

Abstract
Mitochondrial diseases are potentially severe, incurable diseases resulting from dysfunctional mitochondria. Several important mitochondrial diseases are caused by mutations in mitochondrial DNA (mtDNA), the genetic material contained within mitochondria, which is maternally inherited. Classical and modern therapeutic approaches exist to address the inheritance of mtDNA disease, but are potentially complicated by the fact that cellular mtDNA populations evolve according to poorly-understood dynamics during development and organismal lifetimes. We review these therapeutic approaches and models of mtDNA dynamics during development, and discuss the implications of recent results from these models for modern mtDNA therapies. We particularly highlight mtDNA segregation—differences in proliferative rates between different mtDNA haplotypes—as a potential and underexplored issue in such therapies. However, straightforward strategies exist to combat this and other potential therapeutic problems. In particular, we describe haplotype matching as an approach with the power to potentially ameliorate any expected issues from mtDNA incompatibility.
Keywords: mitochondrial DNA, haplotype matching, mtDNA segregation, development, preventing mtDNA disease
Introduction: mitochondrial DNA disease
Mitochondria are vital energy-producing organelles in eukaryotic cells. Mitochondrial diseases are pathologies in which the ability of mitochondria to produce energy and fulfill their normal cellular roles is compromised. These diseases are relatively common, but diagnosed infrequently, because the majority of patients exhibit only very mild symptoms (Manwaring et al., 2007). The range of symptomatic severity associated with mitochondrial disease leads to variability in reported prevalence rates: for example, one mitochondrial pathology (resulting from the m.3243A>G mutation discussed below) has quoted prevalence rates of between 1 in 300 (Manwaring et al., 2007) and 1 in 14 000 (Chinnery et al., 2000).
Mitochondria have their own DNA (henceforth mtDNA), which is the only DNA outside the nucleus in humans. MtDNA is unique in having its own genetic code and subtype of ribosomes. It codes for a minority of subunits required for the respiratory chain, the array of multimeric proteins that form the production line for energy on the inner mitochondrial membrane, and for transfer and ribosomal RNA. The majority of essential respiratory chain proteins are encoded by the nucleus, as well as many proteins required for mtDNA maintenance and replication. Therefore, mutations in either the mitochondrial or nuclear DNA of the cell may cause pathological loss of function in mitochondria and lead to mitochondrial disease (Taylor and Turnbull, 2005; Greaves et al., 2012). In this review, we will focus on diseases resulting from mtDNA mutation. MtDNA is maternally inherited, apparently because sperm contribute almost no cytoplasm to the zygote, and paternal mitochondria are ubiquitinated (Sutovsky et al., 1999, 2000) and targeted for destruction (Cummins et al., 1998; Shitara et al., 2000; Al Rawi et al., 2011; Sato and Sato, 2011; DeLuca and O'Farrell, 2012) as soon as fertilization has taken place, persisting only in abnormal embryos or inter-species crosses (Gyllensten et al., 1991; St John et al., 2000). MtDNA is possibly even eliminated prior to fertilization (Luo et al., 2013).
Diseases resulting from mutations in mtDNA have unique characteristics of onset, severity and inheritance, largely due to the fact that there are thousands of copies of mtDNA in a typical nucleated cell (Lightowlers et al., 1997; Wallace, 1999). In most normal individuals these are effectively genetically identical (a situation termed ‘homoplasmy’). In mtDNA disease there may be a number of different, mutated mtDNA molecules, giving rise to ‘heteroplasmy’ (more than one mtDNA type coexisting in the same cell).
MtDNA is maternally inherited, producing a characteristic disease distribution down the maternal line. MtDNA haplotypes can modulate the pathological effects of mutated nuclear encoded genes (Strauss et al., 2013), and the same mtDNA variation can be deleterious or beneficial depending on its mtDNA background and environment (Ji et al., 2012). Many mtDNA diseases are heteroplasmic, that is, both mutated and wild-type mtDNA co-exist in affected individuals. In most of these cases a dosage effect is observed (Jeppesen et al., 2006), with the proportion, copy number and distribution of mtDNA mutants influencing tissue function (Petruzzella et al., 1994). The ‘threshold’ above which mtDNA disease shows clinical symptoms is around 70% mutated mtDNA in the commonest disorder (Jeppesen et al., 2006). This dependence on mutant load is important because intracellular populations of mtDNA, and thus the proportional presence of mutant mtDNA, can change during development, according to dynamics which are currently poorly characterized.
The developmental modulation of mtDNA populations means that patients with mtDNA disease frequently exhibit progressive symptoms, as mutant mtDNA accumulates in affected tissues (Poulton et al., 1995; Weber et al., 1997). For instance, children with Pearson's syndrome may present with severe anemia and lactic acidosis in early infancy. The characteristic mutation is a single mtDNA deletion of about 5 kilobases, encompassing both protein and tRNA coding regions. Affected children initially have high levels of mutant mtDNA in all tissues. As the disease progresses, the level of mutant in blood drops and their anemia resolves. However, if they survive to adolescence they may develop a myopathy as the proportion of mutant mtDNA in muscle increases (McShane et al., 1991). While the shifting of mtDNA populations is less extreme in most maternally inherited heteroplasmic mtDNA disease, in almost all cases the level of mutant mtDNA is lower in blood than in post-mitotic tissues such as muscle and brain (Rahman et al., 2001). This example illustrates a potential diagnostic problem: as mutant load changes with time, blood levels of mutant mtDNA cannot easily be used to advise patients on their prognosis or transmission risks.
Mitochondrial diseases are often clinically heterogeneous. While many patients do not fit into specific clinical syndromes, well known examples of mtDNA diseases include MIDD (mitochondrially inherited diabetes and deafness) (van den Ouweland et al., 1992), MELAS (mitochondrial myopathy, encephalomyopathy, lactic acidosis, stroke-like symptoms) (Goto et al., 1990), MILS (maternally inherited Leigh's syndrome) (Holt et al., 1990), MERRF (myoclonic epilepsy with ragged red fibers) (Wallace et al., 1988b) and LHON (Leber's hereditary optic neuropathy) (Wallace et al., 1988a; Howell and McCullough, 1990; Johns et al., 1992). Muscle dysfunction is an important feature of MELAS (Ciafaloni et al., 1992) and MERRF, both of which can cause cognitive decline, ataxia, epilepsy, cardiomyopathy and deafness (Chinnery et al., 1997). Diabetes is a common feature of MELAS (van den Ouweland et al., 1992). MILS mainly involves the central nervous system with psychomotor delay, visual and hearing impairment (Degoul et al., 1995). LHON is usually a non-syndromic optic neuropathy and most patients are homoplasmic for mutant mtDNA (Howell and McCullough, 1990; Johns et al., 1992). Specific mutations in mtDNA are known to give rise to these diseases: for example, the m.3243A>G mutation most often causes MIDD, but in more severe cases MELAS (Goto et al., 1990), the m.8344A>G mutation can cause MERRF (Wallace et al., 1988b), and m.11778G>A (Wallace et al., 1988a), m.3460G>A (Howell and McCullough, 1990) and m.14484T>C (Johns et al., 1992) mutations can cause LHON. However other mtDNA mutations also give rise to these diseases. A review of clinical features and a morbidity map of mtDNA mutations can be found in Chinnery and Hudson (2013) and DiMauro et al. (2013).
Another striking feature of mtDNA disease inheritance involves the observed large shifts of heteroplasmy between mother and offspring. For example, it is possible for a phenotypically healthy mother, harboring 50% mutated mtDNA, to produce both healthy and severely affected children (Larsson et al., 1992). The reason for this shift between generations is the so-called ‘bottleneck’ effect, whereby heteroplasmy levels in offspring are remarkably variable with respect to the maternal heteroplasmy, while the average heteroplasmy across many offspring is often comparable to that of the mother (Jenuth et al., 1996). The mechanism underlying this effect is hotly debated (Carling et al., 2011), with some suggesting that responsibility lies with a pronounced decrease in mtDNA copy number in the germline (Cree et al., 2008), others proposing random partitioning of clusters of mtDNA at cell divisions (Cao et al., 2007, 2009), and others proposing replication of a subset of mtDNAs during development (Wai et al., 2008) (Fig. 1).
Figure 1.

The mitochondrial DNA bottleneck during development. (A) A fertilized oocyte has a given heteroplasmy (mutant load) value. During gestation, the female embryo/fetus develops primordial germ cells that develop into oocytes. The heteroplasmy in these oocytes shows high variance due to the bottleneck effect, whose proposed mechanisms are shown in (B)–(D). (B) A reduction of mitochondrial DNA (mtDNA) copy number in the primordial germ cells and consecutive reamplification during oocyte development accelerates random drift and increases variance. (C) Random partitioning of clusters of mtDNAs at each cell division during primordial germ cell development could powerfully increase heteroplasmy variance. (D) Allowing only a small random subset of mtDNAs to replicate (here two instances are depicted with circles and squares)—either a specifically selected set or through restricted random turnover—can increase heteroplasmy variance through imposing a lower effective population size.
In the most extreme examples of the bottleneck, there may be rapid switching from near homoplasmy in one mtDNA type to near homoplasmy for another between mother and child. Using a heteroplasmic length variant in noncoding mtDNA (Marchington et al., 1997) one of the current authors (J.P.) found evidence for this switching in oocytes from women and from mice. This switching was also apparent in oocytes from women who were heteroplasmic for pathogenic mtDNA mutants (Blok et al., 1997; Marchington et al., 1998; Brown et al., 2001). While further work is needed to elucidate this mechanism (Carling et al., 2011), the bottleneck is a clear example of how developmental phenomena mold the statistics of intracellular mtDNA populations.
