

Abstract
Gene therapy strategies for congenital myopathies may require repeat administration of adeno-associated viral (AAV) vectors due to aspects of the clinical application, such as: (i) administration of doses below therapeutic efficacy in patients enrolled in early phase clinical trials; (ii) progressive reduction of the therapeutic gene expression over time as a result of increasing muscle mass in patients treated at a young age; and (iii) a possibly faster depletion of pathogenic myofibers in this patient population. Immune response triggered by the first vector administration, and to subsequent doses, represents a major obstacle for successful gene transfer in young patients. Anti-capsid and anti-transgene product related humoral and cell-mediated responses have been previously observed in all preclinical models and human subjects who received gene therapy or enzyme replacement therapy (ERT) for congenital myopathies. Immune responses may result in reduced efficacy of the gene transfer over time and/or may preclude for the possibility of re-administration of the same vector. In this study, we evaluated the immune response of a Pompe patient dosed with an AAV1-GAA vector after receiving Rituximab and Sirolimus to modulate reactions against ERT. A key finding of this single subject case report is the observation that B-cell ablation with rituximab prior to AAV vector exposure results in non-responsiveness to both capsid and transgene, therefore allowing the possibility of repeat administration in the future. This observation is significant for future gene therapy studies and establishes a clinically relevant approach to blocking immune responses to AAV vectors.
Introduction
Pompe disease is a progressive and often fatal neuromuscular disorder resulting from mutation in the gene for acid alpha-glucosidase (GAA), an enzyme necessary for the degradation of lysosomal glycogen within the lysosome. The condition is characterized by a spectrum of disease severity resulting from variable levels of GAA and possibly differential cellular rates of glycogen synthesis. The result of GAA deficiency is extensive glycogen accumulation in all tissues, especially striated muscle, smooth muscle and the central nervous system (CNS).1–3 The range of disease severity encompasses the fatal early-onset form presenting in infancy to a milder adult-onset of disease symptoms. The disease prevalence has been estimated to be ~4,000 patients in the developed world and the incidence is 1 per 40,000 births.4,5 The phenotypic continuum is directly related to the extent of residual enzyme deficiency2,6 with complete or near complete deficiency of functional GAA protein in early-onset disease and up to 20% of wild-type activity in late-onset patients.7,8
Respiratory and skeletal muscle weakness is a key progressive feature of Pompe disease. Respiratory muscle weakness often leads to the need for assisted ventilation and is the principal cause of mortality in early and late onset Pompe patients.9,10 Skeletal muscle weakness differentially affects the lower limbs and results in loss of ambulation and wheelchair dependency.11 Generally, GAA activity <1% of wild-type level correlates with presentation in infancy, and 2–20% GAA activity is seen in later-onset disease.4,5 Approximately 25% of infants with <1% GAA activity have no detectable GAA protein by Western blot analysis and are considered cross-reactive immunologic material (CRIM)-negative.12 In CRIM-positive patients, the presence of residual GAA protein usually correlates with a lack of neutralizing antibodies (Nabs) against GAA after initiation of enzyme replacement therapy (ERT). In contrast, CRIM-negative patients lack tolerance to GAA protein and produce a robust humoral immune response to ERT, reducing the efficacy of treatment. CRIM-negative patients receiving ERT have a poor prognosis and diminished survival if not managed with immunosuppression.13–16
ERT has extended survival for early onset patients; however, a successful gene therapy strategy may provide additional long-term benefits and improvement in quality of life. Recombinant adeno-associated viral vectors (rAAV) are widely used gene therapy agents for the treatment of genetic diseases. AAV has been used in several clinical trials for the treatment of different conditions including Leber’s congenital amaurosis,17,18 hemophilia B,19,20 Pompe disease,21 Sanfilippo syndrome,22 lipoprotein lipase deficiency,23,24 Alpha-1 antitrypsin deficiency,25 and Limb-girdle muscular dystrophy.26,27 A critical challenge for the success of gene therapy is the host immune responses to both the vector capsid and transgene product. These immune responses raise concerns regarding the safety and longevity of gene expression. In addition, induction of antibodies by natural exposure to AAV is frequent early in life and may influence the use of AAV as a gene therapy vector.28,29 This may be critical in developing effective therapeutic strategies for congenital myopathies that may require repeat administration of AAV vectors. Repeat AAV administration may be necessary because low doses, or doses below optimal therapeutic threshold were provided in early phase studies. Furthermore, many subjects may require re-dosing later in life, as increasing muscle mas or loss of copy number with age may reduce transgene expression. Therefore, potent humoral and cellular memory responses to AAV may compromise the subsequent use of the same vector.28,29 For these reasons, we are developing strategies to manage these immune responses as a sustainable approach to deliver safe and long-term expression of the therapeutic gene by AAV-mediated gene therapy.
