

Significance
A better understanding of intratumor heterogeneity is required to more fully dissect the events which mediate cancer formation and treatment resistance. We used a novel experimental and computational single-cell sequencing approach to directly measure the clonal structures of childhood ALL samples at diagnosis. This approach enabled us to determine the mutation segregation patterns within a single sample and to reconstruct the tumor’s clonal structures with rigorously validated quantitative analysis. We then identified features of each leukemia sample that were shared across patients, including multiple dominant clonal populations at varied stages in differentiation arrest, clone-specific punctuated cytosine mutagenesis, and the late acquisition of proliferative oncogenic point mutations. Together, these findings provide a high-resolution view of the development of childhood ALL.
Keywords: single-cell genomics, acute lymphoblastic leukemia, intratumor heterogeneity, clonal evolution, cytosine mutagenesis
Abstract
Many cancers have substantial genomic heterogeneity within a given tumor, and to fully understand that diversity requires the ability to perform single cell analysis. We performed targeted sequencing of a panel of single nucleotide variants (SNVs), deletions, and IgH sequences in 1,479 single tumor cells from six acute lymphoblastic leukemia (ALL) patients. By accurately segregating groups of cooccurring mutations into distinct clonal populations, we identified codominant clones in the majority of patients. Evaluation of intraclonal mutation patterns identified clone-specific punctuated cytosine mutagenesis events, showed that most structural variants are acquired before SNVs, determined that KRAS mutations occur late in disease development but are not sufficient for clonal dominance, and identified clones within the same patient that are arrested at varied stages in B-cell development. Taken together, these data order the sequence of genetic events that underlie childhood ALL and provide a framework for understanding the development of the disease at single-cell resolution.
A more comprehensive understanding of how malignancies develop could facilitate the rational development of novel anticancer treatment and prevention strategies. Large projects that aim to comprehensively characterize somatic mutations in cancer samples have cataloged many of the recurrent genomic lesions in a wide variety of tumors (1). However, these studies do not measure the correlated cooccurrence of genomic lesions between different cells, which is required for understanding the clonal structure of a tumor as well as for rigorously determining temporal ordering of mutation acquisition. Other studies have provided some temporal resolution of mutation segregation patterns from diagnosis to disease recurrence, allowing for post hoc inference of intratumor clonal heterogeneity at diagnosis (2–5). However, approaches that rely on mutant allele frequencies to determine clonal structure require multiple samples from the same patient and are unable to resolve clones with mutations present at similar frequencies, which is a prerequisite to unambiguously determine the clonal structure and delineate the evolution of the disease (3–5). In principle, single cell genomics provides the most rigorous method to determine the clonal heterogeneity of tumors; as discussed below, there have been recent advances in this approach, but technical limitations have until now prevented it from fully addressing the questions of interest.
Studies of pediatric acute lymphoblastic leukemias (ALL) have provided a limited ordering of the genetic events that underlie childhood leukemogenesis by studying prediagnostic samples. For example, ETV6 -RUNX1 translocations, which occur in about a third of patients under 10 y of age, have been shown to occur in utero by tracking the translocation back to neonatal blood spots (6, 7). In addition, a recent report suggests that ETV6-RUNX1 translocations stall B-cell development so that subsequent recombination-activating gene (RAG)–mediated genomic rearrangements become drivers of the creation of polyclonal structures (8). Furthermore, all of the ALL samples evaluated in this large study had acquired single nucleotide variants (SNVs) during disease progression, suggesting ETV6-RUNX1 translocations and the genomic structural variation in those cells are not sufficient for leukemogenesis (7, 8). However, the order in which each of these mutations are acquired and actual clonal structure of childhood ALL at diagnosis are unknown. It is therefore of paramount interest to develop a detailed understanding of patient-specific tumor clonal structure and evolutionary history both for fundamental understanding of the pathogenesis of childhood ALL, as well as for the design of new therapeutic and prevention strategies.