MtDNA diseases are currently not directly curable, despite several promising approaches to shift the amount of mutated mtDNA in patients affected by heteroplasmic diseases to lower, less pathogenic levels. For example, specifically designed nucleases (including so-called mitoTALENs (Bacman et al., 2013) and zinc-finger nucleases (Gammage et al., 2014)) can cut mutated mtDNA at the site of the mutation. In the absence of clinically available cures of mtDNAs disease, strategies to prevent their transmission to the next generation to allow (subclinically) affected women to have healthy children (or at least highly increase the chances thereof) are extremely important (Fig. 2). We designate those therapeutic strategies that are in current clinical practice as ‘Classical’ and those that have not yet been approved for use in humans as ‘Modern’. Classical strategies aim to either replace the affected oocyte from the patient altogether, or monitor embryo/fetal heteroplasmy. Modern strategies aim at replacing the affected mtDNA. These new strategies have become prominent in the scientific literature and media alike. The appealing catchphrases ‘like changing a laptop battery’ (referring to the replacement of dysfunctional mtDNA) and associated ‘three-parent babies’ (referring to the presence of a third-party's mtDNA in an embryo; see below) have captured the imagination of many involved in communicating science to the general public, and several of these recently proposed therapies for mtDNA disease inheritance are currently on the verge of clinical application.
Figure 2.
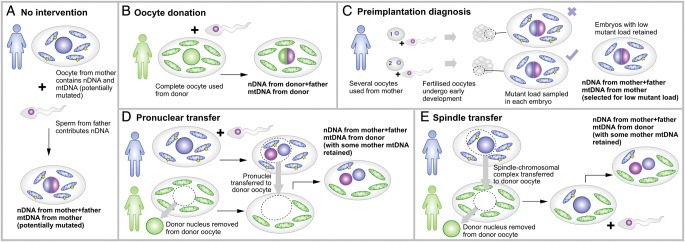
MtDNA disease inheritance and therapeutic approaches. (A) A mother with mtDNA harboring a pathological mutation is at risk of transmitting the associated disease to her offspring. (B) Oocyte donation uses an oocyte from a third-party donor who does not carry the mtDNA mutation. (C) Preimplantation diagnosis involves sampling mutant load in cells after conception. As the mother's oocytes may exhibit a wide range of heteroplasmy levels, some concepti may inherit acceptably low mutant loads: these are retained. (D) Pronuclear transfer involves the removal of the nucleus from a third-party oocyte with unaffected mtDNA, then the transfer of two pronuclei (from the mother's egg and father's sperm) onto this healthy background. (E) Spindle transfer involves the replacement of a third-party oocyte nucleus with the chromosomal complex from the mother, prior to fertilization with the father's sperm.
Classical options in reproductive management of mtDNA disease
Three notable strategies have existed, at least in concept (Sauer and Kavic, 2006), since before the first maternally inherited mtDNA disease was described 25 years ago (Wallace et al., 1988b), to address the issue of potential inheritance of mutant mtDNA in families at risk of transmitting diseases (Fig. 2A).
Oocyte donation (Fig. 2B) is a simple way to completely eliminate the risk of transmitting heteroplasmic mutant mtDNA from mother to child. This approach involves using the oocytes from a third-party donor rather than the mother, thus losing any genetic inheritance from the mother, but representing the only strategy guaranteeing to intercept the transmission of the disease.
Alternative strategies exploit the difference in mutant load between a potential mother's oocytes, arising largely from the aforementioned mtDNA bottleneck, and selected concepti with a low mutant load. This strategy can only be applied in heteroplasmic disease. Selection of low risk concepti was initially carried out during established pregnancy (chorionic villus sampling) (Harding et al., 1992). Here, heteroplasmy in chorionic villi is analyzed at the end of the first trimester, with a view to terminating fetuses displaying high heteroplasmies and thus at high risk of inheriting mtDNA disease. This approach is useful to address mtDNA disease inheritance in disorders where there is a good relationship between phenotype and mutant load (White et al., 1999). It is not suitable for homoplasmic diseases, nor for those in which the mtDNA mutant load is a poor predictor of phenotype, like LHON (Black et al., 1996). Furthermore, if the load of mutant mtDNA in trophoblast is a poor representation of that in the rest of the conceptus, the efficacy of this approach is decreased. This poor representation can be the case when segregation starts early, as described in the next paragraph.
A third therapeutic strategy involves selecting low risk early (‘cleavage’) embryos, through the use of preimplantation genetic diagnosis (PGD) ((Fig. 2C). After fertilization of an oocyte and limited subsequent development, a small fraction (1–2 cells) of the cleavage embryo is sampled to determine the mutant load. At this stage, the variation in mutant load between individual blastomeres is small. The mutant load in an embryo can then be used to estimate the risk of the individual developing symptoms of a mtDNA disorder post-natally (Poulton et al., 2010). This strategy is currently the mainstay of modern clinical practice, being used successfully to help families with mtDNA disease (Steffann et al., 2006; Monnot et al., 2011). However, when PGD is carried out on blastocysts, a later embryonic stage, samples may not adequately reflect post-natal heteroplasmy. This problem arose when a variant of the technique, blastocyst biopsy, was used for prenatal screening of an embryo carrying the m.3243A>G mutation (Treff et al., 2012). In this instance, the mutant load in trophoblast cells (12%; Treff et al. (2012)) was substantially lower than in some samples of the child (47% blood, 52% urine; Wallace and Chalkia (2013) and Mitalipov et al. (2014)). It is currently unknown whether the difference in heteroplasmy occurred between trophoblast and inner cell mass, or whether the heteroplasmy levels changed during gestation. However, this case shows the considerable residual risk of this method. Generally, cell-to-cell heteroplasmy and copy number variation are likely to develop as cells develop down the specific functional lineages found in the blastocyst. Such variation could be further exacerbated by a proposed rapid mtDNA segregation in preimplantation embryos (Lee et al., 2012). However, it needs to be clarified whether this is a general phenomenon, or a result of the merging of two distinct cytoplasts that segregate independently in ‘artificially generated embryos’. If the latter is true, the effect will be of importance in all techniques that include some degree of cytoplasmic transfer, including karyoplast transfer (Steffann et al., 2014). In conclusion, PGD on cleavage stage embryos seems robust, but on blastocysts may be unreliable.
New developments: modern treatments for mtDNA disease
The above approaches to address the inheritance of mitochondrial disease have several shortcomings. Oocyte donation has the disadvantage that none of the mother's nuclear DNA content is retained in the offspring. PGD of blastocysts (but not of cleavage stage embryos) and chorionic villus sampling run the risk that differences between tissues and individual cells may lead to an inaccurate inference of heteroplasmy levels, and thus erroneous risk estimation. PGD of cleavage stage embryos however appears to be robust and accurate (Monnot et al., 2011).
Two recently proposed therapies, pronuclear transfer and chromosomal spindle transfer, are designed to circumvent these problems. Both these approaches aim to transfer the nuclear genome of a parent ‘donor’ oocyte to a healthy ‘recipient’ oocyte with no nucleus and healthy mtDNA. Specifically, pronuclear transfer (Fig. 2D) involves transferring the two pronuclei (from mother and father) of a fertilized donor oocyte to an enucleated recipient oocyte. Chromosomal spindle transfer (Fig. 2E) involves transferring the chromosomal spindle from a donor oocyte into an enucleated recipient oocyte prior to fertilization. Thus, the nuclear genome is transferred to an environment with functional mitochondria, i.e. the defective ‘batteries’ of the cells are replaced with working ones; resulting in a healthy embryo and definitely interrupting inheritance of the pathological mtDNA (Poulton and Oakeshott, 2012). These therapies aim to achieve zero mutant load, consistent across all cells, and allow the mother and father both to contribute nuclear DNA, thus addressing the shortcomings of traditional therapies.
Pilot studies of these techniques have proved their feasibility, but also highlighted the potentially important and currently unavoidable phenomenon of mtDNA carryover. Ideally, pronuclear and spindle transfer both should lead to a complete lack of donor mtDNA in the recipient, but technical limitations currently make this unfeasible (St John and Campbell, 2010). Currently, no method has been shown to reproducibly eliminate 100% of the unwanted donor mtDNA carryover. While all techniques tend to reduce carryover to below 1%, comparison between the studies is hampered by varying detection limits of donor mtDNA, between 0.01 and 2%. In five out of nine human embryos created by pronuclear transfer (Craven et al., 2010) the average carryover of donor mtDNA was 1.68%. Observed carryovers in other studies include 0.5–0.6% from human spindle transfer (Tachibana et al., 2013); 0.31% in human nuclear transfer (Paull et al., 2013); and 1% in rhesus monkeys with spindle transfer (Tachibana et al., 2009; Lee et al., 2012). Therefore, a low-level heteroplasmy in the resulting embryo cannot be excluded.
Such low amounts are generally insufficient to cause disease (Craven et al., 2011). The mtDNA bottleneck may conceivably lead to amplification in the next generation of this amount in female embryos: however, it was recently shown that for carryover <5%, this is no concern also for the following generations, and both methods should therefore be safe in this respect (Samuels et al., 2013).
Potential issues with modern treatments
While these modern treatments are largely deemed sufficiently safe for use in the clinic, several uncertainties exist regarding the behavior of mtDNA populations after treatment. Until these uncertainties are addressed, clinical applications of modern treatments should arguably be limited to cases where no good alternatives exist. Families with severe phenotypes and a homoplasmic mtDNA mutant of proven pathogenicity are the best candidates, because the only classical option from which they benefit is oocyte donation. However, such families are relatively rare, largely because it is essential but difficult to prove that a homoplasmic mutation is causative. Correct selection of the first families for treatment will therefore be imperative.
