

Abstract
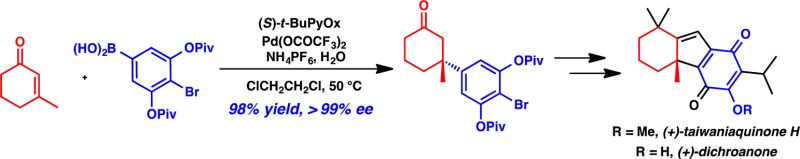
A catalytic, enantioselective formal synthesis of (+)-dichroanone and (+)-taiwaniaquinone H is reported. The all-carbon quaternary stereocenter was constructed by asymmetric conjugate addition catalyzed by a palladium(II) (S)-tert-butylpyridinooxazoline complex. The unexpected formation of a [3.2.1] bicyclic intermediate required the identification of a new route. Analysis of the Hammett constants for para-substituted arenes enabled the rational design of a highly enantioselective conjugate addition substrate that led to the completion of the formal synthesis.
First isolated in 1995, the taiwaniaquinoid natural products are a family of tricyclic diterpenoids with a unique [6,5,6]-abeo-abietane skeleton (Figure 1, 1–7).1 Since their isolation, the taiwaniaquinoids have attracted significant attention from the synthetic community, resulting in a multitude of total and formal syntheses.2 Interest in these compounds stems from their purported biological activity,3 in addition to their unique architecture containing a benzylic quaternary stereocenter. Due to the limited number of methodologies capable of installing benzylic quaternary stereocenters, only four catalytic, enantioselective syntheses of taiwaniaquinoids have been published to date.2e,2m,2q,2t
Figure 1.

Taiwaniaquinoid natural products.
Our laboratory has a long-standing interest in the asymmetric synthesis of quaternary stereocenters. Previously, our group reported the first catalytic, enantioselective total synthesis of (+)-dichroanone (2) via asymmetric palladium-catalyzed allylic alkylation (Figure 2).2e This work featured a linear synthetic sequence, elaborating allylic alkylation product 9 to bicyclic enone 8 by Wacker oxidation and subsequent aldol condensation. The final ring was appended by another aldol condensation, and a novel series of oxidations provided the natural product in only 11 steps.
Figure 2.
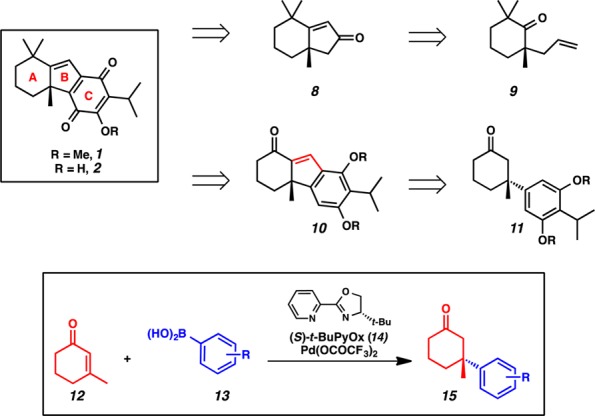
Comparative retrosynthetic analysis of (+)-taiwaniaquinone H (1) and (+)-dichroanone (2).
Recently, we developed a new catalyst for the enantioselective conjugate addition of arylboronic acids (Figure 2, 13) to cyclic enones to form β-quaternary ketones (15) in high yields and enantioselectivities.4 We observed that these β-aryl ketone products closely resembled the core of the taiwaniaquinoids, and we envisioned that a more convergent synthesis of these natural products could be achieved by employing β-aryl ketone 11 as the key intermediate. This modified retrosynthetic analysis facilitates a highly convergent, catalytic, enantioselective key step that joins together 13 of the 19 core carbons of the taiwaniaquinoid tricyclic core, including the quaternary stereocenter, in a single chemical transformation. Herein, we present the rational design of high-yielding and highly enantioselective conjugate addition reactions of arylboronic acids to 3-methyl-2-cyclohexenone that facilitated the catalytic, asymmetric formal synthesis of (+)-dichroanone and (+)-taiwaniaquinone H in greater than 99% enantiomeric excess.