Results
Here we used microfluidic automation to perform whole genome amplification (WGA) of nearly 1,500 single cells from six patients. We used bulk sequencing data to identify regions in the bulk tumor sample with genomic heterogeneity. We then performed targeted single-cell sequencing of these regions to identify SNVs, large deletions, and IgH sequences in each cell before reconstructing the evolution and clonal structure of the sample. Six patients, described in detail in SI Appendix, Table S1, underwent paired tumor and normal tissue exome sequencing with capture oligos that enriched for transcribed regions of the genome, including both coding and noncoding locations. We focused on samples from children with near normal karyotypes to simplify variant calling and interpretation of allele dropout (ADO), and this resulted in five of the six samples harboring ETV6-RUNX1 translocations. As seen in Fig. 1C, we confirmed an average of 46 variants per patient, with significant variability seen between patients (range 10–105) (Dataset S1). Consistent with previous reports, we identified a striking enrichment for cytosine mutations (8). Further evaluation of the neighboring bases did not reveal a WRCY motif that would implicate activation-induced deaminase, but did find a preference for a TC motif, suggesting that an apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (APOBEC) cytosine deaminase family member could be the underlying etiology (8, 9). In addition, we identified several mutations that may describe alterations in previously unidentified biological processes that contribute to leukemogenesis, including a nonsense mutation in the core histone HIST1H2AG, as well as missense mutations in the scaffolding protein PLEC in two of the six patients.
Fig. 1.

Overview of approach and bulk exome sequencing data. (A) Overview of experimental approach where bulk sequencing is used to identify mutations, followed by single-cell interrogations of those loci to reconstruct tumor phylogenies. (B) Overview of computational methods to use the single-cell mutation profiles to determine clonal structures (E–M, expectation maximization). (C) Number and classes of bulk mutations acquired in each patient. (D) Mutation allele frequency distributions for all confirmed bulk mutations. (E) Types of base changes observed in leukemia samples. (F) Evaluation of neighboring bases of C->T and C->G mutations reveal a strong preference for T preceding C.
Analysis of the bulk exome data revealed two distinct distributions of mutant allele frequencies (Fig. 1D). Nine of 10 confirmed mutations for patient 6 were present near a frequency of 50%, suggesting a single dominant clone under the assumption that all mutations were heterozygous. In all other patients, a subset of the mutations had an allele frequency near 50%, whereas a separate group of mutations were present at or below 25%, suggesting the presence of clonal heterogeneity.
To segregate the lower frequency mutations into distinct clones, single cells were captured into physically separated chambers, followed by automated cell lysis and multiple displacement WGA. We then used three approaches to estimate the percentage of the genomes of each cell that had been lost during the WGA, known as the ADO rate: (i) Taqman-based genotyping of 46 loci commonly heterozygous across populations as previously described (10), (ii) targeted resequencing of 96 loci that are spread throughout the genome and commonly heterozygous across populations in the 1,000 genomes data (11), and (iii) determining the fraction of wild-type alleles lost each time a mutation was called. As seen in SI Appendix, Table S2, using the germ-line data as a reference for each patient, the PCR and wild-type dropout methods concordantly estimate the median overall ADO rate to be between 23% and 24%, whereas the targeted resequencing method is somewhat higher at 33%. This difference is likely due to technical limitations of multiplexed assays for the nontumor mutant loci in the resequencing approach, which had higher rates of ADO. When a 30% threshold for ADO is applied, the median ADO rate is reduced to 20% over the remaining cells based on all three methods. The ADO rate in the primary tumor cells was modestly higher than the median ADO rate of 15.6% observed in a control lymphoblastoid cell line and consistent with previous reports showing higher ADO rates in primary patient samples (12). These rates are also consistent with other single-cell primary sample cancer sequencing approaches using MDA and lower than approaches that have used PCR-based genome amplification (12–15). The ADO data and percent of cells removed from further analyses as a result of these quality control measures are summarized in SI Appendix, Fig. S1.