At the current time most possible and ethically justifiable pilot tests have been performed both in animal models and (abnormal) human embryos, with positive results. However, some questions regarding the subsequent behavior of mtDNA populations in offspring produced using these treatments remain, which have been flagged by researchers and noted in the literature. These issues are not fatal flaws in the concept of mtDNA therapies, but do represent areas of uncertainty associated with these therapies. All of them are connected with the mtDNA haplotype of the third-party ‘recipient’ providing an oocyte with healthy mtDNA. In a random pairing, it is likely that the donor and recipient haplotypes vary considerably: pairwise comparisons in human mtDNAs show up to 130 single-nucleotide polymorphism (SNP) differences ((Blanco et al., 2011), with those located in the protein-coding regions of the mtDNA leading to up to 20 amino acid changes (Craven et al., 2011)). On average two Europeans or two Africans will differ at 29.3 and 78.3 sites, respectively (Lippold et al., 2014). Consequently, a rather complex mixture of nuclear DNA and different mtDNAs can arise: nuclear DNA from the patient (‘donor’) and the father, the majority of mtDNA from the enucleated recipient oocyte with presumed wild-type mtDNA (haplotype B) and a small amount of the carryover patient mtDNA (haplotype A; which may be either mutant or wild type if the donor is heteroplasmic). So a maximum of three different mtDNAs can be present in the embryo, with the healthy haplotype B constituting the vast majority, around 99%. This new haplotype is alien to both the patient (maternal) and paternal nuclear DNA, and the implications of this combination are currently largely unexplored.
The first potential issue concerns nuclear-mitochondrial interaction (Fig. 3A). Energy production is dependent on extensive cross-talk between genes from the nucleus and mtDNA (Johnson et al., 2001; Reinhardt et al., 2013). Usually the offspring's mtDNA is inherited with a haploid maternal genome. This co-inheritance is thought to facilitate nuclear-mitochondrial interaction. However, during karyoplast transfer this co-transmission is interrupted, and the mtDNA is confronted with a completely ‘unknown’ nuclear DNA. This situation may well lead to complications, as several physiological parameters like respiration, performance ((Nagao et al., 1998), inter-species and/inter-subspecies heteroplasmy) and learning ((Roubertoux et al., 2003), intra-subspecies heteroplasmy) were reduced in mtDNA-nuclear mismatches in male mice. Males are particularly at risk as maternal inheritance of mtDNA implies that the relevant aspects of natural selection only work directly on females, i.e. accumulation of mtDNA mutations that are harmful to males is facilitated, as discussed with LHON. However, recently arguments have been brought forward that male excess in LHON might have other causes (e.g. lower estrogen levels, as estrogen seems to ameliorate mitochondrial dysfunction in LHON (Giordano et al., 2011)), and studies in macaques and mouse models support the view that nuclear-mitochondrial interaction will have limited effect, if at all, on modern treatments (Chinnery et al., 2014). It was recently found that mtDNA haplotypes define gene expression patterns in mouse embryonic stem cells (Kelly et al., 2013), so clearly nuclear DNA (nDNA)–mtDNA interaction does depend on the mtDNA haplotype. However, in vivo experiments with xenomitochondrial mice show that the nuclear-mitochondrial system seems to be able to compensate for a high level of diversity. In these xenomitochondrial mice harboring Mus terricolor mtDNA on a Mus musculus background virtually no negative in vivo effects were found (Cannon et al., 2011). In contrast, in conplastic strains that harbored a range of different mtDNAs (M. m. domesticus, but also other subspecies of M. musculus) and the nucleus of M. m. domesticus, behavioral differences and varying susceptibility to experimental autoimmune encephalomyelitis were found (Roubertoux et al., 2003; Yu et al., 2009). Some of these effects might however be caused by a single mutation present in a specific mtDNA haplotype, New Zealand Black (NZB), used in that study (and several others) (Moreno-Loshuertos et al., 2006, 2013). In somatic cell nuclear cloning it was found that a certain mtDNA genetic difference between donor cell and its recipient oocyte can even be beneficial for development. A difference in copy number ratio of mtDNA to mitochondrial mRNA between the respective haplotypes might argue for a necessity for homoplasmy on the mRNA level (Bowles et al., 2008). Thus, while slightly deleterious consequences of nDNA-mtDNA mismatch have been observed in several studies, it seems likely that cells are flexible enough to deal with mismatch situations of limited heteroplasmy and genetic difference.
Figure 3.

Potential issues associated with mixed mtDNA populations resulting from modern therapies. (A) Incompatibilities may exist between the nuclear DNA (from mother and father) and mtDNA (from a third party), as these genomes have not necessarily co-evolved. Such incompatibilities may conceivably manifest as, for example, dysfunctional protein products or signaling pathways. (B) The mixture of two mtDNA types within a cell has been found to cause detrimental physiological effects, for unknown reasons. (C) Segregation is the proliferation of one mtDNA haplotype over another in a cellular mixture, potentially causing changes in the population fraction of one mtDNA haplotype. If one mtDNA haplotype experiences a proliferative advantage over another, it may come to dominate the cellular population over time. If some mtDNAs of this haplotype harbor a pathological mutation, this mutation may thus become amplified even if the pathological mutation itself does not affect segregation.
The second potential issue concerns mtDNA–mtDNA interaction (Fig. 3B). As described above, up to three different mtDNAs can be present in the embryo (excluding low-level microvariation (He et al., 2010; Ye et al., 2014)). Different human mtDNA types show differences in oxidative phosphorylation (OXPHOS), potentially triggered by adaption to various climates or energy demands during evolution, a controversial topic that is still discussed (reviewed in (Wallace and Chalkia, 2013)). What happens if such divergent haplotypes are mixed, even at low levels as after karyoplast transfer? In mice, the mixture of two mtDNA haplotypes of the same subspecies led to physiological changes (e.g. hypertension, changed body mass, blood parameters (Acton et al., 2007)) and altered behavior (Sharpley et al., 2012), while mice carrying 100% of either haplotype stayed healthy. It is likely that low heteroplasmies <5% are insufficient to produce strong manifestations of these effects, but further studies are needed to define the exact heteroplasmy thresholds of importance for mismatching.
The third potential issue concerns mtDNA segregation, that is, the process by which one mtDNA type comes to dominate over another within a cell. The simplest example of this is if mutated mtDNA experiences a proliferative advantage over non-mutated mtDNA, and hence even a small initial amount of mutant mtDNA could eventually come to dominate the cell. As we review below, the evidence to support this positive segregation of mutant mtDNA is scanty, even in diseases where it is known to occur. However, the segregation of non-pathological mtDNAs may be a more pertinent issue in mtDNA therapies. If the mtDNA haplotype of the affected woman (‘donor’) experiences a proliferative advantage over that from the ‘recipient’ healthy oocyte (irrespective of the presence of pathological mutations), an arbitrarily small amount of carryover donor mtDNA could subsequently come to dominate the cellular population (Fig. 3C).
In this scenario, the amplification of donor mtDNA is due to haplotypic differences alone, without any segregation specifically arising from pathological mutations. This mechanism can lead to the amplification of a pathological mutation even if that mutation does not affect segregation. Specifically, if the proliferating donor mtDNA haplotype is associated with a pathological mutation, the amplification resulting from haplotype segregation will lead to a concomitant amplification of the mutation, which ‘hitchhikes’ upon the proliferating haplotype as illustrated in Fig. 3C, potentially reaching pathological levels both in the offspring and subsequent generations.
We focus on segregation effects in particular, for two reasons. First, the aforementioned phenomenon of mtDNA carryover potentially creates a situation in which segregation has to be taken into account: that is, where several different mtDNAs are present within a cell, a situation that has so far rarely been described (St John and Schatten, 2004). Second, as we will subsequently describe, experimental evidence exists to suggest that segregation between different mtDNA haplotypes, though rarely commented upon, may be a significant effect, while evidence regarding the other two issues is more sparse.
Segregation of pathological mutations
Due to the potentially dramatic physiological implications of mtDNA mutations and progressive segregation documented in tissue culture (Hayashi et al., 1991; Dunbar et al., 1995; Emmerson et al., 2001), one may expect that pathological mutations would lead to extreme segregation effects. While the topic is controversial (Craven et al., 2010), there are good examples of segregation of disease-related mutations in humans (Larsson et al., 1990; Poulton et al., 1995; Weber et al., 1997). To our knowledge, the level of mutant mtDNA is always lower in blood than in post-mitotic tissues such as muscle and brain (Ciafaloni et al., 1992; Larsson et al., 1992; Rahman et al., 2001). A possible cause is the replacement of defective cells (i.e. cells with high levels of mutant mtDNA) with healthy cells in tissues with rapid turnover (Rahman et al., 2001).
In mice containing a mixture of wild-type mtDNA and mtDNA with a 4696-bp deletion (denoted Δ mtDNA) that leads to lethal renal failure, the Δ mtDNA was observed to preferentially accumulate in several tissues over time (e.g. heart, skeletal muscle, kidney, liver, testis and ovary) (Sato et al., 2007). This model, however, is complex and does not clearly recapitulate human disease for three reasons. Firstly, renal failure is uncommon in human mtDNA disease and has rarely, if ever, been reported in mtDNA deletions. Secondly, this rearrangement was maternally inherited, and included mtDNA duplications as well as deletions, both of which are uncommon in human mtDNA disease. Thirdly, the level of mutant mtDNA in the female germline declined with age, a trend that does not reflect the findings in humans (Chinnery et al., 2004). Nevertheless, the model does recapitulate the accumulation of mutant mtDNA in post-mitotic tissues that appears to be the rule in human mtDNA disease.