Our preliminary strategic disconnections of (+)-dichroanone (2) and (+)-taiwaniaquinone H (1) involved late stage introduction of the gem-dimethyl functionality and oxidation of the C-ring of enone 10 to the quinone (Figure 2).5,2f The B-ring of tricycle 10 was envisioned to be established through ortho-formylation of the phenol 11 and subsequent aldol condensation. We anticipated that the all-carbon quaternary center could be constructed by palladium-catalyzed asymmetric conjugate addition of 3-methyl-2-cyclohexenone (12) with an appropriate arylboronic acid 13.4
We first selected para-acetylphenylboronic acid 19 as our conjugate addition substrate. In our previous work we noted that electron-withdrawing substituents at the para position of the arylboronic acid often afforded highly enantioenriched β-quaternary ketone products (15).4 A plot of the enantiomeric ratio versus the Hammett value (σp) for a variety of para-substituted phenylboronic acids (15) demonstrates a strong positive linear correlation, R2 = 0.92 (Figure 3).6 The positive value of ρ (0.81) suggests that the difference in energy between the diastereomeric transition states leading to the enantiomeric (S) and (R) products increases as the boronic acid becomes increasingly electron deficient. Thus, the best selectivity in the conjugate addition reaction is achieved with electron-withdrawing substituents in the para-position. Therefore, we chose to mask the isopropyl group as a methyl ketone to achieve a selective conjugate addition reaction.
Figure 3.
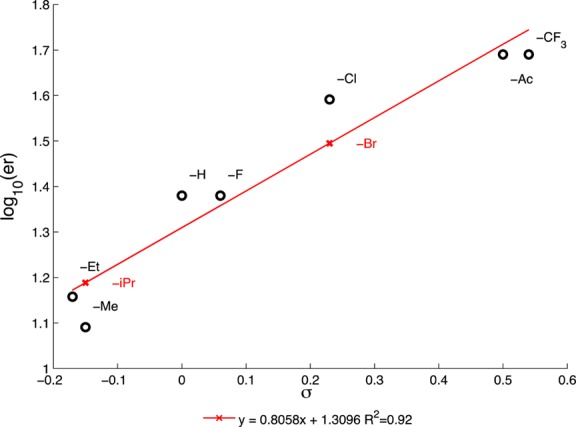
Hammett plot of log10(er) vs σp for select boronic acids in the palladium-catalyzed conjugate addition reaction.
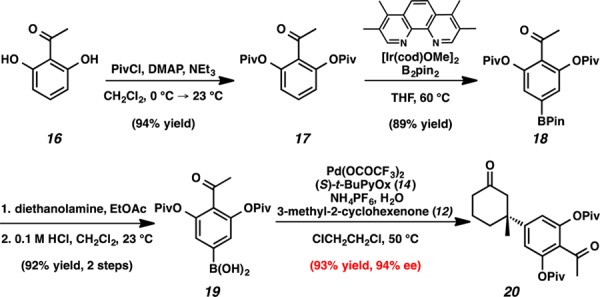
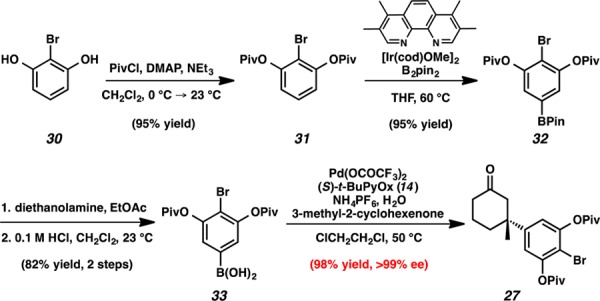
Access to para-acetylphenylboronic acid 19 (Scheme 1) began with acylation of 2,6-dihydroxyacetophenone (16) with pivaloyl chloride to provide arene 17. Installation of the boryl substituent was accomplished through an Ir-catalyzed C–H borylation.7 This reaction provided pinacol boronate ester 18 as the exclusive product in 89% yield. Empirically, we identified that protecting the meta-hydroxyl substituents with pivalate groups allowed for the highest yields in this borylation chemistry. After exhaustive exploration of deprotection conditions, we found that treatment with diethanolamine and subsequent acid-catalyzed hydrolysis of the transesterified intermediate afforded arylboronic acid 19 in 92% yield.8 Boronic acid 19 was treated with 3-methyl-2-cyclohexenone in the presence of Pd(OCOCF3)2, (S)-t-BuPyOx, and NH4PF6 at 50 °C to afford enantioenriched ketone 20, in 93% yield and 94% ee.4b
Scheme 1. Synthesis of Acetyl Conjugate Addition Product 20.
