

Abstract
Current efforts to grow the tropical oilseed crop Jatropha curcas L. economically are hampered by the lack of cultivars and the presence of toxic phorbol esters (PE) within the seeds of most provenances. These PE restrict the conversion of seed cake into animal feed, although naturally occurring ‘nontoxic’ provenances exist which produce seed lacking PE. As an important step towards the development of genetically improved varieties of J. curcas, we constructed a linkage map from four F2 mapping populations. The consensus linkage map contains 502 codominant markers, distributed over 11 linkage groups, with a mean marker density of 1.8 cM per unique locus. Analysis of the inheritance of PE biosynthesis indicated that this is a maternally controlled dominant monogenic trait. This maternal control is due to biosynthesis of the PE occurring only within maternal tissues. The trait segregated 3 : 1 within seeds collected from F2 plants, and QTL analysis revealed that a locus on linkage group 8 was responsible for phorbol ester biosynthesis. By taking advantage of the draft genome assemblies of J. curcas and Ricinus communis (castor), a comparative mapping approach was used to develop additional markers to fine map this mutation within 2.3 cM. The linkage map provides a framework for the dissection of agronomic traits in J. curcas, and the development of improved varieties by marker-assisted breeding. The identification of the locus responsible for PE biosynthesis means that it is now possible to rapidly breed new nontoxic varieties.
Keywords: Jatropha curcas, linkage mapping, oilseed, phorbol esters, plant breeding
Introduction
Jatropha curcas is being developed as a perennial oilseed crop for cultivation in tropical and subtropical climates (King et al., 2009). As a member of the Euphorbiaceae (spurge family), it is also related to a number of other agronomically important crops including cassava (Manihot esculenta), rubber (Hevea brasiliensis), castor (Ricinus communis) and a number of minor oilseed crops including tung (Aleurites fordii) and Chinese tallow (Triadica sebifera). Although there is a history of small-scale cultivation of J. curcas, particularly on the Cape Verde islands (da Silveira, 1934), economical cultivation of J. curcas has currently not reached its full potential due to a number of factors, including lack of knowledge of best agronomic practices and the lack of available purpose-bred cultivars. This has resulted in unpredictable seed yields, ranging from 0.3 to 3.9 tonnes per hectare (Kalannavar, 2008; Mohapatra and Panda, 2011; Yang et al., 2010).
Another current disadvantage of using J. curcas as an oilseed crop is that most provenances produce toxic seed. The exception is the ‘nontoxic’ provenances which occur naturally. Within Mexico, seeds from these nontoxic provenances are consumed by the local population (after roasting) (King et al., 2009). Metabolite analysis has revealed that the only major difference between the toxic and nontoxic seeds of J. curcas is the presence of phorbol esters (PE) within the seeds (He et al., 2011; Makkar et al., 1998). These PE are not fully degraded by the heat processing steps normally used in the conversion of raw seed meal to animal feed (Aregheore et al., 2003; Makkar et al., 1998). The production of animal feed from J. curcas seed therefore requires an additional solvent extraction step to remove the PE (Brooker, 2009). Concerns have also been raised about handling J. curcas products containing PE, as these compounds have also been shown to be cocarcinogens (Hirota et al., 1988)—although not carcinogenic on their own, PE can promote tumour formation caused by exposure to some chemical carcinogens (Goel et al., 2007). The commercialization of seeds lacking PE is therefore desirable, as the seed meal could more readily be converted to animal feed using traditional processes, and any potential risks from handling PE-containing products are removed.
Compared with more established oilseed crops which have seen significant increases in seed yield through breeding and agronomy (Vollmann and Rajcan, 2010), research and development of J. curcas is still at a very early stage. However, our knowledge of the plant has improved vastly in recent years. The genetic diversity of J. curcas is now better understood; a number of studies have revealed that meso-America is the centre of genetic diversity for the species, and there is very little genetic variation in material cultivated outside of this region (Basha et al., 2009; He et al., 2011; Sun et al., 2008; Yi et al., 2010). These studies on genetic diversity have provided useful guidance into sources of material for breeding programmes. Perhaps the most significant advance is in the amount of DNA sequence information which has become available. As well as a published draft genome sequence (Hirakawa et al., 2012; Sato et al., 2011), a number of transcriptomic studies have also been conducted (Costa et al., 2010; King et al., 2011; Natajaran and Parani, 2011). Despite these advances in our understanding of the molecular biology and genetic diversity of J. curcas, molecular breeding for rapid improvement of desirable traits such as seed oil yield and nontoxic seed has been severely hampered by the lack of a genetic linkage map. At present, the only map available for Jatropha sp. is from an interspecific cross between J. curcas and J. integerrima (Wang et al., 2011). Here, we report a high-density genetic linkage map of J. curcas, created from four separate mapping populations, and containing over 500 codominant (SSR and SNP) markers distributed over 11 linkage groups. As a first step in demonstrating the utility of this map, we have identified a locus responsible for the synthesis of the toxic phorbol esters. This work lays the foundation for more rapid crop improvement by marker-assisted breeding and the creation of new nontoxic varieties of J. curcas.
Results and discussion
Development of codominant markers and the construction of an integrated linkage map from four F2 mapping populations
For the construction of linkage maps, codominant markers such as single nucleotide polymorphisms (SNP) and simple sequence repeats (SSR) are the most useful as they provide information on both alleles present in a diploid species such as J. curcas. At the commencement of this study, very few codominant markers were available for J. curcas and these were mainly in the form of SSR (Basha et al., 2009; Phumichai et al., 2011). To provide sufficient markers for the construction of a linkage map, additional SNP markers were obtained from genomic DNA using the CRoPS® technique (File S1). This is a complexity reduction technique that is used for obtaining reduced representation genomic DNA libraries and is an adaptation of the AFLP technique (van Orsouw et al., 2007). SNP markers were also mined from seed transcriptome sequence produced from both toxic (King et al., 2011) and nontoxic [A.J. King and I.A. Graham, unpublished data] developing seeds (File S2). SSR markers were developed using the FIASCO protocol (Zane et al., 2002), or by mining J. curcas genome sequence data (Hirakawa et al., 2012; Sato et al., 2011) using either WebSat (Martins et al., 2009) or Imperfect SSR Finder (Stieneke and Eujayl, 2007). The SSR sequences and the primers used are detailed in File S3. In order to allow the numbering of chromosomes to remain consistent with the previously published map of the J. curcas × J. integerrima interspecific cross (Wang et al., 2011), we included a number of markers which could be used as bridges between these maps (File S4). This was achieved by using markers which had previously been mapped on the interspecific cross, or physical mapping of markers from both maps onto scaffolds of the J. curcas genome sequence (Hirakawa et al., 2012; Sato et al., 2011).