In a mouse model harboring a slightly deleterious tRNA mutation (m.3875delC), the mutational load was reduced through a mechanism acting at the cellular or organelle level in the developing embryo (Freyer et al., 2012), consistent with observations in inter-subspecies cattle ooplasm transfer described in more detail below (Ferreira et al., 2010). In the mouse model this effect is dependent on the initial heteroplasmy of the mother: offspring from mothers with higher heteroplasmy had lower average heteroplasmy than their mothers. The authors argue that this shift has to take place during gestation, at the cellular or organelle level. However, as no oocytes with >80% of mutated mtDNA could be found, it is possible that this effect (additionally) works on oocyte development.
These findings are consistent with the presence of purifying selection in the germ line, a mechanism acting to eliminate highly deleterious mutations, particularly those located in protein-coding regions. Purifying selection has been directly observed in heteroplasmic mice harboring mtDNA with a severe ND6 mutation along with the wild-type mtDNA. In these mice, the mutation was selectively eliminated during oogenesis within four generations, while a milder cytochrome oxidase 1 (COI) mutation was retained (Fan et al., 2008).
Very recently, in heteroplasmic Drosophila melanogaster that harbored a COI mutation that resulted in temperature-sensitive mitochondrial malfunction, it was shown that one possible mechanism of purifying selection is selective propagation of fit mitochondria on the organelle level (Hill et al., 2014).
There is thus some evidence for segregation of pathological mtDNA in animal models, particularly involving selection against mtDNA with demonstrable deleterious effects.
Segregation of genetically different mtDNA haplotypes
In order to elucidate the mechanisms that govern segregation between non-pathological mtDNA haplotypes, ooplasm transfer and blastomere/cytoplast fusion have been used to create various heteroplasmic animal models using naturally occurring haplotypes that do not specifically harbor a pathological mutation. The best-known example is the heteroplasmic mouse line containing a mixture of NZB mtDNA and a common laboratory mouse strain (CIS) mtDNA (Table I). Laboratory mouse strains show very little variation in mtDNA (Goios et al., 2007), with the NZB strain one of the very few that show considerable genetic difference to the common CIS mtDNA.
Table I.
Heteroplasmic animal models with physiological mitochondrial DNA (mtDNA) that show in vivo segregation and/or physiological changes.
| Species | mtDNA/species | Method | Tissue | Physiology | Reference |
|---|---|---|---|---|---|
| Mouse | NZB + BALB/cByJ M. m. domesticus |
Cytoplast fusion | Liver, kidney, blood, spleen | n.a. | Jenuth et al. (1997) |
| Mouse | NZB +129S6 M. m. domesticus |
Cytoplast fusion | Liver, kidney, spleen, pancreas | Altered behavior | Sharpley et al. (2012) |
| Mouse | NZB + BALB/cByJ M. m. domesticus |
Cytoplast fusion | n.a. | Altered blood parameters, hypertension | Acton et al. (2007) |
| Mouse | Wild-derived mice + C57/BL6N 3×M. m. domesticus 1×M. m. musculus |
Ooplasm transfer | 12 different tissues (various segregation regimes) | n.a. | Burgstaller et al. (2014) |
| Mouse | RR + C57BL/6 M. m. molossinus/M. m. domesticus |
Blastomere fusion | All; relative to post-mitotic tissue | n.a. | Takeda et al. (2000) |
| Mouse | JF1+ C57BL/6 M. m. molossinus/M. m. domesticus |
Nuclear transfer | Liver; relative to brain | n.a. | Inoue et al. (2004) |
| Pig | Meishan + Landrace S. vittatus S. scrofa |
Nuclear transfer | Liver; relative to spleen, ear, blood | n.a. | Takeda et al. (2006) |
| Cattle | zebu + taurine cattle B. p. indicus/B. p. taurus |
Ooplasm transfer | Fetus; (B. p. indicus mtDNA reduced during gestation) | n.a | Ferreira et al. (2010) |
In the NZB/CIS model, the mixture of two naturally occurring but genetically different haplotypes (belonging to the same subspecies) leads to tissue-specific segregation effects: the proportion of NZB mtDNA increases with time in liver and kidney and decreases in blood and spleen. This mixture of mtDNAs leads to detrimental physiological (Acton et al., 2007) and behavioral consequences as described above (Sharpley et al., 2012), although both mtDNAs are regarded free of pathological mutations. Interestingly, the offspring showed a considerable reduction of NZB mtDNA compared with their mothers (Sharpley et al., 2012). The difference was already visible in the oocytes of the mother, but it is likely that the drift also occurs during gestation. This argues for a directed segregation effect operating in the germ line in addition to the aforementioned bottleneck effect.
The basic mechanisms of these segregation effects are largely unknown. One nuclear gene has been found to influence segregation in blood (Gimap3) (Jokinen et al., 2010), and one of the 91 SNPs between the two mtDNA haplotypes was proposed, and hotly discussed, as being responsible via its influence on reactive oxygen species (ROS) production (an ‘A’ track polymorphism in the DHU loop of the tRNAArg (Moreno-Loshuertos et al., 2006)). Perhaps the most convincing explanation for why heteroplasmy is detrimental is an evolutionary one. Co-evolution of minor differences of in trans protein reading frames between divergent haplotypes ensures that multimeric enzyme complexes maintain a high efficiency. However these minor changes will impair efficiency of the complexes when heteroplasmy for divergent haplotypes is present. Of note, the NZB mouse strain generates more ROS than other haplotypes. Even if this ROS production itself is not physiologically deleterious, a deleterious underlying mechanism may be responsible for this difference, driving the tendency of offspring to reduce NZB mtDNA levels – probably on the oocyte and (partly) cellular level (Wallace and Chalkia, 2013).
Ooplasm transfer studies of segregation in other model organisms are limited. In cattle, inter-subspecies ooplasm transfer (Bos primigenius taurus/B. p. indicus) has revealed segregation effects during blastocyst development and during gestation, with the B. p. indicus mtDNA being removed over time (Ferreira et al., 2010). Also in two inter-subspecific mouse models effects were observed (M. m. musculus/ M. m. domesticus, Table I).
However, the NZB model has remained the dominant heteroplasmic model utilizing naturally occurring mtDNA for almost 20 years. A large proportion of our knowledge about mtDNA segregation is based on this most prominent and best-studied heteroplasmic model, yet it is unknown whether its segregational effects represent an exception or a rule, and whether other combinations of mtDNA haplotypes may present different results.
Very recently, to address this question, we produced four mouse models by ooplasm transfer, placing various naturally occurring mtDNA haplotypes from mice captured from the wild in Europe onto a common laboratory mouse mtDNA and nuclear background (C57BL/6N). The wild-derived haplotypes we used display a spectrum of genetic differences with C57BL/6N, thus enabling us to control genetic distance (from very similar haplotypes, to haplotypes that differ in a comparable number of sites to two randomly chosen human mtDNAs, as described above). We also developed a mathematical framework to facilitate the direct comparison of many of these mice. We found that tissue-specific segregation was very common (including within post-mitotic tissue types), with the magnitude of segregation increasing with the genetic distance between the mtDNA haplotypes, and identified several contrasting mechanisms related to mtDNA turnover and organismal age by which this segregation occurred (Burgstaller et al., 2014). This study suggests that segregation between naturally occurring haplotypes may be the rule rather than exception, particularly with genetically diverse mtDNA pairings.
Heteroplasmy also exists, and has been studied, after nuclear transfer; but most studies have investigated the effects of the transfer process itself rather than focusing on mtDNA heteroplasmy. Nevertheless, these studies cover several species (cattle, sheep, pig, mouse), and cover inter- and intra-species heteroplasmy (reviewed in detail in St John et al. (2010)), and provide a body of data demonstrating co-existence of two mtDNA haplotypes in several species in vivo. Several studies report aberrance from expected donor mtDNA amounts that could be caused by segregation bias (Hiendleder et al., 1999; Takeda et al., 2003; Burgstaller et al., 2007). In particular, one study systematically analyzing cloned pigs and their offspring demonstrated powerful segregation effects between mtDNA from the genetically distant Meishan and Landrace breeds, which represent two subspecies of Sus scrofa. In these animals, the Meishan mtDNA significantly increased in liver, relative to spleen, ear and blood ((Takeda et al., 2006), Table I). Another inter-species study in mouse (M. m. molossinus/M. m. domesticus), also found segregation in liver, with the M. m. molossinus mtDNA increasing (measured relative to brain (Inoue et al., 2004)).
It is notable that in studies observing many animals over a substantial amount of time, or over several generations, segregation between different mtDNA types is often observed (Table I). Interestingly, in all studies of post-natal animals, liver is the tissue with the highest segregation effect. We can only speculate why this might be, but note that liver tissue has a high energy demand combined with high mtDNA turnover. Liver mtDNA half-lives are estimated at between 2 (Miwa et al., 2008, 2010) and 9 days (Gross et al., 1969; Menzies and Gold, 1971; Korr et al., 1998) when compared with, for example, skeletal muscle (reports from 18 (Korr et al., 1998) to 700 days (Collins et al., 2003)). The fast turnover time of mtDNA in liver and potentially strong selective pressure for energy production may underlie the rapid segregation observed in this tissue.
Implications
We have reviewed classical and modern approaches to address the inheritance of mtDNA disease. Modern approaches—pronuclear transfer and spindle transfer—have the potential to ameliorate mtDNA disease without the unsatisfactory genetic features of classical approaches. However, we have noted that several uncertainties are currently associated with the post-therapy behavior of embryos created using these techniques. These issues include mtDNA-mtDNA and mtDNA-nDNA mismatches, which could be important at high heteroplasmies, but are likely dampened by the ability of modern therapies to guarantee <1–2% donor carryover. Segregation of pathological mtDNA is potentially damaging. A key argument for nuclear transfer is therefore that current evidence suggests such segregation is either of very low magnitude or acts in such a way to remove pathological mutation (or both), and hence is likely not a key issue in mtDNA therapies (Craven et al., 2010). While further work is required to satisfactorily characterize these phenomena, the evidence suggests that they might not pose immediate issues in the application of genetic therapies.
