

Summary
Interleukin-17A (IL-17A) is a proinflammatory cytokine linked to rapid malignant progression of colorectal cancer (CRC) and therapy resistance. IL-17A exerts its pro-tumorigenic activity through its type A receptor (IL-17RA). However, how IL-17RA engagement promotes colonic tumorigenesis is unknown, as IL-17RA is expressed in many cell types in the tumor microenvironment, including hematopoietic, fibroblastoid and epithelial cells. Here we show that IL-17RA signals directly within transformed colonic epithelial cells (enterocytes) to promote early tumor development. IL-17RA engagement activates ERK, p38 MAPK and NF-κB signaling and promotes the proliferation of tumorigenic enterocytes who just lost expression of the APC tumor suppressor. Although IL-17RA signaling also controls production of IL-6, this mechanism makes only a partial contribution to colonic tumorigenesis. Combined treatment with chemotherapy, which induces IL-17A expression, and an IL-17A neutralizing antibody enhanced the therapeutic responsiveness of established colon tumors. These findings establish IL-17A and IL-17RA as therapeutic targets in colorectal cancer.
Introduction
A link between inflammation and cancer has long been suspected, but direct experimental evidence linking the two pathological processes has only become available in recent decades. Chronic inflammation associated with infection and autoimmune disease increases cancer risk and accelerates progression of many malignancies, including stomach, liver and colon cancers (Balkwill and Mantovani, 2001; Grivennikov et al., 2010). Pro-inflammatory cytokines and tumor infiltrating myeloid and immune cells play critical roles in almost every stage of tumorigenesis, from initiation and tumor promotion to malignant progression and metastatic spread. Even in cancers that do not arise in the context of underlying inflammation, a tumor-evoked inflammatory response plays an important promoting role in malignant progression (Grivennikov et al., 2012).
Amongst inflammatory cytokines that promote tumor development, the interleukin-17 (IL-17) family, which includes IL-17A, B, C, D, E and F (Dungan and Mills, 2011), occupies an important position in both mouse models and human cancer. IL-17A and F are the closest members of this family, and both bind to IL-17 receptors A (IL-17RA) and C (IL-17RC), whose engagement activates mitogen-activated protein kinases (MAPK), nuclear factor-kappa B (NF-κB) and CCAAT-enhancer binding protein (C/EBP) signaling pathways through the adaptor proteins Act1 and TRAF6 (Iwakura et al., 2011; Reynolds et al., 2010). IL-17A and F are produced by Th17 cells, γδT cells, natural killer T (NKT) cells, and subsets of innate lymphoid cells (ILCs) (Reynolds et al., 2010; Sutton et al., 2012; Zou and Restifo, 2010). Initial evidence for involvement of IL-17 cytokines in cancer development came from studies of mouse colonic tumorigenesis. Using the ApcMin model it was shown that infection of mice with the human enterotoxigenic Bacteroides fragilis (ETBF) bacteria triggers colitis and accelerates tumor development that is dependent on IL-17A (Wu et al., 2009). Neutralization of IL-17A with a specific antibody prevented ETBF-induced acceleration of colonic tumorigenesis (Wu et al., 2009). Retrospective clinical studies revealed that high IL-17A expression in stage I or II human colorectal tumors are associated with rapid progression to lethal metastatic disease, thus serving as a strong indicator of poor clinical outcome (Tosolini et al., 2011). Subsequent studies demonstrated that IL-17A also enhances development of colitis associated cancer (CAC) induced by the pro-carcinogen azoxymethane (AOM) and the irritant dextran sulphate sodium (DSS) (Hyun et al., 2012; Tanaka et al., 2003; Tong et al., 2012). Although IL-17A and IL-17F are related and signal through the same receptors and effector mechanisms, IL-17F was reported to attenuate CAC development (Tong et al., 2012). The divergent roles of IL-17A and IL-17F in CAC may be explained by their distinct functions in autoimmune- and chemically-induced inflammation, which is a critical step in CAC induction (Yang et al., 2008). Other studies, however, have shown that genetic ablation of either IL-17A or IL-17F attenuates tumor development in ApcMin mice, although the effect of IL-17A is much more pronounced (Chae and Bothwell, 2011; Chae et al., 2010).
A useful mouse model of colorectal tumorigenesis is provided by the so-called CPC-APC mouse in which one allele of the Apc tumor suppressor gene is deleted in the colon and subsequent Apc loss-of-heterozygocity (LOH) results in development of large colonic adenomas that progress to invasive carcinomas (Hinoi et al., 2007). Using this model, we found that early colonic adenomas exhibit substantial upregulation of IL-23 expression by tumor associated macrophages (TAM) due to loss of protective mucin expression and tight junctions between intestinal epithelial cells (IEC), which result in invasion of the barrierless adenomas by components of the microbiome (Grivennikov et al., 2012). A similar process may occur in human colonic adenomas, which also exhibit loss of mucins and junctional adhesion molecules. IL-23 induces tumoral expression of IL-17A and ablation of IL-17RA inhibited colon tumor development and progression in CPC-APC mice. These results established the protumorigenic function of a cytokine cascade in which IL-23 produced by TAM controls IL-17A production by Th17 and other lymphoid cells within the tumor microenvironment and IL-17A stimulates tumor development through IL-17RA (Grivennikov et al., 2012). More recently it was shown that IL-17C also stimulates tumor development in ApcMin mice (Song et al., 2014), presumably through IL-17RA as well. However, the mechanism by which IL-17RA engagement promotes CRC development is not clear, as the cell type in which it acts has not been identified. Whereas IL-23 receptor is expressed on hematopoietic cell types and not on adenoma epithelial cells (Grivennikov et al., 2012), many cell types in the tumor microenvironment express IL-17RA, including epithelial cells, hematopoietic cells and fibroblasts (Iwakura et al., 2011; Reynolds et al., 2010).
Here we show that IL-17RA mainly signals within transformed IEC that have lost APC expression to promote their growth and survival. IL-17RA signaling was required for outgrowth of aberrant crypt foci (ACF) into colonic tumors during the early phase of CRC development. Immune neutralization of IL-17A could inhibit tumor progression and sensitize established tumors to chemotherapy.
