

Abstract
Juvenile hormone (JH) is an insect hormone containing an α,β unsaturated ester consisting of a small alcohol and long, hydrophobic acid. JH degradation is required for proper insect development. One pathway of this degradation is through juvenile hormone esterase (JHE), which cleaves the JH ester bond to produce methanol and JH acid. JHE is a member of the functionally divergent α/β-hydrolase family of enzymes, and is a highly efficient enzyme that cleaves JH at very low in vivo concentrations. We present here a 2.7 Å crystal structure of JHE from the tobacco hornworm Manduca sexta (MsJHE) in complex with the transition state analog inhibitor 3-octylthio-1,1,1-trifluoropropan-2-one (OTFP) covalently bound to the active site. This crystal structure, the first JHE structure reported, contains a long, hydrophobic binding pocket with the solvent inaccessible catalytic triad located at the end. The structure explains many of the interactions observed between JHE and its substrates and inhibitors, such as the preference for small alcohol groups and long hydrophobic backbones. The most potent JHE inhibitors identified to date contain a trifluoromethyl ketone (TFK) moiety and have a sulfur atom beta to the ketone. In this study, sulfur-aromatic interactions were observed between the sulfur atom of OTFP and a conserved aromatic residue in the crystal structure. Mutational analysis supported the hypothesis that these interactions contribute to the potency of sulfur-containing TFK inhibitors. Together these results clarify the binding mechanism of JHE inhibitors and provide useful observations for the development of additional enzyme inhibitors for a variety of enzymes.
Keywords: carboxylesterase, juvenile hormone esterase, trifluoromethyl ketone, crystal structure, inhibitor
Juvenile hormone (JH) (Figure 1A) is a sesquiterpenoid insect hormone that controls a wide range of biological processes. JH and/or its metabolites are known to have effects on insect life cycle processes such as development, metamorphosis, reproduction, diapause, migration, polyphenism and metabolism (1, 2). Perturbation of this system can have deleterious consequences for the insect, a fact which has been exploited for pest control. For example, the JH analog methoprene is a significant chemical line of defense against the larvae of mosquitoes that carry West Nile Virus (3). JH can be metabolized through cleavage of either its epoxide or ester moiety. The relative importance of these two pathways, both catalyzed by α/β-hydrolase enzymes (4–6), varies with insect species, development stage and tissue. Normal lepidopteran metamorphosis, for example, requires the degradation of JH via ester hydrolysis. Inhibition of ester hydrolysis leads to abnormally large larvae and delayed pupation (7). The enzyme responsible for this ester hydrolysis is juvenile hormone esterase (JHE), which is found in the hemolymph and other tissues of insects at key times in development.
Figure 1.

Chemical structures of JHE substrates and inhibitors of JHE. A) Structure of JH 0, I, II, and III. For all JH’s, both unsaturated bonds are in the E isomeric form. All JH’s have a chiral center at carbon 10, and this is the R isomer for all. In addition, JH 0, I, and II have a chiral center at carbon 11. For all three, this is the S isomer. B) Structures of OTFP and TFDK. In aqueous solution the ketone to gem-diol equilibrium is shifted towards the diol based upon inhibitor potency, with potent inhibitors (e.g. OTFP) favoring the gem-diol form. The Greek letters α, β and γ refer to atom position relative to the carbonyl carbon.
Insects have significant effects on human health, as vectors for diseases such as malaria and virus induced encephalitis, and in agriculture as pests and pollinators. It is therefore important to study JHE’s function and specificity, which plays a key role in insect development. Understanding the specificity of the enzyme for various substrates and inhibitors will increase our knowledge of insect development. Results of this work are also generally applicable to the study of other carboxylesterases, some of which are important in the hydrolysis of a number of pharmaceuticals and other exogenous esters (8, 9). Additionally, information gained from our study of the inhibitor’s interactions with the binding pocket may prove useful in improving the potency of protein binding small molecules, such as enzyme inhibitors and receptor agonists and antagonists.
Four major forms of JH (Figure 1A) have been identified, all of which have an α,β unsaturated methyl ester and a terpenoid or homoterpenoid backbone with an epoxide distal to the ester. The α,β unsaturation produces a particularly stable ester bond, due to resonance stabilization between the unsaturated bond and the carbonyl of the ester. Most lepidopterans produce predominantly JH I and JH II. The tobacco hornworm, Manduca sexta, which produces the JHE described in this paper, produces JH0, JHI, JH II and JH III (10–12).
MsJHE is a very efficient enzyme, catalyzing the hydrolysis of JH III with a kcat/Km of at least 3 × 107 M−1s−1 (13). The enzyme is optimized to catalyze the reaction with a moderate kcat of 1.5 s−1, but has a Km of approximately 50 nM. As a member of the α/β-hydrolase family of enzymes, MsJHE catalyzes this hydrolysis in a multiple-step reaction. First, the catalytic serine, acting as a nucleophile, attacks the ester carbonyl carbon, releasing the alcohol and forming a covalent acyl-enzyme intermediate, via a tetrahedral transition state intermediate. Next, water attacks the carbonyl carbon of the acyl-enzyme intermediate, releasing the acid portion of the ester and regenerating the free, active enzyme. As with the first portion of this reaction, this step proceeds via a tetrahedral transition state intermediate.
Inhibition of esterase activity can have important biological consequences. Carboxylesterases, for example, are essential for activation and detoxification of many pharmaceutical compounds and other exogenous esters (8, 9). Inhibition of JHE activity is also ultimately toxic for insects and a number of potent inhibitors of JHE have been developed (14–18). These compounds include the trifluoromethyl ketone (TFK) inhibitors (Figure 1B), which are transition state analogs that form reversible covalent adducts with esterases. TFKs were first reported as inhibitors of acetylcholine esterase (19), and shortly thereafter as inhibitors of both general esterase activity and JHE activity (19–22). Subsequently, they have been investigated as inhibitors of other esterases including mammalian esterases (23) and insect pheromone degrading esterases (24–26). They have proven to be very useful compounds for studying insect development (7). TFK inhibitors function by binding to the catalytic serine, which attacks the electrophilic ketone carbon of the TFK. The resulting acyl-enzyme complex has tetrahedral geometry, consisting of the trifluoromethyl group, the protein (via the serine), a hydroxyl (from the ketone oxygen) and an alkyl chain. However, because the inhibitor does not contain a cleavable residue (the trifluoromethyl group is a very poor leaving group) the enzyme is inhibited. It is possible for the reaction to proceed in the reverse direction, and in fact, OTFP can be removed by exhaustive dialysis (7). However, since the equilibrium favors the formation of the enzyme inhibitor complex, TFKs are potent inhibitors of JHE.
A significant amount of work has been done to understand the structure activity relationships (SAR) of these inhibitors (16–18, 20, 23, 27–31). In general, the potency of aliphatic TFK’s exhibits a strong positive correlation with lipophilicity, with a maximum potency corresponding to compounds of intermediate lipophilicity (logP of ~4). However, inhibitor potency can be increased by specific substitutions that do not strongly affect the overall lipophilicity or shape of the inhibitor. For example, substitution of the beta-carbon with a sulfur atom leads to significantly increased inhibitor potency (21). The sulfur atom was initially incorporated into TFK inhibitors to serve as a bioisostere of the olefin in juvenile hormone. It was thought that the electronic environment of a sulfur atom was an appropriate mimic of a double bond. The resulting increase in compound potency was greater than expected and led to a number of questions regarding the role of the sulfur atom in inhibitor potency. The initial hypothesis that the sulfur atom was mimicking the α,β unsaturated ester present in JH did not explain observed increases in the potency of sulfur-containing inhibitors that was observed with some other carboxylesterases (23). A second hypothesis proposed that the hydration extent of the TFK moiety was directly related to inhibitor potency, with inhibitors whose extent of hydration was shifted to the gem-diol exhibiting greater potency (Figure 1B). The biological activity of the sulfur atom was partially explained by this theory (31), but there were remaining questions regarding its exact role in inhibition.
An additional theory hypothesized that the sulfur atom could potentially form pi-stacking interactions with aromatic residues in the active site of the enzyme. Pi-stacking or sulfur-aromatic interactions in general have been shown to occur extensively in proteins between amino acid residues (32–34). However, very few reports have examined these effects in small molecules (35). Interactions of this type in proteins (e.g. between a cysteine thiol and a phenylalanine phenyl), are often characterized as hydrogen bonding in nature (36). However, there are several different types of favorable sulfur-aromatic interactions that may occur, including hydrogen bonding to the aryl hydrogens, SH-pi interactions, sulfur-pi interactions, electrostatic interactions, van der Waals and hydrophobic interactions (33). Determining the exact factors leading to an energetically favorable sulfur-aromatic interaction is not trivial. In this particular case, the lack of a free hydrogen on a thiol (such as that present in cysteine) simplifies the possible interactions because direct hydrogen bonding can be ruled out. Therefore, any potential interactions between the sulfur atom of OTFP and amino acid residues of the enzyme would involve divalent sulfur. Consequently, dispersion interactions are more suitable given the highly polarizable nature of both the benzene pi-electron cloud and the sulfur empty d-orbitals (37). However, Zauhar et al. (35) report that while sulfur-aromatic energies tend to include a large van der Waals contribution, the electrostatic component is still of significant size, suggesting that sulfur-aromatic interactions may have significant hydrogen bond character. In addition it has been reported (33) that sulfur-aromatic interactions in a β-hairpin, which placed a methionine side chain in close proximity to an aryl side chain, were predominantly due to hydrophobic effects. It is therefore most likely that the observed effects represent cumulative contributions from multiple interactions, with dispersion or van der Waals forces playing a dominant role.