The remaining phenomenon, segregation of non-pathological mtDNA haplotypes, is possibly the most important unaddressed question associated with modern mtDNA therapy, due to the potential consequent amplification of pathological mutations associated with one haplotype in the offspring resulting from genetic therapy, and in subsequent generations.
A key clinical consideration is whether segregation, and subsequent potential amplification of pathological mutations, could occur in post-mitotic tissues in which mtDNA diseases are most often manifest (Reeve et al., 2008). In organs where cells are constantly renewed (for example, skin and intestine), cells with damaged OXPHOS systems are probably replaced by functioning ones (Rahman et al., 2001). Organs particularly at risk for mtDNA disease are the post-mitotic tissues of heart, and skeletal muscle and brain; and liver and kidney which show high (liver) (Michalopoulos and DeFrances, 1997) and rather limited (kidney) (Little, 2006) regenerative potential. Our recent study using wild-derived mouse mtDNA has demonstrated haplotypic segregation in heart and skeletal muscle (Burgstaller et al., 2014). Liver and kidney are both tissues that show mtDNA segregation bias in the famous heteroplasmic mouse model described by the Shoubridge group described above (Jenuth et al., 1997).
An important question when considering the implications of animal models is whether phenomena observed in animals also occur in humans. Investigation of mtDNA dynamics during human development is practically limited for clear reasons, and the mouse NZB model remains by far the best-studied model of mtDNA haplotype segregation. However, reports of coexistence between two mtDNA haplotypes, and reports of mtDNA segregation effects, are present across several species, suggesting that these effects may be shared by all mammals. Further studies are however needed to confirm this assumption, and, importantly, elucidate the mechanisms on which these effects are based.
Based on the current available evidence, we believe that segregation between different naturally occurring mtDNA haplotypes may potentially influence the post-therapy behavior of intracellular mtDNA populations in offspring produced through modern gene therapies. To recap, these therapies involve recruiting a ‘recipient’ oocyte to serve as a healthy mitochondrial background for nuclear DNA resulting from fertilization. However, experimental limitations mean that some of the original mother's ‘donor’ mtDNA will inevitably be present in embryos produced in this way. If donor mtDNA proliferates over recipient mtDNA, the donor mtDNA will become amplified during development and during the lifetime of the offspring. If the donor mtDNA is associated with a pathological mutation, even if this mutation does not affect mtDNA proliferation, its ‘hitchhiking’ on the proliferating mtDNA may cause its amplification to potentially pathological levels in the offspring. We note that this worst-case haplotype segregation will not, in itself, cause additional harm to offspring beyond that expected from mtDNA disease inheritance; rather, it has the potential to nullify the beneficial effects of genetic therapy by re-establishing the original mtDNA mixture that was present in the donor oocyte, possibly in a tissue-specific way. The possibility also exists that the amplification of one mtDNA type through segregation may affect the behavior resulting from the aforementioned mtDNA-mtDNA mismatch, which is very likely suppressed at low heteroplasmies.
It is notable that all potential mtDNA segregation issues (indeed, all three of the potential issues we note) associated with modern mtDNA therapies can be ameliorated by employing a simple ‘haplotype matching’ protocol: that is, ensuring that the donor and recipient mtDNA haplotypes are as similar as possible. This approach will minimize nDNA-mtDNA mismatching (as the donor nucleus will have co-evolved with donor mtDNA, very similar to recipient mtDNA); mtDNA-mtDNA mismatch (due to the genetic similarity); and mtDNA segregation (as two very similar haplotypes are expected to show very little segregation). The ideal recipient would be of the same haplotype as the donor (minus the pathological mutation), for example, from a healthy maternal relative. Alternatively (or in addition), further research on the segregation of different pairs of mtDNA haplotypes could be used to choose suitable recipients for a given donor, in order to minimize segregation effects.
Experts in the field of karyoplast transfer have noted that ‘it is possible to match mitochondrial haplotype between the mother and the mitochondrial donor to avoid any concern, even though the evidence says it should not be needed’ (Chinnery et al., 2014). We think that the somewhat overlooked issue of mtDNA segregation currently constitutes a reason that merits this safety precaution, which would solve all potential concerns reviewed here. Additionally, in the case of exact haplotype matching, offspring mtDNA would have a complete genetic identity with the mother's mtDNA, possibly going some way towards alleviating the ethical issues associated with ‘three-parent babies’; that is, offspring with genetic material from mother, father and a third party.
While uncertainties exist regarding the behavior of mixed intracellular mtDNA populations, and animal models of mtDNA mixtures during development suggest that segregation potentially requires further study and consideration in therapeutic contexts, it should firmly be noted that these recent mtDNA-replacement strategies hold the promise to eliminate transmission of mtDNA diseases for good, and in so doing dramatically improve the lives of families carrying mtDNA disease. The potential advantages of these therapies seem to, in general, substantially outweigh their known risks. The unknown risks must thus be balanced against the certainties of classical genetic management. Hence potential patients for the first treatment trials will be from the rare homoplasmic families at proven high recurrence risk of severe phenotypes, for whom classical genetic management has least to offer. Initiating clinical trials is the only way to evaluate the presently unknown risks and future hopes for families brought by modern mtDNA therapies.
Authors' roles
J.P.B and I.G.J. compiled the content and produced the manuscript; J.P. edited the manuscript and provided clinical input.
Funding
I.G.J. gratefully acknowledges funding from the UK Medical Research Council; J.P. is supported by the Medical Research Council (MR/J010448/1) and the Wellcome Trust (WT0948685MA). Funding to pay the Open Access publication charges for this article was provided by the Medical Research Council.
Conflict of interest
None declared.
References
- Acton BM, Lai I, Shang X, Jurisicova A, Casper RF. Neutral mitochondrial heteroplasmy alters physiological function in mice. Biol Reprod. 2007;77:569–576. doi: 10.1095/biolreprod.107.060806. [DOI] [PubMed] [Google Scholar]
- Al Rawi S, Louvet-Vallee S, Djeddi A, Sachse M, Culetto E, Hajjar C, Boyd L, Legouis R, Galy V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science. 2011;334:1144–1147. doi: 10.1126/science.1211878. [DOI] [PubMed] [Google Scholar]
- Bacman SR, Williams SL, Pinto M, Peralta S, Moraes CT. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat Med. 2013;19:1111–1113. doi: 10.1038/nm.3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black GC, Morten K, Laborde A, Poulton J. Leber's hereditary optic neuropathy: heteroplasmy is likely to be significant in the expression of LHON in families with the 3460 ND1 mutation. Br J Ophthalmol. 1996;80:915–917. doi: 10.1136/bjo.80.10.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco R, Mayordomo E, Montoya J, Ruiz-Pesini E. Rebooting the human mitochondrial phylogeny: an automated and scalable methodology with expert knowledge. BMC Bioinformatics. 2011;12:174. doi: 10.1186/1471-2105-12-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blok RB, Gook DA, Thorburn DR, Dahl HH. Skewed segregation of the mtDNA nt 8993 (T–>G) mutation in human oocytes. Am J Hum Genet. 1997;60:1495–1501. doi: 10.1086/515453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowles EJ, Tecirlioglu RT, French AJ, Holland MK, St John JC. Mitochondrial DNA transmission and transcription after somatic cell fusion to one or more cytoplasts. Stem Cells. 2008;26:775–782. doi: 10.1634/stemcells.2007-0747. [DOI] [PubMed] [Google Scholar]