Results
IL-17RA signals in transformed epithelial cells
IL-17 cytokines promote tumor development in mouse models of colitis-associated or sporadic colorectal cancer by binding to IL-17RA (Chae and Bothwell, 2011; Chae et al., 2010; Grivennikov et al., 2012; Hyun et al., 2012; Song et al., 2014; Tanaka et al., 2003; Tong et al., 2012; Wu et al., 2009). However, it is not known whether IL-17RA engagement exerts its protumorigenic effect directly within epithelial cells, or akin to IL-23R (Grivennikov et al., 2012), acts by controlling production of other protumorigenic cytokines by myeloid cells or cancer-associated fibroblasts (CAF), which also express IL-17RA (Iwakura et al., 2011; Korn et al., 2009). To understand how IL-17RA promotes colorectal tumorigenesis, we employed the CPC-APC model (Hinoi et al., 2007) as previously described (Grivennikov et al., 2012). Absence of IL-17RA in all cells of CPC-APC mice resulted in a marked reduction in tumor cell proliferation and increased apoptotic adenoma cell death (Figure 1A, B). Loss of IL-17RA also resulted in reduced IL-6 expression and consequently decreased signal transducer and activator of transcription (STAT3) activation in adenoma epithelial cells (Figure 1A-C, S1), whereas stimulation of CAF isolated from CPC-APC tumors with recombinant IL-17A resulted in IL-6 mRNA induction (Figure 1D). Activation of Akt and to a lesser extent ERK signaling in tumors was also reduced after IL-17RA ablation (Figure 1E). Expression of other inflammatory cytokines in tumors, including IL-21 and TNF, was elevated upon loss of IL-17RA, although expression of the anti-inflammatory and anti-tumorigenic cytokines IL-10 and transforming growth factor-β1 (TGFβ1) (Becker et al., 2004; Mumm et al., 2011) was also elevated (Figure S1). IL-17C, and the IL-17-related chemokines CXCL1 and CXCL2 were significantly elevated in tumors compared to normal colon tissue, and were reduced in Il17ra-null mice (Figure S1). IL-17A, IL-17F and IL-11 were barely affected by Il17ra ablation (Figure S1). The mRNA amounts of IL-17RC and IL-17RE, which interact with IL-17RA to mediate IL-17A, C and F signaling, were upregulated in Il17ra ablated tumors, possibly due to compensation (Figure S1). Expression of several cytotoxic response markers was elevated in the absence of IL-17RA (Figure S2A). Loss of IL-17RA resulted in decreased infiltration of myeloid cells into tumors, but had no observable impact on angiogenesis, lymphatic vessel formation, or recruitment of CAF (Figure S2B, C). The mRNAs for Lgr5, Sox9 and Msi1, markers of intestinal stem cells and targets of Wnt signaling, were upregulated in colonic tumors but were not affected by the absence of IL-17RA, although tumoral expression of Bmi1 mRNA was slightly elevated after IL-17RA ablation (Figure S2D).
Figure 1. IL-17RA exerts its protumorigenic activity within radio-resistant cells.

(A–D) 5-months old CPC-APC mice heterozygous (+/−) or null (−/−) for the Il17ra gene were sacrificed for cryosectioning and immunostaining (A, B), ELISA (C) or immunoblot analysis (E) of colonic tumors. Data in B were determined by counting tumor epithelial cells positive for the indicated markers in 5 high magnification fields (HMF), AU: artificial unit. (D) CAF isolated from colonic tumors were stimulated with 50 ng/ml recombinant IL-17A for 4 hrs and analyzed by Q-RT-PCR analysis for IL-6 mRNA expression (n=5). (F) CPC-APC mice heterozygous (HE) or null (KO) for Il17ra were lethally irradiated at 6 weeks of age and transplanted with bone marrow of the indicated Il17ra genotypes. At 5 months of age the mice were sacrificed and tumor number and size (diameter) were measured. Tumor load is the sum of all tumor diameters. N=4. Data represent averages ± S.E.M. * p < 0.05. Scale bar = 100 μm. See also Figures S1–S3.
To test the role of IL-17RA signaling in hematopoietic cells during CRC development, we adoptively transferred heterozygous or Il17ra−/− bone marrow into irradiated CPC-APC mice. Absence of IL-17RA in hematopoietic cells had no effect on colonic tumor development (Figure S3A). In contrast, reciprocal bone marrow transfer into Il17ra−/− CPC-APC mice showed that Il17ra ablation in radio-resistant cells significantly decreased tumor development (Figure 1F). After adoptive bone marrow transfer, at least 90% of the CD45+ cells were donor derived, ruling out the possibility that residual recipient cells were masking the effect of the IL-17RA deficiency within donor hematopoietic cells (Figure S3B). Consistent with whole body Il17ra ablation, tumors from CPC-APC mice receiving Il17ra−/− bone marrow also showed increased expression of interferon-γ (IFNγ), perforin, granzyme B and IL-10 (Figure S3C). IL-21 and IL-22 mRNAs were also higher in tumors of mice lacking IL-17RA in hematopoietic cells (Figure S3C).
IL-17RA is expressed in colonic epithelial cells, CAF and different subsets of immune and myeloid cells (Figure S4A). To test if IL-17RA signals directly within colonic epithelial cells to promote CRC development, we generated a conditional Il17ra knockout mouse (Figure S4B). Insertion of a LacZ cassette allows expression of β-galactosidase under control of the Il17ra promoter. FLP-mediated deletion of the LacZ cassette generated an Il17raF allele with two Loxp sites flanking exons 3 and 4. Cre-mediated deletion generated a frame shift in exon 5, resulting in expression of a non-functional, truncated N-terminal IL-17RA fragment (Figure 2A, S4B). Cdx2Cre-mediated ablation of the Il17raF allele in mice containing one Il17ra null allele resulted in nearly complete depletion of IL-17RA mRNA in isolated IEC, which showed a marked decrease in IL-17A induced ERK phosphorylation (Figure 2B, C, S4C). The residual response to IL-17A exhibited by these cells is likely to be due to their contamination with other cell types that retain IL-17RA expression. Consistent with the results of the reciprocal bone marrow transfer described above, deletion of the Il17ra in intestinal epithelial and tumor cells resulted in a marked decrease in colon tumor number, size and load (Figure 2D, E). The strong effect on tumor multiplicity suggested that IL-17A acts early in the tumorigenic process.
Figure 2. IL-17RA signals within APC-deficient enterocytes to promote tumor development and activate ERK, p38 MAPK and NF-κB signaling.

(A) Agarose gel image of PCR-mediated genotyping of the floxed and wildtype (WT) Il17ra alleles (upper panel), and the Cre-mediated deletion product (KO, lower panel). The Il17raF allele came from the newly generated Il17raF/+ strain, whereas the null allele (−) was from a whole body Il17ra ablation (Ye et al., 2001). (B, C) Colonic IECs isolated by EpCAM-biotin staining and magnetic bead enrichment were stimulated with 50 ng/ml IL-17A for indicated periods before immunoblot (B) and Q-RT-PCR analysis (C, n=4). (D) Tumor number, size and load in CPC-APC mice (Cdx2Cre ApcF/+) harboring a specific deletion of IL-17RA in APC-deleted cells (F/−). Il17raF/+ CPC-APC mice were used as controls. N=6. (E) Representative images of CPC-APC tumor-bearing colons that either express (F/+) or do not express (F/−) IL-17RA in APC-deleted enterocytes. (F, G) APC-deleted intestinal organoids were stimulated with 50 ng/ml IL-17A for the indicated times. Whole-cell-lysates (F), cytosol and nuclear extract (G) were collected, gel separated and immunoblotted for the indicated proteins. Data are averages ± S.E.M. * p < 0.05. See also Figure S4.
To better examine the downstream signals that may mediate IL-17RA effects on cancer cell survival and proliferation, we stimulated Apc-deleted organoids derived from cultured intestinal crypts (Sato et al., 2009) of VillinCre-ERT2 ApcF/F mice (el Marjou et al., 2004) with recombinant IL-17A. 4-OH-tamoxifen-induced deletion of Apc, via Cre-ERT2-mediated recombination, resulted in a change in organoid morphology to a perfect spherical structure in 3-D culture (Figure S4D). Apc-deleted organoids grew faster than WT organoids and were resistant to withdrawal of growth factors from the culture medium (Figure S4D). Stimulation of the transformed organoids with IL-17A strongly and rapidly activated ERK, p38 MAPK and NF-κB signaling but had only a modest and delayed effect on STAT3 and JNK (Figure 2F, G). IL-17A did not affect nuclear β-catenin in transformed organoids (Figure 2G). In summary, IL-17RA signaling seems to mainly affect the ERK, p38 and NF-κB pathways.
