

Abstract
The death receptor (DR) ligand TRAIL is being evaluated in clinical trials as an anti-cancer agent; however, many studies have found that TRAIL also enhances tumor progression by activating the NF-κB pathway in apoptosis-resistant cells. Although RIP1, cFLIP and caspase-8 have been implicated in TRAIL-induced JNK and NF-κB activation, underlying mechanisms are unclear. By examining the kinetics of pathway activation in TRAIL-sensitive lymphoma cells wild-type or deficient for RIP1, TRAF2, cIAP1/2 or HOIP, we report here that TRAIL induces two phases of JNK and NF-κB activation. The early phase is activated by TRAF2- and cIAP1-mediated ubiquitination of RIP1, whereas the delayed phase is induced by caspase-dependent activation of MEKK1 independent of RIP1 and TRAF2 expression. cFLIP overexpression promotes the early phase but completely suppresses the delayed phase of pathway activation in lymphoma cells, whereas Bcl-2 overexpression promotes both the early and delayed phases of the pathways. In addition, stable overexpression of cFLIP in RIP1- or TRAF2-deficient cells confers resistance to apoptosis, but fails to mediate NF-κB activation. HOIP is not essential for, but contributes to, TRAIL-induced NF-κB activation in cFLIP-overexpressing cells. These findings not only elucidate details of the mechanisms underlying TRAIL-induced JNK and NF-κB activation, but also clarify conflicting reports in the field.
Keywords: TRAIL, cFLIP, caspase-8, RIP1, NF-κB, apoptosis
1. Introduction
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and agonistic antibodies against TRAIL receptor 1 and 2 [TRAILR1/2; also known as death receptor 4 and 5 (DR4/5)] are considered a potential anti-cancer agents, as they show selective high cytotoxicity toward tumor cells with little or no toxicity against normal cells [1]. Consequently, an optimized version of recombinant human TRAIL and humanized agonistic monoclonal antibodies directed at TRAILR1 and TRAILR2 are currently being tested in clinical trials [2]. However, TRAIL has also been found to act as a tumor promoter in certain contexts, increasing cancer cell proliferation and metastasis by activating the NF-κB and c-Jun N-terminal kinase (JNK) pathways in apoptosis-resistant cells [3, 4]. Therefore, defining the mechanisms that permit TRAIL to activate JNK and NF-κB is critical for the development of strategies that maximize the potential effectiveness of TRAIL in clinical applications.
A subset of the TNF receptor (TNFR) superfamily members, such as TNFR1, Fas and TRAILR1/2, contain a death domain (DD) in their cytoplasmic tails [5]. Ligation of TNFR1 by TNFα leads to the recruitment of TNFR-associated death domain (TRADD), receptor interacting protein 1 (RIP1), TNFR-associated factor 2 (TRAF2) and cellular inhibitor of apoptosis 1 and 2 (cIAP1/2) to trigger activation of the pro-survival NF-κB and JNK pathways. On the other hand, activated TRAILR1/2 and Fas directly recruit Fas-associated death domain (FADD) and caspase-8 to activate the pro-apoptotic pathways in many types of cancer cells [1]. However, in apoptosis-resistant cancer cells, TRAIL and FasL can induce NF-κB and JNK activation and cell proliferation [1, 6]. Although the factors that determine which of the opposing responses (apoptosis or proliferation) predominates are not fully understood, overexpression of cellular FLICE-like inhibitory protein (cFLIP) has been shown to inhibit apoptosis and mediate JNK and NF-κB activation following TRAILR crosslinking [7, 8].
The long form of cFLIP (cFLIPL; hereafter referred to as cFLIP) resembles caspase-8 structurally; however, it lacks caspase activity owing to the substitution of critical amino acids in its caspase-like domain [1]. cFLIP can form a heterodimer with caspase-8, and is able to trigger limited caspase-8 activation while inhibiting its complete cleavage to the fully active p20/p10 dimer. Concomitantly, partially activated caspase-8 cleaves cFLIP at D376 to generate a p43cFLIP fragment, and this p43cFLIP has been reported to be essential for the recruitment of TRAF2 and RIP1 to TRAILR and activation of MAPK and NF-κB [7]. However, several independent studies have demonstrated that cFLIP inhibits TRAIL- and FasL-induced JNK and NF-κB activation [8-10]. In addition, caspase-8 inhibitors have been shown to inhibit or promote JNK and NF-κB activation following TRAIL or FasL treatment, depending on cell type [3, 11-13]. For example, Muhlenbeck et al. reported that zVAD-fmk inhibits TRAIL-induced JNK activation in HeLa cells but promotes this activation in Kym-1 cells [11]. Moreover, Grunert et al. reported that neither RIP1 nor cFLIP is required for TRAIL-induced NF-κB activation [14]. One of the reasons for the accumulation of these conflicting conclusions in the field is that most studies examined JNK and NF-κB activation at limited (either early or late time) time points following TRAIL or FasL stimulation.
In this study, we stably transduced cFLIP or an empty vector in lymphoma cell lines wild-type or deficient for RIP1, TRAF2, cIAP1/2 or HOIP, and analyzed the kinetics of JNK and NF-κB activation following TRAIL stimulation. Our data demonstrate that TRAIL activates two phases of JNK and NF-κB activation; the early phase is dependent on RIP1, TRAF2, cIAP1/2 and HOIP, and the delayed phase is dependent on caspase-8 and MEKK1. Notably, cFLIP plays dual roles in TRAIL signaling, promoting the early phase while suppressing the delayed phase of JNK and NF-κB activation. These findings not only provide detailed mechanisms underlying TRAIL-induced JNK and NF-κB activation, but also clarify conflicting data in the field.
2. Material and Methods
2.1. Cell lines, plasmids and reagents
RIP1+/+ and RIP1-/- Jurkat lymphoma lines were cultured in IMDM supplemented with 10% FCS and antibiotics, and the A20.2J and multiple myeloma cell lines were cultured in RPMI 1640 supplemented with 10% FCS and antibiotics. Antibodies (Abs) and reagents were purchased as follows: anti-TRAF2 (C-20) and anti-DR5 (N-19) Abs and an siRNA pool targeting human MEKK1 from Santa Cruz Biotechnology (Santa Cruz, CA); anti-cFLIP (NF6) Ab from Alexis (San Diego, CA); pan-specific anti-cIAP1/2 (MAB3400) from R&D System (Minneapolis, MN); anti-phospho-IκBα (14D4, #2859), anti-IκBα (#9242), and anti-caspase-8 (1C12) Abs from Cell Signaling (Danvers, MA); anti-RIP1 (#610459) Ab from BD Bioscience (San Jose, CA); recombinant human TRAIL from Roche (Indianapolis, IN); anti-phospho JNK Ab from Promega (Madison, WI); inhibitors of TBK1 (BX795), TAK1 (5Z-7-oxozeaenol) and AKT (124005) from Calbiochem (San Diego, CA); N-ethylmaleimide (NEM) from Sigma-Aldrich (St Louis, MO, USA); zVAD-fmk from BioMol (Plymouth Meeting, PA, USA); and cocktail inhibitors of proteases and phosphatases from Pierce (Rockford, IL). The pBabe-puro-cFLIP retroviral plasmid was generated by PCR amplification and insertion of the amplicons into the pBabe-puro vector. Retroviral pMSCV-IRES-GFP and -Bcl-2 plasmids were kindly provided by Dr. Michael Knudson (University of Iowa). All plasmids were sequenced from both sites to confirm the ligations and mutations.