As summarized in Fig. 1B, after acquiring a mutation profile for each cell, we performed two complementary approaches to determine the clonal structures. First, we developed a probabilistic modeling-based approach where we first execute an expectation maximization algorithm on a multivariate Bernoulli model (SI Appendix, Fig. S2) (16–18). The number of clones was then estimated using Akaike information criterion. The relationships between clones were visualized using multiple correspondence analysis and heatmaps with cells clustered by the output of the expectation maximization algorithm (Fig. 2). As has been done in previous studies to formally determine clonal structure, we also computed the statistical significance of the detected clones using an approach that is analogous to the χ2 statistic on the multisample Bernoulli model (SI Appendix, Table S3) (19). In parallel, we clustered both cells and mutations using Jaccard distance followed by clone number estimation using the within sum of square error. After identifying clones using both methods, the consensus genotypes of the clones were used to generate directed minimum spanning trees that capture the temporal ordering of the clones. The relationships between clones were then visualized, where the size of each clone is proportional to the relative abundance of each population and the length of each edge is proportional to the number of new mutations acquired in that clone. We then determined the effect of relaxing the ADO criteria on the clonal structures, where we found that adding additional cells with higher measured ADO rates did not change the clonal structures. However, we did begin to produce clusters of low quality cells that did not fall into any clones at the higher ADO rates, which were removed when constructing the minimum spanning trees (SI Appendix, Fig. S3).
Fig. 2.
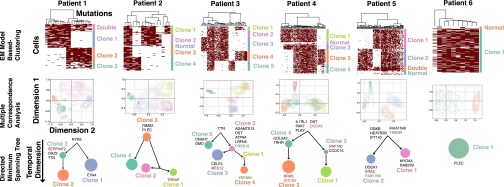
Clone structures determined using expectation maximization algorithm on the multivariate Bernouli distribution model. Cells were visualized on the y-axes and mutations clustered by Jaccard distance on the x-axes. Mutation calls are represented by maroon boxes. The identification of statistically significant groups of cells by the expectation maximization on the multivariate Bernouli distribution model is visualized using multiple correspondence analysis. Interclonal distances and undetectable ancestors are quantitated and visualized using a directed minimum spanning trees. The size of each clone is proportional to its relative abundance, and the length of edges is proportional to the Jaccard distance between clones. Recurrently mutated genes in ETV-RUNX1 leukemias are shown in the clones where they were acquired; green genes are mutated more than once in the same clone, whereas orange genes are mutated more than once in the same patient, but in different clones. Genes that have been colored red have been implicated in ALL by the Cancer Genome Census, suggesting they could be providing increased fitness to those clones (22).
We validated these approaches by performing simulations with randomly generated data with varied ADO to determine the relationship between estimated clone number and the number of mutations measured per cell. As expected, high levels of ADO (>0.3) and low number of mutations per sample (<10) underestimate the number of clones and hamper determination of clonal structures (SI Appendix, Fig. S4). All of our experiments had an ADO rate < 0.3, and all but one patient had at least 20 mutations, confirming we measured a sufficient number of mutations per cell and had a low enough ADO rate to confidently determine the true number of clones. We then simulated the number of cells needed to identify a clone at different frequencies while varying the numbers of mutations interrogated. We did not find a strong effect of mutation number after reaching 30, and determined that we could roughly detect a 1% prevalence clone with 200 cells, 2% with 75 cells, and 4% with 50 cells. Thus, on average, we would roughly need to identify at least 2–3 distinct cells from the same clone to accurately detect that population (SI Appendix, Fig. S5).
Five of six patients had at least two high frequency clones that comprised at least 25% of the cells in that sample (Fig. 2). Both the probabilistic and distance-based approaches identified similar clonal structures, although there were several instances where the hierarchical clustering method separated out clones that were not statistically supported by the probabilistic models (SI Appendix, Fig. S6). In addition, the probabilistic approach enabled a fourth method of ADO estimation by independently estimating the intraclonal variant call dropout rate (SI Appendix, Table S4). As seen in SI Appendix, Fig. S7, there was strong concordance between the measured and inferred dropout rates after subsetting the data for increasing measured ADO thresholds.
We then demonstrated that it is not possible to resolve the cells in each sample into distinct clones based on the bulk allele frequency data alone (SI Appendix, Fig. S8). To determine how representative the single cell mutation calls are of the bulk sample, we also plotted the allele frequency of each mutation measured in the bulk exome sequencing sample and determined how well it correlated to the percent of cells with that mutation called in the single cells (multiplied by 0.5 to correct for the heterozygous state of all measured mutations). As seen in SI Appendix, Fig. S9, there is a strong correlation between the allele frequencies measured in the bulk sample to the percent of single cells found to contain that mutation when corrected for the ADO rate, showing that the single-cell data accurately represent the observed genomic heterogeneity in the bulk samples. In addition, when the bulk exome and single-cell sequencing data are compared, the mutation groups that had been independently determined when clustering the data to construct the mutation maps in SI Appendix, Fig. S6 tightly cluster at the same allele frequencies, further validating our approach for generating the clonal structures.