IL-17RA controls IL-6 but IL-6 does not control tumoral IL-17A expression
As described above, IL-17RA ablation decreases expression of IL-6, a cytokine that contributes to the development of CAC (Bollrath et al., 2009; Greten et al., 2004; Grivennikov and Karin, 2008). We therefore checked the contribution of IL-6 to colonic tumorigenesis in CPC-APC mice. Il6 gene ablation in CPC-APC mice also resulted in reduced colonic tumor load (Figure 3A), but its effect was milder than the effect of the IL-17RA deficiency. Furthermore, expression of IL-17A and RORγt, the transcription factor required for IL-17A production, was barely affected by the loss of IL-6 in the tumors, suggesting that IL-6 acts downstream of IL-17 during colonic tumor development (Figure 3B, S5). Deletion of IL-6, however, resulted in increased expression of IL-1α, IL-1β, TNF and IFNγ mRNAs in tumors (Figure S5). Notably, IL-1β and IL-21 are also known to promote IL-17A production in both Th17 cells and γδT cells (Cua and Tato, 2010), suggesting that in the absence of IL-6 they may induce expression of IL-17A in tumor-associated lymphocytes. Consistent with the notion that IL-6 inhibits development of regulatory T (Treg) cells, the Treg marker Foxp3 was elevated in colonic tumors of IL-6-deficient mice (Figure S5). Thus, IL-17RA signaling in non-epithelial cells modulates the expression of several cytokines, including IL-6, whose pro-tumorigenic activity is less potent than that of IL-17A.
Figure 3. IL-6 contributes to the development of colonic tumors but not to IL-17A expression.
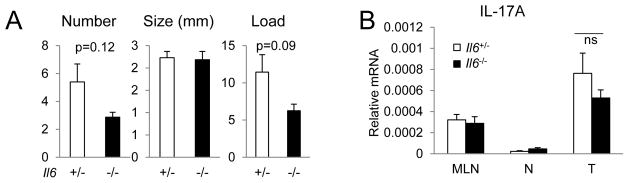
CPC-APC mice heterozygous (+/−) or null (−/−) for the Il6 gene were sacrificed at 5 months of age and analyzed for tumor parameters (A, n=5) or IL-17A mRNA by Q-RT-PCR of mesenteric lymph node (MLN), normal colon (N) and tumor (T) tissues (B, n=9). Data represent averages ± S.E.M. * p < 0.05. See also Figure S5.
IL-17RA signaling is required for growth of aberrant crypt foci
Disruption of the intestinal epithelial barrier during early tumorigenic progression due to loss of mucins and junctional adhesion proteins is followed by localized “tumor-elicited inflammation” that promotes colonic adenoma growth (Grivennikov et al., 2012). To test if IL-17RA signaling is required for early stage tumor development, we employed a model of synchronized intestinal tumorigenesis based on tamoxifen treatment of Cdx2Cre-ERT2 ApcF/F mice (Feng et al., 2011; Feng et al., 2013). One week after tamoxifen-induced bi-allelic Apc deletion, the resulting ACF lesions showed increased β-catenin expression and cell proliferation, lost expression of MUC2, and colon tissues showed upregulation of IL-17A (Figure 4A, B, S6A). Expression of IL-17C and F mRNAs was also increased starting at two weeks since tumor induction (Figure S6B). Ablation of Il17ra in this model led to a significant reduction in the number of visible tumors detected 4 weeks after tamoxifen injection (Figure 4C). To confirm a role for IL-17A signaling in early tumor development, we injected Cdx2Cre-ERT2 ApcF/F mice with an IL-17A neutralizing antibody after the last round of tamoxifen administration and still observed significantly reduced colonic tumor numbers (Figure 4D). Consistent with the notion that IL-17RA mainly signals in transformed colonic epithelial cells to promote CRC development, ablation of IL-17RA in hematopoietic cells by adoptive bone marrow transfer 3.5 months prior to tamoxifen-induced tumorigenesis resulted in no significant change in tumor multiplicity in this model (Figure S6C). Ablation of IL-17RA signaling in Apc deleted mice had no significant effect on apoptosis (caspase 3 cleavage), but significantly reduced cell proliferation within ACF lesions defined by regions with upregulation of β-catenin and loss of goblet cells (Figure 4E, F). The change in cell proliferation was restricted to transformed cells, as Cdx2Cre-ERT2 negative mice showed no signs of reduced normal crypt cell proliferation upon IL-17RA ablation (Figure S6D). Consistent with the ability of IL-17RA to activate NF-κB, ACF in Il17ra−/− Cdx2Cre-ERT2 ApcF/F mice showed reduced nuclear (activated) NF-κB p65 staining in transformed cells identified by upregulation of β-catenin and loss of mucin production in ACF lesions (Figure 4E, F). These data suggest that IL-17RA signaling stimulates the progression of early ACF lesions to adenomas by promoting the proliferation of transformed cells.
Figure 4. IL-17RA signaling is required for the growth of aberrant crypt foci.

Cdx2Cre-ERT2ApcF/F mice were injected with tamoxifen for intestinal tumor induction. (A) Q-RT-PCR analysis of IL-17A mRNA in colon tissues following tamoxifen injection. N=8. (B) Immunostaining of MUC2 in colon cryosections 1 week after tamoxifen injection. Tamoxifen-injected Cdx2Cre-ERT2 negative mice were used as controls. Arrows indicate areas of MUC2 loss. Representative images of 3 slides of each specimen are shown. (C) Cdx2Cre-ERT2 ApcF/F mice heterozygous (+/−) or null (−/−) for Il17ra were sacrificed for colon tumor count 4 weeks after tamoxifen injection. n=5. (D) Cdx2Cre-ERT2 ApcF/F mice were given tamoxifen via i.p. injection. Starting one day after the last tamoxifen dose, mice received i.p. injections of 500 ug of isotype control or anti-IL-17A antibody on a weekly basis. Colon tumors were counted 4 weeks after last tamoxifen injection. N=7. (E, F) Immunostaining of colon cryosections from Cdx2Cre-ERT2 ApcF/F mice heterozygous (+/−) or null (−/−) for Il17ra 2 weeks after tamoxifen injection. (F) Quantification of Ki-67 (n=4) and phospho-NF-κB p65 (P-p65; n=7) positive transformed cells in HMF of tamoxifen-induced ACF lesions. Data shown in arbitrary units (AU) represent averages ± S.E.M. * p < 0.05. Scale bar = 100 μm. See also Figure S6.
IL-17A neutralization potentiates the response to chemotherapy
We examined if IL-17A neutralization can be used to block the growth of colonic tumors. Intraperitoneal injection of IL-17A neutralizing antibody into CPC-APC mice with established colonic tumors resulted in reduced NF-κB activation and cancer cell proliferation (Figure 5A, B). Long-term antibody treatment starting at 2 months of age reduced tumor load compared to CPC-APC mice receiving isotype control (Figure 5C, D). These data suggest that IL-17A neutralization can inhibit the development, progression and survival of colonic tumors.
Figure 5. IL-17A neutralizing antibody reduces colonic tumor load.

(A–C) 5-month-old CPC-APC mice were injected with isotype control or anti-IL-17A antibodies for 2 weeks at 500 ug/injection/week. (A) Immunostaining of cryosections of colonic tumors. (B) Quantitation of P-p65 (n=10) and Ki-67 (n=6) positive cells in HMF from (A). (C, D) 2-month-old CPC-APC mice were injected weekly with isotype control or anti-IL-17A antibodies and sacrificed at 5 months of age for bright field imaging (C) and analysis of tumor parameters (D, n=6). Data are averages ± S.E.M. * p < 0.05. Scale bar = 100 μm.