2.2. Protein extraction and Western Blotting
The cells were mock treated or treated with TRAIL, harvested in ice-cold PBS, and then lysed in TNE lysis buffer R (20 mM HEPES pH 7.4, 1.0 % Triton X-100, 350 mM NaCl, 2 mM EDTA, 30 % glycerol, 1 mM DTT, 0.2 mM PMSF, 1× cocktail inhibitors of protease and phosphatases) on ice for 30 min with gentle agitation, followed by centrifugation at 12,500×g for 15 min at 4°C. 20 μg of protein samples were separated by SDS-PAGE and transferred onto nitrocellulose membranes. For analysis of IκBα and JNK phosphorylation, blots were blocked in 3% BSA/TTBS for 4 hrs before incubation with phosphoantibodies overnight at 4°C. For analysis of the expression of other proteins, blots were blocked in 5% fat-free Milk/T/TBS for 2 hrs, and then incubated with the indicated antibodies overnight at 4°C. Protein expression was then detected using ECL solution. IκBα phosphorylation blots were quantified by densitometry using integrated intensity values (ImageJ program), and the ratios of IκBα phospho-signal to the total protein were calculated and normalized to untreated 0 min signal. The values from three independent experiments were presented as mean ± SE.
2.3. Preparation of retroviral supernatants and infection of Jurkat T and MDA-MB-231 cells
293T cells at 60-70% confluency were co-transfected with 2 μg of pMD.OGP (encoding gag-pol), 2 μg of pMD.G (encoding vesicular stomatitis virus G protein) and 2 μg of pBabe-puro-cFLIP, pMSCV-IRES-GFP or -Bcl-2 by the standard calcium phosphate precipitation method. 48 hrs after transfection, the viral supernatant was collected and filtered through a 0.45 μm filter. The retroviral supernatants were then used immediately for the infection of the cells in the presence of 4 μg/ml polybrene for 24 hrs (Jurkat T cells) or 6 hrs (MDA-MB-231 cells). 48 hrs after infection, pBabe-puro-cFLIP transduced Jurkat T cells were selected with puromycin (2.0 μg/ml) for 14 days, and resistant cells were pooled. In the case of pMCSV-IRES-GFP/Bcl-2 transduced MDA-MB-231 cells, the cells were cultured for five days after infection, and then sorted for GFP positive cells using Becton Dickinson FACS DiVa. After an additional five days of culture, the GFP positive cells were FACS sorted again, and then used for the experiments within three weeks.
2.4. Real time RT-PCR
Real time RT-PCR was carried out as described previously [15, 16]. Briefly, the cells were untreated or treated with TRAIL as indicated, and total RNA was extracted using the RNeasy Mini Kit (QIAGEN). 5 μg of total RNA was treated with RQ1 RNase-free DNase for 30 min at 37 °C, and then reverse transcribed using an oligo dT-primer. 5% of resulting cDNA was then subjected to quantitative real-time PCR using the Power SYBR Green AB Master Mix and an ABI Prism 7700 Sequence Detector (Applied Biosystems). GAPDH-specific primers were used to generate an internal control, and the average threshold cycle (CT) for samples in triplicate was used in the subsequent calculations. Relative expression level of IP-10 was calculated as a ratio relative to the GAPDH expression level. The mean ± S.E. of four independent experiments was considered to be statistically significant at p < 0.05.
2.5. Cell viability assay
Cells (5.0×104/well in100 ul) were plated on 96-well plates in 2% FBS/phenol red-free RPMI, incubated for 24 hrs, and then treated with TRAIL as indicated. At 24 hrs after treatment, MTT at 0.25 mg/mL was added to the plates, and incubation continued for another 4 hrs at 37°C. After which, the 96-well plates were spun down at 1,500 rpm for 10 min, the supernatants (80 μl from each well) were carefully removed, and then 100 μl of DMSO was added to dissolve the formazan crystals. The absorbance of the solubilized product at 570 nm was measured with a 96-well plate reader. All determinations were confirmed in at least three identical experiments.
2.6. Smac-mimetic- and siRNA-mediated gene knockdown
RIP1-/- Jurkat T cells were treated with Smac-mimetic (SM; 200 ng/ml) for 4 hrs to deplete cIAP1/2. For siRNA-mediated knockdown of MEKK1 in MDA-MB-231 cells, cells were transfected with a siRNA pool to human MEKK1 (40 nM) using Lipofectamine RNAiMAX reagent (Invitrogen) and Opti-MEM (Gibco) according to the manufacturer's instruction. 48 hrs after transfection, cells were treated with TRAIL. For RIP1-/- Jurkat T cells, 1 × 107 cells were transduced with the siRNA pool to human MEKK1 (200 nM) by electroporation in serum-free Opti-MEM media with a Gene Pulser Xcell (Bio-Rad; 960-μF/230 V), and then cultured in RPMI-1640 supplemented with 10% FBS for 72 hrs before treatment with TRAIL.
3. Results
3.1. TRAIL can activate the JNK and NF-κB pathways in RIP1-deficient Jurkat T cells
RIP1 expression and cFLIP overexpression have been believed to be essential for TRAIL- and FasL-induced JNK and NF-κB activation [10, 17, 18]. Jurkat T cells and their derivative line deficient for RIP1 express cFLIP at low levels, and are sensitive to TRAIL-induced apoptosis [17]. We found that TRAIL cannot induce JNK and IκBα phosphorylation within 60 min of stimulation in either RIP1+/+ or RIP1-/-Jurkat cells, but that it can efficiently trigger JNK and IκBα phosphorylation in both cell lines at 2 hrs post-stimulation (referred to as the delayed phase of pathway activation hereafter). Notably, this delay in JNK and IκBα phosphorylation correlated with the activation of caspase-8 and -3 and cleavage of MEKK1 and cFLIP (Fig. 1A). These data suggest that TRAIL can activate the JNK and NF-κB pathways through a RIP1-independent pathway in the absence of cFLIP overexpression.
Fig. 1.
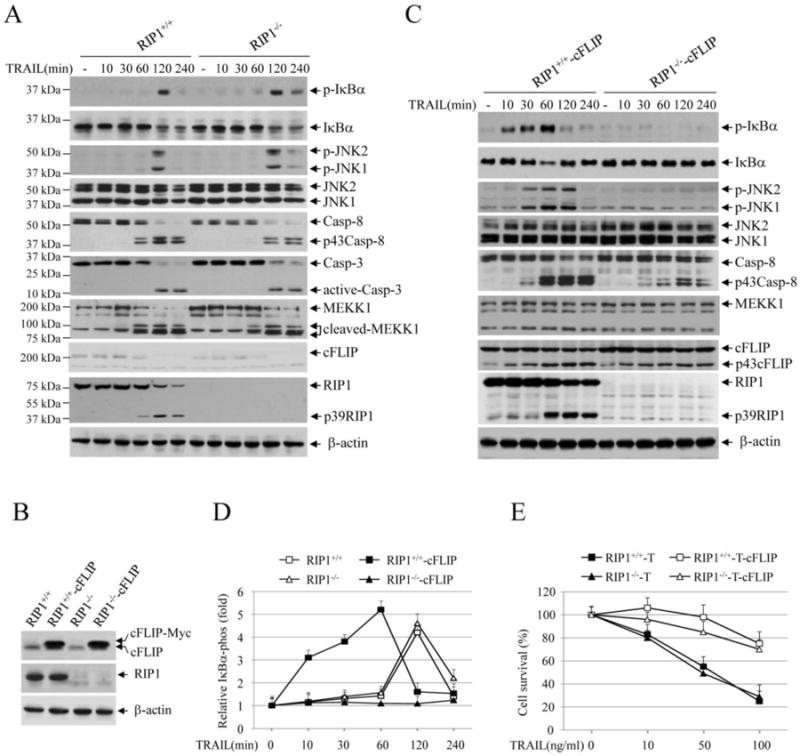
TRAIL induces IKK and JNK activation through RIP1-dependent and -independent pathways. (A) RIP1+/+ and RIP1-/- Jurkat T cells were treated with TRAIL (100 ng/ml) as indicated, and phosphorylation of IκBα and JNK, cleavage of caspase-8/3, MEKK1, cFLIP and RIP1 were examined by Western blotting. (B) RIP1+/+ and RIP1-/- Jurkat T cells were stably transduced with pBabe-puro-cFLIP (RIP1+/+-cFLIP and RIP1-/- -cFLIP), and the expression of cFLIP was then confirmed by Western blotting. (C) RIP1+/+-cFLIP and RIP1-/- -cFLIP Jurkat T cells were treated with TRAIL, and the activation of the downstream pathways was analyzed by Western blotting as in A. (D) IκBα phosphorylation blots from RIP1+/+, RIP1-/-, RIP1+/+-cFLIP and RIP1-/- -cFLIP Jurkat T cells treated with or without TRAIL was quantified by densitometry, and the ratios of IκBα phospho-signal over non-phospho-signal were normalized to 0 min signal. The relative values from three independent experiments were then presented as mean ± SE. (E) RIP1+/+, RIP1-/-, RIP1+/+-cFLIP and RIP1-/- -cFLIP Jurkat T cells were treated with TRAIL as indicated, and 24 hours after treatment, cell viability was assessed by MTT assays. Data shown are the mean ± SE of three experiments.