With ketone 20 in hand, we turned our attention to the installation of the final carbon of the tricyclic core and the completion of the B-ring. Regrettably, attempts to formylate arene 20 were unsuccessful. We rationalized that the sterically congested environment surrounding the arene C–H bonds prohibited installation of a functional group handle. Moreover, deprotection of the hydroxyl groups of ketone 20 was not facile and required treatment with LiSEt to cleanly remove both pivaloyl groups, affording what we predicted to be resorcinol 21 (Scheme 2). Though HRMS data matched the molecular formula of desired resorcinol 21, the 1H NMR spectra did not match that of a typical β-quaternary ketone conjugate addition product. We suspected that an unproductive cyclization may have occurred and sought to crystallize derivatives of ketone 23 to confirm the new structure by single crystal X-ray diffraction. Bromination and methylation of compound 23 provided bromoarene 24 as a white crystalline solid. As we suspected, a cyclization had occurred and the structure proved to be [3.2.1]bicycle 24. Thus, we were able to properly assign the structures of cyclization product 23 and arene bromination adduct 24. This X-ray structure also confirms the absolute stereochemistry imparted in asymmetric conjugate addition reactions of arylboronic acids to cyclic conjugate acceptors facilitated by the catalyst derived from the combination of (S)-t-BuPyOx and Pd(OCOCF3)2.
Scheme 2. Unexpected Cyclization of Phenolic Intermediate.
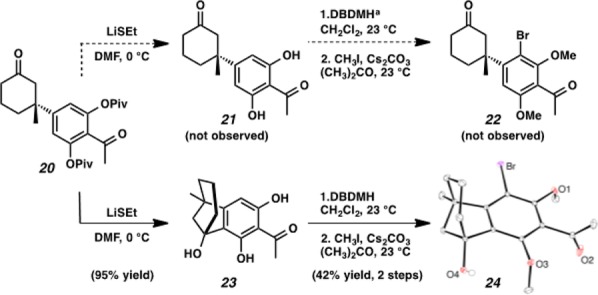
DBDMH = 1,3-dibromo-5,5-dimethylhydantoin.
Cyclizations of β-aryl ketones to form [3.2.1] bicycles are rare; the few other reports of this transformation require treatment with strong Lewis9 or Brønsted acids10 at elevated temperatures. These reactions presumably operate via an electrophilic aromatic substitution mechanism. While it is possible that our noted cyclization proceeds through a similar mechanism, we did not observe cyclization with substrates bearing protected phenols, which suggests that the hydroxyl group or phenoxide may be involved in the cyclization mechanism. This observation led us to propose that this unexpected cyclization may instead occur through a carbonyl ene or lithium phenoxide aldol reaction pathway.