Large deletions are characteristic of ETV6-RUNX1 leukemias (2, 8). To determine the timing of deletions during the development of ALL, as well as further delineate the clonal architecture of the samples using both subclonal SNVs and deletions, we developed a method to detect deletions in both bulk exome sequencing data, as well as single cells. To accomplish this task, we first identified regions with contiguous loss of heterozygosity in the bulk sample based on the frequency of the less abundant allele at all heterozygous sites in the leukemia sample compared with the germ-line sample for each patient. Those putative deletions were then confirmed in the bulk samples using targeted resequencing of the heterozygous loci within those regions. As seen in Fig. 3B, we identified an average of 3.2 deletions per patient with an ETV6-RUNX1 translocation, with a predicted size range of 25 Kb to the entire X chromosome (155.3 Mb). The number of deletions we identified is less than the mean of 6.0 seen in a previous study of ETV6-RUNX1 patients using SNP arrays and 12.3 using whole genome sequencing, suggesting that the exome approach is slightly less sensitive than genome-wide techniques (8, 20). We then interrogated each of the single cells for the presence of deletions identified in the bulk samples, again using targeted resequencing of the heterozygous locations in the deleted region. As seen in Fig. 3D, based on the allele frequency in the tumor, 13 out of 16 deletions were detected in all clones at a level significantly higher than the ADO rate and all patients had at least one clonal deletion, suggesting the underlying cause of the large deletions, such as aberrant RAG activity, had been active before the SNVs in the later clones had been acquired. Patient 4 did have a subclonal deletion of chromosome 16, and further analyses revealed the deletion was only present in one branch of the phylogenetic tree, showing that deletions could continue to be acquired in later clones (Fig. 3 A and C).
Fig. 3.

Overview of deletions detected in bulk exome and single cell data. (A) View of allele frequency of less abundant allele across chromosomes 12 and 16 in patient 4. Regions with a contiguous decrease in the allele frequency in the leukemia compared with the germ line represent deletions (black boxes). The allele frequency for the deletion in chromosome 12 (which includes ETV6) approaches 0%, suggesting it is clonal. Chromosome 16 deletion is near 25%, suggesting it is subclonal. (B) Number and size of deletions detected across all six patients using this approach. (C) Segregation of deletions across clones in patient 4. Chromosome 12 deletion is present in all clones, as predicted in A. Chromosome 16 deletion segregated down one branch of the tree, with a much lower level of deletion detection in other clones due to ADO leading to false calls. (D) Most deletions are detected in all clones, suggesting that the process that produces the deletions occurs before mutations are acquired.
To identify mutations that could be promoting increased fitness of specific clones, we mapped the newly acquired recurrent mutations present in each clone; recurrence was defined by more than one mutation in a gene after combining our data with that from a recent study (8). After analyzing the specific patterns of mutation acquisition, we identified subclonal KRAS mutations in two patients. KRAS mutations are known to be central drivers of tumorigenesis in a number of malignancies (21). However, in both patients in our cohort, KRAS mutations were predicted to be a subclonal event based on the allele frequency in the bulk sample. Further evaluation showed that they were restricted to a single most evolved clone in each sample, both of which had other codominant clones. These findings suggest that KRAS mutations were acquired late in disease development where they drove the expansion of one of the later clones, but did not provide sufficient fitness to outcompete all of the other clones in those patients. In addition, patient 4 acquired a mutation in the ras-related protein RAB27B in the other codominant clone, which may have provided sufficient fitness to compete with the KRAS-mutant clone. To more systematically determine if there were clone-specific “driver” mutations, we then mapped coding mutations in genes determined to be important for the pathogenesis of ALL by the Catalogue of Somatic Mutations in Cancer Census (22). We found that all of the putative “driver” mutations segregated to specific branches of the phylogenetic tree, further suggesting there may be specific genetic lesions that are determining the relative fitness of each clone in the tumors.