Chemotherapy is often employed for CRC that cannot be resected or in combination with surgical removal of primary tumors (Andre et al., 2004). However, chemotherapy also causes mucositis and induces inflammation that curtails its anti-tumor effect by upregulating cytokines that promote tumor growth and survival (Bruchard et al., 2013). In our model of sporadic colonic tumorigenesis, treatment of CPC-APC mice with 5-FU, a commonly used chemotherapeutic agent in CRC, resulted in increased expression of IL-17A and F mRNAs in tumors (Figure 6A). By itself, this treatment regimen did not result in statistically significant tumor shrinkage (Figure 6B). To test if IL-17A neutralization during chemotherapy can potentiate the response to 5-FU, we treated CPC-APC mice with established colonic tumors with anti-IL-17A and 5-FU for 6 weeks starting at 3.5 months of age. This combination resulted in a reduction in tumor size and load compared to mice treated with either agent alone (Figure 6B). Neutralization of IL-17A in 5-FU-treated mice also decreased expression of IL-6 mRNA in tumors (Figure 6C). The combined treatment with IL-17A antibody and 5-FU led to a small decrease in cell proliferation and enhanced apoptosis in established colonic tumors (Figure 6D, E). These results suggest that inhibition of IL-17RA signaling may be used to potentiate the response to chemotherapy, especially agents such as 5-FU, which induce IL-17A expression.
Figure 6. Combined treatment with anti-IL-17A and 5-FU results in a stronger therapeutic effect.

(A) CPC-APC mice were treated with 5-FU (50 mg/kg) for two consecutive days each week for 6 weeks. PBS was used as a control. Colonic tumors were analyzed by Q-RT-PCR for IL-17A and IL-17F mRNAs. (B, C) 3.5-month-old CPC-APC mice were treated with isotype control (Con), anti-IL-17A (Ab), 5-FU + isotype control (5-FU) or 5-FU + anti-IL-17A (Ab + 5-FU) for 6 weeks and sacrificed for analysis of tumor parameters (B, n=8) and Q-RT-PCR analysis of IL-6 mRNA (C, n=21). (D, E) 5-month-old CPC-APC mice were treated with isotype control (Con), anti-IL-17A (Ab), 5-FU + isotype control (5-FU) or 5-FU + anti-IL-17A (Ab + 5-FU) for 2 weeks and sacrificed. Colonic tumors were embedded in paraffin blocks and analyzed for Ki-67 (D) and cleaved caspase 3 (E). 8 HMF images were used for quantification in D and E. Data are averages ± S.E.M. * p < 0.05. * p < 0.05. Scale bar = 100 μm.
Discussion
The IL-23-IL-17 axis plays a key role in tumor-associated inflammation and cancer development (Alshaker and Matalka, 2011; Martin-Orozco and Dong, 2009; Zou and Restifo, 2010). The numbers of Th17 cells and expression of IL-17A are increased in human glioma (Hu et al., 2011) and esophageal cancer (Chen et al., 2012). Increased expression of IL-23, IL-17A and TGF-β1 signals adverse prognosis and predicts poor response to chemotherapy in pancreatic carcinoma (Vizio et al., 2012). Th17 cell abundance and IL-17 expression are also elevated in gastric cancer and indicate poor prognosis (Yamada et al., 2012). In gastric cancer, CD8+ T cells also produce IL-17A (Tc17 cells) and predict reduced overall and disease-free survival (Zhuang et al., 2012). TAM in gastric cancer promote Tc17 development by producing IL-6, IL-23 and IL-1β, and IL-17A induces cancer cells to produce CXCL12, which may attract so-called myeloid-derived suppressor cells (MDSC) or immature myeloid cells that seem to impede CD8+-dependent tumor cytotoxicity (Zhuang et al., 2012). In human patients with stage I/II colorectal cancer, a high “Th17 signature” confers drastically reduced disease-free survival after resection of primary tumors (Tosolini et al., 2011). Despite the ample evidence for the likely involvement of IL-17 family members in cancer, the exact cell type within which IL-17RA engagement promotes tumor development and progression has not been identified in any cancer. It is also not clear, especially in cancers that are not induced by underlying inflammation or infection, such as sporadic CRC, at what stage of the tumorigenic process IL-17RA engagement stimulates cancer development and progression.
Mechanistic proof that elevated IL-17 expression promotes cancer development came from mouse model studies. In a mouse model of CAC induced by pathogen (ETBF) infection of ApcMin mice, antibody-mediated IL-17A neutralization blocked ETBF-induced colitis and tumorigenesis (Wu et al., 2009). IL-17A is also important for tumorigenesis in the AOM-DSS model of CAC (Hyun et al., 2012; Tanaka et al., 2003; Tong et al., 2012). However, in both of these models malignant progression is driven by deliberately induced inflammation. In sporadic colonic tumorigenesis in ApcMin mice, both IL-17A and F promote tumor development, which does not depend on deliberate induction of inflammation (Chae and Bothwell, 2011; Chae et al., 2010). Ablation of Il17ra also reduced tumorigenesis in the CPC-APC model of sporadic colorectal tumorigenesis, in which CRC development depends on LOH at the Apc locus (Grivennikov et al., 2012). IL-17RA, which dimerizes with IL-17RC and RE, can be activated by IL-17A, C, F and IL-25 (Iwakura et al., 2011). Il17a−/− and Il17ra−/− mice are also resistant to chemically-induced skin cancer (He et al., 2012; Wang et al., 2010) and IL-17A promotes development of lung cancer in mice (Chang et al., 2014). Microbiota-driven up-regulation of IL-17C also contributes to colonic tumor development through autocrine induction of Bcl-2 and Bcl-xL in malignant enterocytes (Song et al., 2014). In our model, however, IL-17A is mainly produced by tumor infiltrating Th17 cells and ILCs, whose expansion depends on IL-23 that is produced by TAM that were activated by microbial products (Grivennikov et al., 2012). IL-23R is not expressed on adenoma epithelial cells and its protumorigenic activity is exerted via IL-17RA-activating cytokines. By contrast, IL-17RA is expressed on a much broader range of cells, that in addition to hematopoietic cells include epithelial cells and fibroblasts. IL-17RA signaling in any of these cell types can potentially promote tumor development. The results described above identify transformed epithelial cells that had just lost APC expression as the main cellular site for the protumorigenic activity of IL-17RA. These conclusions are supported both by adoptive transfer experiments as well as by the specific ablation of IL-17RA in IEC that have lost at least one Apc allele. Although IL-17RA signaling also controls the expression of IL-6 within colorectal tumors, whole body Il6 gene ablation had only a minor effect on IL-17A expression and reduced tumor development to a much lower extent than IL-17RA ablation. This stands in marked contrast to the AOM+DSS CAC model, where inflammation (DSS)-induced IL-6 signaling plays a major role in tumorigenesis along with IL-11 (Bollrath et al., 2009; Grivennikov et al., 2009; Putoczki et al., 2013). Importantly, in the CPC-APC model, IL-6 does not play a critical role in induction of IL-17A expression and its function is likely to be replaced by IL-21 or IL-23 (Korn et al., 2009). Furthermore, IL-11 expression is not regulated by IL-17RA signaling and given the strong effect of Il17ra ablation, it seems that IL-11 makes little contribution to tumor development in CPC-APC mice.