Humoral responses to ERT observed in Pompe disease include activation of antigen-specific CD4+ T helper cells and production of neutralizing (NAbs) and non-neutralizing (non-NAbs) antibodies. NAbs have been the focus of gene therapy immunology studies because of their effect on diminishing the efficacy of AAV-mediated gene therapy. NAbs bind to the AAV capsid and may block or reduce the transduction of target cells. Additionally, anti-transgene antibodies may develop against the therapeutic protein or may serve as co-activating factors for cell-mediated immunity, possibly leading to elimination of transduced cells.28,29
One of the strategies to overcome the presence of Nabs in rAAV-mediated gene therapy is pharmacological modulation of the humoral immune response. Rituximab is a monoclonal antibody that induces B-cell depletion by binding CD20 found on the surface of B-cells. Rituximab is FDA approved for the treatment of chronic lymphocytic leukemia, non-Hodgkin’s lymphoma, and rheumatoid arthritis. Recent studies also showed that Rituximab is able to improve immune tolerance induction in patients with hemophilia.30–32 In addition, Rituximab was shown to be successful in reduction of existing anti-rAAVNAbs in humans receiving rituximab for rheumatoid arthritis33 and in non-human primates.19,34 Rituximab’s mechanism of action is by complement-mediated cell lysis, antibody-dependent cell-mediated cytotoxicity and direct induction of apoptosis by binding to CD20.35
Rituximab may be used in combination with a B-cell and T-cell agent, such as Sirolimus.36 Sirolimus inhibits the response to interleukin-2 by binding the FK-binding protein 12 (FKBP12). The Sirolimus-FKBP12 complex acts as an inhibitor of the mammalian target of rapamycin (mTOR) pathway and has been used in preclinical studies for gene therapy.37–39 Therefore, we believe that immunomodulatory regimen with Rituximab and Sirolimus may be beneficial to control expression of immune responses as a consequence of AAV-mediated gene therapy in humans.
Elder et al.13,15 showed that an immune suppression protocol based on B-cell depletion with Rituximab and T-cell immune modulation with Sirolimus before initiation of ERT in CRIM-negative infants with early-onset Pompe disease was able to block GAA antibody responses and impact clinical outcomes. In this article, we describe the immunological and clinical consequences of B-cell depletion and T-cell immune modulation in a cross-reacting immunologic material-negative (CRIM-) child with Pompe disease, who was receiving Rituximab and Sirolimus prior to and during enrollment of a phase I-II clinical trial for intramuscular administration of an rAAV1-CMV-hGAA vector.21
Results
Subject characteristics
The subject of this case study is a CRIM- female child who was diagnosed with Pompe disease (nonsense mutation exon 10, p.W516X; exon 18, p.G828_N882del) at 5.5 months of age. CRIM status was evaluated at the Powell Gene Therapy Center (Gainesville, FL) by Western blot analysis of skin fibroblasts. The patient presented a severe phenotype with GAA activity <1%, cardiac hypertrophy and ultimately required assisted ventilation. Immediately after diagnosis she was enrolled in a study for which she received ERT and immune modulation for management of antibody response to ERT as part of the clinical treatment regimen. Details of this study are presented in Elder et al.13 At 45 months of age, the patient was enrolled in another study evaluating the effect of rAAV1-CMV-hGAA intramuscular gene transfer to the diaphragm. Cohort I data from this study are presented in Smith et al.21 Figure 1 summarizes the time course of events in this study.
Figure 1.
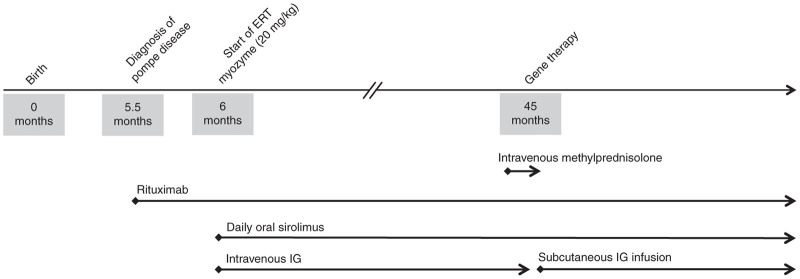
Timeline: At 5.5 months of age the patient received an induction dose of Rituximab followed by daily oral Sirolimus. A monthly dose of IV immunoglobulin (IVIG) was also given. The patient continued the B-cell depletion regimen with Rituximab at 12 weeks intervals while receiving Sirolimus daily. At 6 months of age, following completion of Rituximab, the patient started on enzyme replacement therapy (ERT) at a dose of 20 mg/kg every 7–10 days. A brief course of methylprednisolone was given prior to and following AAV1-GAA intramuscular gene transfer to the diaphragm at 45 months of age.