3.2. cFLIP overexpression exerts opposite effects on the early and delayed phases of JNK and NF-κB activation in response to TRAIL stimulation
The role of cFLIP in death ligand-induced JNK and NF-κB activation has been controversial. For example, Kataoka et al. reported that cFLIP overexpression is essential for TRAIL-induced NF-κB activation, whereas Kreuz et al. showed that cFLIP inhibits FasL-induced NF-κB activation [7, 10]. To assess the role of cFLIP overexpression in TRAIL-induced JNK and NF-κB activation, we stably overexpressed cFLIP in RIP1+/+ and RIP1-/- Jurkat cells (Fig. 1B). Interestingly, cFLIP overexpression enabled RIP1+/+ but not RIP1-/- Jurkat cells to activate the JNK and NF-κB pathways within 30 min of TRAIL stimulation (referred to as the early phase of pathway activation); however, this cFLIP overexpression completely suppressed the delayed phase of JNK and NF-κB activation observed in RIP1-/- Jurkat cells (Fig. 1C). We repeated the Western blot analysis three times in these Jurkat cells following TRAIL stimulation, quantified IκBα phosphorylation blots by densitometry, and obtained similar results (Fig. 1D). Consistent with previous publications [7, 8], cFLIP overexpression did not completely inhibit caspase-8 activation and RIP1 processing in response to TRAIL stimulation, indicating that cFLIP allows moderate activation of caspase-8. Notably, although cFLIP overexpression failed to initiate JNK and NF-κB activation in RIP1-/- Jurkat cells in response to TRAIL stimulation, it nevertheless conferred resistance to TRAIL-induced apoptosis in both RIP1+/+ and RIP1-/- Jurkat cells to a similar extent (Fig. 1E). In fact, several independent studies have demonstrated that while FADD- or caspase-8-deficient Jurkat cells are resistant to TRAIL-induced apoptosis, RIP1-deficient Jurkat cells exhibit a similar level of sensitivity to TRAIL compared to wild-type (WT) counterparts [17, 19, 20]. These data suggest that cFLIP has dual functions in the regulation of TRAIL-induced JNK and NF-κB activation: whereby, it promotes the early but suppresses the delayed phase of pathway activation.
3.3. TRAF2 is required for the early but not the delayed phase of JNK and NF-κB activation
Full activation of NF-κB by TNFα requires RIP1 ubiquitination through the K63- and M1-linkages that are mediated by two E3 ligase complexes, TRAF2/cIAP1 and HOIP/HOIL-1, respectively [15, 21]. However, the roles of these E3 ligases in TRAIL-induced JNK and NF-κB activation have not been studied in TRAF2 or HOIP knockout (KO) lymphoma cells that are sensitive to TRAIL. Our analyses revealed that, similar to the data observed in RIP1+/+ and RIP1-/- Jurkat cells, TRAIL efficiently induced JNK and IκBα phosphorylation at the later time point in A20.2J mouse B-cell lymphoma cells and their derivative lines deficient for TRAF2 or HOIP (Fig. 2A). Next, we stably overexpressed cFLIP in these cells (Fig. 2B), and then examined JNK and NF-κB activation. As shown in Fig. 2C, cFLIP overexpression promoted TRAIL-induced early phase JNK and IKK activation in WT (A20-WT) but not in TRAF2-KO cells. In HOIP-KO lymphoma cells overexpressing cFLIP, we were unable to clearly detect reduced JNK or IκBα phosphorylation compared to WT counterparts (Fig. 2D).). We repeated the Western blot analysis three times in these A20.2J lymphoma cells following TRAIL stimulation, quantified IκBα phosphorylation blots by densitometry, and obtained similar results (Fig. 2E). As expected, cFLIP overexpression also conferred overall resistance to TRAIL-induced cell death in all three lines regardless of the status of JNK and NF-κB activation (Fig. 2F). These data suggest that TRAF2 expression is also required for the activation of the early but not of the delayed phase of JNK and NF-κB activation in response to TRAIL stimulation, and that HOIP is dispensable for the activation of both phases.
Fig. 2.
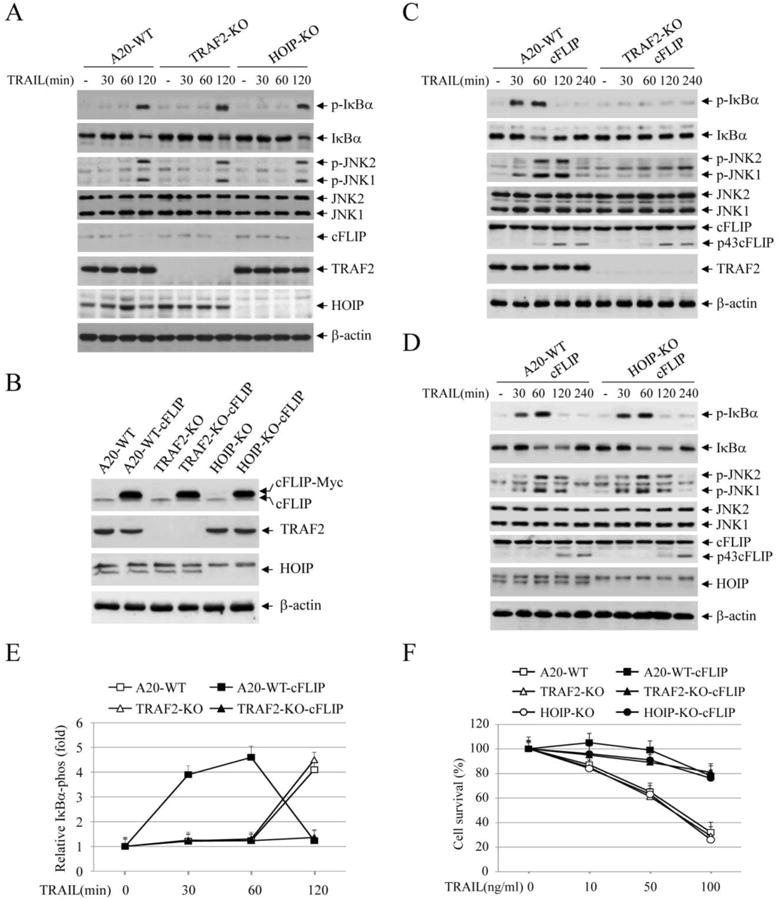
TRAIL induces IKK and JNK activation through TRAF2-dependent and -independent pathways. (A) A20.2J lymphomas cells (A20-WT) and their derivative lines deficient for TRAF2 (TRAF2-KO) or HOIP (HOIP-KO) were treated with TRAIL (100 ng/ml) as indicated, and phosphorylation of IκBα and JNK was then examined by Western blotting. (B) A20-WT, TRAF2-KO and HOIP-KO lymphoma cells were stably transduced with pBabe-puro-cFLIP (A20-WT-cFLIP, TRAF2-KO-cFLIP and HOIP-KO-cFLIP), and the expression of cFLIP was monitored by Western blotting. (C) A20-WT-cFLIP and TRAF2-KO-cFLIP lymphomas cells were treated with TRAIL (100 ng/ml), and the activation of the downstream pathways was analyzed by Western blotting as in A. (D) A20-WT-cFLIP and HOIP-KO-cFLIP lymphomas cells were treated with TRAIL and pathway activation was analyzed by Western blotting as in A. (E) IκBα phosphorylation blots from A20-WT, TRAF2-KO, A20-WT-cFLIP and TRAF2-KO-cFLIP cells treated with or without TRAIL was quantified by densitometry, and the ratios of IκBα phospho-signal over non-phospho-signal were normalized to 0 min signal. The relative values from three independent experiments were then presented as mean ± SE. (F) A20.2J lymphoma cells and their derivative lines indicated were treated with TRAIL at three different concentrations, and 24 hours after treatment, cell viability was assessed by MTT assays. Data shown are the mean ± SE of three experiments.