Identification of the site of action of IL-17RA had also revealed how IL-17A drives tumor development. In the premalignant enterocyte that had lost expression of the Apc tumor suppressor gene, IL-17RA signaling is required for induction of cell proliferation that drives the progression of premalignant ACF lesions into adenomas. In the same cells, IL-17RA engagement activates ERK, p38 and NF-κB, signaling molecules that contribute to cell proliferation and survival. IL-17RA engagement, however, does not lead to direct STAT3 activation. Antibody mediated neutralization of IL-17A in 2 months old CPC-APC mice, which already harbor several ACF lesions and small adenomas (Hinoi et al., 2007), resulted in a marked inhibition of tumor progression. These results indicate that although IL-17A neutralization may enhance colitis-associated mucosal injury (Yang et al., 2008) and therefore may not be suitable for the treatment of IBD or CAC, which occurs in the context of ulcerative colitis, IL-17A neutralization is suitable for the treatment and prevention of sporadic CRC, in which the inflammatory response is limited to the tumor microenvironment and does not affect the surrounding normal tissue. These results also demonstrate that IL-17RA signaling stimulates malignant progression already at the very early ACF stage, acting in cells that had just lost Apc expression.
Elevated expression of IL-17 cytokines has been linked to resistance to both classical cytotoxic drugs and targeted therapeutics, such as the VEGF neutralizing antibody Avastin (Chung et al., 2013). Our results indicate that treatment of mice with 5-FU, a cytotoxic drug used in CRC treatment, results in elevated IL-17A expression within colonic tumors, which appears to attenuate the therapeutic response. Notably, even a relatively short treatment with an IL-17A neutralizing antibody, which on its own was insufficient to affect tumor development, synergized with 5-FU treatment to elicit a reduction in tumor load. These results suggest that IL-17A neutralizing or IL-17RA blocking antibodies (secukinumab, ixekizumab and brodalumab), already proven effective in autoimmune disorders such as psoriasis, psoriatic arthritis and ankylosing spondylitis (Patel et al., 2013), may be used as neo-adjuvants in the treatment of early, non-metastatic, CRC. Although 5 year survival rates in stage I and II CRC approach 90%, it seems likely that the 10% of stage I and II patients that progress to incurable metastatic disease are those whose tumors display high IL-17 expression (Tosolini et al., 2011). These patients, with extremely poor prognosis and reduced overall survival, are the ones who are likely to benefit the most from anti-IL-17A-IL-17RA neo-adjuvant therapy.
Experimental Procedures
Generation of Il17raF/F mice
ES cells (clones EPD0570_2_H07 and H08), of a C57BL/6 background, harboring an Il17raLacZ-Flox targeted allele were obtained from the Knockout Mouse Project (KOMP) Repository at the University of California, Davis. ES cells were injected into C57BL/6-albino blastocysts and transferred to pseudopregnant females. Chimeric offsprings were crossed to C57BL/6-albino mice and F1 generation were selected for transmission of the targeted allele according to coat color and genotype. Il17raLacZ-Flox allele-positive mice were crossed to a FLP deleter mouse (Rodriguez et al., 2000) to remove the LacZ and neo cassettes, resulting in Il17raF strain where two loxP sites flank exons 3 and 4 of the Il17ra gene. Cre-mediated recombination of these sites results in a frame shift and early termination of protein translation in exon 5 (Figure S4B). Genotyping of Il17ra WT and flox allele was performed by P1 (5′-GGGGTTTTTGTTGTTGTTGG -3′) and P2 (5′-GCAGCTGTTCTCAACCTTCC -3′). Ablated Il17ra allele was detected by P1 and P3 (5′-GGCCAGGATCTACCACAAAG-3′).
Mouse models
C57BL/6 control mice were obtained from Charles River Laboratories. Il17ra−/− mice were from Amgen (Ye et al., 2001). VillinCre-ERT2 mice were obtained from Dr. Sylvie Robine (el Marjou et al., 2004). ApcF/F, Cdx2Cre (CPC) and Cdx2Cre-ERT2 mice were described (Feng et al., 2011; Feng et al., 2013; Hinoi et al., 2007). Il6−/− (Kopf et al., 1994) and CD45.1 (Shen et al., 1985) mice were obtained from the Jackson Laboratory. To generate conditional ablation of IL-17RA in colonic epithelial and tumor cells of CPC-APC mice, Il17raF/F ApcF/F mice were crossed to Cdx2Cre Il17ra+/− mice, where the null Il17ra allele is derived from the whole body knockout strain (Ye et al., 2001). All mice were maintained in filter-topped cages on autoclaved food and water at UCSD and all experiments were performed in accordance with UCSD and NIH guidelines and regulations. All experiments used co-housed littermates to ensure consistency of common microflora.
Analysis of spontaneous CRC tumorigenesis was described (Grivennikov et al., 2012). For tamoxifen-inducible tumorigenesis, CDX2Cre-ERT2 ApcF/F mice were given 70 mg/kg body weight tamoxifen (Sigma, dissolved in 5% ethanol, 95% corn oil) i.p. on a daily basis for 3 consecutive days. Mice were sacrificed 1 to 4 weeks after the last dose of tamoxifen for histological analysis and tumor statistics.
Bone marrow transplantation
Six- to eight-week-old recipient mice were irradiated twice during one day to achieve a lethal dose (2 x 600 rad) and intravenously injected with single-cell suspension of 107 donor bone marrow cells. Recipients were co-housed littermates, which were transplanted with both gene-deficient and wild-type bone marrow for comparison. After transplantation the recipients were placed on sulphamethoxazole and trimethoprime in drinking water for two weeks, followed by transfer to cages from the same room/rack with dirty bedding to restore microflora. Mice were sacrificed and analyzed for tumor development 3–4 months after transplantation.
Antibody and 5-FU treatment in mice
For spontaneous CRC model, IL-17A neutralizing antibody or isotype control antibody (Bio X Cell) were i.p. injected at a dose of 500 ug on a weekly basis until sacrifice. For the tamoxifen inducible model of tumorigenesis, antibodies (500 ug) were injected starting one day after the last tamoxifen dose, on a weekly basis until sacrifice. 5-FU was given i.p. at 50 mg/kg body weight each day for 2 consecutive days, and the scheme was repeated every week. For combined treatment of spontaneous CRC model with antibody and 5-FU, antibody and 5-FU were injected on the first day of the week, while the second dose of 5-FU was given on the second day of the week. Treatment was repeated weekly. In all cases isotype antibody was used as control and PBS was used as vehicle control for 5-FU.
Antibodies and stains
Fluorescent-labelled antibodies for flow cytometry were from eBioscience. Immunoblot analysis and immunohistochemistry were performed with antibodies to Ki-67, MUC2 (GeneTex), active caspase 3, ERK1/2, phospho-ERK1/2, phospho-p38, phospho-JNK, phospho-NF-κB p65, phospho-STAT3, phosphor-Akt S473, phosphor-Akt T308 (Cell Signaling), β-catenin, HDAC1, NF-κB p65, p38 (Santa Cruz Biotechnology), CD11b, gp38, Lyve1, EpCam (eBioscience), CD31 (BD), α-smooth muscle actin (Abcam) and α-tubulin (Sigma). Secondary antibodies (host: donkey) for fluorescent microscopy labelled with Alexa 488 or 594 were from Life Technology. For ELISA analysis of IL-6, colonic tumors were cultured in DMEM containing 10% FBS for overnight and supernatant was analyzed by IL-6 ELISA kit from eBioscience. Cytokine concentration was normalized to the weight of tumors in each well.