To build a linkage map of J. curcas, we used four mapping populations created from parental lines displaying differences in a range of traits as shown in Table1. Genotyping assays were performed using both SNP and SSR markers according to the Experimental procedures section. The linkage maps for each mapping population were built individually using CRI-MAP 2.503 (www.animalgenome.org), which uses the multipoint likelihood to calculate genetic distances (Lander et al., 1987). Each map contained 11 linkage groups, which is consistent with cytological evidence showing J. curcas is diploid with 22 chromosomes (n = 11) (Dehgan and Webster, 1979). After completion and error checking of the individual maps, the genotype files were merged and then used to build an integrated linkage map (Figures1 and 2). The total genetic distance of this integrated map was 717.0 cM, with an average marker density of 1.5 and 1.8 cM for all and unique loci, respectively. There are relatively few gaps within the integrated map, with only two pairs of loci separated by more than 15 cM, seven pairs of loci by more than 10 cM, and 30 pairs of loci by more than 5 cM. A summary of the map size, number of markers, unique loci and average marker density for each of the individual maps is shown in Table2. The individual maps are presented in File S5. Although marker densities and numbers were lower for the individual maps, the average density and length were in each case still high compared to first generation maps for most species. The least populated map (G51 × CV) still contained 253 markers and had a mean density of 3.3 cM based on unique loci. The marker coverage on all four maps is sufficient for interval mapping, where an interval of 10 cM is regarded as adequate (Mayer, 2005).
Table 1.
F2 mapping populations used in this study
| Mapping population parents and origin |
|||
|---|---|---|---|
| Maternal parent ♀ | Pollen parent ♂ | size | Primary traits |
| G33 (Guatemala) | G43 (Guatemala) | 320 | Seed toxicity G33 = toxic, G43 = nontoxic |
| G51 (Guatemala) | CV (Cape Verde) | 214 | Branching G51 = open branching, CV = closed branching Oil content G51 = 36.9% oil, CV = 26.0% oil |
| QV-JAT03 (Cape Verde) | QV-JAT02 (Mexico) | 220 | Oil content QV-JAT03 = 34.7% oil, QV-JAT02 = 36.5% oil |
| QV-JAT02 (Mexico) | QV-JAT01 (India) | 220 | Fatty acid composition QV-JAT02 = 34.4% oleic acid, QV-JAT01 = 42.1% oleic acid |
Figure 1.

Integrated genetic map for linkage groups 1–6 of Jatropha curcas produced from four mapping populations. Linkage group positions are indicated in cM (Kosambi).
Figure 2.

Integrated genetic map for linkage groups 7–11 of Jatropha curcas produced from four mapping populations. Linkage group positions are indicated in cM (Kosambi).
Table 2.
Summary of linkage group size marker number and density for the integrated map and four F2 populations
| Linkage group |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Map | 01 | 02 | 03 | 04 | 05 | 06 | 07 | 08 | 09 | 10 | 11 | Total |
| Integrated map | ||||||||||||
| Markers | 38 | 34 | 58 | 45 | 60 | 52 | 37 | 62 | 21 | 36 | 59 | 502 |
| Unique loci | 29 | 29 | 47 | 32 | 50 | 40 | 32 | 46 | 20 | 29 | 44 | 399 |
| Distance, cM | 50.9 | 67.3 | 69.2 | 62.7 | 64.4 | 82.1 | 76.8 | 67.1 | 66.3 | 53.6 | 54.9 | 717.0 |
| Mean density (all), cM | 1.4 | 2.0 | 1.2 | 1.4 | 1.1 | 1.6 | 2.1 | 1.1 | 3.3 | 1.5 | 1.0 | 1.5 |
| Mean density (unique), cM | 1.8 | 2.4 | 1.5 | 2.0 | 1.3 | 2.1 | 2.5 | 1.5 | 3.5 | 1.8 | 1.2 | 1.8 |
| QV-JAT03 × QV-JAT02 | ||||||||||||
| Markers | 27 | 15 | 48 | 33 | 41 | 35 | 32 | 51 | 15 | 34 | 51 | 382 |
| Unique loci | 21 | 13 | 33 | 24 | 32 | 29 | 26 | 38 | 13 | 23 | 30 | 283 |
| Distance, cM | 44.2 | 53.9 | 60.1 | 61.2 | 54.1 | 65.2 | 76.6 | 63.9 | 67.5 | 51.2 | 52.4 | 650.3 |
| Mean density (all), cM | 1.7 | 3.9 | 1.3 | 1.9 | 1.4 | 1.9 | 2.5 | 1.3 | 4.8 | 1.6 | 1.0 | 1.8 |
| Mean density (unique), cM | 2.2 | 4.5 | 1.9 | 2.7 | 1.7 | 2.3 | 3.1 | 1.7 | 5.6 | 2.3 | 1.8 | 2.4 |
| QV-JAT02 × QV-JAT01 | ||||||||||||
| Markers | 28 | 14 | 50 | 30 | 42 | 37 | 32 | 50 | 14 | 34 | 49 | 380 |
| Unique loci | 22 | 13 | 38 | 20 | 30 | 29 | 27 | 37 | 13 | 24 | 30 | 283 |
| Distance, cM | 52.9 | 54.2 | 56.2 | 56.9 | 53.0 | 65.1 | 74.5 | 64.3 | 50.6 | 46.1 | 57.3 | 631.1 |
| Mean density (all), cM | 2.0 | 4.2 | 1.1 | 2.0 | 1.3 | 1.8 | 2.4 | 1.3 | 3.9 | 1.4 | 1.2 | 1.7 |
| Mean density (unique), cM | 2.5 | 4.5 | 1.5 | 3.0 | 1.8 | 2.3 | 2.9 | 1.8 | 42 | 2.0 | 2.0 | 2.3 |
| G33 × G43 | ||||||||||||
| Markers | 26 | 23 | 11 | 26 | 30 | 35 | 19 | 27 | 18 | 18 | 28 | 261 |
| Unique loci | 18 | 20 | 9 | 20 | 25 | 26 | 15 | 17 | 17 | 16 | 23 | 206 |
| Distance, cM | 48.7 | 72.5 | 36.9 | 67.0 | 59.4 | 77.2 | 52.4 | 65.4 | 52.2 | 60.5 | 52.2 | 644.2 |
| Mean density (all), cM | 1.9 | 3.3 | 3.7 | 2.7 | 2.0 | 2.3 | 2.9 | 2.5 | 3.1 | 3.6 | 1.9 | 2.6 |
| Mean density (unique), cM | 2.9 | 3.8 | 4.6 | 3.5 | 2.5 | 3.1 | 3.7 | 4.1 | 3.3 | 4.0 | 2.4 | 3.3 |
| G51 × CV | ||||||||||||
| Markers | 23 | 7 | 31 | 35 | 35 | 23 | 18 | 34 | 5 | 21 | 21 | 253 |
| Unique loci | 17 | 6 | 25 | 22 | 25 | 16 | 12 | 28 | 5 | 10 | 15 | 181 |
| Distance, cM | 48.9 | 5.4 | 58.5 | 57.5 | 50.9 | 57.1 | 74.6 | 67.9 | 59.6 | 34.9 | 45.9 | 561.2 |
| Mean density (all), cM | 2.2 | 0.9 | 2.0 | 1.7 | 1.5 | 2.6 | 4.4 | 2.1 | 14.9 | 1.7 | 2.3 | 2.3 |
| Mean density (unique), cM | 3.1 | 1.1 | 2.4 | 2.7 | 2.1 | 3.8 | 6.8 | 2.5 | 14.9 | 3.9 | 3.3 | 3.3 |