- Brown DT, Samuels DC, Michael EM, Turnbull DM, Chinnery PF. Random genetic drift determines the level of mutant mtDNA in human primary oocytes. Am J Hum Genet. 2001;68:533–536. doi: 10.1086/318190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgstaller JP, Schinogl P, Dinnyes A, Muller M, Steinborn R. Mitochondrial DNA heteroplasmy in ovine fetuses and sheep cloned by somatic cell nuclear transfer. BMC Dev Biol. 2007;7:141. doi: 10.1186/1471-213X-7-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgstaller JP, Johnston IG, Jones NS, Albrechtova J, Kolbe T, Vogl C, Futschik A, Mayrhofer C, Klein D, Sabitzer S, et al. mtDNA segregation in heteroplasmic tissues is common in vivo and modulated by haplotype differences and developmental stage. Cell Rep. 2014;7:2031–2041. doi: 10.1016/j.celrep.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon MV, Dunn DA, Irwin MH, Brooks AI, Bartol FF, Trounce IA, Pinkert CA. Xenomitochondrial mice: investigation into mitochondrial compensatory mechanisms. Mitochondrion. 2011;11:33–39. doi: 10.1016/j.mito.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Shitara H, Horii T, Nagao Y, Imai H, Abe K, Hara T, Hayashi J, Yonekawa H. The mitochondrial bottleneck occurs without reduction of mtDNA content in female mouse germ cells. Nat Genet. 2007;39:386–390. doi: 10.1038/ng1970. [DOI] [PubMed] [Google Scholar]
- Cao L, Shitara H, Sugimoto M, Hayashi J, Abe K, Yonekawa H. New evidence confirms that the mitochondrial bottleneck is generated without reduction of mitochondrial DNA content in early primordial germ cells of mice. PLoS Genet. 2009;5:e1000756. doi: 10.1371/journal.pgen.1000756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling PJ, Cree LM, Chinnery PF. The implications of mitochondrial DNA copy number regulation during embryogenesis. Mitochondrion. 2011;11:686–692. doi: 10.1016/j.mito.2011.05.004. [DOI] [PubMed] [Google Scholar]
- Chinnery PF, Hudson G. Mitochondrial genetics. Br Med Bull. 2013;106:135–159. doi: 10.1093/bmb/ldt017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery PF, Howell N, Lightowlers RN, Turnbull DM. Molecular pathology of MELAS and MERRF. The relationship between mutation load and clinical phenotypes. Brain. 1997;120(Pt 10):1713–1721. doi: 10.1093/brain/120.10.1713. [DOI] [PubMed] [Google Scholar]
- Chinnery PF, Johnson MA, Wardell TM, Singh-Kler R, Hayes C, Brown DT, Taylor RW, Bindoff LA, Turnbull DM. The epidemiology of pathogenic mitochondrial DNA mutations. Ann Neurol. 2000;48:188–193. [PubMed] [Google Scholar]
- Chinnery PF, DiMauro S, Shanske S, Schon EA, Zeviani M, Mariotti C, Carrara F, Lombes A, Laforet P, Ogier H, et al. Risk of developing a mitochondrial DNA deletion disorder. Lancet. 2004;364:592–596. doi: 10.1016/S0140-6736(04)16851-7. [DOI] [PubMed] [Google Scholar]
- Chinnery PF, Craven L, Mitalipov S, Stewart JB, Herbert M, Turnbull DM. The challenges of mitochondrial replacement. PLoS Genet. 2014;10:e1004315. doi: 10.1371/journal.pgen.1004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciafaloni E, Ricci E, Shanske S, Moraes CT, Silvestri G, Hirano M, Simonetti S, Angelini C, Donati MA, Garcia C, et al. MELAS: clinical features, biochemistry, and molecular genetics. Ann Neurol. 1992;31:391–398. doi: 10.1002/ana.410310408. [DOI] [PubMed] [Google Scholar]
- Collins ML, Eng S, Hoh R, Hellerstein MK. Measurement of mitochondrial DNA synthesis in vivo using a stable isotope-mass spectrometric technique. J Appl Physiol. 2003;94:2203–2211. doi: 10.1152/japplphysiol.00691.2002. [DOI] [PubMed] [Google Scholar]
- Craven L, Tuppen HA, Greggains GD, Harbottle SJ, Murphy JL, Cree LM, Murdoch AP, Chinnery PF, Taylor RW, Lightowlers RN, et al. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature. 2010;465:82–85. doi: 10.1038/nature08958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven L, Elson JL, Irving L, Tuppen HA, Lister LM, Greggains GD, Byerley S, Murdoch AP, Herbert M, Turnbull D. Mitochondrial DNA disease: new options for prevention. Hum Mol Genet. 2011;20:R168–R174. doi: 10.1093/hmg/ddr373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cree LM, Samuels DC, de Sousa Lopes SC, Rajasimha HK, Wonnapinij P, Mann JR, Dahl HH, Chinnery PF. A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat Genet. 2008;40:249–254. doi: 10.1038/ng.2007.63. [DOI] [PubMed] [Google Scholar]
- Cummins JM, Wakayama T, Yanagimachi R. Fate of microinjected spermatid mitochondria in the mouse oocyte and embryo. Zygote. 1998;6:213–222. doi: 10.1017/s0967199498000148. [DOI] [PubMed] [Google Scholar]
- Degoul F, Diry M, Rodriguez D, Robain O, Francois D, Ponsot G, Marsac C, Desguerre I. Clinical, biochemical, and molecular analysis of a maternally inherited case of Leigh syndrome (MILS) associated with the mtDNA T8993G point mutation. J Inherit Metab Dis. 1995;18:682–688. doi: 10.1007/BF02436757. [DOI] [PubMed] [Google Scholar]
- DeLuca SZ, O'Farrell PH. Barriers to male transmission of mitochondrial DNA in sperm development. Dev Cell. 2012;22:660–668. doi: 10.1016/j.devcel.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMauro S, Schon EA, Carelli V, Hirano M. The clinical maze of mitochondrial neurology. Nat Rev Neurol. 2013;9:429–444. doi: 10.1038/nrneurol.2013.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbar DR, Moonie PA, Jacobs HT, Holt IJ. Different cellular backgrounds confer a marked advantage to either mutant or wild-type mitochondrial genomes. Proc Natl Acad Sci USA. 1995;92:6562–6566. doi: 10.1073/pnas.92.14.6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmerson CF, Brown GK, Poulton J. Synthesis of mitochondrial DNA in permeabilised human cultured cells. Nucleic Acids Res. 2001;29:E1. doi: 10.1093/nar/29.2.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Waymire KG, Narula N, Li P, Rocher C, Coskun PE, Vannan MA, Narula J, Macgregor GR, Wallace DC. A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science. 2008;319:958–962. doi: 10.1126/science.1147786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira CR, Burgstaller JP, Perecin F, Garcia JM, Chiaratti MR, Meo SC, Muller M, Smith LC, Meirelles FV, Steinborn R. Pronounced segregation of donor mitochondria introduced by bovine ooplasmic transfer to the female germ-line. Biol Reprod. 2010;82:563–571. doi: 10.1095/biolreprod.109.080564. [DOI] [PubMed] [Google Scholar]
- Freyer C, Cree LM, Mourier A, Stewart JB, Koolmeister C, Milenkovic D, Wai T, Floros VI, Hagstrom E, Chatzidaki EE, et al. Variation in germline mtDNA heteroplasmy is determined prenatally but modified during subsequent transmission. Nat Genet. 2012;44:1282–1285. doi: 10.1038/ng.2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammage PA, Rorbach J, Vincent AI, Rebar EJ, Minczuk M. Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol Med. 2014;6:458–466. doi: 10.1002/emmm.201303672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano C, Montopoli M, Perli E, Orlandi M, Fantin M, Ross-Cisneros FN, Caparrotta L, Martinuzzi A, Ragazzi E, Ghelli A, et al. Oestrogens ameliorate mitochondrial dysfunction in Leber's hereditary optic neuropathy. Brain. 2011;134:220–234. doi: 10.1093/brain/awq276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goios A, Pereira L, Bogue M, Macaulay V, Amorim A. mtDNA phylogeny and evolution of laboratory mouse strains. Genome Res. 2007;17:293–298. doi: 10.1101/gr.5941007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348:651–653. doi: 10.1038/348651a0. [DOI] [PubMed] [Google Scholar]
- Greaves LC, Reeve AK, Taylor RW, Turnbull DM. Mitochondrial DNA and disease. J Pathol. 2012;226:274–286. doi: 10.1002/path.3028. [DOI] [PubMed] [Google Scholar]
- Gross NJ, Getz GS, Rabinowitz M. Apparent turnover of mitochondrial deoxyribonucleic acid and mitochondrial phospholipids in the tissues of the rat. J Biol Chem. 1969;244:1552–1562. [PubMed] [Google Scholar]
- Gyllensten U, Wharton D, Josefsson A, Wilson AC. Paternal inheritance of mitochondrial DNA in mice. Nature. 1991;352:255–257. doi: 10.1038/352255a0. [DOI] [PubMed] [Google Scholar]
- Harding AE, Holt IJ, Sweeney MG, Brockington M, Davis MB. Prenatal diagnosis of mitochondrial DNA8993 T—G disease. Am J Hum Genet. 1992;50:629–633. [PMC free article] [PubMed] [Google Scholar]
- Hayashi J, Ohta S, Kikuchi A, Takemitsu M, Goto Y, Nonaka I. Introduction of disease-related mitochondrial DNA deletions into HeLa cells lacking mitochondrial DNA results in mitochondrial dysfunction. Proc Natl Acad Sci USA. 1991;88:10614–8. doi: 10.1073/pnas.88.23.10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Wu J, Dressman DC, Iacobuzio-Donahue C, Markowitz SD, Velculescu VE, Diaz LA, Jr, Kinzler KW, Vogelstein B, Papadopoulos N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature. 2010;464:610–614. doi: 10.1038/nature08802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiendleder S, Schmutz SM, Erhardt G, Green RD, Plante Y. Transmitochondrial differences and varying levels of heteroplasmy in nuclear transfer cloned cattle. Mol Reprod Dev. 1999;54:24–31. doi: 10.1002/(SICI)1098-2795(199909)54:1<24::AID-MRD4>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Hill JH, Chen Z, Xu H. Selective propagation of functional mitochondrial DNA during oogenesis restricts the transmission of a deleterious mitochondrial variant. Nat Genet. 2014;46:389–392. doi: 10.1038/ng.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt IJ, Harding AE, Petty RK, Morgan-Hughes JA. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am J Hum Genet. 1990;46:428–433. [PMC free article] [PubMed] [Google Scholar]