Whatever their exact nature, these associations would serve to increase the binding strength of the inhibitor and could account for the observed potency. In the case of OTFP, the hypothesis that there are sulfur-aromatic interactions between the inhibitor and MsJHE was supported by the observation that substitution of a sulfur atom in the γposition (Figure 1B) did not result in equivalent increases in inhibitor potency as substitution in the β-position (16). This hypothesis was tested in the current study, which provides information on the role of the sulfur atom beta to the TFK moiety in inhibitor binding.
We present here a crystal structure of the JHE from Manduca sexta with the TFK inhibitor OTFP covalently bound to the catalytic serine. This structure contains a substrate binding pocket that is unusual for α/β-hydrolase proteins. The shape and physical properties of this binding pocket help explain the enzyme’s selectivity for JH and also help explain the SAR for JHE inhibitors. Furthermore, the crystal structure shows the presence of an aromatic residue (Phe259) that interacts with the sulfur atom in OTFP. Mutational analysis demonstrates that this phenylalanine accounts for some, though not all, of the increased potency of the sulfur substituted TFKs.
Materials and Methods
Protein Expression and Purification
Recombinant JHE from Manduca sexta was produced in insect High 5 cells cultured in ESF-921 medium (Expression Systems, LLC ) by the recombinant baculovirus AcMS7JHE as previously described (13)(38). The recombinant MsJHE for use in determining the crystal structure was affinity purified using MBTFP (3-(4-mercaptobutylthio)-1,1,1-trifluoropropan-2-one) linked to Sepharose beads and eluted with the inhibitor OTFP (3-octylthio-1,1,1-trifluoropropan-2one) in a buffer containing 0.05% Triton X-100, as previously described (7, 27). Anion exchange chromatography was done to remove the Triton and excess OTFP from the sample. An HQ (Poros 20 HQ, PerSeptive Biosystems, Framingham, MA) column was loaded with JHE in 50 mM Tris/50 mM bis-Tris propane buffer, pH 8.4, and protein was eluted with a 0 – 450 mM NaCl gradient in the same buffer. Surprisingly, this procedure produced two protein peaks of comparable size (with the Triton coming out in the void volume), though an SDS-PAGE analysis of the original sample showed only a single JHE band. The two peaks did not inter-convert or re-equilibrate after three days at 4 °C. Thus, the two peaks were separated and exchanged into 10 mM Tris buffer, pH 7.4. Each sample was concentrated to roughly 10 mg/ml. N-Terminal sequencing of the resulting protein showed that protein in both peaks begin with the sequence RIPSTE (i.e. starting at amino acid 23 of the full length protein, which is the expected product after removal of the signal sequence during processing of the protein).
JHE with selenomethionine was produced as described above, except that 24 hours post-infection the ESF-921 medium was replaced with L-methionine free ESF-921medium (Expression Systems, LLC) supplemented with 100 mg/L of selenomethionine (Acros Organics). The resulting protein was affinity purified as described above, except that OTFP elution buffer did not contain Triton X-100. This method produced a dramatically lower protein yield.
Crystallization and Structure Determination
Each sample was concentrated to roughly 10 mg/ml in 10 mM Tris, pH 7.4 and screened by the hanging drop method, using 1 μl of protein mixed with 1 μl of precipitation buffer. Peak 1 protein formed crystals in 100 mM pH 5.5 citrate with 20% (w/v) PEG 3000 but these crystals were not readily reproducible. Ultimately, more crystals were generated in 14% PEG 3000, 80 mM sodium citrate, 20 mM citric acid. These crystals were used to generate the structure described here. Peak 2 protein initially crystallized into poorly formed crystals in 100 mM pH 5.5 citrate with 2.0 M ammonium sulfate. Well formed crystals of peak 2 selenomethionine containing MsJHE were made in 100 mM pH 5.5 citrate with 0.7 M ammonium sulfate.
Data on a selenomethionine Peak 2 crystal were collected at the Stanford Synchrotron Radiation Laboratory, beamline 9–2. Unit cell dimensions were determined to be a = 98.7 Å, b= 98.7 Å, c = 165.7 Å, and α = β = γ = 90°, with a spacegroup of P41212. The crystal produced reflections at 2.4 Å spacing, however, Rsym increased dramatically at resolutions greater than 2.7 Å, and thus data were refined only to 2.7 Å. Indexing, integration and scaling of reflection data were carried out using Denzo and Scalepack (39). Despite having a significant absorbance peak at selenium edge wavelengths, neither SOLVE (40) nor CNS (41) was able to find a phasing solution with an acceptable figure of merit or interpretable electron density. A synthesis map was made using phases calculated from the partial Peak 1 model (see below) and applied to Peak 2 reflections. Contouring the 2Fo-Fc density to 5 sigma produced strong peaks, 3 of which centered on the sulfur atoms of methionines in conserved regions showing good electron density in the composite omit maps. Not all methionines in the model, and none of the cysteines in the model showed density at 5 sigma, suggesting that these peaks were not due to model bias. In addition, further refinement and model building did not significantly alter the positions of these atoms, further confirming their correct identification. However, even using these positions as seeds for MAD phasing, neither SOLVE nor CNS was able to find a phasing solution.
A crystal from peak 1 native protein diffracted to 2.7 Å resolution using 0.95 Å radiation at SSRL. Analysis of the diffraction pattern gave unit cell dimensions of a = 96.8 Å, b= 96.8 Å, c = 165.4 Å, and α = β = γ = 90° and a space group of P41212. The Matthew’s coefficient of 3.1 Å3/dalton indicated that there is one molecule per asymmetric unit.
Molecular replacement, using EPMR (42), with acetylcholinesterase from Torpedo californica (23% identity and 36% similarity to JHE, as calculated by the AlignX module of Vector NTI Suite 9 software from Invitrogen) as a search model (43), gave a solution that was used as an initial model. Using this model as a starting point, multiple rounds of model building by hand using the program O (44), with composite omit, 2Fo-Fc and solvent flattened maps as a guide to correct amino acid placement, and positional refinement, energy minimization and B-factor refinement using CNS (41) were carried out. The strong electron density peaks in the maps from the seleno-methionine data set were used to identify the positions of methionines, which was a useful guide in determining the register of the amino acid chains.
Final model building brought Rfree to around 30%, at which point the OTFP inhibitor was clearly visible in both Fo-Fc and 2Fo-Fc maps. Libraries for OTFP were generated using the PRODRG website (45), and inhibitor was modeled in. Nucleophilic attack of the catalytic serine on the carbonyl carbon of OTFP produces a tetrahedral covalent adduct. This tetrahedral adduct is chiral, with the chirality determined by the position of the inhibitor during the nucleophilic attack. Each enantiomer was modeled in separately, with comparable results in Rfree, and both fitting comparably within the density. Thus, it was impossible to distinguish between the enantiomers based on the crystal structure data. The enantiomer with the oxyanion pointing toward the putative α/β hydrolase oxyanion hole was chosen for final refinement, because this reflects the expected form based on the generally accepted mechanism of ester hydrolysis. However, it is possible that the other enantiomer, or some mixture of the two enantiomers were actually present in the crystals.
Geometry and side chain placement were adjusted by hand, using 2Fo-Fc density as a guide. Water molecules were added automatically, using CNS software, and then checked manually. The final model of the native Peak1 protein had an Rcryst of 20.4% and an Rfree of 25.2%. Ramachandran statistics were calculated using the program Procheck (46). Interactions between inhibitor and enzyme were calculated with the program Ligplot (47).
Modeling of Substrate Binding
Atomic coordinate, topology and parameter files for JH II (with the correct 2E, 6E,10R, 11S stereochemistry) were generated using the PRODRG website. JH II was initially placed in the active site tunnel by hand, using the program O, with OTFP as a guide to placement of JH II. The initial fit was improved and steric clashes removed by using energy minimization in the program CNS. For the energy minimization step, the OTFP-Ser-226 covalent adduct was replaced by a serine that had been aligned by hand with the Ser-226 portion of the adduct.
Site Directed Mutagenesis
The wild type MsJHE gene (Genbank accession number AF327882) in the baculovirus transfer vector pAcUW21 was subjected to site-directed mutagenesis in order to generate MsJHE with Phe259 replaced by isoleucine. Site-directed mutagenesis was performed using the primers: 5′-ggtaccagttcatctgctatctttaccacgaacccgg -3′ and 5′-ccgggttcgtggtaaagatagcagatgaactggtacc -3′ using a QuickChange Site-Directed Mutagenesis kit (Stratagene). The presence of the mutation was confirmed by DNA sequencing of the recombinant baculovirus transfer vector plasmid (pAcUW21-F259I). A recombinant baculovirus, AcMsJHE F259I, expressing the mutated MsJHE (MsJHE F259I) was generated by the transfection of pAcUW21-F259I with linearized BacPAK6 viral DNA (Clontech) following standard procedures (48). MsJHE F259I protein was expressed in High5 cells as described above for the wild type MsJHE. For the inhibitor studies, MsJHE F259I and the wild type MsJHE were purified by anion exchange chromatography using a Q-Sepharose column basically as described previously (method #2, (13)). The majority of the target protein eluted in the 350 mM NaCl fraction which was desalted and concentrated using a Centricon 30 filtration device (Millipore).
Inhibitors and substrates
OTFP was synthesized as previously described (31). TFDK was synthesized as previously described (49). NMR and MS for TFDK conformed to expected values. After purification TLC analysis yielded a single spot. Purity was also measured by HPLC analysis and was determined to be greater than 95% using UV detection at 214 nm. The HPLC analysis was performed using A: 0.2% formic acid in water and B: 0.2% formic acid in acetonitrile with a gradient that ran from 30% to 90% B over 30 min on a Waters Atlantis C18 column (2.1 × 100 mm). Mass spectral analysis was performed on a ThermoFinnigan LCQ Deca mass spectrometer.
