

Abstract
Pigs have experienced dramatic selection due to domestication, which has led to many different phenotypes when compared to their wild counterparts, especially in the last several decades. Currently, genome-wide scans in both cattle and humans showing positive selection footprints have been investigated. However, few studies have focused on porcine selection footprints, particularly on a genome-wide scale. Surveying for selection footprints across porcine genomes can be quite valuable for revealing the genetic mechanisms of phenotypic diversity. Here, we employed a medium sequencing depth (5–20x/site per individual, on average) approach called genotyping by genome reducing and sequencing (GGRS) to detect genome-wide selection signatures of two domestic pig breeds (Yorkshire and Landrace) that have been under intensive selection for traits of muscle development, growth and behavior. The relative extended haplotype homozygosity test, which identifies selection signatures by measuring the characteristics of haplotypes’ frequency distribution within a single population, was also applied to identify potential positively selected regions. As a result, signatures of positive selection were found in each breed. However, most selection signatures were population specific and related to genomic regions containing genes for biological categories including brain development, metabolism, growth and olfaction. Furthermore, the result of the gene set enrichment analysis indicated that selected regions of the two breeds presented a different over-representation of genes in the Gene Ontology annotations and Kyoto Encyclopedia of Genes and Genomes pathways. Our results revealed a genome-wide map of selection footprints in pigs and may help us better understand the mechanisms of selection in pig breeding.
Keywords: genome reducing and sequencing, pig genome, REHH test, selective sweep
Introduction
Yorkshire and Landrace, two commercial breeds of pigs used worldwide, have been subject to intensive selection for particular production attributes in the last decades. With this in mind, identifying genomic loci under selection that correlate with these production attributes would be beneficial for future pig breeding as well as for the identification of porcine genes related to biological processes and traits of interest.
The advent of high-throughput and cost-effective genotyping techniques allows us to thoroughly explore the patterns of genetic variation in domestic animals at the genome level. One of the strategies for studying genetic variation has been carried out of the genome to phenotypes and aims mainly to detect selection signatures based on identified patterns of linkage disequilibrium (LD; Ennis 2007), which are inconsistent with the hypothesis of genetic neutrality. As the concept of a selective sweep was first proposed by Smith & Haigh (1974) to detect selection signatures, it has been validated by other researchers (Barton 1995; Durrett & Schweinsberg 2004; Przeworski et al. 2005; Pennings & Hermisson 2006). It was assumed that, under the conditions of selection, the frequency of a beneficial allele rises and the allele is driven to fixation within a short period of time in a random-mating population of constant size. So far, different statistics have been established to detect different kinds of selection signatures, for instance, extended haplotype homozygosity (EHH; Sabeti et al. 2002) for recent selection and Tajimas’ D (Tajima 1989) for relatively ancient selection. Although many methods, such as Tajima's D (Tajima 1989), the Hudson–Kreitman–Aguade (HKA) test (Hudson et al. 1987) and Fay and Wu's H test (Fay & Wu 2000), were developed to detect selection signatures; they were not designed for locating genome-wide SNPs. Some other methods, including FST (Wright 1965), EHH (Sabeti et al. 2002) and iHS (integrated haplotype score; Voight et al. 2006), are suitable for detecting selection signatures at the genome level. Unfortunately, their applications are limited as the high-throughput genotyping techniques were not widely used in domestic animals at that time. Recently, the development of sequencing techniques and SNP chips provides us with high-density markers and the ability to identify selection signatures genome wide.
For example, genome-wide scans of selection signatures have been well studied in cattle (Prasad et al. 2008; Flori et al. 2009; Gautier et al. 2009). However, most of these studies applied SNP chips for the identification of SNP genotypes. Although SNP chips are widely used, this technique also brings some bias because of the small size of samples, which leads to a frequency-specific distortion in detecting SNPs. Additionally, Amaral et al.'s (2011) study showed that sequencing of genomic pools by next-generation sequencing was a cost-effective approach to identifying selection signatures without the effect of ascertainment bias. Consequently, next-generation sequencing provides us with the opportunity to estimate the genome-wide genetic diversity of a breed. To date, we have devised a new method for SNP genotyping, called genotyping by genome reducing and sequencing (GGRS), which is cost-effective, can genotype outbred species and is highly reproducible (Chen et al. 2013). This method can identify more than 70 000 SNPs for only $80 (USD)/sample. Among various statistics used in discovering positive selection signatures from the SNP data, iHS and REHH (relative extended haplotype homozygosity) are two of the most widely used tools (Enard et al. 2014; Fagny et al. 2014; Garke et al. 2014). Unfortunately, iHS requires both the genotype of the selected mutation and a known ancestor allele, which makes it difficult to apply in many cases. We therefore used the REHH test for our analysis (Walsh et al. 2006; Zhang et al. 2006).
Yorkshire and Landrace may have selection footprints in their genome resulting from intensive selection for production attributes over the past few decades. To date, some research involving selective sweep analyses in pigs has revealed strong signatures of selection affecting genomic regions that harbor genes underlying economic traits such as body length, disease resistance, pork yield, muscle development and fertility (Amaral et al. 2011; Li et al. 2014). Some specific genes related to coat color (Johansson Moller et al. 1996; Fang et al. 2009), growth (Van Laere et al. 2003), RNA processing and regulation (Groenen et al. 2012), olfaction and hypoxia (Li et al. 2013) have also been found previously to show a correlation between domestication and selection. Therefore, identification of the regions that have been subjected to selective breeding would be beneficial for identifying genes related to traits of interest and biological processes.