To physically map each of the molecular markers used in this study, a blastn search was conducted against build 4.5 of the J. curcas draft genome sequence (www.kazusa.or.jp/jatropha) (Hirakawa et al., 2012). All except one of the markers used produced at least one hit against this database (File S6). Most of the markers aligned only with a single scaffold, and in some cases, multiple markers were located on the same scaffold. Using this approach, it was possible to determine the location of 3077 of the 39 277 predicted gene elements predicted within this genome assembly, and 17 Mbp of the 297 Mbp of sequence (Hirakawa et al., 2012). Comparative mapping of each of the J. curcas scaffolds against the R. communis draft genome was also conducted using the promer command of the MUMmer software (Chan et al., 2010; Kurtz et al., 2004). For the current assembly of the J. curcas genome, the mean and N50 scaffold lengths are 7.6 and 16.0 kbp, respectively (Hirakawa et al., 2012). The R. communis genome sequence currently has mean and N50 scaffold lengths of 14.0 and 496.5 kbp, respectively (Chan et al., 2010), with the mean increasing to 93.0 kbp when scaffolds less than 2 kbp are excluded. The R. communis genome therefore currently comprises fewer and larger scaffolds than the J. curcas genome. Our analysis indicated a number of large syntenic blocks on each of the chromosomes (File S6). This synteny mapping approach proved useful for fine mapping of locus controlling seed toxicity (see below). Further improvement in the scaffold lengths of the either the J. curcas or R. communis genome, or an increase in marker density of the J. curcas linage map would allow a more in-depth analysis of synteny between these genomes.
Previously, the only linkage map available for J. curcas was a map produced for an interspecific cross between J. curcas and J. integerrima (Wang et al., 2011). Although interspecific maps are useful tools for the location of specific genes, or the identification of beneficial traits which may be introgressed from outside the species, their potential for identifying useful traits within J. curcas germplasm is limited, as no assessment of the potential of alleles within the species can be made using these maps. This first intraspecific J. curcas map therefore represents a significant advance for this crop and will be a useful tool for marker-assisted breeding. The maps for the individual crosses have good coverage, although some arms are not mapped in two of the populations (File S5). In some instances, it will be possible to close these gaps by developing additional markers at specific locations on the map by searching for additional polymorphisms in flanking regions of the J. curcas genome. We have demonstrated this approach on linkage group 8 for mapping population G33 × G43 (see below). In some cases, the gaps on maps are likely to be due to identity by descent. For example, in an attempt to map the upper arm of linkage group 3 in the G33 × G43 mapping population (File S5), we screened an additional 48 markers using this approach. None were polymorphic in this population (data not shown).
The total map length of 717.0 cM is much smaller than the previously published interspecific map distance of 1440.9 cM. Based on the high number of markers we have placed on the consensus map, it is unlikely that the difference in map length is caused by a lack of markers at the ends of the linkage groups. The map distances, distances between loci and order of loci were also very consistent between each of the four mapping populations used in this study. It is therefore likely that the map distance observed in this study can be mainly attributed to differences in the error rates of the genotyping data in the two studies. Uncorrected genotyping errors can drastically increase the calculated map lengths (Hackett and Broadfoot, 2003).
Cytological studies of the J. curcas genome have revealed that the estimated genome size for this species is 416 Mbp (Carvalho et al., 2008), which is consistent with the size of the assembled genome data, which is currently 297 Mbp (Hirakawa et al., 2012). Based on these studies, 1 cM of this J. curcas map should correspond to a mean physical distance of around 0.5 Mbp.
This is the first intraspecific genetic linkage map that has been published for J. curcas, a plant which, although showing much promise, requires significant improvement before sustainable economic cultivation can become a reality. For a first generation map, the density is more than sufficient for trait analysis by interval mapping. This map therefore provides a valuable resource for the development of better varieties for the economic production of renewable oil.
Phorbol ester biosynthesis in the seeds of J. curcas is a maternally controlled trait encoded by the nuclear genome
As a prerequisite to mapping the locus (or loci) responsible for seed toxicity, the inheritance of the PE biosynthesis trait was studied. Our previous investigation into the distribution of PE within the seeds of J. curcas revealed that although most of the PE was present within the endosperm of mature seeds, the highest concentration of these diterpenoids was present within the maternally derived inner layer of the seed coat referred to as the tegmen. The distribution of phorbol esters within the endosperm is also not uniform; much higher concentrations were observed in the outer (seed coat facing) endosperm layers than the inner (embryo facing) layers (He et al., 2011). Sujatha et al., 2005 have demonstrated previously that nontoxic plants pollinated by toxic plants bear nontoxic seed, and vice versa. Together, these observations suggest that the PE biosynthesis may only occur within the tegmen. This maternal tissue is crushed during the final stages of seed development, and the PE may then diffuse into the endosperm due to their hydrophobicity.
