency in IFNAR1) at the indicated embryonic (E) and postnatal (P) days.