Artificial selection is not only an important aspect reflecting the domestication process, but also still important for the ongoing improvement of particular traits of value to humans during breed formation. Therefore, a scan of genome-wide selection signatures will help us identify porcine genes related to biological processes and traits of interest and, as well, allow us to better understand the mechanisms of selection in pig breeding. Here, we investigated patterns of selection in these two pig breeds and found several regions related to the traits of growth, muscle development and disease resistance. By analyzing nucleotide diversity, we aimed to identify genomic regions exhibiting signatures of selection and candidate genes reported in proximity to the genomic positions showing the most significant indications of selection and to gain further insight into the genome-wide footprints of pig selection. The functions associated with the putative genes under selection were also investigated by gene set enrichment analysis of Gene Ontology (GO) annotations and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways.
Materials and methods
DNA samples collection and sequencing data preparation
Unrelated or distantly related pigs were chosen according to pedigree information so as to have as few related individuals as possible in the sample set. A total of 70 DNA samples of Yorkshire (n = 34) and Landrace (n = 36) were obtained from breeding farms in Xiangxin, Shanghai, China. The protocol of acquiring each individual genotype, that is, DNA extraction, sequencing and SNP calling, was carried out using GGRS (http://klab.sjtu.edu.cn/GGRS/) (Chen et al. 2013), and the missing genotypes were imputed by iBLUP (http://klab.sjtu.edu.cn/iBLUP/) (Yang et al. 2014). The sequencing library (fragments ranging from 200 to 300 bp) was sequenced by an Illumina Hiseq2000 instrument with a paired-end (2 × 100 bp) pattern; the sequencing process is given in detail by the manufacturer (Illumina). GGRS is one approach of next-generation sequencing technology and can genotype species cost-effectively and with high reproducibility, especially for outbred species with a large genome size, for example, in the pig. Compared with SNP chips, it is not only able to identify novel SNPs, rather than genotypes of ascertainment, but also to discover high-density SNPs with lower cost. These advantages are a benefit to identifying selection signatures without the effect of ascertainment bias (Amaral et al. 2011). iBLUP is a genotype imputation method that imputes missing genotypes using identity-by-descent and linkage disequilibrium information. This method can impute missing genotypes with greater accuracy than can other common imputation methods, for example, beagle. Even at a high missing rate of 70%, it retained an accuracy of 0.95, whereas that of beagle is lower at 0.82 (Yang et al. 2014).
Approximately 347 million 100-bp reads were generated from DNA samples. The raw reads with a base average quality score of at least 20 (error rate of base calling of 1 in 100) and the first 65 bp of at least 30 (error rate of base call of 1 in 1000) were aligned to the pig genome reference. A total of approximately 2.01% of the porcine genome met the alignment quality parameters (see GGRS, Chen et al. 2013), and the average sequencing depth of the whole genome was 5.97 × 102 170 SNPs, which could be identified in more than 30 samples included in the final analysis, with an average sequencing depth of more than 5×. The distance between approximately 88% pairs of adjacent SNPs was <50 Kb. The percentage of those with a distance of more than 150 Kb was <5% (Fig. S1). After imputing the missing genotypes using iBLUP, those genotyped SNPs were phased by fastphase (Scheet & Stephens 2006) for further positive selection analysis.
REHH test
The REHH test, which was first proposed by Sabeti et al. (2002), was used to detect the recent positive selection signatures by evaluating how LD decays across the genome. Under the pressure of positive selection, regions of selection present unusually rapid rises in allele frequency and long-range LD over a short period of time. Thus, Sabeti et al. (2002) defined a region of interest in the genome as the ‘core region’, which contains a set of ‘core haplotypes’ and has a strong LD among SNPs. Their general tests for selection are based on comparing a core haplotype with both higher frequency and higher EHH with other core haplotypes at the same locus. According to selection signatures theory, the core haplotypes harboring the beneficial allele would have a higher frequency due to the hitchhiking effect (Sabeti et al. 2002). For this reason, we discarded core haplotypes with a frequency <0.25. However, the long-range haplotype may be due to low local recombination rates rather than the recent positive selection. We therefore applied the REHH test (Sabeti et al. 2002), which corrects for local variation in recombination rate, to detect selection signatures.
As fully phased haplotype data were required for the analysis, we first reconstructed haplotypes for every chromosome in the two breeds separately using the default parameters of fastphase (Scheet & Stephens 2006). Then, fully phased haplotype data were further analyzed using sweep v.1.1 (http://www.broadinstitute.org/mpg/sweep/index.html) with the default parameter marker H = 0.04 (one type of distance to match) to identify core regions. Furthermore, we placed the data in order, according to the frequency of all the haplotypes, into 20 bins to calculate the significance of the REHH value, obtaining P-values by log-transforming the REHH in the bin to reach normality and calculating the mean and standard deviation. Finally, core haplotypes with extreme REHH values (threshold level: P < 0.01) were regarded as significant.
Identifying QTL overlapping with selection signatures
The candidate regions (the top five with lowest P-values) of selection were regarded as overlapping if their locations were included within the QTL for porcine traits. We wrote a Perl script to identify the distribution of the candidate regions of selection in QTL using PigQTLdb (Hu et al. 2013).
Functional gene set enrichment analysis (FGSEA)
Our FGSEA of KEGG pathways and GO terms was performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID; Dennis et al. 2003; Huang da et al. 2009). First, we retrieved the Ensembl IDs of the genes, which were considered to be overlapping if their positions were contained inside the boundaries of 250 kb upstream or downstream of the candidate regions of selection, based on the annotation for Sus scrofa assembly 10.2 available from the Ensembl database (http://www.ensembl.org/index.html). Because of the incomplete GO annotation for the pig, corresponding human orthologous Ensembl IDs were retrieved using a Perl script and were used to get enrichment function categories. The DAVID was used to analyze enrichment in the KEGG pathways and the GO terms (http://www.genome.jp/kegg/, http://www.geneontology.org/). Finally, the enriched pathways with P-values <0.05 and GO terms with P-values <0.01 [with a false discovery rate (FDR) of <25%] were used for further analysis in our study.