The phorbol ester content of seed produced from reciprocal crosses between toxic and nontoxic varieties of J. curcas was analysed (Table3). Genotyping of the endosperm from F1 to F2 seeds was also conducted using a subset of SSR markers to confirm that crosses were genuine (data not shown). PE−ve maternal plants cross-pollinated with pollen from PE+ve plants produced only seeds lacking PE, whereas PE+ve maternal plants cross-pollinated with the pollen from PE−ve produced seeds contain PE. Self-pollination of F1 plants derived from both PE−ve♀ × PE+ve ♂ and PE+ve♀ × PE−ve ♂ crosses resulted in F2 seed all of which contained PE. Open pollinated seeds were collected from F2 plants. Thirty one of 120 plants analysed produced seeds lacking phorbol esters (File S7). The trait did not segregate within seeds collected from an individual plant. These observations are consistent with a genetic locus causing seeds to lack phorbol esters being encoded on the nuclear genome, and the phorbol esters being synthesized only within maternal tissues. The 3 : 1 segregation also confirms that PE biosynthesis is a dominant monogenic trait.
Table 3.
Maternal control of phorbol ester biosynthesis
| Maternal parent ♀ | Pollen parent ♂ | Seeds | Phorbol ester content of seed |
|---|---|---|---|
| PE+ve | PE+ve | Self | Present |
| PE−ve | PE−ve | Self | Absent |
| PE+ve | PE−ve | F1 | Present (n = 6) |
| PE−ve | PE+ve | F1 | Absent (n = 12) |
| PE+ve ♀ x PE−ve ♂ F1 | PE+ve ♀ x PE−ve ♂ F1 | F2 | Present (n = 8) |
| PE−ve ♀ x PE+ve ♂ F1 | PE−ve ♀ x PE+ve ♂ F1 | F2 | Present (n = 8) |
| PE+ve ♀ PE−ve ♂ F2 | Open pollinated | Individual plants exclusively bear seeds that either do or do not contain PE. PE were absent in seeds from 31/120 plants |
|
The maternal control of phorbol ester biosynthesis has some important implications for cultivation of this crop. These data on the inheritance of phorbol ester biosynthesis indicate that as the embryo/endosperm genome does not control PE biosynthesis, there is no risk of maternal nontoxic plants bearing seed containing phorbol esters due to cross-pollination by a toxic plant. This observation means that guaranteeing production of nontoxic seeds should be feasible even in locations where genotypes producing toxic seed are growing.
Identification of a locus controlling phorbol ester biosynthesis on linkage group 8
To conduct linkage analysis for loci controlling phorbol ester biosynthesis, the data from the 120 F2 plants in mapping population G33 × G43 were analysed. Although the presence of phorbol esters is a qualitative trait, the data were scored quantitatively as the actual concentration of PE present may be influenced by whether the plant is heterozygous at the PE locus. The QTL analysis was performed using GridQTL and resulted in the identification of a single locus at 41 cM on linkage group 8 of the G33 × G43 linkage map (Figure3a). The 95% confidence intervals for the QTL position were 31–49 cM (18 cM length). On the map for this population, the two flanking markers were positioned at 34.8 cM (JCT3) and 47 cM (1401433|12335072), respectively, which is a gap of 12.2 cM (File S5). Further analysis of the data confirmed that all F2 individuals which were homozygous for the G43 (nontoxic) alleles at both these positions yielded seeds which did not contain PE (Figure3b). This confirms that the mutation responsible for the lack of PE within nontoxic seeds must reside between these two markers. The mean values for the PE content of the F2 plants which were heterozygous at these two alleles were also significantly less than the plants which were homozygous for the G33 (toxic) alleles (P < 0.01).
Figure 3.
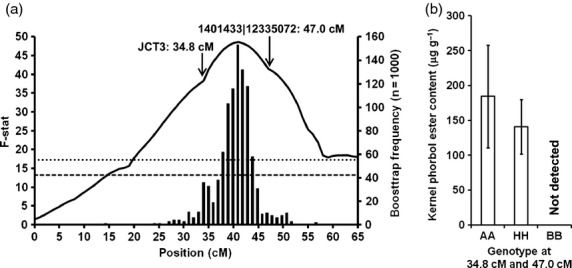
(a) QTL analysis of phorbol ester content based on seeds collected from 120 F2 plants in mapping population G33 × G43 for linkage group 8. Left y axis: The solid black line shows the F-stat score obtained using GridQTL. The horizontal lines show the genome-wide significance thresholds at P = 0.05 (lower, dashed) and P = 0.01 (upper, dotted) based on 1000 iterations. Right axis: The vertical bars show the QTL distribution calculated using bootstrap analysis (n = 1000). (b) Kernel phorbol ester content of seeds collected from F2 plants which were homozygous at two markers flanking the seed toxicity locus (‘AA’, G33 allele), heterozygous (‘HH’), or homozygous (‘BB’, G43). The error bars represent the standard deviations.
The identification of a locus controlling phorbol ester biosynthesis also permits the ‘nontoxic’ seed trait, which occurs naturally within only a limited set of germplasm, to be introgressed into other genetic backgrounds. The ability to improve the economics of J. curcas cultivation by allowing the meal to be converted into animal feed is also a significant advance in J. curcas breeding.
Finer mapping of the locus affecting phorbol ester biosynthesis by marker transfer and comparative mapping
Although variations in PE content for seeds from plants homozygous and heterozygous for the PE+ve alleles were observed, seed toxicity can still be considered a dominant and qualitative trait as only plants containing two copies of the PE+ve allele produced seed containing no phorbol esters. This permits the fine mapping of this trait without the problems encountered with large confidence intervals for the quantitative polygenic traits (Mangin and Goffinet, 1997). To conduct finer mapping of the locus controlling phorbol ester biosynthesis, two strategies were used. First, blastn searches were performed against the J. curcas genome sequence (Hirakawa et al., 2012; Sato et al., 2011) using the markers which mapped in at least one of the other populations in this study or had mapped in the interspecific cross produced previously (Wang et al., 2011). The scaffolds identified from these blastn searches were then mined in-silico for SSR sequences (Table4). Those sequences found to be polymorphic were genotyped in mapping population G33 × G43 then added to the linkage map. As a second approach to increasing the map density in the region containing the locus responsible for PE biosynthesis, a comparative mapping approach was used which exploited the microsynteny (colinearity) between the J. curcas and R. communis genomes. Where synteny exists between chromosomal regions of these two species, it is possible to infer the relative positions of J. curcas genes based on the larger R. communis assemblies. The synteny analysis revealed a number of syntenic blocks on linkage group 8 of J. curcas (Figure4, Table4 and File S6). In particular, the R. communis scaffold 30 174 mapped to two regions of J. curcas linkage group 8, one of which was located near the locus for PE biosynthesis. A number of additional SSR markers were created from J. curcas scaffolds which were syntenic with this R. communis scaffold (Table4). The result from the mapping of 16 additional markers in the region containing the locus is shown in Figure4. Analysis of the seed PE content data with the genotyping data indicated that the genetic locus conferring the absence of PE biosynthesis resides between makers NG285A and G273A. The genetic distance of this region is 2.3 cM. Our ability to map this trait to less than this distance is limited by a breakdown in synteny to the castor genome in this region (Table4 and File S6) and a lack of further progeny with informative recombination events in this region. Our future work on the mapping of this trait will therefore include the analysis of additional F2 and F3 plants to detect additional informative recombination events in this region. A contiguous sequence for this region will also be obtained by BAC sequencing.