Immunosuppression protocol and ERT
At 5.5 months of age, about 20 days before starting ERT, the patient received 375 mg/m2 of rituximab and 10 mg/kg of methylprednisolone intravenously (premedication) weekly for 3 weeks. After the Rituximab induction doses, the patient received daily oral Sirolimus (0.06–1 mg/m2/day). The dosage was adjusted in order to maintain a Sirolimus serum trough level of 3–7 ng/ml. Once the induction dose of Rituximab was completed, the patient started receiving a monthly dose of 500–1,000 mg/kg of IV immunoglobulin (IVIG) (Gamunex; TalecrisBiotherapeutics, Research Triangle Park, North Carolina or Privigen; CSL Behring, King of Prussia, PA). The IVIG dose was adjusted in order to maintain a trough serum IgG level of 700–1,000 mg/dl. The patient continued the B-cell depletion regimen by receiving Rituximab at 12-week intervals along with daily Sirolimus. At 6 months of age, after completion of B cell depletion, the patient started on ERT infusions at a dose of 20 mg/kg every 7–10 days. The ERT infusions interval was subsequently increased to every 10–14 days once clinical improvement was demonstrated.
rAAV-mediated gene therapy
The patient was enrolled in a Phase I/II study to determine the safety and ventilatory outcomes following intramuscular administration of rAAV1-CMV-hGAA (NCT00976352; ClincalTrials.gov). AAV dosing was accomplished at 45 months of age via bilateral diaphragmatic delivery using video-assisted thoracoscopy. Methylprednisolone (10 mg/kg) was given at the time of the procedure and up to post-operative day 3. The vector was injected in each hemidiaphragm in three sites, corresponding to the anterior, lateral and posterior costal regions. Each site received 8.33 × 1011 vg in 0.8 ml. The total dose was 5 × 1012 vg of rAAV1-CMV-hGAA.21
Anti-rAAV1 and anti-hGAA circulating antibody and T-cell–mediated responses
Serum samples were assayed by ELISA for circulating antibodies to the AAV1 capsid proteins. The patient receiving immune modulation was compared to a control group, which consisted of five children with Pompe disease that received 1–5 × 1012 vg/kg of rAAV1-CMV-hGAA but did not receive an immunosuppression regimen. The control group demonstrated at least a 155-fold increase in anti-AAV1Ab titer post exposure to AAV1. The patient of this report had no response to AAV capsid proteins through day 365 of the study (Figure 2). All antibody titers were at least two logs above the limit of detection for the assay.
Figure 2.

A 3.5 year-old Pompe disease subject (solid lines, diamonds) received weekly administration of 375 mg/m2 of rituximab along with 10 mg/kg of methylprednisolone for 3 weeks prior to dosing with 5 × 1012 vg/kg of rAAV1-CMV-hGAA. The patient then received daily oral administration of 0.06–1 mg/m2 of Sirolimus through the study and continued B-cell depletion with Rituximab every 12 weeks. Five Pompe subjects (dash-dot lines, circles), age 2.5–15 years, received 1–5 × 1012 vg/kg of rAAV1-CMV-GAA without immune modulation, served as a control group. The light horizontal lines represent +2 SD above the population mean. (a) Circulating antibodies against AAV1. (b) Circulating antibodies against hGAA. (c) Antigen specific response assay versus AAV1. (d) Antigen specific response assay versus hGAA.
Enzyme linked immunospot (ELISpot) assay and antigen specific response assay (ASR) were used to test CD4+ and CD8+ T-cells for reactivity against both the AAV capsid and the transgene products. ELISPOT assays to AAV1 capsid protein peptide pools and ASR to intact AAV1 capsids were unchanged from baseline through day 90 (Figure 2). Together, these findings demonstrate the potential to block humoral and cellular immune response following exposure to AAV1. The described regimen facilitated successful regional gene transfer with no adverse events and resulted in a clinical benefit.
Clinical pathology laboratory evaluation
Safety laboratory data is shown in Table 1. Serum IgG was maintained above 700–1,000 mg/dl by regular monthly infusions of IVIG. The approach had no impact on WBC, nor was there any evidence of thrombocytopenia or anemia. Serum transaminase levels are commonly elevated in Pompe disease40 and were unchanged in the patient, from baseline through the 90 days following rAAV1-CMV-hGAA dosing. NT-pro BNP, CPK, and CPK-MB were responsive to initiation of ERT and were not influenced by the immune management. There was no impact on CD3 positive lymphocytes. Evaluation of CD20 counts confirmed complete depletion of B-cells by Rituximab.