- Howell N, McCullough D. An example of Leber hereditary optic neuropathy not involving a mutation in the mitochondrial ND4 gene. Am J Hum Genet. 1990;47:629–634. [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Ogonuki N, Yamamoto Y, Takano K, Miki H, Mochida K, Ogura A. Tissue-specific distribution of donor mitochondrial DNA in cloned mice produced by somatic cell nuclear transfer. Genesis. 2004;39:79–83. doi: 10.1002/gene.20029. [DOI] [PubMed] [Google Scholar]
- Jenuth JP, Peterson AC, Fu K, Shoubridge EA. Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nat Genet. 1996;14:146–151. doi: 10.1038/ng1096-146. [DOI] [PubMed] [Google Scholar]
- Jenuth JP, Peterson AC, Shoubridge EA. Tissue-specific selection for different mtDNA genotypes in heteroplasmic mice. Nat Genet. 1997;16:93–95. doi: 10.1038/ng0597-93. [DOI] [PubMed] [Google Scholar]
- Jeppesen TD, Schwartz M, Frederiksen AL, Wibrand F, Olsen DB, Vissing J. Muscle phenotype and mutation load in 51 persons with the 3243A>G mitochondrial DNA mutation. Arch Neurol. 2006;63:1701–1706. doi: 10.1001/archneur.63.12.1701. [DOI] [PubMed] [Google Scholar]
- Ji F, Sharpley MS, Derbeneva O, Alves LS, Qian P, Wang Y, Chalkia D, Lvova M, Xu J, Yao W, et al. Mitochondrial DNA variant associated with Leber hereditary optic neuropathy and high-altitude Tibetans. Proc Natl Acad Sci USA. 2012;109:7391–7396. doi: 10.1073/pnas.1202484109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns DR, Neufeld MJ, Park RD. An ND-6 mitochondrial DNA mutation associated with Leber hereditary optic neuropathy. Biochem Biophys Res Commun. 1992;187:1551–1557. doi: 10.1016/0006-291x(92)90479-5. [DOI] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY, Bykhovskaya Y, Spirina O, Fischel-Ghodsian N. A nuclear-mitochondrial DNA interaction affecting hearing impairment in mice. Nat Genet. 2001;27:191–194. doi: 10.1038/84831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokinen R, Marttinen P, Sandell HK, Manninen T, Teerenhovi H, Wai T, Teoli D, Loredo-Osti JC, Shoubridge EA, Battersby BJ. Gimap3 regulates tissue-specific mitochondrial DNA segregation. PLoS Genet. 2010;6:e1001161. doi: 10.1371/journal.pgen.1001161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly RD, Rodda AE, Dickinson A, Mahmud A, Nefzger CM, Lee W, Forsythe JS, Polo JM, Trounce IA, McKenzie M, et al. Mitochondrial DNA haplotypes define gene expression patterns in pluripotent and differentiating embryonic stem cells. Stem Cells. 2013;31:703–716. doi: 10.1002/stem.1313. [DOI] [PubMed] [Google Scholar]
- Korr H, Kurz C, Seidler TO, Sommer D, Schmitz C. Mitochondrial DNA synthesis studied autoradiographically in various cell types in vivo. Braz J Med Biol Res. 1998;31:289–298. doi: 10.1590/s0100-879x1998000200012. [DOI] [PubMed] [Google Scholar]
- Larsson NG, Holme E, Kristiansson B, Oldfors A, Tulinius M. Progressive increase of the mutated mitochondrial DNA fraction in Kearns-Sayre syndrome. Pediatr Res. 1990;28:131–136. doi: 10.1203/00006450-199008000-00011. [DOI] [PubMed] [Google Scholar]
- Larsson NG, Tulinius MH, Holme E, Oldfors A, Andersen O, Wahlstrom J, Aasly J. Segregation and manifestations of the mtDNA tRNA(Lys) A–>G(8344) mutation of myoclonus epilepsy and ragged-red fibers (MERRF) syndrome. Am J Hum Genet. 1992;51:1201–1212. [PMC free article] [PubMed] [Google Scholar]
- Lee HS, Ma H, Juanes RC, Tachibana M, Sparman M, Woodward J, Ramsey C, Xu J, Kang EJ, Amato P, et al. Rapid mitochondrial DNA segregation in primate preimplantation embryos precedes somatic and germline bottleneck. Cell Rep. 2012;1:506–515. doi: 10.1016/j.celrep.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightowlers RN, Chinnery PF, Turnbull DM, Howell N. Mammalian mitochondrial genetics: heredity, heteroplasmy and disease. Trends Genet. 1997;13:450–455. doi: 10.1016/s0168-9525(97)01266-3. [DOI] [PubMed] [Google Scholar]
- Lippold S, Xu H, Ko A, Li M, Renaud G, Butthof A, Schroeder R, Stoneking M. Human paternal and maternal demographic histories: insights from high-resolution Y chromosome and mtDNA sequences. bioRxiv. 2014 doi: 10.1186/2041-2223-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little MH. Regrow or repair: potential regenerative therapies for the kidney. J Am Soc Nephrol. 2006;17:2390–2401. doi: 10.1681/ASN.2006030218. [DOI] [PubMed] [Google Scholar]
- Luo SM, Ge ZJ, Wang ZW, Jiang ZZ, Wang ZB, Ouyang YC, Hou Y, Schatten H, Sun QY. Unique insights into maternal mitochondrial inheritance in mice. Proc Natl Acad Sci USA. 2013;110:13038–13043. doi: 10.1073/pnas.1303231110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manwaring N, Jones MM, Wang JJ, Rochtchina E, Howard C, Mitchell P, Sue CM. Population prevalence of the MELAS A3243G mutation. Mitochondrion. 2007;7:230–233. doi: 10.1016/j.mito.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Marchington DR, Hartshorne GM, Barlow D, Poulton J. Homopolymeric tract heteroplasmy in mtDNA from tissues and single oocytes: support for a genetic bottleneck. Am J Hum Genet. 1997;60:408–416. [PMC free article] [PubMed] [Google Scholar]
- Marchington DR, Macaulay V, Hartshorne GM, Barlow D, Poulton J. Evidence from human oocytes for a genetic bottleneck in an mtDNA disease. Am J Hum Genet. 1998;63:769–775. doi: 10.1086/302009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShane MA, Hammans SR, Sweeney M, Holt IJ, Beattie TJ, Brett EM, Harding AE. Pearson syndrome and mitochondrial encephalomyopathy in a patient with a deletion of mtDNA. Am J Hum Genet. 1991;48:39–42. [PMC free article] [PubMed] [Google Scholar]
- Menzies RA, Gold PH. The turnover of mitochondria in a variety of tissues of young adult and aged rats. J Biol Chem. 1971;246:2425–2429. [PubMed] [Google Scholar]
- Michalopoulos GK, DeFrances MC. Liver regeneration. Science. 1997;276:60–66. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- Mitalipov S, Amato P, Parry S, Falk MJ. Limitations of preimplantation genetic diagnosis for mitochondrial DNA diseases. Cell Rep. 2014;7:935–937. doi: 10.1016/j.celrep.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa S, Lawless C, von Zglinicki T. Mitochondrial turnover in liver is fast in vivo and is accelerated by dietary restriction: application of a simple dynamic model. Aging Cell. 2008;7:920–923. doi: 10.1111/j.1474-9726.2008.00426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa S, Lawless C, von Zglinicki T. Correction of radiolabel pulse-chase data by a mathematical model: application to mitochondrial turnover studies. Biochem Soc Trans. 2010;38:1322–1328. doi: 10.1042/BST0381322. [DOI] [PubMed] [Google Scholar]
- Monnot S, Gigarel N, Samuels DC, Burlet P, Hesters L, Frydman N, Frydman R, Kerbrat V, Funalot B, Martinovic J, et al. Segregation of mtDNA throughout human embryofetal development: m.3243A>G as a model system. Hum Mutat. 2011;32:116–125. doi: 10.1002/humu.21417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Loshuertos R, Acin-Perez R, Fernandez-Silva P, Movilla N, Perez-Martos A, Rodriguez de Cordoba S, Gallardo ME, Enriquez JA. Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nat Genet. 2006;38:1261–1268. doi: 10.1038/ng1897. [DOI] [PubMed] [Google Scholar]
- Moreno-Loshuertos R, Perez-Martos A, Fernandez-Silva P, Enriquez JA. Length variation in the mouse mitochondrial tRNA(Arg) DHU loop size promotes oxidative phosphorylation functional differences. FEBS J. 2013;280:4983–4998. doi: 10.1111/febs.12466. [DOI] [PubMed] [Google Scholar]
- Nagao Y, Totsuka Y, Atomi Y, Kaneda H, Lindahl KF, Imai H, Yonekawa H. Decreased physical performance of congenic mice with mismatch between the nuclear and the mitochondrial genome. Genes Genet Syst. 1998;73:21–27. doi: 10.1266/ggs.73.21. [DOI] [PubMed] [Google Scholar]
- Paull D, Emmanuele V, Weiss KA, Treff N, Stewart L, Hua H, Zimmer M, Kahler DJ, Goland RS, Noggle SA, et al. Nuclear genome transfer in human oocytes eliminates mitochondrial DNA variants. Nature. 2013;493:632–637. doi: 10.1038/nature11800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petruzzella V, Moraes CT, Sano MC, Bonilla E, DiMauro S, Schon EA. Extremely high levels of mutant mtDNAs co-localize with cytochrome c oxidase-negative ragged-red fibers in patients harboring a point mutation at nt 3243. Hum Mol Genet. 1994;3:449–454. doi: 10.1093/hmg/3.3.449. [DOI] [PubMed] [Google Scholar]
- Poulton J, Oakeshott P. Nuclear transfer to prevent maternal transmission of mitochondrial DNA disease. BMJ. 2012;345:e6651. doi: 10.1136/bmj.e6651. [DOI] [PubMed] [Google Scholar]
- Poulton J, O'Rahilly S, Morten KJ, Clark A. Mitochondrial DNA, diabetes and pancreatic pathology in Kearns-Sayre syndrome. Diabetologia. 1995;38:868–871. doi: 10.1007/s001250050366. [DOI] [PubMed] [Google Scholar]