Table 4.
Development of additional markers for fine mapping
| Position (cM) | Jatropha curcas marker | Bridge marker used |
J. curcas genome sequence |
R. communis genome sequence |
||
|---|---|---|---|---|---|---|
| Scaffold | Marker position | Scaffold | Syntenic region | |||
| 34.8 | NG291 | N/A | Jcr4S04633 | 10 436–10 735 | 30 174 | 2 724 054–2 749 754 |
| 34.8 | JCT3 | N/A | Jcr4S01892 | 18 299–18 382 | 30 174 | 2 695 280–2 722 645 |
| 35.1 | G284 | N/A | Jcr4S00160 | 23 304–23 696 | 30 174 | 2 663 765–2 686 270 |
| 35.4 | NG289B | N/A | Jcr4S03474 | 13 586–13 985 | 30 174 | 2 642 521–2 666 724 |
| 36.0 | NG288C | N/A | Jcr4S00541 | 36 116–36 276 | 30 174 | 2 553 645–2 632 854 |
| 36.4 | NG287B | N/A | Jcr4S04231 | 31 626–31 926 | 30 174 | 2 521 238–2 536 532 |
| 36.5 | NG286D | N/A | Jcr4S00188 | 39 908–40 226 | 30 174 | 2 442 869–2 507 050 |
| 36.8 | NG286A | N/A | Jcr4S00188 | 10 227–10 414 | ″ | ″ |
| 37.6 | NG285A | N/A | Jcr4S00012 | 9750–10 085 | 30 174 | 2 278 471–2 410 014 |
| 38.4 | G262 | SNP10542 | Jcr4S05837 | 4261–4517 | 29 996 | 51 566–62 602 |
| 38.4 | NG331C | N/A | Jcr4S01963 | 25 834–26 225 | 29 996 | 62 985–63 609 |
| 38.8 | G270B | 1404867|12356283 | Jcr4S03364 | 6780–6962 | 29 747 | 274 981–367 675 |
| 38.8 | G270D | 1404867|12356283 | Jcr4S03364 | 17 347–17 502 | ″ | ″ |
| 39.1 | G282B | SNP5294 | Jcr4S01263 | 23 937–24 259 | Low level of synteny | |
| 39.9 | G273A | 1400435|12286473 | Jcr4S08292 | 1249–1548 | Low level of synteny | |
| 40.2 | G268 | 1401665|12362006 | Jcr4S01025 | 42 898–43 155 | 28 266 | 19 280–78 337 |
| 42.2 | G269 | eSNP0195 | Jcr4S01910 | 8711–8987 | Low level of synteny | |
| 47.0 | 1401433|12335072 | N/A | Jcr4S02123 | 25 922–25 718 | Low level of synteny | |
Figure 4.
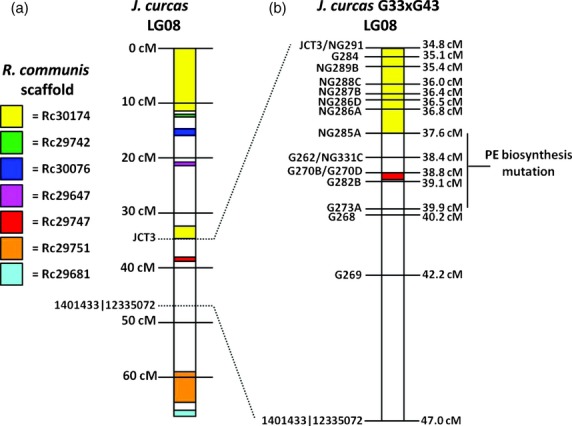
(a) Identification of syntenic regions on J. curcas linkage group 8 by comparison of scaffolds of the J. curcus and R. communis genome sequence. (b) – finer mapping of the region of linkage group 8 of mapping population G33 × G43 containing the QTL for phorbol ester biosynthesis.
The mapping of the mutation responsible for the lack of PE biosynthesis to within a relatively small region of the genome means that it will now be possible to breed new high oil yielding varieties of J. curcas which lack phorbol esters. This 2.3 cM region is likely to represent less than 1% of the entire 717.0 cM genome, which means it will be possible to remove large portions of the remaining genome of the nontoxic variety, which may contain many undesirable alleles, by backcrossing.
We have also demonstrated the utility of exploiting the existence of syntenous colinear regions within genomes to develop markers targeted to specific regions of a genome. Although this comparative mapping has been used previously in organisms such as Brassica sp. (Lagercrantz et al., 1996) and Triticum aestivum (Liu and Anderson, 2003), this approach has typically relied on the availability of a near-complete genome sequence (i.e. Arabidopsis thaliana and Oryza sativa, respectively). In this study, we compared genomic sequences of J. curcas (Hirakawa et al., 2012; Sato et al., 2011) and R. communis (Chan et al., 2010), both of which are in a draft stage, containing thousands of scaffolds. Due to the low cost and rapidity of the shotgun sequencing approach, most genome sequences are currently only assembled as ‘drafts’. This approach to mapping will therefore have utility in other species.
The four populations in this study were also developed from parents with variations in a number of traits including oil content and seed size. We will therefore use these mapping populations to dissect these traits once these perennial plants approach maturity.
Experimental procedures
Mapping populations
Four mapping populations were used in this study, as detailed in Table1. Parental lines G43, Cabo Verde (CV), QV-JAT01, QV-JAT02 and QV-JAT03 were homozygous at all loci, whereas parental lines G33 and G51 were heterozygous at some of the loci scored in this study. To minimize the number of uninformative markers in mapping populations G33 × G43 and G51 × CV, the F2 were created using an F1 intercross. All other mapping populations were created by self-pollinating F1 plants.