Results and Discussion
Marker and core haplotype statistics
A total of 10 932 and 11 185 core regions with 72 989 and 71 354 SNPs (71.4% and 69.8%) spanning 1437 Mb and 1377 Mb (63.5% and 60.9%) of the genome were detected in Yorkshire and Landrace respectively. Their mean lengths of core region were calculated as 131.5 ± 287.6 kb and 123.1 ± 274.4 kb, with a maximum of 4139.2 kb and 4505.4 kb in chromosomes X and 4 respectively. The distance between approximately 72% of pairs of adjacent core regions was <50 Kb, and close to 10% of pairs had a distance of more than 150 Kb. The position information for the core regions for each pig breed is listed in Tables S1 and S2. The distribution of the size and number of SNPs in the core regions is depicted in Fig.1. As shown in Fig.1(a) and (b), for both breeds, there was a high frequency of core regions with a length <50 kb. This is probably because of the advantage of the sequencing technology we used, which can identify novel SNPs. This resulted in regions with a higher density of SNP markers. According to EHH, a region with a high density of SNPs indicates a stronger LD among these SNPs and contributes to a higher possibility for them to form into core regions even with a short length. Therefore, the core regions took up a high percentage of the length and number of SNPs in the genomes. This suggests that the sequencing technique could influence the final numbers of core region because of the high density of SNPs identified compared to SNP chips. This would be beneficial for finding regions under selection, even with a small length. As the maximum number of SNPs in core regions was set to 20 (the core region would retain only 20 SNPs, even if it included more than 20), the frequency of 20 SNPs in a core region was increased, as shown in Fig.1(c) and (d).
Figure 1.
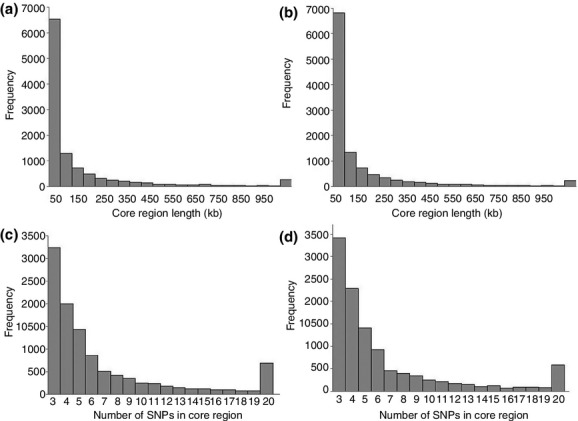
Distribution of length of core regions and the number of SNPs forming the core regions in Yorkshire and Landrace. (a) and (b) show the distribution of length of core regions, separately, in Yorkshire and Landrace respectively. The Y-axis represents frequency, and the X-axis represents the length of core regions. (c) and (d) show the distribution of the number of SNPs forming the core regions, separately, in Yorkshire and Landrace respectively. The Y-axis represents the frequency, and the X-axis represents the number of SNPs.
Genome-wide scanning for selection signatures
To identify outlying core haplotypes potentially representing candidate regions under positive selection, we estimated REHH using sweep. The results of this test are presented in Table1, which also includes the number of candidate regions and SNPs putatively under selection for each chromosome in both breeds. In total, 251 and 269 candidate core regions with lengths >0.5 kb displayed outlying peaks at a threshold level of 0.01 in Yorkshire and Landrace respectively. The total length and number of SNPs in candidate regions of the two breeds were nearly the same, approximately 1.3% and 1.7% respectively of the whole genome. This is not consistent with the previous study of Amaral et al. (2011), who estimated that approximately 7% of the porcine genome has been affected by selection events. The differences come from mainly two aspects: First, the P-valve used in our study (P < 0.01) was stricter than was theirs (P < 0.05), and second, the usage of SNPs and identification methods of selection signatures was different, which may have led to underestimating the proportion.
Table 1.
Summary of significant (P ≤ 0.01) core region (CR) and SNPs distribution in Yorkshire and Landrace.
| Population | Chr | No. CR | CR SNPs1 | CR length2 (kb) | Chr length (Mbp) | CR length/Chr length3 | Chr SNPs (n) | CR SNPs/Chr SNPs4 |
|---|---|---|---|---|---|---|---|---|
| Yorkshire | 1 | 139 | 17 | 3693.918 | 295.5 | 0.013 | 9028 | 0.015 |
| 2 | 190 | 24 | 2169.685 | 140.1 | 0.015 | 9557 | 0.020 | |
| 3 | 130 | 19 | 2832.248 | 123.6 | 0.023 | 7506 | 0.017 | |
| 4 | 136 | 17 | 2089.096 | 136.3 | 0.015 | 5796 | 0.023 | |
| 5 | 31 | 7 | 445.340 | 100.5 | 0.004 | 3736 | 0.008 | |
| 6 | 186 | 34 | 2529.724 | 123.3 | 0.021 | 12 910 | 0.014 | |
| 7 | 168 | 21 | 3167.319 | 136.4 | 0.023 | 5817 | 0.029 | |
| 8 | 17 | 2 | 358.194 | 120 | 0.003 | 3687 | 0.005 | |
| 9 | 184 | 27 | 2978.478 | 132.5 | 0.022 | 5202 | 0.035 | |
| 10 | 70 | 7 | 941.014 | 66.7 | 0.014 | 3148 | 0.022 | |
| 11 | 44 | 8 | 611.479 | 79.8 | 0.008 | 3795 | 0.012 | |
| 12 | 85 | 11 | 773.873 | 57.4 | 0.013 | 4028 | 0.021 | |
| 13 | 79 | 9 | 3345.927 | 145.2 | 0.023 | 4661 | 0.017 | |
| 14 | 113 | 19 | 1084.485 | 148.5 | 0.007 | 7094 | 0.016 | |
| 15 | 48 | 10 | 547.061 | 134.5 | 0.004 | 4968 | 0.010 | |
| 16 | 40 | 6 | 868.423 | 77.4 | 0.011 | 2796 | 0.014 | |