3.4. cIAPs are required for activation of NF-κB but not of JNK
The KMS-28PE multiple myeloma (MM) cell line is somatically deleted of both cIAP1 and cIAP2 [22]. Therefore, utilizing this cell line, we next examined the effect of cIAPs on TRAIL-induced JNK and NF-κB activation in MM cells. As shown in Fig. 3A, MM cell lines exhibited relatively high basal IκBα phosphorylation, and TRAIL stimulation further increased the early phase of IκBα phosphorylation in MM.1S and KMS-11 cells that express cIAPs, but not in cIAPs-deficient KMS-28PE cells. Interestingly, these MM lines displayed different JNK activation profiles in response to TRAIL treatment; JNK phosphorylation was transient in MM.1S and KMS-11 cells but prolonged in KMS-28PE cells. In addition, KMS-11 cells exhibited a high level of TRAF2 expression and elevated basal JNK phosphorylation. Moreover, these MM cells expressed relatively high levels of cFLIP, and failed to elicit delayed phase JNK and IκBα phosphorylation following TRAIL stimulation. To further examine the role of cIAPs in TRAIL-induced JNK and NF-κB activation, we depleted cIAPs in RIP1+/+-cFLIP Jurkat cells with Smac mimetic (SM). As shown in Fig. 3B, depletion of cIAPs resulted in a clear inhibition of IκBα phosphorylation, but only partially suppressed JNK activation. These data suggest that cIAPs are required for TRAIL-induced NF-κB activation, and that they contribute to, but are not essential for, TRAIL-induced JNK activation.
Fig. 3.
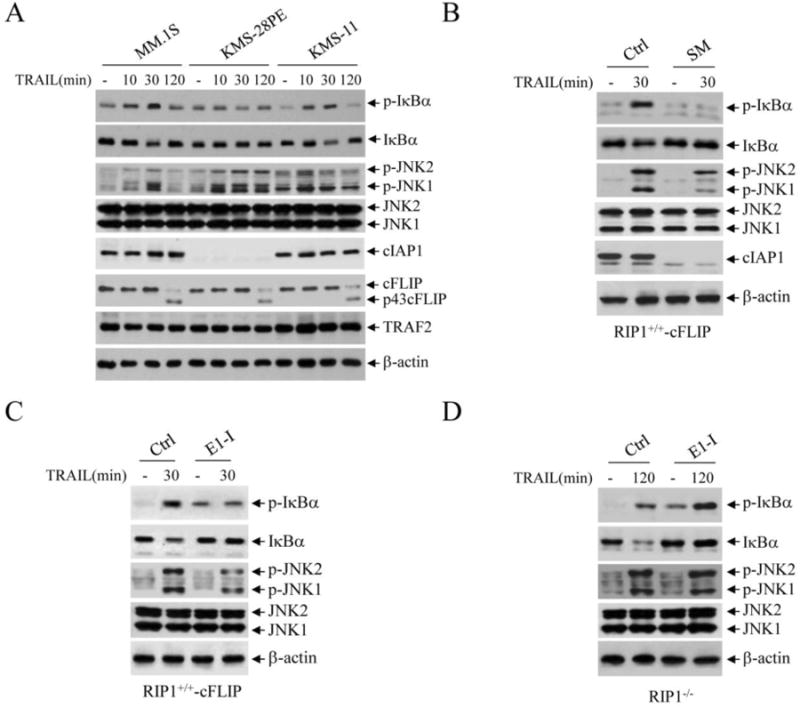
cIAP1/2 are required for TRAIL-induced IKK but not JNK activation in multiple myeloma (MM) cells. (A) MM lines that express cIAPs (MM.1S and KMS-11) or somatically deleted of both cIAP1 and cIAP2 (KMS-28PE) were treated with TRAIL (100 ng/ml) as indicated, and the phosphorylation of IκBα and JNK was examined by Western blotting. (B) RIP1+/+-cFLIP Jurkat T cells were depleted of cIAPs by Smac-mimetic (SM; 200 nM), and then the early phase of IκBα and JNK phosphorylation was monitored by Western blotting. (C) RIP1+/+-cFLIP cells were pretreated with an ubiquitin E1 inhibitor (E1-I; PYR-41, 50 μM) for 30 min, and then TRAIL-induced activation of the early phase of IκBα and JNK phosphorylation was examined by Western blotting. (D) RIP1-/- cells were pretreated with E1-I as in C, and then TRAIL-induced activation of the delayed phase of IκBα and JNK phosphorylation was then monitored by Western blotting.
3.5. The early but not the delayed phase of NF-κB and JNK activation is regulated by ubiquitination
As mentioned above, TNFα-induced NF-κB and JNK activation is dependent on noncanonical ubiquitination of effector proteins [15, 21]. To determine the role of noncanonical protein ubiquitination in TRAIL signaling, we pre-treated RIP1+/+-cFLIP Jurkat cells with an inhibitor of ubiquitin-activation enzyme E1 (PYR-41) at a high concentration (50 μM) known to completely inhibit all protein ubiquitination [23]. Interestingly, this treatment increased basal IκBα phosphorylation but completely inhibited TRAIL-induced activation of the early phase of IκBα phosphorylation (Fig. 3C). Similar to depletion of cIAPs, inhibition of E1 activity in RIP1+/+-cFLIP cells only partially suppressed the early phase of JNK activation. Conversely, the same inhibition of E1 activity in RIP1-/- Jurkat cells had no effect on TRAIL-induced activation of the delayed phase of IκBα and JNK phosphorylation (Fig. 3D). These data suggest that the early phase of IKK activation is regulated by ubiquitination of the effector proteins (e.g. RIP1, TRAF2 and cIAPs), and that early phase JNK activation is modulated by both the ubiquitination-dependent and -independent pathways.
3.6. MEKK1 is responsible for the initiation of the delayed phase of IKK and JNK activation
To understand the possible mechanisms responsible for RIP1-independent delayed phase JNK and IKK activation following TRAIL stimulation, we pre-treated RIP1-/- Jurkat cells with inhibitors of upstream kinases (e.g. TAK1, TBK1, AKT and MEKK1) and caspases (e.g. caspase-8 and -3) that have been previously implicated in TNFα- and/or TRAIL-induced JNK and IKK activation. Notably, although a small molecule inhibitor of MEKK1 is not commercially available, N-ethylmaleimide (NEM) has been shown to specifically inhibit MEKK1 but not other MAP3Ks in cell culture systems [24]. Nevertheless, NEM and zVAD completely inhibited delayed JNK and IκBα phosphorylation in RIP1-/- Jurkat cells induced by TRAIL, whereas other kinase inhibitors had no effect on this phosphorylation (Fig. 4A). Consistent with this result, siRNA-mediated knockdown of MEKK1 in RIP1-/- Jurkat cells almost completely inhibited the delayed phase of JNK and IκBα phosphorylation induced by TRAIL. Genotoxins have been shown to trigger caspase-mediated cleavage of MEKK1 to a constitutively active form (a 91 kDa fragment), and MEKK1 has been shown to directly activate both MKK4/7 (upstream MAP2Ks of JNK) and IKK [25, 26]. These published findings and our data suggest that caspase-mediate activation of MEKK1 is responsible for TRAIL-induced activation of the delayed phase of JNK and IKK in lymphoma cells.