DNA extraction and amplification
DNA from mapping populations G33 × G43 and G51 × CV was extracted using the Qiagen DNEasy Plant Mini kit (Qiagen, Venlo, the Netherlands). DNA from the other mapping populations was extracted using the CTAB protocol (Doyle and Doyle, 1987). DNA was quantified using the DNA binding dye EvaGreen (Biotium, Hayward, CA), using salmon sperm DNA as a standard. (Wang et al., 2006). Where insufficient DNA was obtained, DNA was amplified using the Qiagen RepliG kit.
Single Nucleotide Polymorphism (SNP) marker identification and analysis
SNPs were obtained from two sources. A reduced representation library was produced from toxic and nontoxic provenances of J. curcas using the CRoPS® technique (van Orsouw et al., 2007). SNPs identified by this technique were converted to an Illumina VeraCode assay (File S1). SNPs were also identified from ESTs of developing seeds obtained from pyrosequencing of cDNA from varieties bearing toxic (King et al., 2011) and nontoxic seeds (A.J. King and I.A. Graham, unpublished). The SNPs obtained from the EST databases were assayed using one of two different methods. Capillary electrophoresis based method; an additional three noncomplementary nucleotides were added to the 5′-end of one of the allele-specific primers to permit allele discrimination by amplicon size. An additional mismatch near the 3′-end of each allele-specific primer was also incorporated to increase specificity (Bui and Liu, 2009). An M13 tail sequence (5′-TGTAAAACGACGGCCAGT-3′) was added to the 5′-end of the locus-specific primers to facilitate fluorescent labelling of the PCR products. PCR reactions for the two alleles were performed independently using the Qiagen Multiplex PCR kit. Each 10 μL reaction contained 5 μL of reagent, 2–20 ng of DNA, 0.5 pmol allele-specific primer, 0.5 pmol locus-specific primer and 2 pmol of either FAM (‘low’ allele) or VIC (‘high’ allele) labelled M13 primer. Up to 12 SNPs were multiplexed per PCR reaction. After pooling of the ‘low’ and ‘high’ allele PCR reactions, fragment analysis was performed using an Applied Biosystems 3730 DNA Analyzer (Life Technolgies, Carlsbad, CA) using a LIZ-500 internal standard. KASPar method; the KASPar assay was performed according to the manufacturer's protocol (KBiosciences, Hoddesdon, UK). Allele discrimination was performed using an Applied Biosystems 7300 Real-Time PCR System. Sequences of SNPs and the primers used for their detection by capillary electrophoresis or KASPar assays are detailed in File S2.
Simple Sequence Repeat (SSR) marker identification and analysis
SSR markers were obtained from a number of sources as detailed in File S3. Accession numbers KC344792–KC344818 were obtained by dye-terminator sequencing clones from an AGn-enriched DNA library created according to the FIASCO protocol (Zane et al., 2002). SSRs were also identified by mining of EST data (King et al., 2011) or genomic sequence data (Lee and Sonnhammer, 2003). Finally, a number of publicly available SSR markers were also used. Primers for SSR analysis were designed using Primer3Plus (Untergasser et al., 2007) according to the nearest-neighbour method (SantaLucia, 1998). An M13 tail sequence was appended to 5′-end of the shortest primer to facilitate fluorescent labelling of the PCR products. SSR analysis was conducted using the QIAGEN Type-It Microsatellite PCR kit. Each 10 μL reaction contained 5 μL of reagent, 2–20 ng of DNA, 0.5 pmol forward primer, 0.5 pmol reverse primer and 2 pmol of VIC labelled M13 primer. Thermocycling was conducted according to the manufacturer's recommended protocol, using an annealing temperature of 57 °C. Up to 12 SSRs were multiplexed per PCR reaction. Fragment analysis was performed using an Applied Biosystems 3730 DNA Analyzer (Life Technolgies). A LIZ-500 dye was used as an internal size standard.
Construction of genetic linkage maps
To build linkage maps, CRI-MAP version 2.503 was used (Lander and Green, 1987). Linkage maps for each of the mapping populations were first constructed independently. Linkage groups were first determined using the twopoint function with an LOD cut-off point >4.0. Maps for each linkage group were then constructed using the build function. Orders were checked with the flipsn function. To check for genotyping errors, the chrompic function was used to identify suspect double crossover events—that is, those observed within a short distance (<5 cM). Suspect markers were then checked where possible by reanalysis of the original data and corrected when a manual scoring error was evident. After the initial correction, any markers still found to produce excessive double-crossovers (>2.5% of population) were omitted from the map. The remaining suspect genotypes resulting in double-crossovers were converted to missing data. After completion of the four individual linkage maps, the genotype files were combined manually using Microsoft Excel spreadsheets. An integrated linkage map was then produced by rebuilding a single map using CRI-MAP.
Analysis of phorbol ester content of seeds
The testa of the seeds, which have previously been shown to lack phorbol esters (He et al., 2011), was removed and the kernels were ground to a fine powder in a mortar and pestle. Approximately 200 mg of kernel was transferred to a 2 mL Eppendorf tube. Five micro-liter of 1 mg/mL of phorbol 12-myristate a 13-acetate (Sigma-Aldrich Co Ltd, Poole, Dorset, UK) was added as an internal standard. The seed kernel was then extracted with 1.5 mL of methanol for 1 h. Solvent was recovered after centrifugation of samples at 10 000 g for 2 min. The seed kernel was then extracted twice more for 1 h with 1 mL of methanol. The extracts were combined, and the solvent was removed using a Speedvac. The oily residue was dissolved in 800 μL of hexane. Eight hundred micro-liter of acetonitrile was then added, and the sample was vortexed. After centrifugation of the sample for 1 min at 10 000 g, the lower acetonitrile phase containing the phorbol esters was recovered and filtered through a 0.45 μm PTFE membrane (Millex®-LH). The filtered extract was then evaporated using a Genevac EZ-2 plus evaporator (Genevac Ltd, Ipswich, Suffolk, UK) and dissolved in 200 μL of acetonitrile. HPLC analysis was then performed as described previously (He et al., 2011).
Marker-trait association studies
To determine the position of markers affecting PE biosynthesis, PE content data were scored quantitatively using GridQTL (Seaton et al., 2006). Chromosome-wide permutation studies were also conducted with 1000 iterations to determine significance thresholds. After the identification of the region controlling phorbol ester biosynthesis, further data analyses were performed by scoring of PE biosynthesis as a qualitative trait, by direct comparison of genotype and trait data.