Table 1. Safety data.
| Day −30 | Day −1 | Day 1 | Day 3 | Day 14 | Day 90 | Day 180 | Day 270 | |
|---|---|---|---|---|---|---|---|---|
| IgG (mg/dl) | 674 | – | – | – | – | 994 | – | 1,209 |
| IgA (mg/dl) | <5 | – | – | – | – | <5 | – | <5 |
| WBC (thou/cu mm) | 15.3 | 8.4 | 5.5 | 10.5 | 7.5 | 12.3 | 5.6 | 8.1 |
| Platelet (thou/cu mm) | 481 | 403 | 385 | 491 | 389 | 374 | 304 | 324 |
| Neutrophil (%) | 61 | 16.6 | 24 | 23.8 | 29 | 69.7 | 29 | 48.1 |
| Lymphocyte (%) | 12 | 58.5 | 63 | 60.5 | 49.2 | 19 | 50.9 | 39.8 |
| Monocyte (%) | 5 | 6.6 | 9 | 10.2 | 8 | 7.7 | 13 | 7 |
| Eosinophil (%) | 1 | 16.2 | 3 | 3 | 10.1 | 2.3 | 4.2 | 2.4 |
| Basophil (%) | 0 | 0.4 | 0 | 0.6 | 0.7 | 0.2 | 0.5 | 0.4 |
| RBC (mill/cu mm) | 4.42 | 4.61 | 4.33 | 4.75 | 4.54 | 5.17 | 5.12 | 5.2 |
| AST | 366 | 484 | 445 | 438 | 469 | 496 | 575 | 436 |
| ALT | 246 | 306 | 287 | 316 | 239 | 277 | 426 | 292 |
| CD20% | 0 | – | – | – | – | 0 | – | 4 |
| CD3% | 80 | – | – | – | – | 86 | – | 88 |
| NT-PRO-BNP | 225 | – | – | – | – | 44 | – | – |
| CK (U/L) | 2,549 | 1,967 | 1,518 | 1,170 | 2,433 | 2,858 | 2,613 | – |
| CKMB (ng/ml) | 22.7 | – | – | – | – | 22.8 | – | – |
There were no adverse events identified during the follow up period to date related to the study procedure or immune modulation regimen.
Functional measures
Maximal Inspiratory Pressure (MIP) remained stable at day 90 and increased to 6% above the baseline value by day 180. A global measure of motor function, GMFM-88, increased 2.95% at day 90 and decreased 1.33% at day 180 (Table 2), which are not clinically significant changes.
Table 2. Functional data.
| Day −1 | Day 90 | Day 180 | |
|---|---|---|---|
| Maximal inspiratory pressure (MIP) (cmH20) | 58.75 | 58.75 | 62.45 |
| Gross motor function measure (GMFM - 88) | |||
| Lying and rolling (% score) | 23.53 | 33.33 | 23.53 |
| Sitting (% score) | 28.33 | 33.33 | 21.66 |
| Crawling and kneeling (% score) | 0 | 0 | 0 |
| Standing (% score) | 0 | 0 | 0 |
| Walking, running, and jumping (% score) | 0 | 0 | 0 |
| Total (% score) | 10.37 | 13.32 | 9.04 |
Discussion
The aim of this single subject case report is to describe the observation that B-cell depletion with rituximab prior to AAV vector exposure resulted in immune non-responsiveness to rAAV-CMV-hGAA vector administration, therefore allowing for the possibility of repeat administration of the a vector of the same AAV serotype. This observation is significant for future gene therapy studies and establishes a clinically relevant approach to blocking immune response to AAV vectors. Both eye and the central nervous system are considered immune-privileged compartments because of the limited immune responses.29 However, this approach can be applied to prevent immune response to AAV vectors administered systemically as a means to target the liver, heart, or skeletal muscle.
In a recent review, Mingozzi and High suggested that in muscle and liver gene transfer, the magnitude of T-cell response against the AAV capsid appears to be correlated to the dose of vector administered.29 While it is difficult to compare vector dose and capsid load across studies and vector preparations, we can compare the immune response of the subject who received immune modulation to the control group in this study. In our study, the vector dose for the case subject was fivefold higher than the control cohort, yet no capsid-specific T cell responses were observed. Since the vector dose in this study is not based on body weight it is difficult to compare the impact of the immune modulation regimen on dose in relation to T-cell response. The current immune modulatory regimen, which targets B and T cells, had an effect on both cellular and humoral immunity. It is also possible that the young age of this patient contributed to a lack of memory CD8+ T cells to capsid, which are believed to be the main cause of the previously observed responses.41,42 All subjects were negative for capsid antigen-specific response; therefore we cannot conclude that T-cell non-response is due solely to the immune modulation regimen or the low dose of vector. Nonetheless, the impact of B cells, which function both in antibody production and antigen presentation to T cells, on T-cell response is an aspect of the current observation that warrants further study. In this subject, the ELISPOT assay for both capsid peptides as well as for the transgene were negative at all time points. One consideration is that this subject continues to receive the recombinant ERT that originally led to the immune modulation regimen for clinical management of Pompe disease. Therefore, ongoing rituximab infusions are administered every 4–6 months to maintain the B-cell depletion. If capsid proteins were persistent past the window of B-cell depletion in the event a patient were to receive a single dose of rituximab, then the anti-capsid response would potentially be observed once the CD20+ B-cell counts were reestablished. While we cannot evaluate that possibility in this patient, a related study was done with AAV9 dosing in non-human primates and capsid antigens did not persist past the period of B-cell depletion.43
In addition, the subject received supplemental IVIG to maintain a trough serum IgG level of 700–1,000 mg/dl. We considered the possibility that pooled sera for production of IVIG might contain anti-AAV antibodies represented in the donor population. Interestingly, the IVIG product used in this subject was selected because of the young age of the subject and to accommodate reduced volume administration. We tested an aliquot of the IVIG used in this case and there was no detectable anti-AAV1 antibody.