| 17 | 49 | 8 | 1005.974 | 64.4 | 0.016 | 3079 | 0.016 | |
| 18 | 32 | 3 | 434.741 | 54.3 | 0.008 | 2006 | 0.016 | |
| X | 20 | 2 | 220.651 | 125.9 | 0.002 | 3356 | 0.006 | |
| Total | 1761 | 251 | 30097.630 | 2262.3 | 0.013 | 102 170 | 0.017 | |
| Landrace | 1 | 80 | 11 | 1 823 125 | 295.5 | 0.006 | 9028 | 0.009 |
| 2 | 137 | 24 | 2 477 535 | 140.1 | 0.018 | 9557 | 0.014 | |
| 3 | 81 | 14 | 911 766 | 123.6 | 0.007 | 7506 | 0.011 | |
| 4 | 65 | 12 | 719 409 | 136.3 | 0.005 | 5796 | 0.011 | |
| 5 | 57 | 12 | 1 623 945 | 100.5 | 0.016 | 3736 | 0.015 | |
| 6 | 108 | 18 | 598 300 | 123.3 | 0.005 | 12910 | 0.008 | |
| 7 | 126 | 16 | 1 154 729 | 136.4 | 0.008 | 5817 | 0.022 | |
| 8 | 42 | 7 | 1 025 267 | 120 | 0.009 | 3687 | 0.011 | |
| 9 | 232 | 31 | 4 361 383 | 132.5 | 0.033 | 5202 | 0.045 | |
| 10 | 70 | 14 | 1 188 160 | 66.7 | 0.018 | 3148 | 0.022 | |
| 11 | 94 | 14 | 2 351 956 | 79.8 | 0.029 | 3795 | 0.025 | |
| 12 | 105 | 17 | 960 269 | 57.4 | 0.017 | 4028 | 0.026 | |
| 13 | 73 | 12 | 1 880 105 | 145.2 | 0.013 | 4661 | 0.016 | |
| 14 | 122 | 24 | 1 048 849 | 148.5 | 0.007 | 7094 | 0.017 | |
| 15 | 132 | 21 | 3 050 612 | 134.5 | 0.023 | 4968 | 0.027 | |
| 16 | 17 | 5 | 383 365 | 77.4 | 0.005 | 2796 | 0.006 | |
| 17 | 35 | 8 | 720 063 | 64.4 | 0.011 | 3079 | 0.011 | |
| 18 | 8 | 1 | 13 433 | 54.3 | 0.000 | 2006 | 0.004 | |
| X | 50 | 8 | 541 350 | 125.9 | 0.004 | 3356 | 0.015 | |
| Total | 1634 | 269 | 2 683 3621 | 2262.3 | 0.012 | 102 170 | 0.016 |
Number of SNPs forming significant core regions.
Total length covered by significant core regions.
Proportion of total significant core region lengths on chromosome length.
Proportion of total number of SNPs forming significant core regions on number of SNPs used.
The positional information of the candidate selected regions for the two breeds is listed in Tables S3 and S4. The signals were vastly overrepresented on parts of chromosome 9. This pattern of distribution was similar to bovine (Qanbari et al. 2010). Studies on selection signatures in pigs were also carried out by other researchers who relied on the Porcine60K SNP chip (Wilkinson et al. 2013). Based on comparisons with Wilkinson et al.'s data (five genomic regions showing selection in five or more breeds), we found that the selected region identified in their study (SSC7, 54.00–57.00 Mb) was consistent with our results (SSC7, 53.8–54.00 Mb). The different statistical method used in their study (FST, comparing different populations) and ours (REHH, studying within a population) might account for the low overlap rate. However, the results of Wilkinson et al.'s study may still provide a piece of evidence of the reliability of our study.
Identifying QTL overlapping with positively selected regions
To test whether the selection signatures we detected were a result of human's selection during pig breeding, we explored the pig QTL database (http://www.animalgenome.org/cgi-bin/QTLdb/SS/download?file=bedSS_10.2). We identified any overlapping of the outlying core regions (the top five with lowest P-value) with published QTL in pigs. The overlapping pig QTL for the core regions with lowest P-values (top five) is shown in Table S5. Interestingly, we found that the core regions showing the most significant indications of selection were contained in hundreds of reported QTL related to production, reproduction, health and meat quality. The number of QTL relating to meat quality (such as loin muscle area, average backfat thickness, meat color, pH value and drip loss; details shown in Table S5) is especially greater than others, with a proportion of 71% (Fig. S2). This observation is in accordance with the history of meat quality selection (such as lean meat percentage) in pig breeding programs (Vidal et al. 2005; Amaral et al. 2011). Furthermore, this indicates that selection during pig breeding has left a detectable footprint in the pig genome.
Genes within positively selected regions
We further investigated the genes in candidate regions of selection and identified corresponding genes by comparing their genomic locations with the available annotation of the porcine genome (Sscrofa 10.2). We extended core regions in both directions up to 250 kb. A summary of statistics for the five positively selected core regions with lengths >0.5 kb and with the lowest P-values (top five) using the REHH test is shown in Table2. These top five positively selected regions harbored genes related to muscle growth or immune response, such as BARX2, which is an important regulator of muscle growth, regeneration and maintenance (Meech et al. 2012), and APON, which is speculated to play a role in either the regulation of steroidogenesis or immunosuppression (O'Bryan et al. 2004).
Table 2.