Tritiated JH III was purchased from PerkinElmer Life Sciences, Inc. (Boston, MA), catalog number NET586, specific activity of 17.5 Ci/mmol, concentration of 0.1 mCi/ml in toluene:hexane (4:1). Unlabeled JH III was purchased from Sigma (St. Louis, MO), catalog number J2000.
IC50 Measurements
IC50 measurements of OTFP and TFDK against both wild-type and F259I MsJHE were done essentially as previously described (27, 28). Briefly, enzymes were diluted to concentrations such that they consumed 15% to 20% of JH III substrate in the time of the assay (1:8,000 dilution for wild-type enzyme and 1:10,000 dilution for F259I enzyme). Inhibitors were diluted in ethanol to 100 times final concentration (100×). The enzyme was aliquoted at 100 μl/tube into glass test-tubes, 3 tubes per concentration of inhibitor, on ice, and 1 μl of 100x inhibitor was added. Following a 30 minute pre-incubation on ice, a 1 μl solution of tritiated (5.15 × 10−9 mmoles) and unlabeled (5 × 10−7 mmoles) JH III was added for a final concentration of 5 μM JH III. Samples were mixed by vortexing and then put in a 30 °C shaking water bath for 15 minutes. The reaction was stopped by adding 100 μl per tube of stop solution (methanol, water, ammonium hydroxide at a 10:9:1 V/V ratio) and vortexing. Iso-octane (250 μl) was added per tube, and tubes were vortexed to extract unreacted JH III. Tubes were then spun for 5 minutes at 1,800 × g and 50 μl of the aqueous phase liquid (containing the reaction product JH III acid) was removed using a Hamilton syringe and added to a tube containing 1 ml of liquid scintillation liquid. The tube was then vortexed and radioactivity counted by a Wallac scintillation counter. Four concentrations, closely bracketing the IC50 value, were used for each IC50 determination. IC50’s were measured for both OTFP and TFDK at the same time, using the same stock of enzyme and the same no inhibitor control in order to reduce variability in the measurement of the ratio of IC50’s. Each IC50 experiment was done 3 independent times, and the values averaged to produce the results reported.
Results and Discussion
Overall structure
The final model of MsJHE contains 530 amino acids, 101 water molecules, and an OTFP molecule covalently bound to serine 226. The 4 N-terminal amino acids (23 through 27) are not visible in the electron density, nor are the 8 Cterminal amino acids (566 through 573). Additionally, there is 1 disordered loop in which residues 128 through 135 lacked electron density. MsJHE is a member of the α/β hydrolase family, and maintains the canonical fold of this family (5) (Figure 2A). The MsJHE structure extends the canonical β-sheet core from 8 to 11 strands by adding one strand (β3) to the side of the core nearest the N-terminus and two strands (β13 and β14) to the side nearest the C-terminus. In addition there is a β-sheet composed of 1 long (β2) and 2 short (β1, and β4) β strands that is located near the N-terminal side of the core sheet. The long strand is sandwiched between the two shorter strands on one end of the strand and adjoins the central β-sheet core at the other. The 14 β-strands of JHE overlay closely with those of AChE from Torpedo californica. The structure contains 16 helices. Most of these also overlay well with helices of AChE. The RMSD between 438 Cα’s of MsJHE and AChE (43) is 1.9 Å.
Figure 2.
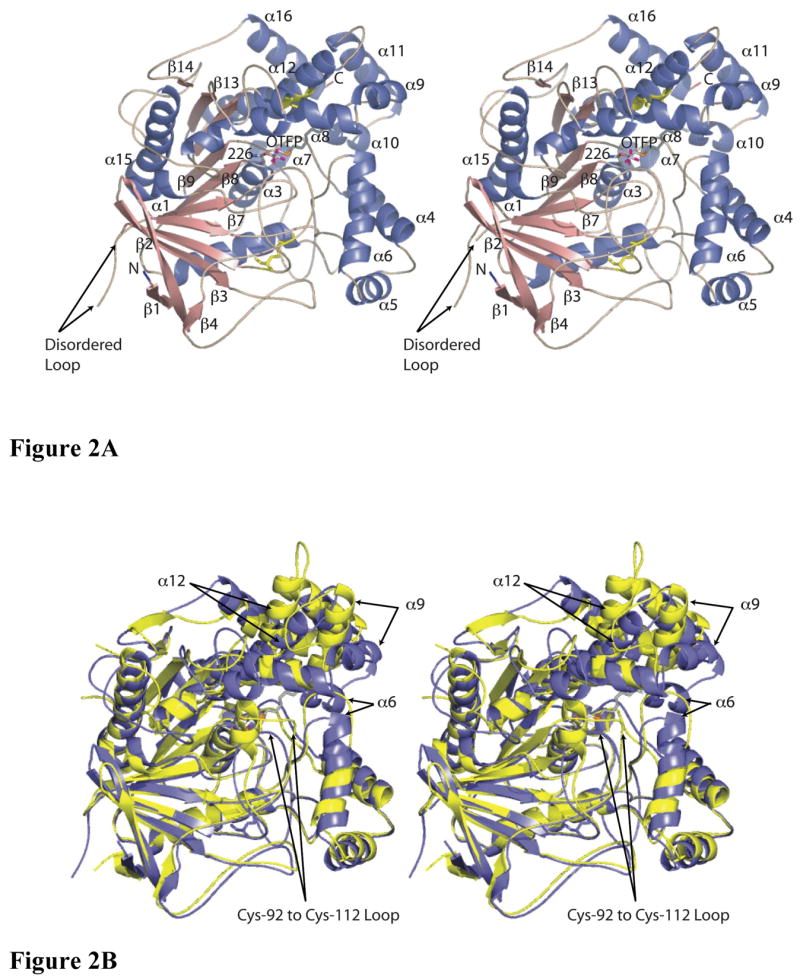
Crystal structures of MsJHE and AChE. A) Stereo view of a cartoon representation of the secondary structure of JHE with OTFP covalently bound. Helices are colored blue, sheets are salmon, and loops and turns are tan. The N-terminus is labeled and is colored blue. The C-terminus is labeled and is colored red. The disulfide bonds are in yellow and are shown with the atoms as stick figures. OTFP and Ser226 (to which OTFP is covalently attached) are shown as a stick representation, with carbons colored grey, oxygen red, nitrogen blue, sulfur orange, and fluorine magenta. Helices and strands that are referred to in the text are labeled. B) Overlay of MsJHE and AChE from Torpedo californica. MsJHE is shown in blue and AChE is shown in yellow. Secondary structural elements that contribute to the binding pocket of MsJHE and which differ significantly from the corresponding elements in AChE are indicated with arrows. The labeling of the arrows indicates the secondary structural elements as found in MsJHE. This figure and all other figures of JHE structure were produced using the program PyMol ((DeLano, W.L., The PyMOL Molecular Graphics System (2002) DeLano Scientific, San Carlos, CA, USA.).
The opening of the binding pocket is formed by helices 4, 6, 9 and 10, and by the loop between helices 9 and 10. The lining of the pocket is formed by loops and helices from a wide range of the primary sequence. These include helices 8 and 12, which are connected by a disulfide linkage between Cys-358 and Cys-420. A long loop between beta strands 3 and 4 reaches up to form one side of the pocket, making this the thinnest wall of the pocket, with solvent on one side of the loop and the tunnel on the other side. This loop also contains a disulfide linkage between cysteine residues 92 and 112, which are somewhat distant from the binding pocket. This disulfide is conserved in AChE, but in the case of AChE, the loop forms part of the opening to the active site, and the loop beyond the disulfide linkage is not well conserved between the two structures (Figure 2B). A loop between beta strand 6 and helix 1 contains Gly-146 and Gly-147, which form part of the oxyanion hole. The nucleophile “elbow” between beta strand 8 and helix 3 contains the catalytic Ser-226 and Ala-227, whose main-chain nitrogen forms the remaining part of the oxyanion hole. Other structural elements that make up the binding pocket include a loop between helices 6 and 7 and also a loop between beta strand 9 and helix 4 (including part of helix 4).
Active site
The binding pocket of JHE is unusual for an α/β hydrolase in that it is a long narrow tunnel that contains the enzyme active site at one end (Figure 3). This structure makes the catalytic residues inaccessible to surface solvent when the substrate is bound. This observation is in contrast to AChE and rabbit carboxylesterase, the two most closely related structures, based on primary sequence analysis, whose crystal structures have been solved (43, 50). In these two structures, the catalytic serine is solvent accessible, even with substrate bound. Many lipases have lids that cover the active site and make them inaccessible to water. However, in these cases, the lids open in the appropriate lipid environment to expose the active site both to substrate and solvent (4).
Figure 3.
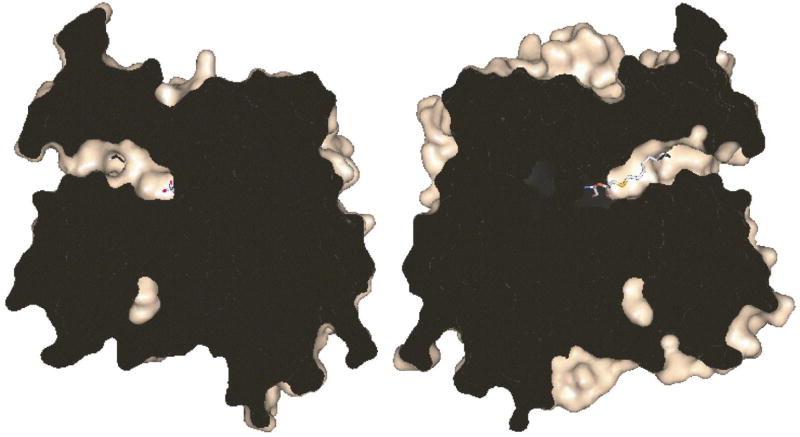
View of the long, narrow active site tunnel. A surface representation of JHE was split vertically through the active site tunnel by a plane perpendicular to the page. The halves produced from the split were rotated 90° in opposite directions along the vertical axis to produce the view shown. OTFP is shown as a stick representation and was split into two parts. The interior of the protein is shown in black. A pocket which holds an internal ordered water is seen near the bottom.