A similar study with non-human primates was done by administering rituximab and cyclosporine A prior to AAV8 factor IX vector administration.34 They analyzed the impact of this drug combination on anti-factor IX antibody titer and also observed a decrease in anti-capsid antibody titer. A second dose of vector was given under these circumstances, however the capsid used was AAV6.34 Changes in anti-AAV titers have also been studied in the use of rituximab for treatment of rheumatoid arthritis.34 Patients with low starting titers had a fall in anti-AAV titers to less than 1:400, however, those with high titer at the start of rituximab therapy had no change. This finding supports the use of preventative therapy as described in this report, since plasma cells can be long-lived. In the situation of pre-existing immunity, it may be necessary to consider plasmaphoresis44 or use of proteasome inhibitors, which would reduce antibody production in plasma cells.45 The clinical vector used in this study was not further fractionated by density gradient to reduce empty capsids, therefore, a greater proportion of the total capsids are empty capsids. Even with high capsid dose, the antibody response was prevented using the current regimen.
Sirolimus was also used to promote the survival and expansion of regulatory T cells while allowing for programmed cell death of activated effector T cells. Our preclinical46 and clinical experience13 supports the combination of Rituximab and Sirolimus as a successful immune modulation approach for this patient population.
While this report is a preliminary finding related to a concomitant medication use for ERT, we believe these findings emphasize the need for additional comprehensive prospective studies. To that end, we have begun an IND-enabling, well-controlled non-human primate (NHP) study, which will evaluate the requirements to establish both primary and subsequent dosing with AAV9 vectors, including dose, route of administration and timing of the immune-modulation. The NHP study is in support of a human study to directly test the safety and utility of this approach by intramuscular or systemic administration routes.
The clinical application of this approach will ultimately be most important in pediatric patients who will be the most likely candidates for multiple dosing events over the course of their lifetime. Three basic principles support this conclusion. First, the body mass will increase up to 20-fold from infancy to adulthood. Second, many of the conditions for which early rAAV-mediated gene therapy is being considered have increased rates of cell division or cell death due to the underlying disease. We have observed loss of vector genomes due to increased cellular turnover as a result of primary myopathy.47 Either of these factors alone or in combination will almost certainly lead to a decline in genome copy number per cell, leading to loss of efficacy over time. Third, early phase studies include subjects in low-dose cohorts and therefore these subjects may not receive a clinically effective vector dose.26,48 This may impact on the need for re-exposure to the therapeutic vector. In order to ethically manage such studies in rare disease populations, it is appropriate to have a strategy in hand to remove the confounding effects of anti-capsid immunity and allow such subjects the chance to enter subsequent studies where a more effective dose is being proposed.
Our findings related to this single patient warrant a more in depth study of the mechanism of both B-cell and T-cell interactions following exposure to AAV capsids in humans. We propose future studies to test this clinical strategy in a prospective trial, thereby allowing for randomization in dose escalation studies or the possibility of AAV vector re-administration.
Materials and Methods
This study was approved by the Institutional Review Board at the University of Florida and all procedures were performed in consideration of the Declaration of Helsinki. All patients gave their written informed consent before participating to this study. The study is registered atclinicaltrials.gov, identifier NCT00976352.
Outcome measures
Safety measures.
Laboratory data and clinical evaluation confirmed safety of the procedure. Other safety laboratory studies included quantitative real-time polymerase chain reaction (PCR) to assess the presence of vector DNA in peripheral blood. Serum samples were assayed by enzyme linked immunosorbent assay (ELISA) for circulating antibodies to the intact AAV1 capsid and hGAA protein.21,49 Anti-AAV1 and anti-hGAAASR and ELISPOT assay were performed as previously described on isolated peripheral blood mononuclear cells.50
Enumeration of B- and T-lymphocyte subpopulations in peripheral blood was done by flow cytometry using monoclonal Abs to CD19 or CD20 for B cells and monoclonal Abs to CD3, CD4, and CD8 for T cells. Sirolimus trough levels, IgG, CD3, CD4, CD8, CD19, CD20, N-terminal pro-brain natriuretic peptide, creatine kinase, creatine kinase-MB, C-reactive protein, platelets, alkaline phosphatase, γ-glutamyl transferase, aspartate aminotransferase, and alanine aminotransferase were measured at regular intervals.
Functional measures
MIP was evaluated using a one-way inspiratory occlusion. A one-way valve permitted exhalation, but prevented inspiratory airflow. A pressure transducer was used to record the negative pressure. The test was repeated three times and the most negative pressure was recorded.