Cell culture and cytokine treatment
Organoid culture of small intestinal crypts was previously described (Sato et al., 2009). Briefly, crypts were isolated from small intestines of VillinCre-ERT2 ApcF/F mice. Organoids were plated in Matrigel (BD Bioscience) and maintained in DMEM/F12 media (Life Technologies) containing B27 and N2 supplements (Life Technologies), 1.25 mM N-acetyl L-cysteine (Sigma), 100 ng/ml noggin (Peprotech), 50 ng/ml mEGF (Biosource), and 10% Rspo1-Fc-conditioned medium. Rspo1-Fc-expressing cell line was a generous gift from Dr. Calvin Kuo (Stanford). APC inactivation in organoids was induced with 1 μM 4-hydroxytamoxifen (Sigma). Two days after APC inactivation, organoids were maintained in DF+++ (DMEM/F12 media supplemented with GlutaMAX, HEPES, and penicillin-streptomycin). To study IL-17 signaling in vitro, APC-inactivated organoids were replenished with fresh DF+++ 3 hrs before treated with 50 ng/ml recombinant human IL-17A for indicated periods and lyzed for Western blotting analysis. CAF were purified from CPC-APC tumors by magnetic bead-mediated depletion of EpCam+ and CD45+ cells and cultured in DMEM containing 10% FBS.
Flow cytometry and cell sorting
Isolated immune cells were stained with labelled antibodies in PBS with 2% FCS and analyzed on BD LSRII or on a Beckman Coulter Cyan ADP flow cytometers. Dead cells were excluded on the basis of staining with Live/Dead fixable dye (eBioscience). IEC were purified by EpCam labeling followed by biotin positive selection with magnetic beads (Stemcell). Purified IEC were subjected to flow cytometic analysis and more than 90% of them were EpCam+.
RT–qPCR analysis
Total RNA was extracted using RNeasy Plus kit (Qiagen) and reverse transcribed using an IScript kit (Biorad). Q-RT-PCR was performed using Soofast EvaGreen supermix (Biorad) on a Biorad CFX96 machine. Expression data were normalized to RPL32 mRNA levels. The data are presented in arbitrary units and were calculated as 2(Ct(RPL32–gene of interest)). Primer sequences are listed in Supplementary Table 1 and generally were obtained from the NIH qPrimerDepot (http://mouseprimerdepot.nci.nih.gov). Whenever possible, primers were intron-spanning, such that amplification is only feasible on complementary DNA.
Statistical analysis
Data are presented as averages +/− S.E.M. and were analyzed by Students’ t-test. P values less than 0.05 were considered significant.
Supplementary Material
Five-month old tumor-bearing CPC-APC mice harboring heterozygous (+/−) or knockout (−/−) Il17ra gene were sacrificed for Q-RT-PCR analysis of the indicated cytokine mRNAs in normal (N) and tumor (T) tissues. N=11. Data represent averages ± S.E.M. * p < 0.05.
(A, D) Q-RT-PCR analysis of the indicated mRNAs in normal (N) and tumor (T) tissues of 5-month old CPC-APC mice. N=11. (B, C) Immunostaining of colon tumors that are heterozygous or null for Il17ra. Six HMF images taken on tumor sections from four mice on each side were used for quantification by measuring immuno-reactive areas (IRA, C). Data represent averages ± S.E.M. * p < 0.05. Scale bar = 100 μm.
(A) Six weeks old CPC-APC mice underwent adoptive transfer of bone marrow from Il17ra heterozygous (+/−) or null (−/−) donors. Mice were sacrificed at 5 months of age and tumor parameters were determined (n=12). (B) Six weeks old CPC-APC mice (C57BL/6 background) were irradiated and transplanted with bone marrow from CD45.1/CD45.2 C57BL/6 mice. Recipient mice were sacrificed at 5 months of age and cells from spleen, mesenteric lymph nodes (MLN), normal colon tissue (Normal) and colonic tumors (Tumor) were collected and stained for CD45.1 and CD45.2 and analyzed by flow-cytometry. Live CD45+ cells were gated on in the graphs. Percentages of donor-originated CD45.1/CD45.2 double positive cells indicate the rate of reconstitution. (C) Q-RT-PCR analysis of mRNAs corresponding to the indicated genes in normal colon (N) and tumor (T) tissues from CPC-APC mice that received heterozygous or Il17ra-null bone marrow (n=18). Data represent averages ± S.E.M. * p < 0.05.
(A) Q-RT-PCR analysis of IL-17RA mRNA in purified cells from colon tumors. CAF: cancer associated fibroblasts; IEC: intestinal epithelial cells. N=4. (B) Scheme for conditional knockout of the Il17ra locus. Il17raLacZ-Flox mice were generated by conventional ES cell targeting and blastocyst injection. Transgene positive mice were crossed to a FLP+ strain for deletion of the FRT-flanked LacZ cassette to generate the Il17raF/+ strain. (C) Quantification on the amount of activated ERK assayed by Western blotting in IL-17A stimulated IEC isolated from control or Cdx2Cre Il17raF/− mice. α-tubulin was used as loading control. N=2 for control, 3 for Il17ra conditional knockout. (D) Bright-field images of WT and Apc-deleted (Apc IEC) small intestinal organoids in 3-D culture, supplied with complete culture medium (left and middle panels). Right panel: Apc-deleted (Apc IEC) small intestinal organoids maintained and propagated in medium without serum and growth factors (DF+++) for more than one month. Scale bar = 100 μm.
CPC-APC mice that are heterozygous (+/−) or null (−/−) for the Il6 gene were sacrificed at 5 months of age for Q-RT-PCR analysis of the indicated genes in mesenteric lymph nodes (MLN), normal (N) colon and tumor (T) tissues. n=9. Data represent averages ± S.E.M. * p < 0.05.
(A) Immunostaining of colon cryosections from ApcF/F (Control) or Cdx2Cre-ERT2 ApcF/F (TAM 1wk) mice 1 week after tamoxifen injection. (B) Q-RT-PCR analysis of IL-17A and IL-17C mRNAs in colon tissues following tamoxifen injection (n=8). (C) Cdx2Cre-ERT2 ApcF/F mice underwent adoptive transfer of bone marrow cells from control (Il17ra+/−) or Il17ra−/− donors. At 5 months of age, the recipient mice were i.p. injected with tamoxifen and sacrificed one month later for analysis of colon tumor parameters (n=5). (D) Immunostaining of cryosections of colons from ApcF/F mice that are heterozygous (+/−) or null (−/−) for Il17ra 1 week after tamoxifen injection. Data represent averages ± S.E.M. * p < 0.05. Scale bar = 100 μm.
Supplemental Table 1: List of primers used in Q-RT-PCR analysis, related to Experimental Procedures.
Acknowledgments
We thank Dr. Shujuan Chen for scientific discussion; eBioscience, GeneTex, Santa Cruz, and Cell Signaling for antibodies; Amgen and Dr. Sylvie Robine for Il17ra−/− and VillinCre-ERT2 mice, respectively. This work was supported by the Croucher Foundation and China Postdoctoral Science Foundation (20110490919) to K.W.; Italian Association for Cancer Research, AIRC Fellowship for Abroad to G.D.C.; Canadian Institutes of Health Research Postdoctoral Fellowship to J. Wong; German Research Foundation Fellowship to S.S.; Shenzhen Science and Technology Project (GJHZ20120616153140827) to J. Wan; Cancer Research Institute Irvington Postdoctoral Fellowship to Z.Z.; A traveling grant NSC-101-2918-I-006-005 by National Science Council in Taiwan to L.W.W.; Postdoctoral Fellowship for Research Abroad, the Research Fellowship for Young Scientists from the Japan Society for the Promotion of Science, and the Uehara Memorial Foundation Fellowship to K.T.; NIH/NIDDK R00DK088589, FCCC-Temple University Nodal grant, AACR-Landon Innovator Award in Tumor Microenvironment, and the Pew Scholar in Biomedical Sciences Program for S.I.G.; and NIH (AI043477; DK035108) and American Association for Cancer Research (07-60-21-KARI) grants to M.K., who is an American Cancer Society Research Professor and the Ben and Wanda Hildyard Chair for Mitochondrial and Metabolic Diseases.