As seen in Figure 4, the surface of the JHE binding pocket is formed largely by hydrophobic residues. At the “floor” of the pocket, there are polar atoms from the oxyanion hole, the catalytic histidine and the catalytic serine. Aside from the catalytic residues, there are also some polar atoms closer to the opening. For example, the hydroxyl group from Thr-314 makes up a part of the surface of the binding pocket, as do the hydroxyls of Tyr-416 and Tyr-424, though those hydroxyls are hydrogen bonded to each other. As with AChE, a number of aromatic residues line the binding pocket. For example, the aromatic face of Phe-259 faces into the binding pocket, while the edge of the phenyl ring of Phe-425 points into the pocket.
Figure 4.

Surface representation of the active site tunnel. The surface is shown in a mesh representation and is color coded by the type of atoms that contribute to making the surface, with carbon atoms in gold, oxygen in red and nitrogen in blue. The bottom panel is a 180° rotation of the top panel about the y-axis.
Inhibitor binding
Covalently bound OTFP is clearly visible in a simulated annealing Fo-Fc omit map of this structure and is seen attached to the catalytic serine, residue 226 (Figure 5). The catalytic histidine 471 is 2.8 Å away from the gamma oxygen of Ser-226, while the catalytic acid, Glu-357 is only 2.5 Å away from His-471. The oxyanion of the covalently bound OTFP is well oriented toward the oxyanion hole, which consists of the backbone nitrogens of Gly-146, Gly-147 and Ala-227. This arrangement of atoms is entirely consistent with the proposed catalytic mechanism of JHE ester hydrolysis (4, 6). The arrangement of the catalytic triad is as expected, with the serine acting as a nucleophile to attack the carbonyl carbon, and the histidine in position to polarize the serine oxygen and catalyze the removal of the proton from the attacking nucleophile. The aspartic acid is found on the opposite side of the histidine from the serine, as expected for its function of orienting the histidine (51). Note that the catalytic histidine provides the hydrogen for the methanol leaving group of JH (5). Consistent with this, His-471 is only about 3.9 Å from the carbon atom of the trifluoromethyl group. This carbon atom is analogous to the alkoxide oxygen atom in the JH ester, which gives rise to the alcohol hydroxyl.
Figure 5.
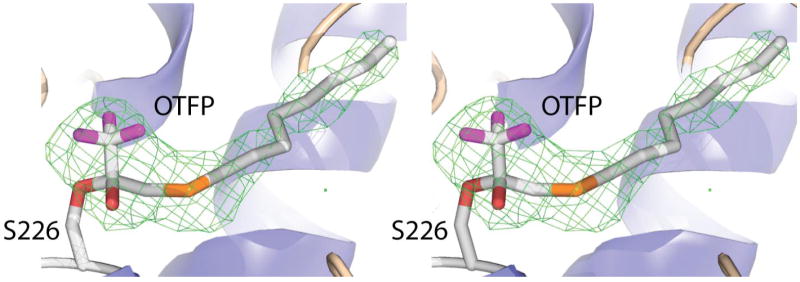
Stereo diagram of a simulated anneal Fo-Fc omit map of JHE contoured at 3σ The map was generated by removing the OTFP portion of the covalent adduct (and leaving the Ser-226 as shown in the structure). The map was drawn around the OTFP using a 2 Å cushion.
The remainder of the OTFP consists of carbon atoms and the one sulfur atom. Of these atoms, only the C3 carbon, as labeled in Figure 1B, and the C11 carbon of the alkyl chain of OTFP (Figure 1B) make contact with polar residues. The C3 carbon is 3.2 Å from His-471, and the C11 carbon is 3.1 Å from the hydroxyl of Tyr-416. The remaining carbon atoms make hydrophobic contacts with mostly hydrophobic residues lining the binding pocket of JHE, including Phe-259, Leu-313, Phe-361, Phe-365, and Ile-368. In addition, there are contacts with non-polar atoms of polar residues, such as with the side chain gamma carbon of Thr-314, and the aromatic ring of Tyr-424 (Figure 6). The sulfur atom of OTFP is roughly 3.7 Å away from the aromatic face of Phe-259 (Figure 7).
Figure 6.
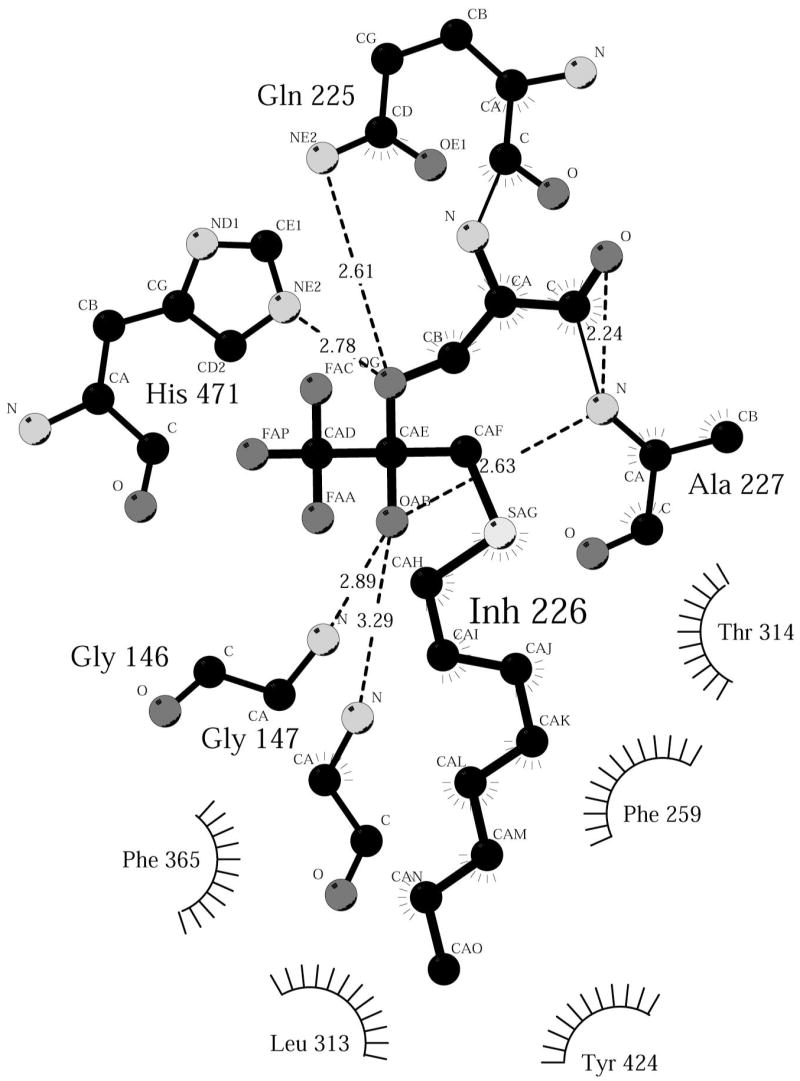
Ligplot diagram (47) showing binding interactions between OTFP and JHE. OTFP forms a covalent adduct with Ser-226 and this adduct is labeled Inh-226.
Figure 7.
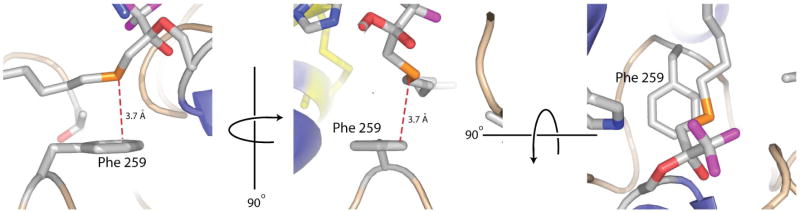
OTFP sulfur atom is positioned close to Phe259. Colors are the same as in Figure 2. The sulfur-phenylalanine interaction is shown in 3 orientations. In the leftmost two orientations, the same carbon (which is closest to the sulfur atom) is labeled by the dashed red line. In the rightmost orientation, this carbon is not visible because it is below the OTFP.
Substrate binding and selectivity in JHE family enzymes
A structure that includes a bound inhibitor facilitates predictions regarding the binding of the JH substrate to the enzyme. Figure 8 shows a model of JH II bound to the active site, prior to the formation of the acyl-enzyme intermediate. The substrate fits remarkably well into the binding pocket. Note that there is a constriction in the binding pocket that is between the substrate epoxide and ester moieties. This constriction is due to Leu-313 side chain, and allows for a tight fit between the substrate and the binding pocket. In particular, the 2E isomer appears to be favored by this geometry, as the 2Z form would likely lead to steric clashes between Leu-313 and the β and γ carbons of JH. Once the substrate was modeled, a multiple sequence alignment was performed to give some hint at determinants for specificity of the enzyme by examining conservation in the active site region. Figure 9 shows a sequence alignment of portions of JHE with 3 other lepidopteran JHEs (Bombyx mori, Heliothis virescens, and Choristoneura fumiferana), one dipteran JHE (Drosophila melanogaster) and one coleopteran JHE (Tenebrio molitor). This alignment contains all of the residues that make up the binding pocket. The catalytic triad and residues making up the oxyanion hole are absolutely conserved in these enzymes. Surprisingly, despite working on the same or very similar substrates, only one non-catalytic residue in the binding pocket is absolutely conserved in all six species, Thr-314. Interestingly, the substrate can be modeled such that the hydroxyl group of this residue is close to the epoxide of JH II. This suggests that a hydrogen bond between this residue and the epoxide may provide some substrate selectivity. Hydrogen bonds to an epoxide are seen in the crystal structure of human epoxide hydrolase, another α/β hydrolase enzyme (52).