Fig. 4.
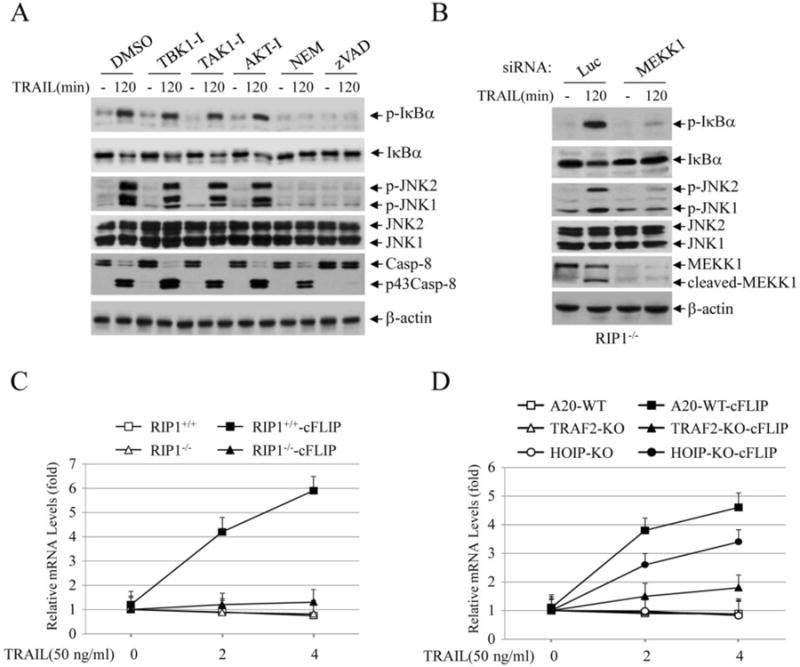
TRAIL activates the delayed phase of IKK and JNK through a MEKK1-dependent pathway. (A) RIP1-/- Jurkat cells were pretreated with inhibitors of TBK1 (TBK1-I; BX795, 1 μM), TAK1 (TAK-I; 5Z-7-oxozeaenol, 0.5 μM), AKT (AKT-I; 124005, 20 μM), MEKK1 (NEM; 0.5 mM) or caspases (zVAD; 40 μM), and then TRAIL-induced phosphorylation of IκBα and JNK at 2 hrs time point was examined by Western blotting. (B) MEKK1 was knocked down by siRNA in RIP1-/- cells, and then TRAIL-induced phosphorylation of IκBα and JNK at 2 hrs time point was monitored by Western blotting. (C) RIP1+/+, RIP1-/-, RIP1+/+-cFLIP and RIP1-/- -cFLIP Jurkat T cells were treated with TRAIL as indicated, and then the expression of IP-10 was assessed by real-time RT-PCR. Data shown are the mean ± SE of three experiments. (D) A20.2J lymphoma cells and their derivative lines indicated were treated with TRAIL for 2 and 4 hrs, and then the expression of IP-10 was monitored by real-time RT-PCR. Data shown are the mean ± SE of three experiments.
3.7. The delayed phase of NF-κB activation is not sufficient for target gene expression in Jurkat cells
To assess the roles of RIP1-dependent and -independent NF-κB activation in target gene expression, we analyzed the expression of IP-10, a well-known NF-κB target gene, in Jurkat and A20.2J cell lines. As shown in Fig. 4C, TRAIL efficiently induced IP-10 expression only in RIP1+/+-cFLIP but not in RIP1-/-, RIP1-/- -cFLIP and RIP1+/+ cells. Similarly, TRAIL also efficiently induced IP-10 expression only in A20-WT-cFLIP but not in A20-WT, TRAF2-KO, TRAF2-KO-cFLIP and HOIP-KO cells (Fig. 4D). Interestingly, TRAIL-induced IP-10 expression was clearly reduced in HOIP-KO-cFLIP cells when compared to A20-WT-cFLIP counterparts, though we were not able to detect a clear difference in IκBα phosphorylation between these two lines (Fig. 2D). It is likely that real-time RT-PCR is more sensitive in assessing the overall activation status of NF-κB. Nevertheless, these data suggest that TRAIL induces the expression of NF-κB target genes in cFLIP-overexpressing lymphoma cells in a RIP1- and TRAF2-dependent manner, and that in the absence of cFLIP overexpression, RIP1/TRAF2-independent NF-κB activation observed at later time points is incapable of inducing target gene expression in Jurkat and A20.2J cells.
3.8. The delayed phase of NF-κB activation can induce target gene expression in MDA-MB-231-Bcl-2 cells
TRAIL-induced apoptosis can be inhibited by Bcl-2 overexpression in Type-II cancer cells [27, 28]. Jurkat T cells are Type-I cells, as Bcl-2 overexpression in these cells is unable to inhibit TRAIL-induced apoptosis [27]. Consistent with this report, we also found that stable overexpression of Bcl-2 in RIP1-/-Jurkat cells neither inhibited TRAIL-induced apoptosis nor conferred upon cells the ability to express NF-κB target genes (data not shown). On the other hand, stable overexpression of Bcl-2 in a breast cancer cell line MDA-MB-231 (MDA-Bcl2) partially inhibited apoptosis when treated with TRAIL at lower concentrations (Fig. 5A); however, when the cells were treated with TRAIL at higher concentrations (75 and 100 ng/ml), no differences in cell death were observed between MDA-Bcl-2 cells and their counterparts expressing GFP (MDA-GFP). Interestingly, the kinetic profiles of JNK and NF-κB activation in MDA-MB-231 cells were different from that observed in Jurkat cells: i) the delayed phase of JNK and IκBα phosphorylation occurred earlier at 60 min and lasted until 180 min in MDA-GFP cells as opposed to the occurrence of such phosphorylation at 120 min in Jurkat cells; ii) the early phase of JNK phosphorylation was transient and weak in MDA-Bcl-2 cells; iii) both the early and delayed phase of IκBα phosphorylation were observed in MDA-Bcl-2 cells; and iv) the cleavage of caspase-8 and caspase-3 to their fully active forms was delayed but not impaired in MDA-Bcl2 cells, and this delayed activation correlated well kinetically with the delayed phase of JNK and IκBα phosphorylation in these cells (Fig. 5B).). We repeated the Western blot analysis three times in these MDA-GFP and MDA-Bcl-2 cells following TRAIL stimulation, and quantification of IκBα phosphorylation blots by densitometry revealed similar results (Fig. 5C). Notably, cFLIP expression was low in MDA-MB-231 cells and its cleavage following TRAIL stimulation was also slightly delayed in MDA-Bcl-2 cells. As expected, knockdown of MEKK1 in MDA-Bcl-2 cells suppressed the delayed phase of IκBα and JNK phosphorylation (Fig. 5D). Importantly, MEKK1 knockdown also significantly suppressed TRAIL-induced IP-10 expression in MDA-Bcl-2 cells (Fig. 5E). Collectively, these data suggest that Bcl-2 overexpression inhibits to some extent TRAIL-induced apoptosis in MDA-MB-231 cells, and that TRAIL can induce NF-κB target gene expression via a MEKK1-dependent pathway in cells like MDA-Bcl-2 that exhibit limited or delayed caspase activation in response to TRAIL stimulation.
Fig. 5.