GMFM-88,51 which is an evaluative tool designed to measure change in the gross motor function of patients over time (i.e., lying/rolling, sitting, crawling/kneeling, standing, and walking/running/jumping) was also tested. MIP and GMFM-88 were collected at baseline, day 90 and day 180.
Acknowledgments
We are grateful to Jeff Kelley for technical assistance. The UF Human Applications Laboratory manufactured rAAV vectors for the clinical trial. This study was supported by grants from the National Institute of Health (NHLBI P01 HL59412-06, the NHLBI Gene Therapy Resource Program GTRP – NHLBI and NICHD-K12HD055929-02 (B.K.S.)).
B.J.B., Johns Hopkins University, and the University of Florida could be entitled to patent royalties for inventions described in this article.
M.C., D.J.F., and B.J.B. are inventors of patents related to this research. B.J.B. is a founder of Applied Genetic Technologies Corporation (AGTC). Any proceeds from this study are donated to the University of Florida.
References
- DeRuisseau LR, Fuller DD, Qiu K, DeRuisseau KC, Donnelly WH, Jr, Mah C. Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc Natl Acad Sci USA. 2009;106:9419–9424. doi: 10.1073/pnas.0902534106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N, Plotz P, Byrne BJ. Acid alpha-glucosidase deficiency (glycogenosis type II, Pompe disease) Curr Mol Med. 2002;2:145–166. doi: 10.2174/1566524024605789. [DOI] [PubMed] [Google Scholar]
- Sidman RL, Taksir T, Fidler J, Zhao M, Dodge JC, Passini MA. Temporal neuropathologic and behavioral phenotype of 6neo/6neo Pompe disease mice. J Neuropathol Exp Neurol. 2008;67:803–818. doi: 10.1097/NEN.0b013e3181815994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne BJ, Falk DJ, Pacak CA, Nayak S, Herzog RW, Elder ME. Pompe disease gene therapy. Hum Mol Genet. 2011;20 R1:R61–R68. doi: 10.1093/hmg/ddr174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne BJ, Kishnani PS, Case LE, Merlini L, Müller-Felber W, Prasad S. Pompe disease: design, methodology, and early findings from the Pompe Registry. Mol Genet Metab. 2011;103:1–11. doi: 10.1016/j.ymgme.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Hirschhorn JN, Sklar P, Lindblad-Toh K, Lim YM, Ruiz-Gutierrez M, Bolk S. SBE-TAGS: an array-based method for efficient single-nucleotide polymorphism genotyping. Proc Natl Acad Sci USA. 2000;97:12164–12169. doi: 10.1073/pnas.210394597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toscano A, Schoser B. Enzyme replacement therapy in late-onset Pompe disease: a systematic literature review. J Neurol. 2013;260:951–959. doi: 10.1007/s00415-012-6636-x. [DOI] [PubMed] [Google Scholar]
- van der Beek NA, Hagemans ML, van der Ploeg AT, Reuser AJ, van Doorn PA. Pompe disease (glycogen storage disease type II): clinical features and enzyme replacement therapy. Acta Neurol Belg. 2006;106:82–86. [PubMed] [Google Scholar]
- Moufarrej NA, Bertorini TE. Respiratory insufficiency in adult-type acid maltase deficiency. South Med J. 1993;86:560–567. doi: 10.1097/00007611-199305000-00015. [DOI] [PubMed] [Google Scholar]
- McKusick VA, Amberger JS, Francomano CA. Progress in medical genetics: map-based gene discovery and the molecular pathology of skeletal dysplasias. Am J Med Genet. 1996;63:98–105. doi: 10.1002/(SICI)1096-8628(19960503)63:1<98::AID-AJMG19>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Leslie N, Tinkle BT.1993. Glycogen storage disease type II (pompe disease). In: Pagon, RA, Adam, MP, Ardinger, HH, Bird, TD, Dolan, CR, Fong, CTGeneReviews. University of Washington; Seattle, WA < < http://www.ncbi.nlm.nih.gov/books/NBK1261/ >. [Google Scholar]
- Bali DS, Tolun AA, Goldstein JL, Dai J, Kishnani PS. Molecular analysis and protein processing in late-onset Pompe disease patients with low levels of acid a-glucosidase activity. Muscle Nerve. 2011;43:665–670. doi: 10.1002/mus.21933. [DOI] [PubMed] [Google Scholar]