Summary statistics for five core haplotypes of two breeds showing the lowest P-value after the relative extended haplotype homozygosity (REHH) test.
| Population | Chr | Position start | Position end | Core length (kb) | REHH P-value | Candidate gene | Function |
|---|---|---|---|---|---|---|---|
| Yorkshire | 16 | 3133480 | 3137443 | 3.963 | 0.00000043 | NA1 | NA |
| 9 | 62313602 | 62547091 | 233.489 | 0.00000135 | BARX2 | Controls cell adhesion and remodeling of the actin cytoskeleton in myoblast fusion and chondrogenesis | |
| SNORD112 | Small nucleolar RNAs (snoRNAs), like SNORD112, guide the formation of 2-prime O-methylation of ribosomal RNA (rRNA) and small nuclear RNAs (snRNAs) through a specific RNA duplex at each modification site | ||||||
| 5 | 4927436 | 4930181 | 2.745 | 0.00000430 | XPNPEP3 | Encodes a protein that belongs to the family of X-pro- aminopeptidases that utilize a metal cofactor and remove the N-terminal amino acid from peptides with a proline residue in the penultimate position | |
| ST13 | The assembly process of glucocorticoid receptor | ||||||
| SLC25A17 | Encodes a peroxisomal membrane protein | ||||||
| RPL31 | Encodes a ribosomal protein | ||||||
| MCHR1 | Encodes an integral plasma membrane protein that binds melanin-concentrating hormone | ||||||
| 6 | 37712730 | 37717184 | 4.454 | 0.00000667 | ZNF507 | Encodes a zinc finger protein | |
| 4 | 123539775 | 123622691 | 82.916 | 0.00000957 | NA | NA | |
| Landrace | 11 | 71519780 | 71559246 | 39.466 | 0.00000037 | NA | NA |
| 14 | 24592939 | 24779049 | 186.11 | 0.00000040 | P2RX2 | Encodes a ligand-gated ion channel receptor | |
| PUS1 | Stabilizes the secondary and tertiary structure of many RNAs | ||||||
| MMP17 | Involved in the breakdown of extracellular matrix in normal physiological processes, such as embryonic development, reproduction and tissue remodeling | ||||||
| 11 | 77297111 | 77299461 | 2.35 | 0.00000158 | ITGBL1 | Encodes a beta-integrin-related protein | |
| 5 | 85123381 | 85141164 | 17.783 | 0.00000294 | U6 | Encodes U6 spliceosomal RNA | |
| 5 | 23288996 | 24074802 | 785.806 | 0.00000541 | APOF | Encodes olipoprotein | |
| APON | Encodes ovarian and testicular apolipoprotein | ||||||
| MIP | Encodes a major intrinsic protein of lens fiber | ||||||
| STAT6 | Encodes a signal transducer and activator | ||||||
| MYL6 | Encodes a myosin light polypeptide | ||||||
| CS | Encodes citrate synthase |
NA, no genes were found.
We screened the subset of genes in all core regions displaying extreme REHH values. Interestingly, some regions overlapped with genes previously detected as being under selection. For example, in Amaral et al.'s (2011) study, they identified a few genes relating to neuron function, growth, muscle development, metabolism and disease, such as MAPK8IP3, L3MBTL2, SLC22A17, ENO2, CACNG7, SPHK and FGFR2, under positive selection, which were also detected in our study. Among them, the MAPK8IP3 gene is involved in the MAPK signaling pathway, which is essential in regulating many cellular processes including inflammation, cell differentiation, cell proliferation and death (Wilkinson & Millar 2000). According to Miyamoto et al.'s (2011) study, SLC22A17, in cooperation with LCN2, is involved in the acquisition of aggressive behavior among endometrial carcinoma cells. In another study (Groenen et al. 2012), the ERI2 gene, which is located around position 26Mb on SSC3 and encodes ERI1 (exoribonuclease family member 2), was detected in a selective sweep region. This was also observed in our study. Although the exact function of ERI2 is unknown, the ERI1 exoribonuclease family members have been shown to be involved in the degradation of mRNA (Kupsco et al. 2006). What's more, the results suggest a large number of genes related to olfaction were within candidate selection regions. This finding is consistent with Amaral et al.'s (2011) study, in which a significant enrichment of genes related to olfaction within positive selection regions was observed. There has been a significant expansion of the olfactory receptor gene family in the porcine genome, and this probably reflects the strong reliance of pigs on their sense of smell while searching for food (Groenen et al. 2012). We speculated that a well-developed sense of smell might spur the pigs’ appetite and increase in their food intake and, as a result, further accelerate their growth. This observation is consistent with the history of the domestic pig breeds currently studied, which have higher growth rates than do the ancestral wild boar populations. In addition, in our study, we observed several genes related to brain and neuron functions overlapped within regions likely experiencing positive selection in pigs. For example, the NRXN2 gene is a member of the neurexin gene family encoding polymorphic presynaptic proteins that are implicated in synaptic plasticity and memory processing (Rozic et al. 2012). It was reported that neurexin genes are related to neurodevelopmental disorders affecting cognition and behavior, such as the diseases of autism spectrum disorder (Kim et al. 2008), intellectual disability (Ching et al. 2010) and schizophrenia (Rujescu et al. 2009). Because it is expected that farmers would have selected for more docile animals, the process of domestication leads to a relative change in behavior (Price 1999). Our results may support the hypothesis that these positive signatures might be a result of domestication (Amaral et al. 2011). This is similar to the dog, which has inferior observational learning skills compared to the wolf (Frank 1980).
Involved biological processes under selection
We then sought to investigate the functions associated with the putative genes undergoing positive selection by analyzing over-represented annotations and pathways using DAVID (Dennis et al. 2003; Huang da et al. 2009). If the P-value was <0.01 for GO annotation and <0.05 for the KEGG pathway (with a FDR of <25%), that was considered significant. The significant GO terms and KEGG pathways of over-represented genes are shown in Table3 for Yorkshire and Landrace.