Footnotes
Author Contributions
K.W. and M.K. conceived the project. K.W., M.K.K, G.D.C, J.W., S.S., J.W., W.Z., Z.Z., E.S.L., L.W.W., and K.T. performed the experiments and analyzed the data. Y.F. and E.F. generated CPC-APC mice and Cdx2Cre-ERT2 mice. E.F. and S.I.G. provided conceptual advice. K.W. and M.K. wrote the manuscript, with all authors contributing to the writing and providing advice.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alshaker HA, Matalka KZ. IFN-gamma, IL-17 and TGF-beta involvement in shaping the tumor microenvironment: The significance of modulating such cytokines in treating malignant solid tumors. Cancer cell international. 2011;11:33. doi: 10.1186/1475-2867-11-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre T, Boni C, Mounedji-Boudiaf L, Navarro M, Tabernero J, Hickish T, Topham C, Zaninelli M, Clingan P, Bridgewater J, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. The New England journal of medicine. 2004;350:2343–2351. doi: 10.1056/NEJMoa032709. [DOI] [PubMed] [Google Scholar]
- Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- Becker C, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A, Burg J, Strand S, Kiesslich R, Huber S, et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004;21:491–501. doi: 10.1016/j.immuni.2004.07.020. [DOI] [PubMed] [Google Scholar]
- Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, Nebelsiek T, Lundgren-May T, Canli O, Schwitalla S, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer cell. 2009;15:91–102. doi: 10.1016/j.ccr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Bruchard M, Mignot G, Derangere V, Chalmin F, Chevriaux A, Vegran F, Boireau W, Simon B, Ryffel B, Connat JL, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nature medicine. 2013;19:57–64. doi: 10.1038/nm.2999. [DOI] [PubMed] [Google Scholar]
- Chae WJ, Bothwell AL. IL-17F deficiency inhibits small intestinal tumorigenesis in ApcMin/+ mice. Biochemical and biophysical research communications. 2011;414:31–36. doi: 10.1016/j.bbrc.2011.09.016. [DOI] [PubMed] [Google Scholar]
- Chae WJ, Gibson TF, Zelterman D, Hao L, Henegariu O, Bothwell AL. Ablation of IL-17A abrogates progression of spontaneous intestinal tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:5540–5544. doi: 10.1073/pnas.0912675107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SH, Mirabolfathinejad SG, Katta H, Cumpian AM, Gong L, Caetano MS, Moghaddam SJ, Dong C. T helper 17 cells play a critical pathogenic role in lung cancer. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:5664–5669. doi: 10.1073/pnas.1319051111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Hu Q, Mao C, Jiao Z, Wang S, Yu L, Xu Y, Dai D, Yin L, Xu H. Increased IL-17-producing CD4(+) T cells in patients with esophageal cancer. Cellular immunology. 2012;272:166–174. doi: 10.1016/j.cellimm.2011.10.015. [DOI] [PubMed] [Google Scholar]
- Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J, Vernes JM, Jiang Z, Meng YG, Peale FV, et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nature medicine. 2013;19:1114–1123. doi: 10.1038/nm.3291. [DOI] [PubMed] [Google Scholar]
- Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nature reviews Immunology. 2010;10:479–489. doi: 10.1038/nri2800. [DOI] [PubMed] [Google Scholar]
- Dungan LS, Mills KH. Caspase-1-processed IL-1 family cytokines play a vital role in driving innate IL-17. Cytokine. 2011;56:126–132. doi: 10.1016/j.cyto.2011.07.007. [DOI] [PubMed] [Google Scholar]
- el Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, Robine S. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- Feng Y, Bommer GT, Zhao J, Green M, Sands E, Zhai Y, Brown K, Burberry A, Cho KR, Fearon ER. Mutant KRAS promotes hyperplasia and alters differentiation in the colon epithelium but does not expand the presumptive stem cell pool. Gastroenterology. 2011;141:1003–1013. e1001–1010. doi: 10.1053/j.gastro.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Sentani K, Wiese A, Sands E, Green M, Bommer GT, Cho KR, Fearon ER. Sox9 induction, ectopic Paneth cells, and mitotic spindle axis defects in mouse colon adenomatous epithelium arising from conditional biallelic Apc inactivation. The American journal of pathology. 2013;183:493–503. doi: 10.1016/j.ajpath.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, Karin M. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov S, Karin M. Autocrine IL-6 signaling: a key event in tumorigenesis? Cancer Cell. 2008;13:7–9. doi: 10.1016/j.ccr.2007.12.020. [DOI] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung KE, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491:254–258. doi: 10.1038/nature11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He D, Li H, Yusuf N, Elmets CA, Athar M, Katiyar SK, Xu H. IL-17 mediated inflammation promotes tumor growth and progression in the skin. PloS one. 2012;7:e32126. doi: 10.1371/journal.pone.0032126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinoi T, Akyol A, Theisen BK, Ferguson DO, Greenson JK, Williams BO, Cho KR, Fearon ER. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer research. 2007;67:9721–9730. doi: 10.1158/0008-5472.CAN-07-2735. [DOI] [PubMed] [Google Scholar]
- Hu J, Mao Y, Li M, Lu Y. The profile of Th17 subset in glioma. International immunopharmacology. 2011;11:1173–1179. doi: 10.1016/j.intimp.2011.03.015. [DOI] [PubMed] [Google Scholar]
- Hyun YS, Han DS, Lee AR, Eun CS, Youn J, Kim HY. Role of IL-17A in the development of colitis-associated cancer. Carcinogenesis. 2012;33:931–936. doi: 10.1093/carcin/bgs106. [DOI] [PubMed] [Google Scholar]
- Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity. 2011;34:149–162. doi: 10.1016/j.immuni.2011.02.012. [DOI] [PubMed] [Google Scholar]
- Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, Zinkernagel R, Bluethmann H, Kohler G. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature. 1994;368:339–342. doi: 10.1038/368339a0. [DOI] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annual review of immunology. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- Martin-Orozco N, Dong C. The IL-17/IL-23 axis of inflammation in cancer: friend or foe? Curr Opin Investig Drugs. 2009;10:543–549. [PubMed] [Google Scholar]
- Mumm JB, Emmerich J, Zhang X, Chan I, Wu L, Mauze S, Blaisdell S, Basham B, Dai J, Grein J, et al. IL-10 elicits IFNgamma-dependent tumor immune surveillance. Cancer cell. 2011;20:781–796. doi: 10.1016/j.ccr.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Patel DD, Lee DM, Kolbinger F, Antoni C. Effect of IL-17A blockade with secukinumab in autoimmune diseases. Annals of the rheumatic diseases. 2013;72(Suppl 2):ii116–123. doi: 10.1136/annrheumdis-2012-202371. [DOI] [PubMed] [Google Scholar]
- Putoczki TL, Thiem S, Loving A, Busuttil RA, Wilson NJ, Ziegler PK, Nguyen PM, Preaudet A, Farid R, Edwards KM, et al. Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer cell. 2013;24:257–271. doi: 10.1016/j.ccr.2013.06.017. [DOI] [PubMed] [Google Scholar]