As we were unable to functionalize ketone 20 without causing the undesired cyclization, we decided to redesign the arylboronic acid substrate and began examining alternative conjugate addition reactions. Removing the acetyl group would obviate the need to differentiate the benzylic carbonyl from the cyclic ketone while installing the requisite isopropyl group in the first-generation conjugate addition product 20. We envisioned that a para-halogenated arylboronic acid derivative would allow for facile installation of the isopropyl group via cross-coupling chemistry. Additionally, based on our Hammett plot analysis, we posited that the para-halide would serve as a necessary para-electron-withdrawing group to impart high enantioselectivity in the Pd/PyOx conjugate addition chemistry (Figure 3).4 Gratifyingly, we found that use of these boronic acids furnished products bearing para-iodo (26), para-bromo (27), and para-chloro (28) arenes in high ee and moderate to high yields (Scheme 3).
Scheme 3. Identification of a Suitable Conjugate Addition System.
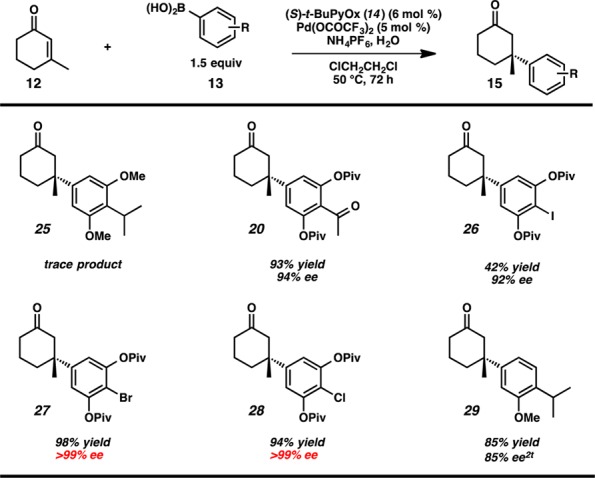
We consequently pursued a revised approach to the natural products via bromoarene 27, selected for both its superior reactivity in palladium-catalyzed conjugate addition chemistry and facile cross-coupling ability. To access this intermediate, 2-bromoresorcinol 30 was converted to para-bromophenyl boronic acid derivative 33 in 73% yield over four steps (Scheme 4). Subsequent catalytic, asymmetric conjugate addition furnished ketone 27 in 98% yield and >99% ee.
Scheme 4. Synthesis of Ketone 27.
Having set the absolute stereochemistry of the quaternary center, we next sought to install the isopropyl group. Attempts to directly cross-couple an isopropyl zinc reagent with bromide 27 gave an inseparable mixture of iso- and n-propyl products.11 The steric hindrance of the nearby pivaloyl groups frustrated our attempts to cross-couple 27 with isopropenyl organometallic reagents, but we ultimately achieved success using Molander’s protocol for Suzuki–Miyaura couplings of potassium isopropenyltrifluoroboronate salts.12 Our optimized conditions (170 °C, 1 h, microwave) gave a crude mixture of both cross-coupled product and monodeprotected cross-coupled product that could be stirred with tetrabutylammonium hydroxide to furnish isopropenyl phenol 34 in 70% yield (Scheme 5).
Scheme 5. Isopropenyl Cross-Coupling.

With a route to monodeprotected arene 34 established, we turned our efforts once more toward the formation of the B-ring. However, despite the successful removal of one pivaloyl group, the system proved resistant to a number of metal and nonmetal mediated ortho-formylations, directed ortho-metalations, and halogenations, regardless of protection of the ketone. We speculate that the failure of these efforts may again be attributed to the formidable steric environment of the arene C–H bonds.
Recognizing that significant steric hindrance would prevent the formation of the B-ring, we sought to diminish the steric environment of the arene by reductive removal of the free phenol. Activation of phenol 34 by exposure to excess perfluorobutanesulfonyl fluoride led to the generation of nonaflate 35 in 80% yield (Scheme 6). Subsequent hydrogenation with Pd/C simultaneously cleaved the nonaflate and reduced the isopropenyl functionality to afford isopropyl arene 36 in 72% yield.13 Finally, the pivaloyl group was replaced with a methyl group by one-pot deprotection and methylation with LiSEt and Me2SO4 to provide ketone 29 in 83% yield. With the less substituted arene 29 in hand, we believed that the necessary tricycle could now be readily formed by ortho-formylation.