Table 3.
Enrichment of Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways among the positively selected regions.
| Population | Category | Term | P-value |
|---|---|---|---|
| Yorkshire | GOTERM_BP_2 | GO:0055114∼oxidation reduction | 0.0083 |
| GOTERM_CC_2 | GO:0031012∼extracellular matrix | 0.0071 | |
| GOTERM_MF_2 | GO:0060589∼nucleoside triphosphatase regulator activity | 0.0042 | |
| Landrace | KEGG_PATHWAY | hsa00590: Arachidonic acid metabolism | 0.0172 |
| GOTERM_BP_2 | GO:0006810∼transport | 0.0034 | |
| GOTERM_BP_2 | GO:0051234∼establishment of localization | 0.0039 | |
| GOTERM_BP_2 | GO:0019725∼cellular homeostasis | 0.0072 |
The results suggest that the two breeds presented a different over-representation of genes with GO annotations and KEGG pathways. The GO terms for Yorkshire included ‘oxidation reduction’, ‘extracellular matrix’ and ‘nucleoside triphosphatase regulator activity’, whereas the Landrace GO terms included ‘transport’, ‘establishment of localization’ and ‘cellular homeostasis’. These results indicate that the two breeds displayed different types of biological processes under the selected regions. Previous studies have reported that, in pigs, the genetic selection for lean, large muscle blocks and fast growth is associated with an increased prevalence of metabolic diseases, such as mulberry heart disease (Rice & Kennedy 1989) and porcine stress syndrome. These diseases are linked to cardiovascular inadequacy, which may result in oxidative stress (Brambilla et al. 2002). Among our findings, genes over-represented in oxidation reduction (P = 0.0083) may relate to selection against the above diseases. Moreover, our results of genes over-represented in transport (P = 0.0034) and establishment of localization (P = 0.0039) revealed genes involved in growth that overlap with positive selection regions, such as TBRG1, which is related to cell growth and differentiation (Garcia-Alai et al. 2010). As pigs with higher growth rates and a higher proportion of muscle are preferred in current breeding programs, they need to have an efficient gene network (such as transport) to support their high growth system.
In addition, the pathway of arachidonic acid metabolism (P = 0.0172) was identified in Landrace. Arachidonic acid, routinely added to infant formula along with docosahexaenoic acid, is a natural component of breast milk. It plays an important role in growth and development during the perinatal period (Innis 2005, 2007). The observation that this particular pathway is also related to porcine growth is in agreement with the history of domestic pig breeds currently studied, which have higher growth rates than do their wild relatives. Yorkshire and Landrace are two of several European domestic pig breeds that are more similar to their ancestors and yet are highly differentiated in terms of genotypes and phenotypes (Amaral et al. 2011). The different phenotypes selected during the breeding of two pigs may leave footprints of selection in different parts of the pig genome. This may be an explanation of why the two breeds presented a different over-representation of genes with GO annotations and KEGG pathways.
However, our dataset included approximately only 2% of the genome, which may have limited the utility of the gene set enrichment analyses. The current annotation of the pig genome has a limited availability of GO terms and genes mapped in the KEGG pathways, further decreasing the sensitivity of the analysis. Therefore, we could provide only suggestive evidence for the over-represented annotations and pathways affected by positive selection.
In conclusion, this study provides a genome-wide map of selection signatures in Yorkshire and Landrace genomes and yields insight into the mechanisms of selection in pig breeding. Our results show that genes related to metabolism, olfaction and nerves may also experience positive selection. Furthermore, there are indications that selection has impacted different genes and pathways in the two breeds studied.
Acknowledgments
This study was supported by the 2011–2013 Animal Germplasm Resources Conservation Project from the Ministry of Agriculture of China, the National Natural Science Foundation of China (grant nos. 31072003, 3100992, 31101706, 31272414 and 31370043) and the National 948 Project of China (2012-Z26, 2011-G2A).
Conflict of interest
The authors declare no conflict of interest.
Authors′ contributions
Y. P. designed the study. Y. P. and Q. W. supervised the study. Z.W. analyzed the data. Z.W. wrote the manuscript. Y. Y. implemented the method in the iBLUP software package with the help of F. H., Z. Z and X. Z. Q. C. developed the GGRS approach for outbred populations with the help of Z.C, R.L and Y.T. X.X and J.Y. assisted pig sample collection. All authors have read and edited the manuscript.
Supporting Information
Additional supporting information may be found in the online version of this article.
Figure S1. Distribution of the distance between pairs of adjacent SNPs on the genome.
Figure S2. Distribution of the number of QTL overlapping with putatively selected core regions displaying the lowest P-values (top five).
Table S1. Core region positions in Yorkshire, including the chromosome number, start position, end position and length.
Table S2. Core region positions in Landrace, including the chromosome number, start position, end position and length.
Table S3. Candidate selected regions positions in Yorkshire, including the chromosome number, start position, end position, length and REHH P-value.
Table S4. Candidate selected regions positions in Landrace, including the chromosome number, start position, end position, length and REHH P-value.
Table S5. Traits and the position of the overlapping pig QTL for the core regions with lowest P-values (top five), including traits information, the position of the core regions and QTL, and the QTL IDs in the pig QTL database.