- Elder ME, Nayak S, Collins SW, Lawson LA, Kelley JS, Herzog RW. B-Cell depletion and immunomodulation before initiation of enzyme replacement therapy blocks the immune response to acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr. 2013;163:847–54.e1. doi: 10.1016/j.jpeds.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banugaria SG, Patel TT, Mackey J, Das S, Amalfitano A, Rosenberg AS. Persistence of high sustained antibodies to enzyme replacement therapy despite extensive immunomodulatory therapy in an infant with Pompe disease: need for agents to target antibody-secreting plasma cells. Mol Genet Metab. 2012;105:677–680. doi: 10.1016/j.ymgme.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banugaria SG, Prater SN, Patel TT, Dearmey SM, Milleson C, Sheets KB. Algorithm for the early diagnosis and treatment of patients with cross reactive immunologic material-negative classic infantile pompe disease: a step towards improving the efficacy of ERT. PLoS ONE. 2013;8:e67052. doi: 10.1371/journal.pone.0067052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani PS, Goldenberg PC, DeArmey SL, Heller J, Benjamin D, Young S. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab. 2010;99:26–33. doi: 10.1016/j.ymgme.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Hauswirth WW, Aleman TS, Kaushal S, Schwartz SB, Boye SL. Vision 1 year after gene therapy for Leber’s congenital amaurosis. N Engl J Med. 2009;361:725–727. doi: 10.1056/NEJMc0903652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Ratnakaram R, Heon E, Schwartz SB, Roman AJ. Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol. 2012;130:9–24. doi: 10.1001/archophthalmol.2011.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani AC, Gray JT, McIntosh J, Ng CY, Zhou J, Spence Y. Safe and efficient transduction of the liver after peripheral vein infusion of self-complementary AAV vector results in stable therapeutic expression of human FIX in nonhuman primates. Blood. 2007;109:1414–1421. doi: 10.1182/blood-2006-03-010181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- Smith BK, Collins SW, Conlon TJ, Mah CS, Lawson LA, Martin AD. Phase I/II trial of adeno-associated virus-mediated alpha-glucosidase gene therapy to the diaphragm for chronic respiratory failure in Pompe disease: initial safety and ventilatory outcomes. Hum Gene Ther. 2013;24:630–640. doi: 10.1089/hum.2012.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardieu M, Zérah M, Husson B, de Bournonville S, Deiva K, Adamsbaum C. Intracerebral Administration of Adeno-Associated Viral Vector Serotype rh.10 Carrying Human SGSH and SUMF1 cDNAs in Children with Mucopolysaccharidosis Type IIIA Disease: Results of a Phase I/II Trial. Hum Gene Ther. 2014;25:506–516. doi: 10.1089/hum.2013.238. [DOI] [PubMed] [Google Scholar]
- Ferreira VG, Nabeshima CK, Marques MM, Paris AF, Gioso MA, dos Reis RS. Tooth bleaching induces changes in the vascular permeability of rat incisor pulps. Am J Dent. 2013;26:298–300. [PubMed] [Google Scholar]
- Kaeppel C, Beattie SG, Fronza R, van Logtenstein R, Salmon F, Schmidt S. A largely random AAV integration profile after LPLD gene therapy. Nat Med. 2013;19:889–891. doi: 10.1038/nm.3230. [DOI] [PubMed] [Google Scholar]
- Brantly ML, Spencer LT, Humphries M, Conlon TJ, Spencer CT, Poirier A. Phase I trial of intramuscular injection of a recombinant adeno-associated virus serotype 2 alphal-antitrypsin (AAT) vector in AAT-deficient adults. Hum Gene Ther. 2006;17:1177–1186. doi: 10.1089/hum.2006.17.1177. [DOI] [PubMed] [Google Scholar]
- Mendell JR, Rodino-Klapac LR, Rosales-Quintero X, Kota J, Coley BD, Galloway G. Limb-girdle muscular dystrophy type 2D gene therapy restores alpha-sarcoglycan and associated proteins. Ann Neurol. 2009;66:290–297. doi: 10.1002/ana.21732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodino-Klapac LR, Lee JS, Mulligan RC, Clark KR, Mendell JR. Lack of toxicity of alpha-sarcoglycan overexpression supports clinical gene transfer trial in LGMD2D. Neurology. 2008;71:240–247. doi: 10.1212/01.wnl.0000306309.85301.e2. [DOI] [PubMed] [Google Scholar]
- Calcedo R, Wilson JM. Humoral Immune Response to AAV. Front Immunol. 2013;4:341. doi: 10.3389/fimmu.2013.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi F, High KA. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood. 2013;122:23–36. doi: 10.1182/blood-2013-01-306647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleem A, Saidu A, Abdulkarim H, Al-Diab AR, Al-Sagheer A, Qayum A. Rituximab as a single agent in the management of adult patients with haemophilia A and inhibitors: marked reduction in inhibitor level and clinical improvement in bleeding but failure to eradicate the inhibitor. Haemophilia. 2009;15:210–216. doi: 10.1111/j.1365-2516.2008.01865.x. [DOI] [PubMed] [Google Scholar]
- Collins PW, Mathias M, Hanley J, Keeling D, Keenan R, Laffan M, UK Haemophilia Centre Doctors’ Organisation Rituximab and immune tolerance in severe hemophilia A: a consecutive national cohort. J Thromb Haemost. 2009;7:787–794. doi: 10.1111/j.1538-7836.2009.03332.x. [DOI] [PubMed] [Google Scholar]