Four genes had more than one exonic mutation in the same patient, all of which were C->T or C->G changes. Patient 4 acquired nonsense mutations in DOCK5 at two different times, suggesting DOCK5 mutations underwent independent positive selection in each of those clones. Conversely, PRSS12, FAM178A, and ZNF880 mutations were acquired in close genomic proximity in the same clone in those patients, suggesting they occurred in a punctuated event. Most strikingly, three different C->G mutations were acquired in the same exon of ZNF880, and all contained a TCA motif, providing further evidence that the underlying etiology is a sequence-specific process that can have focal activity, such as a processive enzyme (SI Appendix, Fig. S10).
We then further dissected the clonal biology by performing intercellular and interclonal correlations of IgH sequence characteristics to other mutation measurements. At the bulk level, three of the six patients had no evidence of VH replacement, which was recently shown to occur at higher levels than previously detectable in some ALL samples using immune repertoire sequencing (23). However, patient 5 had two distinct IgH sequences identified in the bulk samples, and single-cell interrogations detected both sequences in most cells, suggesting that both IgH alleles had been rearranged. In the other three patients, differing levels of VH-replacement were detected. At the clonal level, those three patients had an enrichment of cells with no mutation calls, suggesting some clones are arrested at earlier stages in B-cell development and had not yet undergone VDJ recombination (SI Appendix, Fig. S11). In addition, by looking at V-segment use within clones, we identified a group of cells within clone 1 in patient 1 that uniquely used IGHV3-33*01 and also harbored mutations in the 3′UTR of EYA4, suggesting the EYA4 mutation allowed that clonal precursor B-cell population to progress to a later developmental stage, resulting in the termination of VDJ recombination (Fig. 4C). At the cellular level, we evaluated the VH segments to determine if some of the detected SNVs could be attributed to activation-induced deaminase, which normally mutates cytosine residues in VH segments of mature B cells as a part of somatic hypermutation (24). We found only a few VH segment mutations and no correlation between the percent of mutations that occurred at cytosine residues and the number of VH segment mutations (SI Appendix, Fig. S12). Thus, our evaluations of the IgH sequences revealed that some of the most evolved cells can continue to undergo VDJ recombination, there can be variability in the magnitude of VH replacement between clones in the same patient, and that there is inconsistent detection of recombined IgH sequences between clones from the same patient which may indicate that some clones are arrested at earlier stages in B-cell differentiation.
Fig. 4.

Determination of IgH VDJ recombination across cells and clones clustered by Jaccard distance. (A) Patient 4 represents a pattern seen for three of six patients, with the use of a single VH segment in the rearranged VDJ sequences. (B) Patient 5 had two rearranged alleles detected in both clones. (C) Patient 1 had a significant fraction of VH-replacement clones. In addition, in clone 1 EYA4 mutation closely segregates with VH-segment IGHV3-33*01 (dashed box), suggesting it is a separate clone with a unique IgH sequence. Clone 2 had a much higher rate of VH-replacement, as well as rate of no VDJ calls. (D) Patient 2 clone 4 almost exclusively used IGHV3-64*01, whereas the other clones had high levels of VH replacement and no VDJ sequence calls. Black box represents the VH segment call for the VDJ sequence detected in each cell, whereas the white box represents no call.