- Reynolds JM, Angkasekwinai P, Dong C. IL-17 family member cytokines: regulation and function in innate immunity. Cytokine & growth factor reviews. 2010;21:413–423. doi: 10.1016/j.cytogfr.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, Stewart AF, Dymecki SM. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nature genetics. 2000;25:139–140. doi: 10.1038/75973. [DOI] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- Shen FW, Saga Y, Litman G, Freeman G, Tung JS, Cantor H, Boyse EA. Cloning of Ly-5 cDNA. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:7360–7363. doi: 10.1073/pnas.82.21.7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Gao H, Lin Y, Yao Y, Zhu S, Wang J, Liu Y, Yao X, Meng G, Shen N, et al. Alterations in the microbiota drive interleukin-17C production from intestinal epithelial cells to promote tumorigenesis. Immunity. 2014;40:140–152. doi: 10.1016/j.immuni.2013.11.018. [DOI] [PubMed] [Google Scholar]
- Sutton CE, Mielke LA, Mills KH. IL-17-producing gammadelta T cells and innate lymphoid cells. European journal of immunology. 2012;42:2221–2231. doi: 10.1002/eji.201242569. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer science. 2003;94:965–973. doi: 10.1111/j.1349-7006.2003.tb01386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Z, Yang XO, Yan H, Liu W, Niu X, Shi Y, Fang W, Xiong B, Wan Y, Dong C. A protective role by interleukin-17F in colon tumorigenesis. PloS one. 2012;7:e34959. doi: 10.1371/journal.pone.0034959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, Berger A, Bruneval P, Fridman WH, Pages F, Galon J. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer research. 2011;71:1263–1271. doi: 10.1158/0008-5472.CAN-10-2907. [DOI] [PubMed] [Google Scholar]
- Vizio B, Novarino A, Giacobino A, Cristiano C, Prati A, Ciuffreda L, Montrucchio G, Bellone G. Potential plasticity of T regulatory cells in pancreatic carcinoma in relation to disease progression and outcome. Experimental and therapeutic medicine. 2012;4:70–78. doi: 10.3892/etm.2012.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Yi T, Zhang W, Pardoll DM, Yu H. IL-17 enhances tumor development in carcinogen-induced skin cancer. Cancer research. 2010;70:10112–10120. doi: 10.1158/0008-5472.CAN-10-0775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nature medicine. 2009;15:1016–1022. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y, Saito H, Ikeguchi M. Prevalence and clinical relevance of Th17 cells in patients with gastric cancer. The Journal of surgical research. 2012;178:685–691. doi: 10.1016/j.jss.2012.07.055. [DOI] [PubMed] [Google Scholar]
- Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z, Dong C. Regulation of inflammatory responses by IL-17F. The Journal of experimental medicine. 2008;205:1063–1075. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. The Journal of experimental medicine. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Y, Peng LS, Zhao YL, Shi Y, Mao XH, Chen W, Pang KC, Liu XF, Liu T, Zhang JY, et al. CD8(+) T cells that produce interleukin-17 regulate myeloid-derived suppressor cells and are associated with survival time of patients with gastric cancer. Gastroenterology. 2012;143:951–962. e958. doi: 10.1053/j.gastro.2012.06.010. [DOI] [PubMed] [Google Scholar]
- Zou W, Restifo NP. T(H)17 cells in tumour immunity and immunotherapy. Nature reviews Immunology. 2010;10:248–256. doi: 10.1038/nri2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Five-month old tumor-bearing CPC-APC mice harboring heterozygous (+/−) or knockout (−/−) Il17ra gene were sacrificed for Q-RT-PCR analysis of the indicated cytokine mRNAs in normal (N) and tumor (T) tissues. N=11. Data represent averages ± S.E.M. * p < 0.05.
(A, D) Q-RT-PCR analysis of the indicated mRNAs in normal (N) and tumor (T) tissues of 5-month old CPC-APC mice. N=11. (B, C) Immunostaining of colon tumors that are heterozygous or null for Il17ra. Six HMF images taken on tumor sections from four mice on each side were used for quantification by measuring immuno-reactive areas (IRA, C). Data represent averages ± S.E.M. * p < 0.05. Scale bar = 100 μm.
(A) Six weeks old CPC-APC mice underwent adoptive transfer of bone marrow from Il17ra heterozygous (+/−) or null (−/−) donors. Mice were sacrificed at 5 months of age and tumor parameters were determined (n=12). (B) Six weeks old CPC-APC mice (C57BL/6 background) were irradiated and transplanted with bone marrow from CD45.1/CD45.2 C57BL/6 mice. Recipient mice were sacrificed at 5 months of age and cells from spleen, mesenteric lymph nodes (MLN), normal colon tissue (Normal) and colonic tumors (Tumor) were collected and stained for CD45.1 and CD45.2 and analyzed by flow-cytometry. Live CD45+ cells were gated on in the graphs. Percentages of donor-originated CD45.1/CD45.2 double positive cells indicate the rate of reconstitution. (C) Q-RT-PCR analysis of mRNAs corresponding to the indicated genes in normal colon (N) and tumor (T) tissues from CPC-APC mice that received heterozygous or Il17ra-null bone marrow (n=18). Data represent averages ± S.E.M. * p < 0.05.
(A) Q-RT-PCR analysis of IL-17RA mRNA in purified cells from colon tumors. CAF: cancer associated fibroblasts; IEC: intestinal epithelial cells. N=4. (B) Scheme for conditional knockout of the Il17ra locus. Il17raLacZ-Flox mice were generated by conventional ES cell targeting and blastocyst injection. Transgene positive mice were crossed to a FLP+ strain for deletion of the FRT-flanked LacZ cassette to generate the Il17raF/+ strain. (C) Quantification on the amount of activated ERK assayed by Western blotting in IL-17A stimulated IEC isolated from control or Cdx2Cre Il17raF/− mice. α-tubulin was used as loading control. N=2 for control, 3 for Il17ra conditional knockout. (D) Bright-field images of WT and Apc-deleted (Apc IEC) small intestinal organoids in 3-D culture, supplied with complete culture medium (left and middle panels). Right panel: Apc-deleted (Apc IEC) small intestinal organoids maintained and propagated in medium without serum and growth factors (DF+++) for more than one month. Scale bar = 100 μm.
CPC-APC mice that are heterozygous (+/−) or null (−/−) for the Il6 gene were sacrificed at 5 months of age for Q-RT-PCR analysis of the indicated genes in mesenteric lymph nodes (MLN), normal (N) colon and tumor (T) tissues. n=9. Data represent averages ± S.E.M. * p < 0.05.
(A) Immunostaining of colon cryosections from ApcF/F (Control) or Cdx2Cre-ERT2 ApcF/F (TAM 1wk) mice 1 week after tamoxifen injection. (B) Q-RT-PCR analysis of IL-17A and IL-17C mRNAs in colon tissues following tamoxifen injection (n=8). (C) Cdx2Cre-ERT2 ApcF/F mice underwent adoptive transfer of bone marrow cells from control (Il17ra+/−) or Il17ra−/− donors. At 5 months of age, the recipient mice were i.p. injected with tamoxifen and sacrificed one month later for analysis of colon tumor parameters (n=5). (D) Immunostaining of cryosections of colons from ApcF/F mice that are heterozygous (+/−) or null (−/−) for Il17ra 1 week after tamoxifen injection. Data represent averages ± S.E.M. * p < 0.05. Scale bar = 100 μm.
Supplemental Table 1: List of primers used in Q-RT-PCR analysis, related to Experimental Procedures.