Mapping of markers onto the draft genome sequence of J. curcas and synteny analysis with R. communis
A blastn search for all markers described in this study was performed against build 4.5 of the J. curcas genome (Hirakawa et al., 2012). To identify synteny between the J. curcas and R. communis genomes, the blast2 search data against R. communis, which is provided on the J. curcas genome database, were retrieved. Where synteny was observed for a number of consecutive markers, a more detailed analysis of synteny was obtained using the promer function of MUMmer 3.0 (Kurtz et al., 2004), with the amino acid identity threshold set at 40% and the minimum length set at 100 base pairs.
Acknowledgments
The authors thank Judith Mitchell for administrative support. This work was partly funded by a European Union Framework 7 Program Knowledge Based Bio-Economy grant under contract 245236. The CRoPS® technology is covered by patents and patent applications owned by Keygene N.V. CRoPS® is a registered trademark of Keygene N.V.
Supporting Information
File S1 SNP markers developed from genomic DNA using the CRoPS® technique.
File S2 SNP markers mined from seed transcriptome data.
File S3 SSR markers used in this study.
File S4 Markers used to bridge the maps produced in this study with the previously published map produced from an interspecific cross between J. curcas and J. integerrima (Wang.,).
File S5 Individual linkage maps from the four mapping populations used in this study.
File S6 Location of mapped markers on the genome sequence of J. curcas [build 4.5 (Hirakawa.,)], and syntenic blocks observed with R. communis genome.
File S7 PE concentration measured in open-pollinated seed collected from 120 F2 plants.
References
- Aregheore EM, Becker K, Makkar HPS. Detoxification of a toxic vatiety of Jatropha curcas using heat and chemical treatments, and preliminary nutitional evaluation with rats. South Pac.J Nat Sci. 2003;21:51–56. [Google Scholar]
- Basha SD, Francis G, Makkar HPS, Becker K, Sujatha M. A comparative study of biochemical traits and molecular markers for assessment of genetic relationships between Jatropha curcas L. germplasm from different countries. Plant Sci. 2009;176:812–823. [Google Scholar]
- Brooker J. 2009. Methods for detoxyfying oil seed crops (Patent WO/2010/070264)
- Bui M, Liu Z. Simple allele-discriminating PCR for cost-effective and rapid genotyping and mapping. Plant Methods. 2009;5:1. doi: 10.1186/1746-4811-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CR, Clarindo WR, Praça MM, Araújo FS, Carels N. Genome size, base composition and karyotype of Jatropha curcas L., an important biofuel plant. Plant Sci. 2008;174:613–617. [Google Scholar]
- Chan AP, Crabtree J, Zhao Q, Lorenzi H, Orvis J, Puiu D, Melake-Berhan A, Jones KM, Redman J, Chen G, Cahoon EB, Gedil M, Stanke M, Haas BJ, Wortman JR, Fraser-Liggett CM, Ravel J, Rabinowicz PD. Draft genome sequence of the oilseed species Ricinus communis. Nat Biotechnol. 2010;28:951–956. doi: 10.1038/nbt.1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa G, Cardoso K, Del Bem L, Lima A, Cunha M, de Campos-Leite L, Vicentini R, Papes F, Moreira R, Yunes J, Campos F, Da Silva M. Transcriptome analysis of the oil-rich seed of the bioenergy crop Jatropha curcas L. BMC Genomics. 2010;11:462. doi: 10.1186/1471-2164-11-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silveira JC. Contribution a l'étude du pulghère aux iles du Cap Vert. Anais Inst. Super Agron. (Lisbon) 1934;6:116–126. [Google Scholar]
- Dehgan B, Webster GL. Morphology and infrageneric relationships in Jatropha (Euphorbiaceae) University of California Press; 1979. [Google Scholar]
- Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987;19:11–15. [Google Scholar]
- Goel G, Makkar HPS, Francis G, Becker K. Phorbol esters: structure, biological activity, and toxicity in animals. Int. J. Toxicol. 2007;26:279–288. doi: 10.1080/10915810701464641. [DOI] [PubMed] [Google Scholar]
- Hackett CA, Broadfoot LB. Effects of genotyping errors, missing values and segregation distortion in molecular marker data on the construction of linkage maps. Heredity. 2003;90:33–38. doi: 10.1038/sj.hdy.6800173. [DOI] [PubMed] [Google Scholar]
- He W, King AJ, Khan MA, Cuevas JA, Ramiaramanana D, Graham IA. Analysis of seed phorbol-ester and curcin content together with genetic diversity in multiple provenances of Jatropha curcas L. from Madagascar and Mexico. Plant Physiol. Biochem. 2011;49:1183–1190. doi: 10.1016/j.plaphy.2011.07.006. [DOI] [PubMed] [Google Scholar]
- Hirakawa H, Tsuchimoto S, Sakai H, Nakayama S, Fujishiro T, Kishida Y, Kohara M, Watanabe A, Aizu T, Toyoda A, Fujiyama A, Yamada M, Tabata S, Fukui K, Sato S. Upgraded genomic information of Jatropha curcas L. Plant Biotechnol. 2012;29:123–130. [Google Scholar]
- Hirota M, Suttajit M, Suguri H, Endo Y, Shudo K, Wongchai V, Hecker E, Fujiki H. A new tumor promoter from the seed oil of Jatropha curcas L., an intramolecular diester of 12-deoxy-16-hydroxyphorbol. Cancer Res. 1988;48:5800–5804. [PubMed] [Google Scholar]
- Kalannavar VN. Response of Jatropha curcas to nitrogen, phosphorus and potassium levels in nortern transition zone of Karnataka. Dharwad, India: Dharwad University of Agrigultural Sciences; 2008. 109. [Google Scholar]
- King AJ, Li Y, Graham IA. Profiling the developing Jatropha curcas L. seed transcriptome by pyrosequencing. BioEnergy Research. 2011;4:211–221. [Google Scholar]
- King AJ, He W, Cuevas JA, Freudenberger M, Ramiaramanana D, Graham IA. Potential of Jatropha curcas as a source of renewable oil and animal feed. J. Exp. Bot. 2009;60:2897–2905. doi: 10.1093/jxb/erp025. [DOI] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher A, Smoot M, Shumway M, Antonescu C, Salzberg S. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagercrantz U, Putterill J, Coupland G, Lydiate D. Comparative mapping in Arabidopsis and Brassica, fine scale genome collinearity and congruence of genes controlling flowering time. Plant J. 1996;9:13–20. doi: 10.1046/j.1365-313x.1996.09010013.x. [DOI] [PubMed] [Google Scholar]