References
- Amaral AJ, Ferretti L, Megens HJ, Crooijmans RP, Nie H, Ramos-Onsins SE, Perez-Enciso M, Schook LB, Groenen MA. Genome-wide footprints of pig domestication and selection revealed through massive parallel sequencing of pooled DNA. PLoS ONE. 2011;6:e14782. doi: 10.1371/journal.pone.0014782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton NH. Linkage and the limits to natural selection. Genetics. 1995;140:821–41. doi: 10.1093/genetics/140.2.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla G, Civitareale C, Ballerini A, Fiori M, Amadori M, Archetti LI, Regini M, Betti M. Response to oxidative stress as a welfare parameter in swine. Redox Report. 2002;7:159–63. doi: 10.1179/135100002125000406. [DOI] [PubMed] [Google Scholar]
- Chen Q, Ma Y, Yang Y, et al. Genotyping by genome reducing and sequencing for outbred animals. PLoS ONE. 2013;8:e67500. doi: 10.1371/journal.pone.0067500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching MS, Shen Y, Tan WH, et al. Deletions of NRXN1neurexin-1) predispose to a wide spectrum of developmental disorders. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2010;153B:937–47. doi: 10.1002/ajmg.b.31063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biology. 2003;4:P3. [PubMed] [Google Scholar]
- Durrett R, Schweinsberg J. Approximating selective sweeps. Theoretical Population Biology. 2004;66:129–38. doi: 10.1016/j.tpb.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Enard D, Messer PW, Petrov DA. Genome-wide signals of positive selection in human evolution. Genome Research. 2014;24:885–95. doi: 10.1101/gr.164822.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennis S. Linkage disequilibrium as a tool for detecting signatures of natural selection. Methods in Molecular Biology. 2007;376:59–70. doi: 10.1007/978-1-59745-389-9_5. [DOI] [PubMed] [Google Scholar]
- Fagny M, Patin E, Enard D, Barreiro LB, Quintana-Murci L, Laval G. Exploring the occurrence of classic selective sweeps in humans using whole-genome sequencing datasets. Molecular Biology and Evolution. 2014;31:1850–68. doi: 10.1093/molbev/msu118. [DOI] [PubMed] [Google Scholar]
- Fang M, Larson G, Ribeiro HS, Li N, Andersson L. Contrasting mode of evolution at a coat color locus in wild and domestic pigs. PLoS Genetics. 2009;5:e1000341. doi: 10.1371/journal.pgen.1000341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wu CI. Hitchhiking under positive Darwinian selection. Genetics. 2000;155:1405–13. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flori L, Fritz S, Jaffrezic F, Boussaha M, Gut I, Heath S, Foulley JL, Gautier M. The genome response to artificial selection: a case study in dairy cattle. PLoS ONE. 2009;4:e6595. doi: 10.1371/journal.pone.0006595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank H. Evolution of canine information processing under conditions of natural and artificial selection. Zeitschrift für Tierpsychologie. 1980;53:389–99. doi: 10.1111/j.1439-0310.1980.tb01059.x. [DOI] [PubMed] [Google Scholar]
- Garcia-Alai MM, Allen MD, Joerger AC, Bycroft M. The structure of the FYR domain of transforming growth factor beta regulator 1. Protein Science. 2010;19:1432–8. doi: 10.1002/pro.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garke C, Ytournel F, Sharifi AR, Pimentel EC, Ludwig A, Simianer H. Footprints of recent selection and variability in breed composition in the Gottingen Minipig genome. Animal Genetics. 2014;45:381–91. doi: 10.1111/age.12150. [DOI] [PubMed] [Google Scholar]
- Gautier M, Flori L, Riebler A, Jaffrezic F, Laloe D, Gut I, Moazami-Goudarzi K, Foulley JL. A whole genome Bayesian scan for adaptive genetic divergence in West African cattle. BMC Genomics. 2009;10:550. doi: 10.1186/1471-2164-10-550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenen MA, Archibald AL, Uenishi H, et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature. 2012;491:393–8. doi: 10.1038/nature11622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu ZL, Park CA, Wu XL, Reecy JM. Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Research. 2013;41:D871–9. doi: 10.1093/nar/gks1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Hudson RR, Kreitman M, Aguade M. A test of neutral molecular evolution based on nucleotide data. Genetics. 1987;116:153–9. doi: 10.1093/genetics/116.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innis SM. Essential fatty acid transfer and fetal development. Placenta. 2005;26(Suppl A):S70–5. doi: 10.1016/j.placenta.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Innis SM. Human milk: maternal dietary lipids and infant development. Proceedings of the Nutrition Society. 2007;66:397–404. doi: 10.1017/S0029665107005666. [DOI] [PubMed] [Google Scholar]
- Johansson Moller M, Chaudhary R, Hellmen E, Hoyheim B, Chowdhary B, Andersson L. Pigs with the dominant white coat color phenotype carry a duplication of the KIT gene encoding the mast/stem cell growth factor receptor. Mammalian Genome. 1996;7:822–30. doi: 10.1007/s003359900244. [DOI] [PubMed] [Google Scholar]
- Kim HG, Kishikawa S, Higgins AW, et al. Disruption of neurexin 1 associated with autism spectrum disorder. American Journal of Human Genetics. 2008;82:199–207. doi: 10.1016/j.ajhg.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupsco JM, Wu MJ, Marzluff WF, Thapar R, Duronio RJ. Genetic and biochemical characterization of Drosophila Snipper: a promiscuous member of the metazoan 3′hExo/ERI-1 family of 3′ to 5′ exonucleases. RNA. 2006;12:2103–17. doi: 10.1261/rna.186706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Tian S, Jin L, et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nature Genetics. 2013;45:1431–8. doi: 10.1038/ng.2811. [DOI] [PubMed] [Google Scholar]