Figure 8.
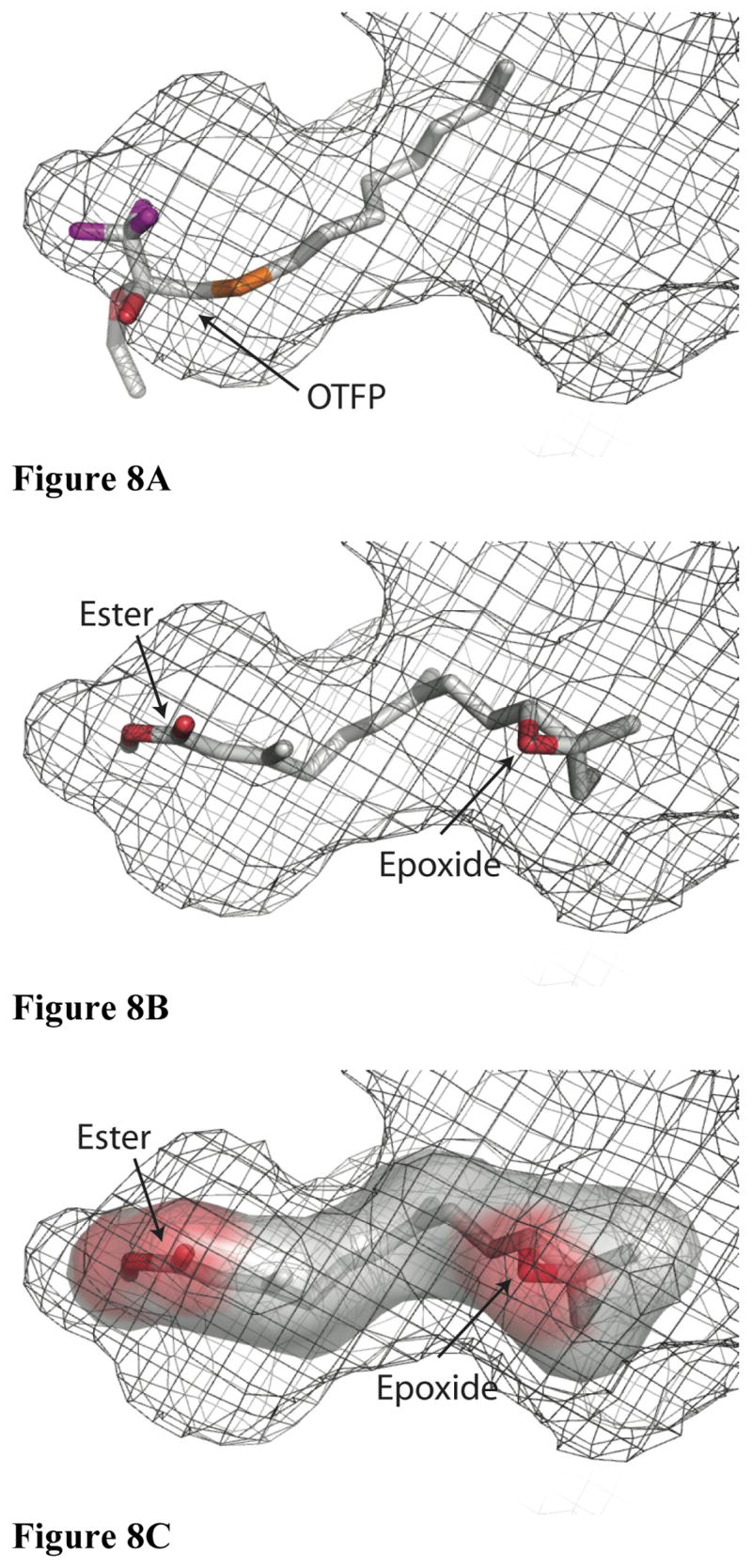
A model of JH binding to substrate. Orientation is identical to the top panel in Figure 4, and the surface is again shown as a mesh representation, though it is not color coded. A) OTFP is shown as a stick representation using the same color scheme as in Figure 2. B) JH II is shown as a stick representation using the same color scheme as in Figure 2 and with the same orientation as in Fig. 8A. C) A surface representation of JH II was produced in the absence of JHE and then shown inside the binding pocket.
Figure 9.
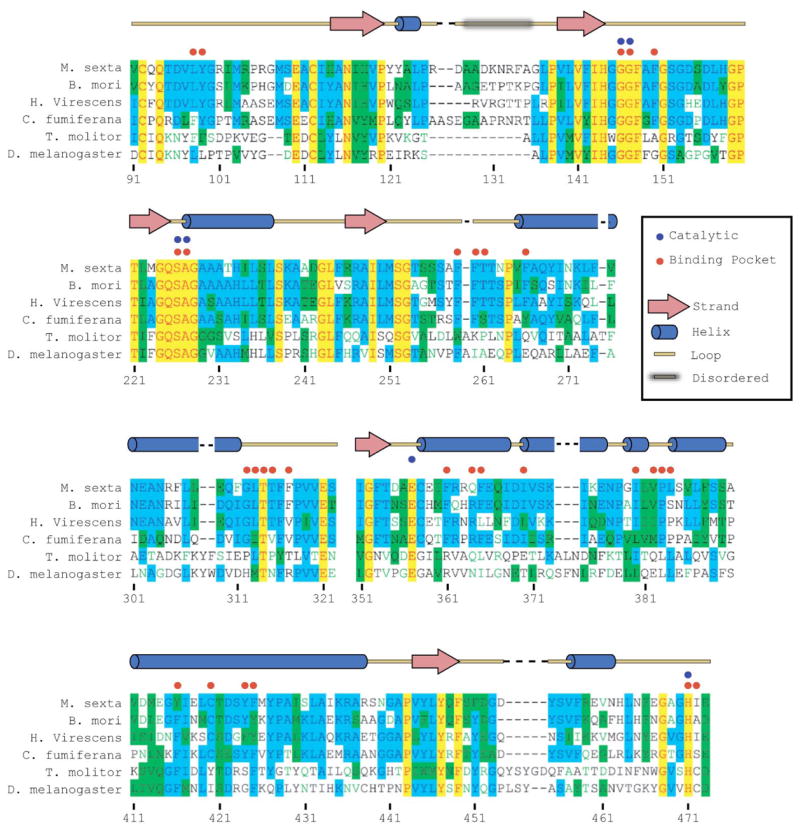
Partial sequence alignment of 6 JHE’s. Sequence alignments showing those portions containing all residues that line the active site pocket of four lepidopteran (top 4) a coleopteran and a dipteran JHE. Absolutely conserved residues are shown in red type with yellow background. Residues conserved in at least 3 of the 6 are shown in blue type with light blue background. Blocks of residues of similar kind are shown black type with green background. Residues showing no conservation are shown in black type with a white background. Number reflects the numbering of JHE. Catalytic residues are indicated with a blue dot, and residues that directly form the active site are indicated with a red dot. Secondary structure of MsJHE is indicated above the alignments.
A JH epoxide hydrolase is found in insects and produces JH diol from JH (53, 54). It is not clear whether this reaction happens before or after hydrolysis of the ester. It is possible, therefore, that the Thr-314 hydrogen bonds to the diol (most likely the alcohol off of the C10) rather than or in addition to the epoxide. The 6E geometry is important for the interaction between the Thr-314 and the epoxide (or diol). The 6Z isomer appears to fit into the binding cavity well, but it appears to prevent the epoxide (or diol) from moving within an easy hydrogen bond distance of the Thr-314 hydroxyl. The 11S stereochemistry is also worth noting. When JH is modeled so that the epoxide interacts with Thr-314, the 11S stereochemistry places the C11 ethyl group of JH II into a fairly large cavity, and away from any interactions with the walls of MsJHE binding tunnel. Thus, one would not expect there to be a large difference in the kcat/Km ratio between JH II, which has a C11 ethyl and JH III, which has a C11 methyl group, as substrates. Although JH II is not available in radiolabeled form, Abdel-Aal (28) found little difference in the ratio with JH I and JH III with MsJHE.
The lack of conservation across orders of residues other than Thr-314 may be a result of the hydrophobic nature of much of the enzyme-substrate interaction. It may be sufficient for the binding pocket to be largely hydrophobic and correctly shaped in order for the substrate to bind with high selectivity. For example, the Leu-313 that forms the constriction in the binding pocket of MsJHE is an isoleucine in C. fumiferana and a methionine in D. melanogaster. Both of those substitutions should produce a similar constriction in the binding pocket.
Although Thr-314 is the only absolutely conserved non-catalytic residue lining the binding pocket, among the lepidopteran JHEs there is substantial conservation of these residues. For example, of the 24 non-catalytic residues lining the binding pocket of MsJHE, 16 (67%) are identical to the corresponding residues in the H. virescens JHE (HvJHE). This strongly suggests that the binding pockets of at least the lepidopteran JHEs (and, considering the absolutely conserved Thr-314, most likely all JHEs) will be very similar. Previously, a homology model of HvJHE had been made using AChE and a lipase as a basis for the model (55). Now that we have a structure of MsJHE and know that HvJHE most likely has a similar binding pocket, we can evaluate the accuracy of the HvJHE homology model. The homology model failed to predict that JHE would have a binding tunnel, and instead inaccurately predicted that it would have a binding cleft. Furthermore, while predictions of the location of the catalytic residues and of over-all topology were correct, prediction of the substrate binding was incorrect. In the HvJHE model, the alpha carbon of JH is modeled in the position where the OTFP trifluoromethyl group is found in MsJHE, and the aliphatic chain extends through what is one wall of the binding tunnel in MsJHE. Consequently, the alcohol portion of JH is modeled in HvJHE where the OTFP aliphatic chain is found in the MsJHE structure. It turns out that the regions making up the binding pocket in MsJHE deviate significantly in their structure from those comparable regions of AChE (Figure 2B) and the lipase. This points to the difficulty of generating accurate homology models of α/β-hydrolases when the hydrolases act on different substrates and suggests that great care should be taken when interpreting such homology models.