- Streif W, Escuriola Ettingshausen C, Linde R, Kropshofer G, Zimmerhackl LB, Kreuz W. Inhibitor treatment by rituximab in congenital haemophilia A - Two case reports. Hamostaseologie. 2009;29:151–154. [PubMed] [Google Scholar]
- Mingozzi F, Chen Y, Edmonson SC, Zhou S, Thurlings RM, Tak PP. Prevalence and pharmacological modulation of humoral immunity to AAV vectors in gene transfer to synovial tissue. Gene Ther. 2013;20:417–424. doi: 10.1038/gt.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masat E, Pavani G, Mingozzi F. Humoral immunity to AAV vectors in gene therapy: challenges and potential solutions. Discov Med. 2013;15:379–389. [PubMed] [Google Scholar]
- Weiner GJ. Rituximab: mechanism of action. Semin Hematol. 2010;47:115–123. doi: 10.1053/j.seminhematol.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitsky J, Mathew JM, Abecassis M, Tambur A, Leventhal J, Chandrasekaran D. Systemic immunoregulatory and proteogenomic effects of tacrolimus to sirolimus conversion in liver transplant recipients. Hepatology. 2013;57:239–248. doi: 10.1002/hep.25579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak S, Sivakumar R, Cao O, Daniell H, Byrne BJ, Herzog RW. Mapping the T helper cell response to acid a-glucosidase in Pompe mice. Mol Genet Metab. 2012;106:189–195. doi: 10.1016/j.ymgme.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak S, Cao O, Hoffman BE, Cooper M, Zhou S, Atkinson MA. Prophylactic immune tolerance induced by changing the ratio of antigen-specific effector to regulatory T cells. J Thromb Haemost. 2009;7:1523–1532. doi: 10.1111/j.1538-7836.2009.03548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak S, Herzog RW. Progress and prospects: immune responses to viral vectors. Gene Ther. 2010;17:295–304. doi: 10.1038/gt.2009.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case L. Pompe disease diagnosis and management guideline. Genet Med. 2006;8:267–288. doi: 10.1097/01.gim.0000218152.87434.f3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi F, Maus MV, Hui DJ, Sabatino DE, Murphy SL, Rasko JE. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat Med. 2007;13:419–422. doi: 10.1038/nm1549. [DOI] [PubMed] [Google Scholar]
- Li H, Lasaro MO, Jia B, Lin SW, Haut LH, High KA. Capsid-specific T-cell responses to natural infections with adeno-associated viruses in humans differ from those of nonhuman primates. Mol Ther. 2011;19:2021–2030. doi: 10.1038/mt.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk DJ, Cloutier DD, Nayak S, Doerfler P, Corti M, Clement N. Prevention of anti-capsid response following systemic AAV9 dosing infant rhesus monkeys. Mol Ther. 2013;21:e1–e46. [Google Scholar]
- Chicoine LG, Montgomery CL, Bremer WG, Shontz KM, Griffin DA, Heller KN. Plasmapheresis eliminates the negative impact of AAV antibodies on microdystrophin gene expression following vascular delivery. Mol Ther. 2014;22:338–347. doi: 10.1038/mt.2013.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karman J, Gumlaw NK, Zhang J, Jiang JL, Cheng SH, Zhu Y. Proteasome inhibition is partially effective in attenuating pre-existing immunity against recombinant adeno-associated viral vectors. PLoS ONE. 2012;7:e34684. doi: 10.1371/journal.pone.0034684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghimi B, Sack BK, Nayak S, Markusic DM, Mah CS, Herzog RW. Induction of tolerance to factor VIII by transient co-administration with rapamycin. J Thromb Haemost. 2011;9:1524–1533. doi: 10.1111/j.1538-7836.2011.04351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacak CA, Conlon T, Mah CS, Byrne BJ. Relative persistence of AAV serotype 1 vector genomes in dystrophic muscle. Genet Vaccines Ther. 2008;6:14. doi: 10.1186/1479-0556-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S. Dystrophin immunity in Duchenne’s muscular dystrophy. N Engl J Med. 2010;363:1429–1437. doi: 10.1056/NEJMoa1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brantly ML, Chulay JD, Wang L, Mueller C, Humphries M, Spencer LT. Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy. Proc Natl Acad Sci USA. 2009;106:16363–16368. doi: 10.1073/pnas.0904514106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauswirth WW, Aleman TS, Kaushal S, Cideciyan AV, Schwartz SB, Wang L. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther. 2008;19:979–990. doi: 10.1089/hum.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hielkema T, Hamer EG, Ebbers-Dekkers I, Dirks T, Maathuis CG, Reinders-Messelink HA. GMFM in infancy: age-specific limitations and adaptations. Pediatr Phys Ther. 2013;25:168–76; discussion 177. doi: 10.1097/PEP.0b013e318288d370. [DOI] [PubMed] [Google Scholar]