- Lander ES, Green P. Construction of multilocus genetic linkage maps in humans. Proc. Natl Acad. Sci. USA. 1987;84:2363–2367. doi: 10.1073/pnas.84.8.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Sonnhammer EL. Genomic gene clustering analysis of pathways in eukaryotes. Genome Res. 2003;13:875–882. doi: 10.1101/gr.737703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Anderson JA. Targeted molecular mapping of a major wheat QTL for Fusarium head blight resistance using wheat ESTs and synteny with rice. Genome. 2003;46:817–823. doi: 10.1139/g03-066. [DOI] [PubMed] [Google Scholar]
- Makkar HPS, Aderibigbe AO, Becker K. Comparative evaluation of non-toxic and toxic varieties of Jatropha curcas for chemical composition, digestibility, protein degradability and toxic factors. Food Chem. 1998;62:207. [Google Scholar]
- Mangin B, Goffinet B. Comparison of several confidence intervals for QTL location. Heredity. 1997;78:345–353. [Google Scholar]
- Martins WS, Lucas DC, Neves KF, Bertioli DJ. WebSat — a web software for microsatellite marker development. Bioinformation. 2009;3:282–283. doi: 10.6026/97320630003282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer M. A comparison of regression interval mapping and multiple interval mapping for linked QTL. Heredity. 2005;94:599–605. doi: 10.1038/sj.hdy.6800667. [DOI] [PubMed] [Google Scholar]
- Mohapatra S, Panda PK. Effects of fertilizer application on growth and yield of Jatropha curcas L.in an aeric tropaquept of Eastern India. Notulae Scientia Biologicae. 2011;3:95–100. [Google Scholar]
- Natajaran P, Parani M. De novo assembly and transcriptome analysis of five major tissues of Jatropha curcas L. using GS FLX titanium platform of 454 pyrosequencing. BMC Genomics. 2011;12:191. doi: 10.1186/1471-2164-12-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phumichai C, Phumichai T, Kongsiri N, Wongkaew A, Sripichit P, Kaveeta R. Isolation of 55 microsatellite markers for Jatropha curcas and its closely related species. Biol. Plant. 2011;55:387–390. [Google Scholar]
- SantaLucia J. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl Acad. Sci. USA. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Hirakawa H, Isobe S, Fukai E, Watanabe A, Kato M, Kawashima K, Minami C, Muraki A, Nakazaki N, Takahashi C, Nakayama S, Kishida Y, Kohara M, Yamada M, Tsuruoka H, Sasamoto S, Tabata S, Aizu T, Toyoda A, Shin-i T, Minakuchi Y, Kohara Y, Fujiyama A, Tsuchimoto S, Kajiyama S, Makigano E, Ohmido N, Shibagaki N, Cartagena JA, Wada N, Kohinata T, Atefeh A, Yuasa S, Matsunaga S, Fukui K. Sequence analysis of the genome of an oil-bearing tree, Jatropha curcas L. DNA Res. 2011;18:65–76. doi: 10.1093/dnares/dsq030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaton G, Hernandez J, Grunchec JA, White I, Allen J, De Koning DJ, Wei W, Berry D, Haley C, Knott S. 2006. Proceedings of the 8th World Congress on Genetics Applied to Livestock Production Brazil Belo Horizonte GridQTL: A grid portal for QTL mapping of compute intensive datasets.
- Stieneke DL, Eujayl I. 2007. Imperfect SSR Finder http://ssr.nwisrl.ars.usda.gov/ )
- Sujatha M, Makkar HPS, Becker K. Shoot bud proliferation from axillary nodes and leaf sections of non-toxic Jatropha curcas L. Plant Growth Regul. 2005;47:83. [Google Scholar]
- Sun QB, Li LF, Li Y, Wu GJ, Ge XJ. SSR and AFLP markers reveal low genetic diversity in the biofuel plant Jatropha curcas in China. Crop Sci. 2008;48:1865–1871. [Google Scholar]
- Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JAM. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007;35:W71–W74. doi: 10.1093/nar/gkm306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Orsouw NJ, Hogers RCJ, Janssen A, Yalcin F, Snoeijers S, Verstege E, Schneiders H, van der Poel H, van Oeveren J, Verstegen H, van Eijk MJT. Complexity reduction of polymorphic sequences (CRoPS™): A novel approach for large-scale polymorphism discovery in complex genomes. PLoS ONE. 2007;2:e1172. doi: 10.1371/journal.pone.0001172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmann J, Rajcan I. Oil Crop Breeding and Genetics. In: Vollmann J, Rajcan I, editors. Handbook of Plant Breeding: Oil Crops. Vol. 4. 2010. pp. 1–31. eds) . In: , Vol. [Google Scholar]
- Wang CM, Liu P, Yi C, Gu K, Sun F, Li L, Lo LC, Liu X, Feng F, Lin G, Cao S, Hong Y, Yin Z, Yue GH. A first generation microsatellite- and SNP-based linkage map of Jatropha. PLoS ONE. 2011;6:e23632. doi: 10.1371/journal.pone.0023632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Chen K, Xu C. DNA quantification using EvaGreen and a real-time PCR instrument. Anal. Biochem. 2006;356:303–305. doi: 10.1016/j.ab.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Yang C, Fang Z, Li B, Liu G, Li J. Breeding of high-oil Jatropha curcas L for biodiesel production. Chin. J. Biotechnol. 2010;26:1514–1525. [PubMed] [Google Scholar]
- Yi C, Zhang S, Liu X, Bui H, Hong Y. Does epigenetic polymorphism contribute to phenotypic variances in Jatropha curcas L.? BMC Plant Biol. 2010;10:1–9. doi: 10.1186/1471-2229-10-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zane L, Bargelloni L, Patarnello T. Strategies for microsatellite isolation: a review. Mol. Ecol. 2002;11:1–16. doi: 10.1046/j.0962-1083.2001.01418.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
File S1 SNP markers developed from genomic DNA using the CRoPS® technique.
File S2 SNP markers mined from seed transcriptome data.
File S3 SSR markers used in this study.
File S4 Markers used to bridge the maps produced in this study with the previously published map produced from an interspecific cross between J. curcas and J. integerrima (Wang.,).
File S5 Individual linkage maps from the four mapping populations used in this study.
File S6 Location of mapped markers on the genome sequence of J. curcas [build 4.5 (Hirakawa.,)], and syntenic blocks observed with R. communis genome.
File S7 PE concentration measured in open-pollinated seed collected from 120 F2 plants.