The structure of MsJHE offers an explanation for the SAR previously seen for JHE substrates and inhibitors. The requirement for a small alcohol portion of the ester (56, 57) is readily explained by the lack of space in the “floor” of the binding pocket. Large alcohols simply lack the room to move deeply enough into the enzyme to allow the catalytic serine to attack the carbonyl carbon of the ester. The good correlation of lipophilicity with inhibitor potency and ability of the thio-ester substrates to be cleaved is also fairly evident from the structure. Smaller alkyl chains are more hydrophilic and thus do not partition as well into the hydrophobic binding pocket. Longer chains become too long for the binding pocket and thus do not fit as well. The preference for straight alkyl chains is likely due to steric constraints in the narrow binding pocket. For example, it is easy to see why a cyclohexyl or phenyl group attached to the OTFP sulfur would not fit as well into the pocket as would a straight hexyl group.
Enzyme-inhibitor sulfur-aromatic interactions
The potency of TFK inhibitors had previously been shown to positively correlate with the extent of ketone hydration (31). However, for some enzymes (especially JHE) this correlation did not appear to fully explain the increased potency. To clarify this discrepancy, it was hypothesized that the sulfur atom beta to the carbonyl (Figure 1B) could engage in pi-stacking interactions with an aromatic residue in the enzyme active site, thereby increasing the affinity of the inhibitor for the enzyme, though no JHE structure had as yet been determined (31). Examination of the JHE structure shows that Phe-259 is 3.7 Å from the OTFP sulfur atom (Figure 7). Pi-stacking interactions of sulfur atoms with aromatic residues occurs at distances of 5.0 Å or less (37, 58). The evidence from the crystal structure was therefore highly suggestive of an energetically favorable interaction between the sulfur of OTFP and the Phe-259 residue. In further support of this hypothesis, examination of the sequence alignment of multiple JHEs shows that aromatic residues are conserved in the Phe-259 position (Figure 9). The conserved residue is a tryptophan in T. molitor and a phenylalanine in all other species examined. Consistent with the role of sulfur in inhibitor binding is the observation that thio-ester JH surrogate substrates have an approximately 4-fold lower Km when sulfur is substituted for the carbon beta to the carbonyl (i.e., analogous to the sulfur position in OTFP) (56).
In order to directly test the hypothesis that the increased potency of sulfur-containing JHE inhibitors was due to sulfur-aromatic interactions, a JHE mutant was generated in which the Phe-259 was replaced by isoleucine. The inhibition potency was measured for both OTFP and an OTFP-analog in which the sulfur atom beta to the ketone had been replaced with a methylene group (TFDK, trifluoromethyl decyl ketone, Figure 1B) for both the wild-type and the F259I mutant. Results showed that OTFP has a 24-fold lower IC50 against the wild-type enzyme than TFDK (IC50’s = 22 ±3 nM and 530 ±32 nM, respectively; Table 2). On the other hand, OTFP has only a 5-fold lower IC50 with the F259I mutant versus with TFDK (IC50’s = 330 ±57 nM and 1,600 ±150 nM, respectively). This increase in the relative potency of OTFP for the wild-type over the mutant enzyme is consistent with the sulfur atom contributing to the binding affinity of the inhibitor. Interestingly, OTFP was still more potent than TFDK with the F259I mutant, indicating that the effects of the beta sulfur atom upon ketone hydration state are also important for inhibitor potency (31). These results suggest that the favorable effects of sulfur inclusion in JHE inhibitors are due to a combination of effects including hydration equilibrium, sulfur-aromatic interactions, as well as other unknown contributions.
Table 2.
IC50 values (nM) of OTFP and TFDK as inhibitors of either wild-type or F259I MsJHE. Values are averages of triplicate IC50 measurements. Standard deviations are shown in parentheses.
| Wild Type | IC50 (nM) | F259I | IC50 (nM) |
|---|---|---|---|
| TFDK | 530 (32) | TFDK | 1,600 (150) |
| OTFP | 22 (3) | OTFP | 330 (57) |
| TFDK:OTFP | 24 | 5 |
The strength of the sulfur-aromatic interaction depends greatly upon the geometry of the interaction. In this structure, the sulfur of OTFP is 3.7 Å from the ε-carbon of Phe-259 and a line connecting these 2 atoms is nearly perpendicular to the plane of the aromatic face of the phenylalanine (Figure 7). It is important to note that this structure shows the covalent attachment of OTFP to MsJHE, and so, except for any interaction that acts to favor the tetrahedral form of OTFP, the sulfur-aromatic interaction does not affect the attachment of OTFP to MsJHE. The sulfur-aromatic interactions are significant in the initial binding that places the ketone carbon near Ser-226 enabling the formation of the covalent interaction, and in maintaining the position of OTFP when the covalent bond reverses, thereby allowing re-formation of the bond. The exact placement of the sulfur atom in the non-covalently bond state will likely be different from the orientation in the covalently bound state since the covalent attachment brings the ketone carbon close to the Ser-226 γ-oxygen and changes the geometry of the ketone carbon (from planar to tetrahedral), thus changing the fit of the inhibitor in the active site. Changes in the placement of the TFK portion of the inhibitor are likely to lead to changes in placement of the sulfur atom, which is only 2 atoms away from the ketone carbon.
In addition to the uncertainties in the location of the sulfur atom in the non-covalent inhibitor-enzyme complex, there is some uncertainty as to the optimal geometry of the sulfur-aromatic interaction. For example, an interaction between sulfur and the face of the phenylalanine benzene ring has been reported to be favored in some studies (59) and unfavored in others (35, 37). Tauer et al. (59) modeled interactions of H2S with benzene and reported that the optimal intermonomer distance was 3.8 Å with an interaction energy of -2.81 kcal mol−1. While these results agree nicely with those observed in the JHE structure, they also report that the H2S-benzene attraction arises from a favorable electrostatic interaction between partially positive hydrogens in H2S with the negatively charged pi-cloud of the benzene. Because the divalent sulfur in these studies contains no hydrogens, it is unlikely that such strong electrostatic interactions are occurring. Other studies have reported that a polarized sulfur atom can interact with the positive hydrogens of the aromatic ring. However, given that sulfur has a low electronegativity (essentially the same as carbon, 2.58); it is difficult to imagine this scenario accounting for all of the observed favorable effects. It is possible that the sulfur lone pairs could interact favorably with the ring hydrogens (35). However this interaction would depend greatly upon the geometry of the bound inhibitor.
Despite uncertainty in the nature of the sulfur-aromatic interaction between OTFP and MsJHE, the combination of the proximity of the OTFP sulfur atom to Phe-259 and the IC50 data of the wild-type and F259I mutant with OTFP or TFDK demonstrate that there is a binding energy benefit to the inclusion of sulfur in inhibitor structure and that a large portion of the effects are due to interactions with the Phe-259. While it is not possible to directly determine the strength of this particular sulfur-aromatic interaction, previously reported studies generally agree on a range of 1–3 kcal mol−1 for sulfur-aromatic interactions in general, placing the interaction between pure van der Waals forces and that of a hydrogen bond (35). This is in general agreement with our data. To get a rough estimation of the free energy advantage of the sulfur-aromatic interaction, we can use the following analysis: Keq = [EI]/[E][I], where [EI] = concentration of enzyme inhibitor complex, [E] = concentration of enzyme unbound, and [I] = concentration of inhibitor unbound. Therefore, if we assume there is excess (constant) inhibitor concentration and we compare the IC50 values of OTFP against wild type and mutant enzymes, we find that Keqmut/KeqW.T. ≈ ([EImut]/[Imut])/([EIW.T.]/[IW.T.]) ≈ IC50mut/IC50W.T. = 330/22 = 15. Since Keq = e(ΔG/RT), we can solve for Δ ΔG and find that Δ ΔG = −RTln(15) = −1.6 kcal/mol. This equation gives us the over all stabilization of the OTFP in the wild type versus the mutant enzyme. However, the wild type IC50 is lower than the mutant even with TFDK as the inhibitor, suggesting a general destabilization due to the mutation. To account for that, we can perform the same calculation for mutant and wild type enzymes with TFDK, and we find that Δ ΔG = −RTln(1600/530) = −0.66 kcal/mol. Thus the overall stabilization (enhancement of potency of OTFP with phenylalanine instead of the mutant minus loss of potency due to the mutation) of the sulfur-aromatic interaction in our structure is roughly 1 kcal/mol. Further studies could attempt to more accurately determine the strength of this sulfur-aromatic interaction. We have demonstrated that these sulfur-aromatic interactions can be important in small molecule binding to protein targets. Therefore increased focus should be placed upon investigating these forces as sulfur-aromatic interactions are not restricted to amino acid residues within the protein.
Conclusions
The increased potency resulting from the sulfur moiety of OTFP appears to be due to a combination of effects including sulfur-aromatic interactions with aromatic residues in the active site as well as contributions to the hydration state of the inhibitor. The improved potency from the sulfur interactions of the OTFP sulfur with the phenylalanine has implications for the development not only of JHE inhibitors but also of pharmaceutical compounds that bind to proteins, especially other carboxylesterases. In cases such as treatment of central nervous system disorders or intracellular enzyme targets, it can be desirable to increase the compound potency without generating a charge or increasing polarity, since this can reduce the ability of a compound to cross the blood-brain barrier or a cell membrane. Provided there is a suitable aromatic residue at the site of compound binding, the addition of a sulfur atom capable of interacting with the aromatic residue may increase compound potency while keeping the compound sufficiently lipophilic to reach its intended destination.