Scheme 6. Synthesis of Key Intermediate 29.
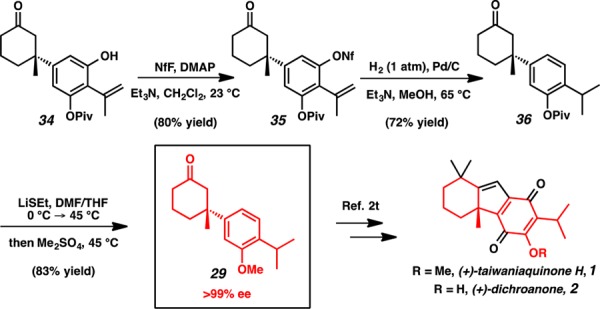
NfF = perfluorobutanesulfonyl fluoride.
Concurrent with our synthetic efforts, Qin and co-workers reported a formal total synthesis of (+)-dichroanone and (+)-taiwaniaquinone H via asymmetric conjugate addition of (4-isopropyl-3-methyloxyphenyl)boronic acid to 3-methyl-2-cyclohexenone (12) in 85% yield and 85% ee (Scheme 3, 29) using the catalyst developed in our laboratory.2t,4a However, Qin was unable to optimize the conjugate addition substrates to achieve yields or enantiomeric excesses over 85%. This result aligns well with our Hammett plot analysis; the determined linear relationship predicts an ee of 88% for the electron-donating isopropyl group (Figure 3). Moreover, this study demonstrated that our common intermediate 29 could be further transformed into the taiwaniaquinoid tricyclic skeleton by ortho-formylation followed by aldol condensation. Therefore, our synthesis of ketone 29 in >99% ee constitutes a formal synthesis of (+)-dichroanone (2) and (+)-taiwaniaquinone H (1) in the highest reported ee to date.
In summary, we have presented the formal catalytic, asymmetric total syntheses of (+)-dichroanone and (+)-taiwaniaquinone H in 35% overall yield, starting from commercially available 2-bromoresorcinol (30). Investigation of electronic effects of arylboronic acid substituents on enantioselectivity enabled the rational design of a highly enantioselective reaction that furnished established intermediate 29 in exceptionally high yield and ee. Additionally, an unexpected cyclization to form a [3.2.1] bicycle permitted the unambiguous determination of the absolute stereochemistry of the quaternary stereocenter installed by the Pd/PyOx-catalyzed conjugate addition reaction by X-ray diffraction analysis. Currently, efforts to apply para-halogen conjugate addition substrates to the synthesis of a range of [6,6,6] and [6,7,6] tricyclic scaffolds are underway in our laboratory.
Acknowledgments
The authors thank NIH-NIGMS (B.M.S., R01 GM080269), Caltech, and the American Chemical Society Division of Organic Chemistry (predoctoral fellowship J.C.H) for financial support. Dr. Scott Virgil is thanked for helpful discussions and assistance with chiral chromatography. Dr. David VanderVelde of the Caltech NMR facility is thanked for invaluable assistance with NMR experiments and helpful discussions. Lawrence Henling and Dr. Michael K. Takase (Caltech) are gratefully acknowledged for X-ray crystallographic structural determination. Dr. Mona Shahgholi is thanked for mass spectroscopy determination. Niklas B. Thompson is thanked for his assistance with crystallographic and Hammett plot analysis. The Bruker KAPPA APEXII X-ray diffractometer was purchased via an NSF CRIF:MU award to Caltech. The Varian 400 MHz NMR spectrometer at Caltech was purchased via an NIH grant (RR027690).