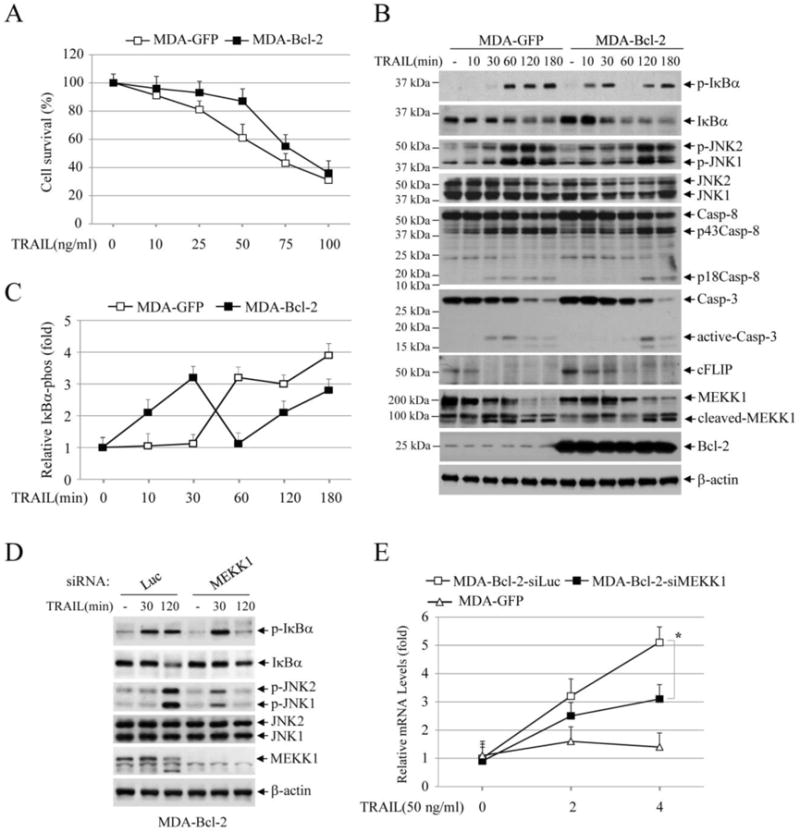
TRAIL activates both the early and delayed phase of IKK in MDA-MB-231-Bcl-2 cells. (A) MDA-MB-231 cells stably expressed with GFP (MDA-GFP) or Bcl-2 (MDA-Bcl-2) were treated with TRAIL as indicated, and 24 hours after treatment, cell viability was assessed by MTT assays. Data shown are the mean ± SE of three experiments. (B) MDA-GFP and MDA-Bcl-2 cells were treated with TRAIL (100 ng/ml) as indicated, and then the activation of the downstream pathways was monitored by Western blotting. (C) IκBα phosphorylation blots from MDA-GFP and MDA-Bcl-2 cells treated with or without TRAIL was quantified by densitometry and the ratios of IκBα phospho-signal over non-phospho-signal were normalized to 0 min signal. The relative values from three independent experiments were then presented as mean ± SE. (D) MEKK1 was knocked down by siRNA in MDA-Bcl-2 cells, and then TRAIL-induced phosphorylation of IκBα and JNK was monitored by Western blotting. (E) MEKK1 was knocked down by siRNA in MDA-Bcl-2 cells, and then TRAIL-induced expression of IP-10 was monitored by real-time RT-PCR. Data shown are the mean ± SE of three experiments.
4. Discussion
RIP1 has been suggested to be essential for TRAIL-induced activation of the JNK and NF-κB pathways [10, 17, 18]; however, Grunert et al. recently reported that RIP1 is not required for JNK and NF-κB activation in response to TRAIL stimulation [14]. These conflicting conclusions are based on the analyses of JNK phosphorylation and/or IκBα degradation at two or three time points after stimulation. By analyzing JNK and IκBα phosphorylation in RIP1+/+, RIP1-/-, RIP1+/+-cFLIP and RIP1-/- -cFLIP Jurkat T cells from 10 min to 4 hrs after TRAIL treatment, we found that TRAIL elicits two phases of JNK and IKK activation; the early phase is regulated by RIP1 in an ubiquitination-dependent manner, and the delayed phase is regulated by MEKK1 in a caspase-dependent manner (Fig. 6A and 6B). Thus, this discovery of the two phases of JNK and IKK activation by TRAIL through two different mechanisms clarifies those conflicting conclusions in the field.
Fig. 6.
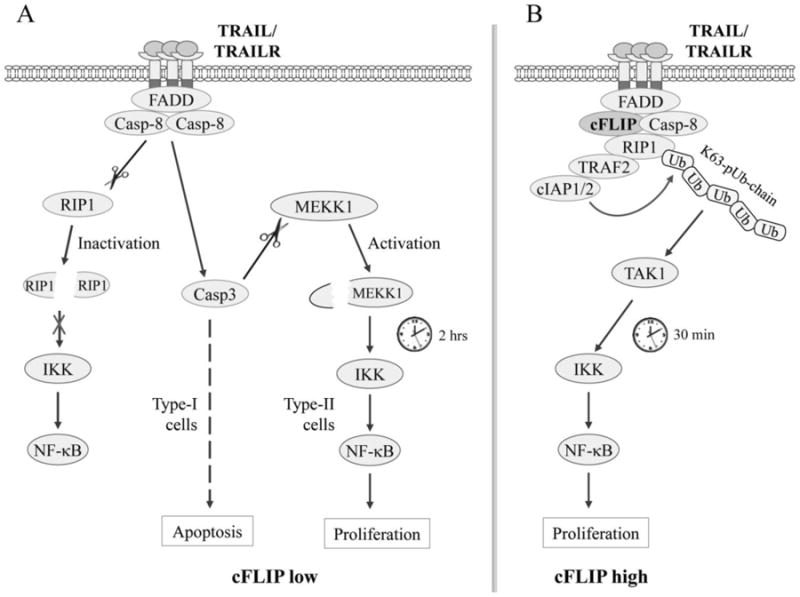
The TRAIL signaling pathways to NF-κB activation in apoptosis-sensitive and -resistant cells. (A) In cells with low cFLIP expression, ligation of TRAILR leads to its recruitment and activation of caspase-8 via FADD, which in turn cleaves RIP1 and impairs RIP1-dependent immediate IKK/NF-κB activation. Simultaneously, caspase-8 cleaves and activates caspase-3, which in turn activates MEKK1 by cleaving its regulatory domain, resulting in MEKK1-dependent activation of IKK/NF-κB at a later time. In Type-I cells, caspase-8-mediated caspase-3 activation is sufficient to trigger apoptosis, and thereby it disrupts MEKK1/IKK/NF-κB-dependent gene expression and cell proliferation, whereas in Type-II cells in which caspase-8-mediated caspase-3 activation is insufficient to trigger apoptosis, caspase-8/3-dependent MEKK1 and IKK/NF-κB activation is able to induce target gene expression and cell proliferation. (B) In cells with high cFLIP expression, cFLIP competes with caspase-8 for binding to FADD and thereby restricts its full activation, allowing the recruitment of RIP1, TRAF2 and cIAP1/2 to the TRAILR complex. TRAF2 and cIAP1/2 then catalyze RIP1 ubiquitination through K63-linkages, resulting in RIP1 ubiquitination-dependent immediate IKK/NF-κB activation.
TRAF2- and cIAP1-mediated RIP1 ubiquitination through the K63-linkage has been well established as a mediator of TNFα-induced NF-κB activation [21]. Although TRAF2 was originally considered to function as an E3 ligase to activate the JNK and NF-κB pathways, recent studies suggest that TRAF2 lacks E3 ligase activity, and that it activates the JNK and NF-κB pathways by recruiting the potent E3 ligases cIAP1 and cIAP2 to the receptor complexes [29-31]. Interestingly, our data reveal that TRAF2 has a cIAP1/2-independent function in regulating JNK activation in response to TRAIL stimulation. In A20-TRAF2-KO lymphoma cells, TRAIL-induced JNK activation was impaired; however, depletion of cIAP1/2 with SM or the inhibition of protein ubiquitination with an E1 inhibitor only partially suppressed JNK activation. The N-terminal RING domain of TRAF2 has been shown to recruit MEKK1 to TNFR1, triggering JNK activation [32]. Thus, these published findings and our data suggest that the early phase of JNK activation is regulated by two events, TRAF2/cIAP1-mediated ubiquitination of RIP1 and TRAF2-dependent recruitment of MEKK1.