Discussion
The development of methods for the physical isolation and WGA of individual cells have begun to allow for the direct measurement of the genomic variation within humans at single-cell resolution (25, 26), and the approach described here of performing whole genome amplification followed by targeted analysis of regions of interest has a carefully calibrated balance in the tradeoff between the amount of data obtained per cell versus the number of cells that can be practically analyzed. Previous studies using single-cell sorting or micropipetting have shown that investigators can use PCR-based whole genome amplification to detect copy number variation (CNV) in cancer cells (9, 14). In addition, two papers established the feasibility of using isothermal single-cell WGA to detect SNVs in cancer samples, although neither study was able to determine clonal structures that were shown to represent the bulk samples (12, 13). Another recent study was unable to determine the clonal structure of myeloid leukemia samples with single-cell data alone, as they evaluated 12 cells per patient and lost an average of 55% of the genomes of those cells during WGA and uneven sequencing after target capture (15). Two other studies used single-cell sorting followed by target-specific preamplification to perform quantitative PCR (qPCR)-based CNV analyses, as well as allele-discriminating qPCR-based SNV detection (8, 27). They were able to identify subclonal populations in those samples, but only evaluated 6 genetic lesions due to the requirement of multiplexing patient and gene-specific allele-discriminating assays. In addition, they did not have a method to differentiate dropout of a mutant allele during the single-cell PCR amplification from absence of a mutation in that cell and did not show that the inferred clonal structures actually represented the bulk samples. With those methods, the authors only identified one major clone in each sample in the first study, but did find evidence for multiple high frequency clones in the two patients interrogated in the subsequent study (8, 27). Hence, previous single-cell cancer sequencing studies were limited by throughput due to challenges with single-cell manipulation and isolation, an inability to query large parts of the genomes from single cells, high ADO rates due to sampling by using target capture rather than target-specific amplification, or a lack of quality control and validation procedures to differentiate true mutations from background genome dropout during single-cell target or genome amplification. More recently, using MDA of sorted tetraploid nuclei, it was shown that some subclonal structure could be detected in a breast cancer sample, although it is unclear how representative that model and those cells are of the actual tumor (28).
In the present study, we leveraged the efficient single-cell capture and WGA of microfluidic devices to obtain amplified genomes from 1,479 ALL cells that then underwent targeted resequencing analysis. We then developed two complementary approaches to remove low-quality cells, accurately determine the number of clones, resolve the clonal populations and their genotypes, and determine the relationships between those populations. These methods enabled us to perform the an accurate evaluation of transcribed regions of single ALL cells, which surprisingly revealed codominant clones in five of six patients. In addition, our bulk and single cell data show that most large deletions occur before cytosine mutagenesis-driven SNV acquisition, and provide further evidence that the majority of the SNVs in these B-cell leukemia patients are caused by an APOBEC protein, as summarized in Fig. 5. In addition to the clonal structure, our approach determines how deletions, IgH sequences, and specific mutations segregate between clones. Using these data, we show that ongoing VDJ recombination can occur in the most evolved clones, which can have variable magnitude between clones in the same patient. In addition, our studies found that some cells may not have detectable IgH sequences, which could have important implication for understanding the heterogeneity in differentiation arrest within a single malignancy and may be an important variable for understanding treatment resistance. Taken together, these data provide an unprecedented view of the events that resulted in the development of each patient’s malignancy.
Fig. 5.

Temporal ordering of events in the development of ALL. ETV6-RUNX1 translocation occurs in utero, followed by preleukemic evolution as a result of further genomic structural variation. The outgrowth of multiple dominant clones is then driven by cytosine mutations causing branching evolution. IgH rearrangement can occur before mutation acquisition, or continue to be ongoing in the most evolved clones.
With the ability to accurately and efficiently resolve clonal populations in a single sample based on single-cell genetic analyses, we have begun to dissect biological phenomena within and between clones, including temporal resolution of mutation acquisition, changes in underlying causes of mutations, and clonal fitness. With the development of these experimental and analysis methods, we have gained a deeper understanding of childhood leukemogenesis, and now have a toolkit to begin to dissect the development of all tumor types at single-cell resolution.
Materials and Methods
Bone marrow samples from six ALL patients underwent exome sequencing, followed by SNV, deletion, and IgH sequence confirmation using microfluidic-based targeted resequencing. A median of 245 single cells from each of the bulk samples were then isolated and lysed, and the DNA was amplified using the C1 Single-Cell Auto Prep System. The amplified DNA then underwent targeted resequencing of the confirmed mutations to determine cell-specific SNVs, deletions, and IgH sequences (Dataset S2). The sequence variants for each cell were then used to construct minimum spanning trees after determining relationships between cells using the expectation maximization algorithm executed on multivariate Bernouli distributions. See SI Appendix for details.