- Li M, Tian S, Yeung CK, et al. Whole-genome sequencing of Berkshire (European native pig) provides insights into its origin and domestication. Scientific Reports. 2014;4:4678. doi: 10.1038/srep04678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meech R, Gonzalez KN, Barro M, Gromova A, Zhuang L, Hulin JA, Makarenkova HP. Barx2 is expressed in satellite cells and is required for normal muscle growth and regeneration. Stem Cells. 2012;30:253–65. doi: 10.1002/stem.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto T, Asaka R, Suzuki A, Takatsu A, Kashima H, Shiozawa T. Immunohistochemical detection of a specific receptor for lipocalin 2 (solute carrier family 22 member 17, SLC22A17) and its prognostic significance in endometrial carcinoma. Experimental and Molecular Pathology. 2011;91:563–8. doi: 10.1016/j.yexmp.2011.06.002. [DOI] [PubMed] [Google Scholar]
- O'Bryan MK, Foulds LM, Cannon JF, et al. Identification of a novel apolipoprotein, ApoN, in ovarian follicular fluid. Endocrinology. 2004;145:5231–42. doi: 10.1210/en.2004-0630. [DOI] [PubMed] [Google Scholar]
- Pennings PS, Hermisson J. Soft sweeps III: the signature of positive selection from recurrent mutation. PLoS Genetics. 2006;2:e186. doi: 10.1371/journal.pgen.0020186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad A, Schnabel RD, McKay SD, Murdoch B, Stothard P, Kolbehdari D, Wang Z, Taylor JF, Moore SS. Linkage disequilibrium and signatures of selection on chromosomes 19 and 29 in beef and dairy cattle. Animal Genetics. 2008;39:597–605. doi: 10.1111/j.1365-2052.2008.01772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price EO. Behavioral development in animals undergoing domestication. Applied Animal Behaviour Science. 1999;65:245–71. [Google Scholar]
- Przeworski M, Coop G, Wall JD. The signature of positive selection on standing genetic variation. Evolution. 2005;59:2312–23. [PubMed] [Google Scholar]
- Qanbari S, Pimentel EC, Tetens J, Thaller G, Lichtner P, Sharifi AR, Simianer H. A genome-wide scan for signatures of recent selection in Holstein cattle. Animal Genetics. 2010;41:377–89. doi: 10.1111/j.1365-2052.2009.02016.x. [DOI] [PubMed] [Google Scholar]
- Rice DA, Kennedy S. Vitamin E, selenium, and polyunsaturated fatty acid concentrations and glutathione peroxidase activity in tissues from pigs with dietetic microangiopathy (mulberry heart disease) American Journal of Veterinary Research. 1989;50:2101–4. [PubMed] [Google Scholar]
- Rozic G, Lupowitz Z, Zisapel N. Exonal elements and factors involved in the depolarization-induced alternative splicing of neurexin 2. Journal of Molecular Neuroscience. 2012;50:221–33. doi: 10.1007/s12031-012-9919-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rujescu D, Ingason A, Cichon S, et al. Disruption of the neurexin 1 gene is associated with schizophrenia. Human Molecular Genetics. 2009;18:988–96. doi: 10.1093/hmg/ddn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti PC, Reich DE, Higgins JM, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–7. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- Scheet P, Stephens M. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. American Journal of Human Genetics. 2006;78:629–44. doi: 10.1086/502802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JM, Haigh J. The hitch-hiking effect of a favourable gene. Genetical Research. 1974;23:23–35. [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Laere AS, Nguyen M, Braunschweig M, et al. A regulatory mutation in IGF2 causes a major QTL effect on muscle growth in the pig. Nature. 2003;425:832–6. doi: 10.1038/nature02064. [DOI] [PubMed] [Google Scholar]
- Vidal O, Noguera JL, Amills M, Varona L, Gil M, Jimenez N, Davalos G, Folch JM, Sanchez A. Identification of carcass and meat quality quantitative trait loci in a Landrace pig population selected for growth and leanness. Journal of Animal Science. 2005;83:293–300. doi: 10.2527/2005.832293x. [DOI] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biology. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh EC, Sabeti P, Hutcheson HB, et al. Searching for signals of evolutionary selection in 168 genes related to immune function. Human Genetics. 2006;119:92–102. doi: 10.1007/s00439-005-0090-0. [DOI] [PubMed] [Google Scholar]
- Wilkinson MG, Millar JB. Control of the eukaryotic cell cycle by MAP kinase signaling pathways. FASEB Journal. 2000;14:2147–57. doi: 10.1096/fj.00-0102rev. [DOI] [PubMed] [Google Scholar]
- Wilkinson S, Lu ZH, Megens HJ, Archibald AL, Haley C, Jackson IJ, Groenen MA, Crooijmans RP, Ogden R, Wiener P. Signatures of diversifying selection in European pig breeds. PLoS Genetics. 2013;9:e1003453. doi: 10.1371/journal.pgen.1003453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S. The interpretation of population structure by F-statistics with special regard to systems of mating. Evolution. 1965;19:395–420. [Google Scholar]
- Yang Y, Wang Q, Chen Q, Liao R, Zhang X, Yang H, Zheng Y, Zhang Z, Pan Y. A new genotype imputation method with tolerance to high missing rate and rare variants. PLoS ONE. 2014;9:e101025. doi: 10.1371/journal.pone.0101025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Bailey DK, Awad T, et al. A whole genome long-range haplotype (WGLRH) test for detecting imprints of positive selection in human populations. Bioinformatics. 2006;22:2122–8. doi: 10.1093/bioinformatics/btl365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Distribution of the distance between pairs of adjacent SNPs on the genome.
Figure S2. Distribution of the number of QTL overlapping with putatively selected core regions displaying the lowest P-values (top five).
Table S1. Core region positions in Yorkshire, including the chromosome number, start position, end position and length.
Table S2. Core region positions in Landrace, including the chromosome number, start position, end position and length.
Table S3. Candidate selected regions positions in Yorkshire, including the chromosome number, start position, end position, length and REHH P-value.
Table S4. Candidate selected regions positions in Landrace, including the chromosome number, start position, end position, length and REHH P-value.
Table S5. Traits and the position of the overlapping pig QTL for the core regions with lowest P-values (top five), including traits information, the position of the core regions and QTL, and the QTL IDs in the pig QTL database.