The demonstration that there are sulfur-aromatic interactions between the OTFP sulfur and Phe-259 suggests that similar interactions occur between the α/β unsaturated bond of JH, which is in roughly the same position as the OTFP sulfur, and this conserved aromatic residue. These interactions could facilitate binding and alignment of the substrate in the active site. These interactions would also reduce the resonance stabilization of the α/β unsaturated bond, effectively lowering the activation energy required to cleave this relatively stable bond.
The crystal structure of MsJHE gives us insights into why it is such an efficient enzyme, with a very low Km. The efficiency is likely due to the tight fit between the enzyme binding pocket and the substrate, along with the hydrophobic nature of the interaction. The substrate is very hydrophobic, and will subsequently readily partition into the enzyme active site. Aside from the shape of the active site, at least two specific residues, Thr-314 and Phe-259, appear likely to interact with the JH epoxide and α,β unsaturated ester bonds, respectively. It seems likely that the slow kcat may be due to a combination of the solvent inaccessibility of the catalytic serine, hydrophobic interactions between the JH and the binding pocket, and the stable α,β unsaturated ester of JH. This would result in a slow cleavage of the acyl-enzyme intermediate and hence a lower kcat.
The examination of the role of the sulfur atom in the mechanism of inhibitor binding has therefore provided important information on the binding of the endogenous substrate to JHE. It is most likely that JHEs have evolved to include an aromatic residue that assists in the substrate binding, which could partly explain the extremely low Km value. This work therefore provides insight into the mechanism by which JHE effectively regulates the biological development of insects.
Table 1. Data collection and structure refinement statistics for OTFP-bound JHE.
Parentheses indicate values for the high resolution shell.
| Data Collection Statistics | |
|---|---|
| Wavelength (Å) | 0.953695 |
| Resolution (Å) (highest shell) | 100-2.7 (2.8-2.7) |
| Reflections (observed/unique) | 202,077/21,455 |
| Completeness (%) | 96.4(97.4) |
| I/σ(I) | 9.3 (2.3) |
| Rmerge | 0.081 (0.418) |
| Refinement Statistics | |
| Resolution (Å) | 30-2.7 |
| Rcryst (%) | 20.4 |
| Rfree (%) | 25.2 |
| r.m.s.d. bond lengths (Å) | 0.011 |
| r.m.s.d. bond angles (°) | 1.61 |
| Ramachandran plot | |
| % in most favorable regions | 83.0 |
| % in allowed regions | 17.0 |
Acknowledgments
The authors thank Dr. Walter Voegtli for his considerable help in solving the crystal structure of MsJHE, Dr. Randy Zauhaur from the University of the Sciences in Philadelphia for useful discussions and Dr. Lynn M. Riddiford for her generous gift of pictures of Manduca sexta.
Footnotes
This project was supported by the National Institutes of Health (GM66135), National Research Initiative of the USDA Cooperative State Research, Education and Extension Service (CSREES), grant # 2003-35302-13499, and National Institute of Environmental Health Sciences grant # R37 ES02710. C.E.W. was supported by a Japanese Society for the Promotion of Science (JSPS) post-doctoral fellowship and NIH post-doctoral training grant T32 DK07355-22. M. W. was supported under a NSF Graduate Research Fellowship and NIH training grant T32 GM070377. The data collection facilities at Stanford Synchrotron Radiation Laboratory are funded by the U.S. Department of Energy and the NIH.
PDB code: 2FJ0
Abbreviations used: AChE, acetyl cholinesterase; JH, juvenile hormone; JHE, juvenile hormone esterase; MsJHE, JHE from Manduca sexta; HvJHE, JHE from Heliothis virescens; OTFP, 3-octylthio-1,1,1-trifluoropropan-2-one; TFK, trifluoromethyl ketone; TFDK, trifluoromethyl decyl ketone.
References
- 1.Truman JW, Riddiford LM. Endocrine insights into the evolution of metamorphosis in insects. Annu Rev Entomol. 2002;47:467–500. doi: 10.1146/annurev.ento.47.091201.145230. [DOI] [PubMed] [Google Scholar]
- 2.Gilbert LI, Granger NA, Roe RM. The juvenile hormones: historical facts and speculations on future research directions. Insect Biochem Mol Biol. 2000;30:617–44. doi: 10.1016/s0965-1748(00)00034-5. [DOI] [PubMed] [Google Scholar]
- 3.Shapiro H, Micucci S. Pesticide use for West Nile virus. CMAJ. 2003;168:1427–30. [PMC free article] [PubMed] [Google Scholar]
- 4.Holmquist M. Alpha/Beta-hydrolase fold enzymes: structures, functions and mechanisms. Curr Protein Pept Sci. 2000;1:209–35. doi: 10.2174/1389203003381405. [DOI] [PubMed] [Google Scholar]
- 5.Ollis DL, Cheah E, Cygler M, Dijkstra B, Frolow F, Franken SM, Harel M, Remington SJ, Silman I, Schrag J, Sussman JL, Verschueren KHG, Goldman A. The Alpha/Beta-Hydrolase Fold. Protein Engineering. 1992;5:197–211. doi: 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
- 6.Heikinheimo P, Goldman A, Jeffries C, Ollis DL. Of barn owls and bankers: a lush variety of alpha/beta hydrolases. Structure Fold Des. 1999;7:R141–6. doi: 10.1016/s0969-2126(99)80079-3. [DOI] [PubMed] [Google Scholar]
- 7.Abdel-Aal YA, Hammock BD. Transition state analogs as ligands for affinity purification of juvenile hormone esterase. Science. 1986;233:1073–6. doi: 10.1126/science.3738525. [DOI] [PubMed] [Google Scholar]
- 8.Satoh T, Hosokawa M. The mammalian carboxylesterases: from molecules to functions. Annu Rev Pharmacol Toxicol. 1998;38:257–88. doi: 10.1146/annurev.pharmtox.38.1.257. [DOI] [PubMed] [Google Scholar]
- 9.Wheelock CE, Shan G, Ottea JA. Overview of carboxylesterases and their role in metabolism of insecticides. J Pestic Sci. 2005;30:75–83. [Google Scholar]
- 10.Bergot BJ, Jamieson GC, Ratcliff MA, Schooley DA. JH Zero - New Naturally-Occurring Insect Juvenile-Hormone from Developing Embryos of the Tobacco Hornworm. Science. 1980;210:336–338. doi: 10.1126/science.210.4467.336. [DOI] [PubMed] [Google Scholar]
- 11.Judy KJ, Schooley DA, Hall MS, Bergot BJ, Siddall JB. Chemical structure and absolute configuration of a juvenile hormone from grasshopper corpora allata in vitro. Life Sci. 1973;13:1511–6. doi: 10.1016/0024-3205(73)90139-2. [DOI] [PubMed] [Google Scholar]
- 12.Nijhout HF. Insect Hormones. Princeton University Press; Princeton: 1994. [Google Scholar]
- 13.Hinton AC, Hammock BD. In vitro expression and biochemical characterization of juvenile hormone esterase from Manduca sexta. Insect Biochem Mol Biol. 2003;33:317–29. doi: 10.1016/s0965-1748(02)00246-1. [DOI] [PubMed] [Google Scholar]
- 14.Sparks TC, Hammock BD. Comparative Inhibition of the Juvenile-Hormone Esterases from Trichoplusia ni, Tenebrio molitor, and Musca domestica. Pesticide Biochemistry and Physiology. 1980;14:290–302. [Google Scholar]
- 15.Hammock BD, Sparks TC, Mumby SM. Selective-Inhibition of JH Esterases from Cockroach Hemolymph. Pesticide Biochemistry and Physiology. 1977;7:517–530. [Google Scholar]
- 16.Linderman RJ, Upchurch L, Lonikar M, Venkatesh K, Roe RM. Inhibition of insect juvenile hormone esterase by α,β-unsaturated and α-acetylenic trifluoromethyl ketones. Pestic Biochem Physiol. 1989;35:291–299. [Google Scholar]
- 17.Wheelock CE, Nakagawa Y, Akamatsu M, Hammock BD. Use of classical and 3-D QSAR to examine the hydration state of juvenile hormone esterase inhibitors. Bioorg Med Chem. 2003;11:5101–16. doi: 10.1016/j.bmc.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 18.Szekacs A, Bordas B, Hammock BD. Transition State Analog Enzyme Inhibitors: Structure-Activity Relationships of Trifluoromethyl Ketones. In: Draber W, Fujita T, editors. Rational Apporaches to Structure, Activity, and Ecotoxicology of Agrochemicals. CRC Press; Boca Raton: 1992. [Google Scholar]
- 19.Brodbeck U, Schweikert K, Gentinetta R, Rottenberg M. Fluorinated aldehydes and ketones acting as quasi-substrate inhibitors of acetylcholinesterase. Biochim Biophys Acta. 1979;567:357–69. doi: 10.1016/0005-2744(79)90122-0. [DOI] [PubMed] [Google Scholar]
- 20.Abdel-Aal YAI, Roe RM, Hammock BD. Kinetic-Properties of the Inhibition of Juvenile-Hormone Esterase by 2 Trifluoromethylketones and O-Ethyl, S-Phenyl Phosphoramidothioate. Pesticide Biochemistry and Physiology. 1984;21:232–241. [Google Scholar]