Supporting Information Available
Experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
† S.E.S. and J.C.H. contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Majetich G.; Shimkus J. M. J. Nat. Prod. 2010, 73, 284. [DOI] [PubMed] [Google Scholar]; b Lin W.-H.; Fang J.-M.; Cheng Y.-S. Phytochemistry 1995, 40, 871. [Google Scholar]; c Lin W.-H.; Fang J.-M.; Cheng Y.-S. Phytochemistry 1996, 42, 1657. [Google Scholar]; d Kawazoe K.; Yamamoto M.; Takaishi Y.; Honda G.; Fujita T.; Sezik E.; Yesilada E. Phytochemistry 1999, 50, 493. [Google Scholar]; e Chang C.-I.; Chien S.-C.; Lee S.-M.; Kuo Y.-H. Chem. Pharm. Bull. 2003, 51, 1420. [DOI] [PubMed] [Google Scholar]; f Chang C.-I.; Chang J.-Y.; Kuo C.-C.; Pan W.-Y.; Kuo Y.-H. Planta Med. 2005, 71, 72. [DOI] [PubMed] [Google Scholar]
- a Banerjee M.; Mukhopadhyay R.; Achari B.; Banerjee A. K. Org. Lett. 2003, 5, 3931. [DOI] [PubMed] [Google Scholar]; b Fillion E.; Fishlock D. J. Am. Chem. Soc. 2005, 127, 13144. [DOI] [PubMed] [Google Scholar]; c Banerjee M.; Mukhopadhyay R.; Achari B.; Banerjee A. K. J. Org. Chem. 2006, 71, 2787. [DOI] [PubMed] [Google Scholar]; d Planas L.; Mogi M.; Takita H.; Kajimoto T.; Node M. J. Org. Chem. 2006, 71, 2896. [DOI] [PubMed] [Google Scholar]; e McFadden R. M.; Stoltz B. M. J. Am. Chem. Soc. 2006, 128, 7738. [DOI] [PubMed] [Google Scholar]; f Liang G.; Xu Y.; Seiple I. B.; Trauner D. J. Am. Chem. Soc. 2006, 128, 11022. [DOI] [PubMed] [Google Scholar]; g Li S.; Chiu P. Tetrahedron Lett. 2008, 49, 1741. [Google Scholar]; h Tang S.; Xu Y.; He J.; He Y.; Zheng J.; Pan X.; She X. Org. Lett. 2008, 10, 1855. [DOI] [PubMed] [Google Scholar]; i Majetich G.; Shimkus J. M. Tetrahedron Lett. 2009, 50, 3311. [Google Scholar]; j Alvarez-Manzaneda E.; Chahboun R.; Cabrera E.; Alvarez E.; Alvarez-Manzaneda R.; Meneses R.; Es-Samti H.; Fernández A. J. Org. Chem. 2009, 74, 3384. [DOI] [PubMed] [Google Scholar]; k Alvarez-Manzaneda E.; Chahboun R.; Cabrera E.; Alvarez E.; Haidour A.; Ramos J. M.; Alvarez-Manzaneda R.; Charrah Y.; Es-Samti H. Org. Biomol. Chem. 2009, 7, 5146. [DOI] [PubMed] [Google Scholar]; l Alvarez-Manzaneda E.; Chahboun R.; Cabrera E.; Alvarex E.; Haidour A.; Ramos J. M.; Alvarez-Manzaneda R.; Hmamouchi M.; Es-Samti H. Chem. Commun. 2009, 592. [DOI] [PubMed] [Google Scholar]; m Node M.; Ozeki M.; Planas L.; Nakano M.; Takita H.; Mori D.; Tamatani S.; Kajimoto T. J. Org. Chem. 2010, 75, 190. [DOI] [PubMed] [Google Scholar]; n Jana C. K.; Scopelliti R.; Gademann K. Synthesis 2010, 2223. [DOI] [PubMed] [Google Scholar]; o Jana C. K.; Scopelliti R.; Gademann K. Chem.—Eur. J. 2010, 16, 7692. [DOI] [PubMed] [Google Scholar]; p Alvarez-Manzaneda E.; Chahboun R.; Alvarez E.; Tapia R.; Alvarez-Manzaneda R. Chem. Commun. 