- Poulton J, Chiaratti MR, Meirelles FV, Kennedy S, Wells D, Holt IJ. Transmission of mitochondrial DNA diseases and ways to prevent them. PLoS Genet. 2010;6:e1001066. doi: 10.1371/journal.pgen.1001066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S, Poulton J, Marchington D, Suomalainen A. Decrease of 3243 A-->G mtDNA mutation from blood in MELAS syndrome: a longitudinal study. Am J Hum Genet. 2001;68:238–240. doi: 10.1086/316930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve AK, Krishnan KJ, Turnbull D. Mitochondrial DNA mutations in disease, aging, and neurodegeneration. Ann N Y Acad Sci. 2008;1147:21–29. doi: 10.1196/annals.1427.016. [DOI] [PubMed] [Google Scholar]
- Reinhardt K, Dowling DK, Morrow EH. Medicine. Mitochondrial replacement, evolution, and the clinic. Science. 2013;341:1345–1346. doi: 10.1126/science.1237146. [DOI] [PubMed] [Google Scholar]
- Roubertoux PL, Sluyter F, Carlier M, Marcet B, Maarouf-Veray F, Cherif C, Marican C, Arrechi P, Godin F, Jamon M, et al. Mitochondrial DNA modifies cognition in interaction with the nuclear genome and age in mice. Nat Genet. 2003;35:65–69. doi: 10.1038/ng1230. [DOI] [PubMed] [Google Scholar]
- Samuels DC, Li C, Li B, Song Z, Torstenson E, Boyd Clay H, Rokas A, Thornton-Wells TA, Moore JH, Hughes TM, et al. Recurrent tissue-specific mtDNA mutations are common in humans. PLoS Genet. 2013;9:e1003929. doi: 10.1371/journal.pgen.1003929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Sato K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science. 2011;334:1141–1144. doi: 10.1126/science.1210333. [DOI] [PubMed] [Google Scholar]
- Sato A, Nakada K, Shitara H, Kasahara A, Yonekawa H, Hayashi J. Deletion-mutant mtDNA increases in somatic tissues but decreases in female germ cells with age. Genetics. 2007;177:2031–2037. doi: 10.1534/genetics.107.081026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer MV, Kavic SM. Oocyte and embryo donation 2006: reviewing two decades of innovation and controversy. Reprod Biomed Online. 2006;12:153–162. doi: 10.1016/s1472-6483(10)60855-3. [DOI] [PubMed] [Google Scholar]
- Sharpley MS, Marciniak C, Eckel-Mahan K, McManus M, Crimi M, Waymire K, Lin CS, Masubuchi S, Friend N, Koike M, et al. Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell. 2012;151:333–343. doi: 10.1016/j.cell.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shitara H, Kaneda H, Sato A, Inoue K, Ogura A, Yonekawa H, Hayashi JI. Selective and continuous elimination of mitochondria microinjected into mouse eggs from spermatids, but not from liver cells, occurs throughout embryogenesis. Genetics. 2000;156:1277–1284. doi: 10.1093/genetics/156.3.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St John JC, Campbell KH. The battle to prevent the transmission of mitochondrial DNA disease: is karyoplast transfer the answer? Gene Ther. 2010;17:147–149. doi: 10.1038/gt.2009.164. [DOI] [PubMed] [Google Scholar]
- St John JC, Schatten G. Paternal mitochondrial DNA transmission during nonhuman primate nuclear transfer. Genetics. 2004;167:897–905. doi: 10.1534/genetics.103.025049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St John J, Sakkas D, Dimitriadi K, Barnes A, Maclin V, Ramey J, Barratt C, De Jonge C. Failure of elimination of paternal mitochondrial DNA in abnormal embryos. Lancet. 2000;355:200. doi: 10.1016/s0140-6736(99)03842-8. [DOI] [PubMed] [Google Scholar]
- St John JC, Facucho-Oliveira J, Jiang Y, Kelly R, Salah R. Mitochondrial DNA transmission, replication and inheritance: a journey from the gamete through the embryo and into offspring and embryonic stem cells. Hum Reprod Update. 2010;16:488–509. doi: 10.1093/humupd/dmq002. [DOI] [PubMed] [Google Scholar]
- Steffann J, Frydman N, Gigarel N, Burlet P, Ray PF, Fanchin R, Feyereisen E, Kerbrat V, Tachdjian G, Bonnefont JP, et al. Analysis of mtDNA variant segregation during early human embryonic development: a tool for successful NARP preimplantation diagnosis. J Med Genet. 2006;43:244–247. doi: 10.1136/jmg.2005.032326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffann J, Gigarel N, Samuels DC, Monnot S, Borghese R, Hesters L, Frydman N, Burlet P, Frydman R, Benachi A, et al. Data from artificial models of mitochondrial DNA disorders are not always applicable to humans. Cell Rep. 2014;7:933–934. doi: 10.1016/j.celrep.2014.05.005. [DOI] [PubMed] [Google Scholar]
- Strauss KA, DuBiner L, Simon M, Zaragoza M, Sengupta PP, Li P, Narula N, Dreike S, Platt J, Procaccio V, et al. Severity of cardiomyopathy associated with adenine nucleotide translocator-1 deficiency correlates with mtDNA haplogroup. Proc Natl Acad Sci USA. 2013;110:3453–3458. doi: 10.1073/pnas.1300690110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutovsky P, Moreno RD, Ramalho-Santos J, Dominko T, Simerly C, Schatten G. Ubiquitin tag for sperm mitochondria. Nature. 1999;402:371–372. doi: 10.1038/46466. [DOI] [PubMed] [Google Scholar]
- Sutovsky P, Moreno RD, Ramalho-Santos J, Dominko T, Simerly C, Schatten G. Ubiquitinated sperm mitochondria, selective proteolysis, and the regulation of mitochondrial inheritance in mammalian embryos. Biol Reprod. 2000;63:582–590. doi: 10.1095/biolreprod63.2.582. [DOI] [PubMed] [Google Scholar]
- Tachibana M, Sparman M, Sritanaudomchai H, Ma H, Clepper L, Woodward J, Li Y, Ramsey C, Kolotushkina O, Mitalipov S. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature. 2009;461:367–372. doi: 10.1038/nature08368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Amato P, Sparman M, Woodward J, Sanchis DM, Ma H, Gutierrez NM, Tippner-Hedges R, Kang E, Lee HS, et al. Towards germline gene therapy of inherited mitochondrial diseases. Nature. 2013;493:627–631. doi: 10.1038/nature11647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Takahashi S, Onishi A, Hanada H, Imai H. Replicative advantage and tissue-specific segregation of RR mitochondrial DNA between C57BL/6 and RR heteroplasmic mice. Genetics. 2000;155:777–783. doi: 10.1093/genetics/155.2.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Akagi S, Kaneyama K, Kojima T, Takahashi S, Imai H, Yamanaka M, Onishi A, Hanada H. Proliferation of donor mitochondrial DNA in nuclear transfer calves (Bos taurus) derived from cumulus cells. Mol Reprod Dev. 2003;64:429–437. doi: 10.1002/mrd.10279. [DOI] [PubMed] [Google Scholar]
- Takeda K, Tasai M, Iwamoto M, Akita T, Tagami T, Nirasawa K, Hanada H, Onishi A. Transmission of mitochondrial DNA in pigs and progeny derived from nuclear transfer of Meishan pig fibroblast cells. Mol Reprod Dev. 2006;73:306–312. doi: 10.1002/mrd.20403. [DOI] [PubMed] [Google Scholar]
- Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treff NR, Campos J, Tao X, Levy B, Ferry KM, Scott RT., Jr Blastocyst preimplantation genetic diagnosis (PGD) of a mitochondrial DNA disorder. Fertil Steril. 2012;98:1236–1240. doi: 10.1016/j.fertnstert.2012.07.1119. [DOI] [PubMed] [Google Scholar]
- van den Ouweland JM, Lemkes HH, Ruitenbeek W, Sandkuijl LA, de Vijlder MF, Struyvenberg PA, van de Kamp JJ, Maassen JA. Mutation in mitochondrial tRNA(Leu)(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat Genet. 1992;1:368–371. doi: 10.1038/ng0892-368. [DOI] [PubMed] [Google Scholar]
- Wai T, Teoli D, Shoubridge EA. The mitochondrial DNA genetic bottleneck results from replication of a subpopulation of genomes. Nat Genet. 2008;40:1484–1488. doi: 10.1038/ng.258. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- Wallace DC, Chalkia D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb Perspect Med. 2013;3:a021220. doi: 10.1101/cshperspect.a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ, II, Nikoskelainen EK. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science. 1988a;242:1427–1430. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- Wallace DC, Zheng XX, Lott MT, Shoffner JM, Hodge JA, Kelley RI, Epstein CM, Hopkins LC. Familial mitochondrial encephalomyopathy (MERRF): genetic, pathophysiological, and biochemical characterization of a mitochondrial DNA disease. Cell. 1988b;55:601–610. doi: 10.1016/0092-8674(88)90218-8. [DOI] [PubMed] [Google Scholar]
- Weber K, Wilson JN, Taylor L, Brierley E, Johnson MA, Turnbull DM, Bindoff LA. A new mtDNA mutation showing accumulation with time and restriction to skeletal muscle. Am J Hum Genet. 1997;60:373–380. [PMC free article] [PubMed] [Google Scholar]
- White SL, Collins VR, Wolfe R, Cleary MA, Shanske S, DiMauro S, Dahl HH, Thorburn DR. Genetic counseling and prenatal diagnosis for the mitochondrial DNA mutations at nucleotide 8993. Am J Hum Genet. 1999;65:474–482. doi: 10.1086/302488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Lu J, Ma F, Keinan A, Gu Z. Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proc Natl Acad Sci USA. 2014;111:10654–10659. doi: 10.1073/pnas.1403521111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Gimsa U, Wester-Rosenlof L, Kanitz E, Otten W, Kunz M, Ibrahim SM. Dissecting the effects of mtDNA variations on complex traits using mouse conplastic strains. Genome Res. 2009;19:159–165. doi: 10.1101/gr.078865.108. [DOI] [PMC free article] [PubMed] [Google Scholar]