MEKK1 has also been shown to directly activate the IKK complex in response to cytokines such as TNFα and stresses such as UV irradiation [26]. However, gene knockout studies revealed that MEKK1 regulates stress-induced JNK activation but is not essential for proinflammatory cytokine-induced activation of JNK and IKK [33, 34]. Nevertheless, these published studies demonstrated that activated MEKK1 is able to activate both the JNK and NF-κB pathways. MEKK1 is a caspase substrate, and is cleaved by caspase-3 at D874 in response to TRAIL and genotoxins, which results in the generation of a constitutively active 91 kDa kinase fragment [12, 25]. We also observed MEKK1 processing to this constitutively active form in both RIP1+/+ and RIP1-/- Jurkat T cells following TRAIL treatment. In addition, knockdown of MEKK1 and inhibition of caspases blocked the delayed phase of JNK and NF-κB activation in RIP1-/- cells. These data suggest that caspase-induced MEKK1 activation is responsible for activation of the delayed phase of JNK and NF-κB in response to TRAIL stimulation. This notion is also indirectly supported by the findings of Muhlenbeck et al. who have demonstrated that TRAIL activates JNK via caspase-dependent and caspase-independent pathways [11].
The role of cFLIP in TRAIL-induced JNK and NF-κB activation has been controversial; for example, cFLIP overexpression has been shown to either inhibit or promote JNK and NF-κB activation, depending on cell type and experimental approach [3, 7-13]. Our data demonstrate that cFLIP overexpression confers upon cells the ability to induce the early phase of JNK and IKK activation while inhibiting caspase-dependent cleavage of MEKK1 and subsequent activation of the delayed phase of JNK and NF-κB. Thus, our identification of the opposing effects of cFLIP on the early and delayed phases of JNK and IKK activation offers an explanation for the conflicting reports in the literature. Notably, cFLIP is also known to exhibit dual effects on caspase-8 activity, as cFLIP overexpression triggers limited caspase-8 activation while suppressing the complete cleavage of caspase-8 to its fully active p20/p10 dimer [1]. Such dual effects of cFLIP on caspase-8 activity may also provide explanation for the slow kinetics of JNK and NF-κB activation in response to TRAIL stimulation: i) cFLIP overexpression allows caspase-8 processing to the moderately active p43caspase-8 fragment which is able to cleave RIP1 but not caspase-3, as such it inhibits apoptosis; and ii) this limited RIP1 cleavage allows for the formation of the RIP1-TRAF2-cIAP1 complex and activation of the JNK and NF-κB pathways while also reducing the amount of full-length RIP1 within the complex to prohibit quick and robust activation of JNK and IKK. In other words, RIP1 cleavage is the rate-limiting event that controls the kinetics of JNK and IKK activation. In apoptosis-sensitive cells, fully activated caspase-8 immediately cleaves RIP1 within the receptor complex, and thus abolishes RIP1-dependent JNK and IKK activation, whereas in cFLIP-overexpressing, apoptosis-resistant cells, suppression of caspase-8 activity to a moderate level permits as well as limits the formation of the RIP1-TRAF2-cIAP1 complex, resulting in slow and weak activation of the JNK and IKK pathways.
The expression of NF-κB target genes is regulated at multiple levels, including negative regulation by caspases that cleave core elements of the NF-κB pathways, such as cIAP1, IKKβ and p65 subunit of NF-κB [21, 35, 36]. Although TRAIL stimulation induced the early phase of NF-κB activation in RIP1+/+-cFLIP and A20-WT-cFLIP cells and the delayed phase of NF-κB activation in RIP1-/- and A20-TRAF2-KO cells, the expression of IP-10 was efficiently induced in RIP1+/+-cFLIP and A20-WT-cFLIP cells, but not in RIP1-/- and A20-TRAF2-KO cells. This suggests that strong activation of caspase-8 and caspase-3 in RIP1-/- and A20-TRAF2-KO cells at the same later time points could lead to proteolytic cleavage of the core elements of the NF-κB pathway as well as other general co-factors required for gene transcription. As such, even though NF-κB is activated at later times, concomitant activation of caspases impairs gene transcription. Notably, although variation in TRAIL-induced IκBα phosphorylation was inconclusive in A20-HOIP-KO-cFLIP cells, IP-10 expression was clearly reduced. Western blotting is a semi-quantitative method, and thus it seems that real-time RT-PCR analysis of gene expression is more sensitive than Western blot analysis of IκBα phosphorylation in assessing overall activation status of NF-κB. Nevertheless, these data suggest that HOIP also contributes to a certain extent to TRAIL-induced expression of NF-κB target genes.
Unlike cFLIP whose overexpression protects nearly all type of tumor cells form death ligand-induced apoptosis, overexpression of Bcl-2 protects Type-II but not Type-I tumor cells from apoptosis triggered by DR ligation [27, 28]. Interestingly, Bcl-2 overexpression in MDA-MB-231 cells partially suppressed TRAIL-induced apoptosis, but did not completely inhibit caspase activation and MEKK1 cleavage. As a consequence, partial inhibition of apoptosis and limited activation of caspases in MDA-Bcl-2 cells allowed RIP1-dependent early and MEKK1-dependent delayed IKK activation, resulting in efficient expression of NF-κB target genes in response to TRAIL stimulation. Importantly, knockdown of MEKK1 significantly reduced the expression of IP-10 in MDA-Bcl-2 cells. These data suggest that processing of a small portion of caspase-8 to a fully active form of p18/p20 and the cleavage of MEKK1 to the constitutively active form of p91MEKK1 have pathophysiological relevance in promoting TRAIL-induced JNK and NF-κB activation and target gene expression in tumor cells in which TRAIL stimulation only activates caspase-8 and caspase-3 to levels that are not sufficient for inducing massive apoptosis. For example, DR ligation-induced activation of the FADD-caspase-8-caspase-3 pathway is not sufficient to trigger apoptosis in Type-II cancer cells.
5. Conclusion
Our study provides evidence for the existence of two phases of JNK and NF-κB activation in cancer cells; the early phase is activated by TRAF2/cIAP1-mediated ubiquitination of RIP1, and the delayed phase is induced by caspase-mediated cleavage and activation of MEKK1 (Fig. 6A and 6B). Notably, cFLIP overexpression exerts opposite effects on the early and delayed phases of the pathway activation, whereas Bcl-2 overexpression partially suppresses caspase activation in some cancer cells to permit activation of both the early and delayed phase of pathway activation. These findings not only elucidate the details of the mechanisms underlying TRAIL-induced JNK and NF-κB activation, but also clarify conflicting reports in the field.
Highlights.
TRAIL induces two phases of JNK and NF-κB activation.
The early phase is activated by TRAF2/cIAP1 -mediated ubiquitination of RIP 1.
The delayed phase is mediated by caspase-dependent activation of MEKK1.
cFLIP exerts opposite effects on the early and delayed phases of pathway activation.
These findings clarify conflicting reports in the field of TRAIL study.
Acknowledgments
We thank Dr. Adrian Ting (Mount Sinai Medical Center, NY) for providing us with RIP1+/+ and RIP1-/- Jurkat T cells, Xiaodong Wang (University of Texas Southwestern Medical Center, Dallas, USA) for Smac-mimetic, Michael Knudson (University of Iowa) for pMSCV-IRES-GFP and -Bcl-2 plasmids, Apollina Goel (University of Iowa) for MM.1S and KMS-28PE cells, and Fenghuang Zhan for KMS-11 cells. Support by NCI grant CA138475 (to HH) is gratefully acknowledged.