Supplementary Material
Acknowledgments
We thank the Stanford Stem Cell Institute Genome Center, including Norma Neff, Ben Passarelli, Gary Mantalas, and Sean Lui, for performing sequencing and providing the necessary computational resources. In addition, we thank Marc Unger, Stephane Boutet, and Suzanne Weaver for their technical assistance in generating the single-cell amplicons. C.G. is funded by a Scholar Award from the American Society of Hematology, Special Fellow Award from the Leukemia and Lymphoma Society, and pilot grant from the Spectrum Child Health Research Institute at Stanford. C.G. would also like to thank the faculty and trainees in the Translational Research Training in Hematology Program for their input on the project. W.K. is supported by A*STAR, agency of science, technology and research, Singapore.
Footnotes
Conflict of interest statement: S.R.Q. is a founder of and consultant for Fluidigm.
Data deposition: The sequences reported in this paper have been deposited in the Sequence Read Archive database (accession no. SRP044380), and the complete R code has been deposited to GitHub, github.com/lianchye/Clonal_Analysis.git.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1420822111/-/DCSupplemental.
References
- 1.Kandoth C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mullighan CG, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322(5906):1377–1380. doi: 10.1126/science.1164266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Welch JS, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walter MJ, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012;366(12):1090–1098. doi: 10.1056/NEJMoa1106968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ding L, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Golub TR, et al. Fusion of the TEL gene on 12p13 to the AML1 gene on 21q22 in acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 1995;92(11):4917–4921. doi: 10.1073/pnas.92.11.4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greaves M. Pre-natal origins of childhood leukemia. Rev Clin Exp Hematol. 2003;7(3):233–245. [PubMed] [Google Scholar]
- 8.Papaemmanuil E, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet. 2014;46(2):116–125. doi: 10.1038/ng.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zong C, Lu S, Chapman AR, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science. 2012;338(6114):1622–1626. doi: 10.1126/science.1229164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan HC, Wang J, Potanina A, Quake SR. Whole-genome molecular haplotyping of single cells. Nat Biotechnol. 2011;29(1):51–57. doi: 10.1038/nbt.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abecasis GR, et al. 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hou Y, et al. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell. 2012;148(5):873–885. doi: 10.1016/j.cell.2012.02.028. [DOI] [PubMed] [Google Scholar]
- 13.Xu X, et al. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell. 2012;148(5):886–895. doi: 10.1016/j.cell.2012.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472(7341):90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hughes AE, et al. Clonal architecture of secondary acute myeloid leukemia defined by single-cell sequencing. PLoS Genet. 2014;10(7):e1004462. doi: 10.1371/journal.pgen.1004462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Myllykangas S, Tikka J, Böhling T, Knuutila S, Hollmén J. Classification of human cancers based on DNA copy number amplification modeling. BMC Med Genomics. 2008;1:15. doi: 10.1186/1755-8794-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dempster AP, Laird NM, Rubin DB. Maximum likelihood from incomplete data via the EM algorithm (with discussion) J R Stat Soc [Ser A] 1977;39:1–38. [Google Scholar]
- 18.Wolfe JW. Pattern clustering by multivariate mixture analysis. Multivariate Behav Res. 1970;5:329–350. doi: 10.1207/s15327906mbr0503_6. [DOI] [PubMed] [Google Scholar]
- 19.Begg CB, Eng KH, Hummer AJ. Statistical tests for clonality. Biometrics. 2007;63(2):522–530. doi: 10.1111/j.1541-0420.2006.00681.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mullighan CG, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446(7137):758–764. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 21.Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7(4):295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- 22.Futreal PA, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4(3):177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gawad C, et al. Massive evolution of the immunoglobulin heavy chain locus in children with B precursor acute lymphoblastic leukemia. Blood. 2012;120(22):4407–4417. doi: 10.1182/blood-2012-05-429811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Longerich S, Basu U, Alt F, Storb U. AID in somatic hypermutation and class switch recombination. Curr Opin Immunol. 2006;18(2):164–174. doi: 10.1016/j.coi.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 25.Wang J, Fan HC, Behr B, Quake SR. Genome-wide single-cell analysis of recombination activity and de novo mutation rates in human sperm. Cell. 2012;150(2):402–412. doi: 10.1016/j.cell.2012.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jan M, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4(149):149ra118. doi: 10.1126/scitranslmed.3004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Potter NE, et al. Single-cell mutational profiling and clonal phylogeny in cancer. Genome Res. 2013;23(12):2115–2125. doi: 10.1101/gr.159913.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512(7513):155–160. doi: 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.