- 21.Hammock BD, Abdel-Aal YAI, Mullin CA, Hanzlik TN, Roe RM. Substituted Thiotrifluoropropanones as Potent Selective Inhibitors of Juvenile-Hormone Esterase. Pesticide Biochemistry and Physiology. 1984;22:209–223. [Google Scholar]
- 22.Hammock BD, Wing KD, Mclaughlin J, Lovell VM, Sparks TC. Trifluoromethylketones as Possible Transition-State Analog Inhibitors of Juvenile-Hormone Esterase. Pesticide Biochemistry and Physiology. 1982;17:76–88. [Google Scholar]
- 23.Ashour MB, Hammock BD. Substituted trifluoroketones as potent, selective inhibitors of mammalian carboxylesterases. Biochem Pharmacol. 1987;36:1869–79. doi: 10.1016/0006-2952(87)90483-7. [DOI] [PubMed] [Google Scholar]
- 24.Duran I, Parrilla A, Feixas J, Guerrero A. Inhibition of Antennal Esterases of the Egyptian Armyworm Spodoptera littoralis by Trifluoromethyl Ketones. Bioorganic & Medicinal Chemistry Letters. 1993;3:2593–2598. [Google Scholar]
- 25.Parrilla A, Guerrero A. Trifluoromethyl Ketones as Inhibitors of the Processionary Moth Sex-Pheromone. Chemical Senses. 1994;19:1–10. doi: 10.1093/chemse/19.1.1. [DOI] [PubMed] [Google Scholar]
- 26.Pophof B. Inhibitors of sensillar esterase block reversibly the responses of moth pheromone receptor cells. Olfaction and Taste Xii. 1998;855:316–319. doi: 10.1111/j.1749-6632.1998.tb10589.x. [DOI] [PubMed] [Google Scholar]
- 27.Abdel-Aal YAI, Hammock BD. 3-Octylthio-1,1,1-Trifluoro-2-Propanone, a High-Affinity and Slow Binding Inhibitor of Juvenile-Hormone Esterase from Trichoplusia ni (Hubner) Insect Biochemistry. 1985;15:111–122. [Google Scholar]
- 28.Abdel-Aal YAI, Hammock BD. Apparent Multiple Catalytic Sites Involved in the Ester Hydrolysis of Juvenile Hormones by the Hemolymph and by an Affinity-Purified Esterase from Manduca sexta Johansson (Lepidoptera, Sphingidae) Archives of Biochemistry and Biophysics. 1985;243:206–219. doi: 10.1016/0003-9861(85)90789-1. [DOI] [PubMed] [Google Scholar]
- 29.Prestwich GD, Eng WS, Roe RM, Hammock BD. Synthesis and Bioassay of Isoprenoid 3-Alkylthio-1,1,1-Trifluoro-2-Propanones - Potent, Selective Inhibitors of Juvenile-Hormone Esterase. Archives of Biochemistry and Biophysics. 1984;228:639–645. doi: 10.1016/0003-9861(84)90033-x. [DOI] [PubMed] [Google Scholar]
- 30.Rosell G, Herrero S, Guerrero A. New trifluoromethyl ketones as potent inhibitors of esterases: F-19 NMR spectroscopy of transition state analog complexes and structure-activity relationships. Biochemical and Biophysical Research Communications. 1996;226:287–292. doi: 10.1006/bbrc.1996.1347. [DOI] [PubMed] [Google Scholar]
- 31.Wheelock CE, Colvin ME, Uemura I, Olmstead MM, Sanborn JR, Nakagawa Y, Jones AD, Hammock BD. Use of ab initio calculations to predict the biological potency of carboxylesterase inhibitors. J Med Chem. 2002;45:5576–93. doi: 10.1021/jm020072w. [DOI] [PubMed] [Google Scholar]
- 32.Reid KSC, Lindley PF, Thornton JM. Sulphur-aromatic interactiions in proteins. FEBS Lett. 1985;190:209–213. [Google Scholar]
- 33.Tatko CD, Waters ML. Investigation of the nature of the methionine-pi interaction in beta-hairpin peptide model systems. Protein Sci. 2004;13:2515–22. doi: 10.1110/ps.04820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Viguera AR, Serrano L. Side-chain interactions between sulfur-containing amino acids and phenylalanine in alpha-helices. Biochemistry. 1995;34:8771–9. doi: 10.1021/bi00027a028. [DOI] [PubMed] [Google Scholar]
- 35.Zauhar RJ, Colbert CL, Morgan RS, Welsh WJ. Evidence for a strong sulfur-aromatic interaction derived from crystallographic data. Biopolymers. 2000;53:233–48. doi: 10.1002/(SICI)1097-0282(200003)53:3<233::AID-BIP3>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 36.Duan G, Smith VH, Weaver DF. Characterization of aromatic-thiol p-type hydrogen bonding and phenylalanine-cysteine side chain interactions through ab initio calculations and protein database analyses. Mol Phys. 2001;99:1689–1699. [Google Scholar]
- 37.Pranata J. Sulfur-aromatic interactions: A computational study of the dimethyl sulfide-benzene complex. Bioorg Chem. 1997;25:213–219. [Google Scholar]
- 38.Kamita SG, Hinton AC, Wheelock CE, Wogulis MD, Wilson DK, Wolf NM, Stok JE, Hock B, Hammock BD. Juvenile hormone (JH) esterase: why are you so JH specific? Insect Biochem Mol Biol. 2003;33:1261–73. doi: 10.1016/j.ibmb.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 39.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Vol. 276. Academic Press; New York: 1997. [DOI] [PubMed] [Google Scholar]
- 40.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt 4):849–61. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54(Pt 5):905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 42.Kissinger CR, Gehlhaar DK, Fogel DB. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt 2):484–91. doi: 10.1107/s0907444998012517. [DOI] [PubMed] [Google Scholar]
- 43.Raves ML, Harel M, Pang YP, Silman I, Kozikowski AP, Sussman JL. Structure of acetylcholinesterase complexed with the nootropic alkaloid, (−)-huperzine A. Nat Struct Biol. 1997;4:57–63. doi: 10.1038/nsb0197-57. [DOI] [PubMed] [Google Scholar]
- 44.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallographica Section A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 45.van Aalten DM, Bywater R, Findlay JB, Hendlich M, Hooft RW, Vriend G. PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules. J Comput Aided Mol Des. 1996;10:255–62. doi: 10.1007/BF00355047. [DOI] [PubMed] [Google Scholar]
- 46.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 47.Wallace AC, Laskowski RA, Thornton JM. Ligplot - a Program to Generate Schematic Diagrams of Protein Ligand Interactions. Protein Engineering. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 48.Merrington CL, King LA, Possee RD. Baculovirus Expression Systems. In: Higgins SJ, Hames BD, editors. Protein Expression a Practical Approach. Oxford University Press; Oxford: 1999. [Google Scholar]
- 49.Parrilla A, Villuendas I, Guerrero A. Synthesis of trifluoromethyl ketones as inhibitors of antennal esterases of insects. Bioorg Med Chem. 1994;2:243–52. doi: 10.1016/s0968-0896(00)82167-7. [DOI] [PubMed] [Google Scholar]
- 50.Bencharit S, Morton CL, Howard-Williams EL, Danks MK, Potter PM, Redinbo MR. Structural insights into CPT-11 activation by mammalian carboxylesterases. Nat Struct Biol. 2002;9:337–42. doi: 10.1038/nsb790. [DOI] [PubMed] [Google Scholar]
- 51.Dodson G, Wlodawer A. Catalytic triads and their relatives. Trends Biochem Sci. 1998;23:347–52. doi: 10.1016/s0968-0004(98)01254-7. [DOI] [PubMed] [Google Scholar]
- 52.Gomez GA, Morisseau C, Hammock BD, Christianson DW. Structure of human epoxide hydrolase reveals mechanistic inferences on bifunctional catalysis in epoxide and phosphate ester hydrolysis. Biochemistry. 2004;43:4716–23. doi: 10.1021/bi036189j. [DOI] [PubMed] [Google Scholar]
- 53.Mullin CA, Wilkinson CF. Insect Epoxide Hydrolase - Properties of a Purified Enzyme from the Southern Armyworm (Spodoptera eridania) Pesticide Biochemistry and Physiology. 1980;14:192–207. [Google Scholar]
- 54.Mullin CA, Wilkinson CF. Purification of an Epoxide Hydrolase from the Midgut of the Southern Army Worm (Spodoptera eridania) Insect Biochemistry. 1980;10:681–691. [Google Scholar]
- 55.Thomas BA, Church WB, Lane TR, Hammock BD. Homology model of juvenile hormone esterase from the crop pest, Heliothis virescens. Proteins. 1999;34:184–96. [PubMed] [Google Scholar]
- 56.McCutchen BF, Uematsu T, Szekacs A, Huang TL, Shiotsuki T, Lucas A, Hammock BD. Development of surrogate substrates for juvenile hormone esterase. Arch Biochem Biophys. 1993;307:231–41. doi: 10.1006/abbi.1993.1584. [DOI] [PubMed] [Google Scholar]
- 57.Grieneisen ML, Mok A, Kieckbusch TD, Schooley DA. The specificity of juvenile hormone esterase revisited. Insect Biochemistry and Molecular Biology. 1997;27:365–376. [Google Scholar]
- 58.Morgan RS, McAdon JM. Predictor for sulfur-aromatic interactions in globular proteins. Int J Peptide Protein Res. 1980;15:177–180. doi: 10.1111/j.1399-3011.1980.tb02566.x. [DOI] [PubMed] [Google Scholar]
- 59.Tauer TP, Derrick ME, Sherrill CD. Estimates of the ab initio limit for sulfur-π interactions: The H2S-benzene dimer. J Phys Chem A. 2005;109:191–196. doi: 10.1021/jp046778e. [DOI] [PubMed] [Google Scholar]