Abbreviations
- cIAP
cellular inhibitor of apoptosis
- cFLIP
cellular caspase-8 (FLICE)-like inhibitory protein
- cIAP1
cellular inhibitor of apoptosis 1
- DR
death receptor
- FADD
Fas-associated death domain
- IκB
inhibitor of κB
- IKK
IκB kinase
- JNK
c-Jun N-terminal kinase
- KO
knockout
- MEKK1
mitogen-activated kinase kinase (MEK) kinase 1
- NF-κB
nuclear factor κB
- RIP1
receptor interacting protein 1
- siRNA
small interfering RNA
- SM
Smac mimetic
- TAK1
transforming growth factor β-activated kinase 1
- TBK1
TANK-binding kinase 1
- TNFα
tumor necrosis factor α
- TNFR
TNF receptor
- TRADD
TNFR associated death domain
- TRAFs
TNFR associated factor2
- TRAIL
Tumor necrosis factor-related apoptosis-inducing ligand
- WT
wild-type
Footnotes
None of the authors have conflicts of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gonzalvez F, Ashkenazi A. Oncogene. 2010;29:4752–4765. doi: 10.1038/onc.2010.221. [DOI] [PubMed] [Google Scholar]
- 2.Stuckey DW, Shah K. Trends Mol Med. 2013 doi: 10.1016/j.molmed.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trauzold A, Siegmund D, Schniewind B, Sipos B, Egberts J, Zorenkov D, Emme D, Roder C, Kalthoff H, Wajant H. Oncogene. 2006;25:7434–7439. doi: 10.1038/sj.onc.1209719. [DOI] [PubMed] [Google Scholar]
- 4.Ishimura N, Isomoto H, Bronk SF, Gores GJ. Am J Physiol Gastrointest Liver Physiol. 2006;290:G129–136. doi: 10.1152/ajpgi.00242.2005. [DOI] [PubMed] [Google Scholar]
- 5.Aggarwal BB. Nat Rev Immunol. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 6.Lamkanfi M, Declercq W, Vanden Berghe T, Vandenabeele P. J Cell Biol. 2006;173:165–171. doi: 10.1083/jcb.200509092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kataoka T, Tschopp J. Mol Cell Biol. 2004;24:2627–2636. doi: 10.1128/MCB.24.7.2627-2636.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Siegmund D, Mauri D, Peters N, Juo P, Thome M, Reichwein M, Blenis J, Scheurich P, Tschopp J, Wajant H. J Biol Chem. 2001;276:32585–32590. doi: 10.1074/jbc.M100444200. [DOI] [PubMed] [Google Scholar]
- 9.Iyer AK, Azad N, Talbot S, Stehlik C, Lu B, Wang L, Rojanasakul Y. J Immunol. 2011;187:3256–3266. doi: 10.4049/jimmunol.1002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kreuz S, Siegmund D, Rumpf JJ, Samel D, Leverkus M, Janssen O, Hacker G, Dittrich-Breiholz O, Kracht M, Scheurich P, Wajant H. J Cell Biol. 2004;166:369–380. doi: 10.1083/jcb.200401036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muhlenbeck F, Haas E, Schwenzer R, Schubert G, Grell M, Smith C, Scheurich P, Wajant H. J Biol Chem. 1998;273:33091–33098. doi: 10.1074/jbc.273.49.33091. [DOI] [PubMed] [Google Scholar]
- 12.Sun BK, Kim JH, Nguyen HN, Oh S, Kim SY, Choi S, Choi HJ, Lee YJ, Song JJ. Oncol Rep. 2011;25:537–544. doi: 10.3892/or.2010.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi C, Kutsch O, Park J, Zhou T, Seol DW, Benveniste EN. Mol Cell Biol. 2002;22:724–736. doi: 10.1128/MCB.22.3.724-736.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grunert M, Gottschalk K, Kapahnke J, Gundisch S, Kieser A, Jeremias I. Cell Death Dis. 2012;3:e414. doi: 10.1038/cddis.2012.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blackwell K, Zhang L, Workman LM, Ting AT, Iwai K, Habelhah H. Mol Cell Biol. 2013;33:1901–1915. doi: 10.1128/MCB.01416-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin Y, Devin A, Cook A, Keane MM, Kelliher M, Lipkowitz S, Liu ZG. Mol Cell Biol. 2000;20:6638–6645. doi: 10.1128/mcb.20.18.6638-6645.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ofengeim D, Yuan J. Nat Rev Mol Cell Biol. 2013;14:727–736. doi: 10.1038/nrm3683. [DOI] [PubMed] [Google Scholar]
- 19.Braeuer SJ, Buneker C, Mohr A, Zwacka RM. Mol Cancer Res. 2006;4:715–728. doi: 10.1158/1541-7786.MCR-05-0231. [DOI] [PubMed] [Google Scholar]
- 20.Ting AT, Pimentel-Muinos FX, Seed B. EMBO J. 1996;15:6189–6196. [PMC free article] [PubMed] [Google Scholar]
- 21.Workman LM, Habelhah H. Cell Signal. 2013;25:1654–1664. doi: 10.1016/j.cellsig.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, Van Wier S, Tiedemann R, Shi CX, Sebag M, Braggio E, Henry T, Zhu YX, Fogle H, Price-Troska T, Ahmann G, Mancini C, Brents LA, Kumar S, Greipp P, Dispenzieri A, Bryant B, Mulligan G, Bruhn L, Barrett M, Valdez R, Trent J, Stewart AK, Carpten J, Bergsagel PL. Cancer Cell. 2007;12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Y, Kitagaki J, Dai RM, Tsai YC, Lorick KL, Ludwig RL, Pierre SA, Jensen JP, Davydov IV, Oberoi P, Li CC, Kenten JH, Beutler JA, Vousden KH, Weissman AM. Cancer Res. 2007;67:9472–9481. doi: 10.1158/0008-5472.CAN-07-0568. [DOI] [PubMed] [Google Scholar]
- 24.Cross JV, Templeton DJ. Biochem J. 2004;381:675–683. doi: 10.1042/BJ20040591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Widmann C, Gerwins P, Johnson NL, Jarpe MB, Johnson GL. Mol Cell Biol. 1998;18:2416–2429. doi: 10.1128/mcb.18.4.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee FS, Hagler J, Chen ZJ, Maniatis T. Cell. 1997;88:213–222. doi: 10.1016/s0092-8674(00)81842-5. [DOI] [PubMed] [Google Scholar]
- 27.Suliman A, Lam A, Datta R, Srivastava RK. Oncogene. 2001;20:2122–2133. doi: 10.1038/sj.onc.1204282. [DOI] [PubMed] [Google Scholar]
- 28.Fulda S, Meyer E, Debatin KM. Oncogene. 2002;21:2283–2294. doi: 10.1038/sj.onc.1205258. [DOI] [PubMed] [Google Scholar]
- 29.Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, Arora V, Mak TW, Lacasse EC, Waring J, Korneluk RG. Proc Natl Acad Sci U S A. 2008;105:11778–11783. doi: 10.1073/pnas.0711122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K, Fairbrother WJ, Vucic D. J Biol Chem. 2008;283:24295–24299. doi: 10.1074/jbc.C800128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yin Q, Lamothe B, Darnay BG, Wu H. Biochemistry. 2009;48:10558–10567. doi: 10.1021/bi901462e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baud V, Liu ZG, Bennett B, Suzuki N, Xia Y, Karin M. Genes Dev. 1999;13:1297–1308. doi: 10.1101/gad.13.10.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yujiri T, Ware M, Widmann C, Oyer R, Russell D, Chan E, Zaitsu Y, Clarke P, Tyler K, Oka Y, Fanger GR, Henson P, Johnson GL. Proc Natl Acad Sci U S A. 2000;97:7272–7277. doi: 10.1073/pnas.130176697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yujiri T, Sather S, Fanger GR, Johnson GL. Science. 1998;282:1911–1914. doi: 10.1126/science.282.5395.1911. [DOI] [PubMed] [Google Scholar]
- 35.Hayden MS, Ghosh S. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 36.Lamkanfi M, Festjens N, Declercq W, Vanden Berghe T, Vandenabeele P. Cell Death Differ. 2007;14:44–55. doi: 10.1038/sj.cdd.4402047. [DOI] [PubMed] [Google Scholar]