2010, 46, 9244. [DOI] [PubMed] [Google Scholar]; q Liao X.; Stanley L. M.; Hartwig J. F. J. Am. Chem. Soc. 2011, 133, 2088. [DOI] [PMC free article] [PubMed] [Google Scholar]; r Tapia R.; Guardia J. J.; Alvarez E.; Haidöur A.; Ramos J. M.; Alvarez-Manzaneda R.; Chahboun R.; Alvarez-Manzaneda E. J. Org. Chem. 2012, 77, 573. [DOI] [PubMed] [Google Scholar]; s Deng J.; Li R.; Luo Y.; Li J.; Zhou S.; Li Y.; Hu J.; Li A. Org. Lett. 2013, 15, 2022. [DOI] [PubMed] [Google Scholar]; t Li L.-Q.; Li M.-M.; Chen D.; Liu H.-M.; Geng H.-C.; Lin J.; Qin H.-B. Tetrahedron Lett. 2014, 55, 5960. [Google Scholar]
- a Iwamoto M.; Ohtsu H.; Tokuda H.; Nishino H.; Matsunaga S.; Tanaka R. Bioorg. Med. Chem. 2001, 9, 1911. [DOI] [PubMed] [Google Scholar]; b Minami T.; Iwamoto M.; Ohtsu H.; Ohishi H.; Tanaka R.; Yoshitake A. Planta Med. 2002, 68, 742. [DOI] [PubMed] [Google Scholar]; c Katoh T.; Akagi T.; Noguchi C.; Kajimoto T.; Node M.; Tanaka R.; Nishizawa M.; Ohtsu H.; Suzuki N.; Saito K. Bioorg. Med. Chem. 2007, 15, 2736. [DOI] [PubMed] [Google Scholar]
- a Kikushima K.; Holder J. C.; Gatti M.; Stoltz B. M. J. Am. Chem. Soc. 2011, 133, 6902. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Holder J. C.; Zou L.; Marziale A. N.; Liu P.; Lan Y.; Gatti M.; Kikushima K.; Houk K. N.; Stoltz B. M. J. Am. Chem. Soc. 2013, 135, 14996. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Holder J. C.; Marziale A. N.; Gatti M.; Mao B.; Stoltz B. M. Chem.—Eur. J. 2013, 19, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reetz M. T.; Westermann J.; Steinbach R. J. Chem. Soc., Chem. Commun. 1981, 237. [Google Scholar]
- Hansch C.; Leo A.; Taft R. W. Chem. Rev. 1991, 91, 165. [Google Scholar]; Red crosses in Figure 3 are interpolated from the linear least square regression using the known Hammett values (σp) for para-Br and para-iPr.
- Ishiyama T.; Takagi J.; Ishida K.; Miyaura N.; Anastasi N. R.; Hartwig J. F. J. Am. Chem. Soc. 2002, 124, 390. [DOI] [PubMed] [Google Scholar]
- Sun J.; Perfetti M. T.; Santos W. L. J. Org. Chem. 2011, 76, 3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston M. D.; Shapiro B. L.; Shapiro M. J.; Proulx T. W.; Godwin A. D.; Pearce H. L. J. Am. Chem. Soc. 1975, 97, 542. [Google Scholar]
- Takeda M.; Inoue H.; Noguchi K.; Honma Y.; Kawamori M.; Tsukamoto G.; Yamawaki Y.; Saito S. Chem. Pharm. Bull. 1976, 24, 1514. [DOI] [PubMed] [Google Scholar]
- Han C.; Buchwald S. L. J. Am. Chem. Soc. 2009, 131, 7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molander G. A.; Rivero M. R. Org. Lett. 2002, 4, 107. [DOI] [PubMed] [Google Scholar]
- Saá J. M.; Dopico M.; Martorell G.; García-Raso A. J. Org. Chem. 1990, 55, 991. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.