

Background: Tpl2 kinase plays an essential, non-redundant role in activating ERK during TLR signaling.
Results: TLRs 2, 4, and 7 directly induce IKKβ-Tpl2-ERK signaling; TLRs 3 and 9 activate ERK indirectly via autocrine ROS signaling.
Conclusion: Tpl2-dependent ROS generation drives ERK phosphorylation during TLR 3 and 9 signaling.
Significance: The different contributions of Tpl2 to TLR signaling pathways influences early host defense mechanisms.
Keywords: Extracellular-signal-regulated Kinase (ERK), Mitogen-activated Protein Kinase (MAPK), Reactive Oxygen Species (ROS), Signal Transduction, Toll-like Receptor (TLR)
Abstract
Signal transduction via NFκB and MAP kinase cascades is a universal response initiated upon pathogen recognition by Toll-like receptors (TLRs). How activation of these divergent signaling pathways is integrated to dictate distinct immune responses to diverse pathogens is still incompletely understood. Herein, contrary to current perception, we demonstrate that a signaling pathway defined by the inhibitor of κB kinase β (IKKβ), MAP3 kinase tumor progression locus 2 (Tpl2/MAP3K8), and MAP kinase ERK is differentially activated by TLRs. TLRs 2, 4, and 7 directly activate this inflammatory axis, inducing immediate ERK phosphorylation and early TNFα secretion. In addition to TLR adaptor proteins, IKKβ-Tpl2-ERK activation by TLR4 is regulated by the TLR4 co-receptor CD14 and the tyrosine kinase Syk. Signals from TLRs 3 and 9 do not initiate early activation of IKKβ-Tpl2-ERK pathway but instead induce delayed, NADPH-oxidase-dependent ERK phosphorylation and TNFα secretion via autocrine reactive oxygen species signaling. Unexpectedly, Tpl2 is an essential regulator of ROS production during TLR signaling. Overall, our study reveals distinct mechanisms activating a common inflammatory signaling cascade and delineates differences in MyD88-dependent signaling between endosomal TLRs 7 and 9. These findings further confirm the importance of Tpl2 in innate host defense mechanisms and also enhance our understanding of how the immune system tailors pathogen-specific gene expression patterns.
Introduction
Toll-like receptors (TLRs)2 are a major class of pattern recognition receptors that specifically detect conserved pathogen-associated molecular patterns (PAMPs) and alarm the host of an infection. TLRs are expressed either on the cell surface or within specific intracellular compartments. Cell surface TLRs (TLR1, 2, 4, 5, and 6) detect outer membrane components of microbes, whereas endosomal TLRs (TLR3, 7, 8, and 9) sense microbial nucleic acids (1). Signals emanating from TLRs activate various intracellular signaling cascades including NFκB, mitogen-activated protein (MAP) kinases, and interferon regulatory factors that collectively induce the secretion of host protective proinflammatory cytokines and interferons (1). The magnitude and quality of this early response also regulates the initiation of adaptive responses (2). Despite extensive research, the precise molecular mechanisms that dictate specific cellular responses to TLRs are still incompletely understood.
NFκB and MAP kinase pathways are the two major signaling cascades initiated after recognition of specific PAMPs by TLRs (3). Engagement of all TLRs activates both of these pathways, and cross-talk between them coordinates the cellular responses to external stimuli (3, 4). One of the key regulatory molecules known to coordinate the activation of both NFκB and MAP kinase pathways is the inhibitor of κB kinase β (IKKβ). IKKβ is activated in response to proinflammatory stimuli, including TLRs and cytokines, and it regulates activation of NFκB and MAP kinases by phosphorylating IκBα, NFκB1p105, and the MAP3 kinase, Tumor progression locus 2 (Tpl2) (5, 6).
Tpl2 is a serine-threonine kinase originally identified as a proto-oncogene and expressed in both hematopoietic and non-hematopoietic compartments (7). Differential translation initiation of Tpl2 mRNA gives rise to 52 and 58 kDa isoforms expressed in equimolar levels in macrophages (8). In unstimulated cells, Tpl2 is constitutively associated with NFκB1p105, and this interaction is necessary for Tpl2 stability but blocks Tpl2 kinase activity (9). Phosphorylation of p105 by IKKβ leads to Tpl2 release (10). IKKβ also mediates phosphorylation of Tpl2 at threonine 290 and serine 400, which regulates Tpl2 kinase activity (5, 10–12). Once phosphorylated, Tpl2 transiently transduces signals but is unstable and undergoes rapid proteosomal degradation (9, 13). The p58 isoform is preferentially released and degraded in LPS treated macrophages, since only this isoform undergoes IKKβ-mediated Thr290 phosphorylation (5, 10).
Early studies on Tpl2 signaling established the non-redundant role of Tpl2 in LPS-mediated activation of ERK1/2 (14). Tpl2−/− mice are resistant to endotoxin-induced shock due to defective ERK-dependent TNFα secretion. Further studies demonstrated a cell type- and stimulus-specific role for Tpl2 in transducing signals leading to the production of a variety of immune mediators, including IL-1β, IL-10, IL-12, and COX-2 (15–18). Because of its role in regulating expression, secretion and signaling of proinflammatory cytokines like TNFα and IL-1β, Tpl2 is considered an attractive target for immunotherapy of inflammatory conditions. Several studies have examined Tpl2 regulation of signal transduction and cellular responses to diverse TLR ligands (19). Tpl2 kinase activity and Tpl2-dependent ERK phosphorylation were demonstrated in macrophages in response to ligands of TLR2, 3, 4, 7, and 9 (19). Moreover, ERK phosphorylation in response to LPS, TNFα, CpG, Pam3CSK, poly I:C, flagellin, and R848 were blocked in nfkb1SSAA macrophages which express a p105 mutant that cannot be phosphorylated by IKKβ (20). From these studies, it has been concluded that all TLRs similarly activate the Tpl2-ERK signaling pathway.
To better understand the molecular mechanisms utilized by different TLRs to distinguish their cellular responses, we examined the induction of proinflammatory genes and signal transduction events by diverse TLR ligands, focusing on Tpl2 signaling. Contrary to prevailing thought, we demonstrate that the signaling pathway defined by IKKβ, Tpl2, and ERK, which helps to initiate and influence the nature of the innate immune response, is differentially regulated by TLRs. Among the MyD88-coupled TLRs, TLR4 uniquely requires CD14 and the tyrosine kinase Syk for Tpl2-ERK activation. TLRs 3 and 9 do not induce Tpl2-p58 phosphorylation or early ERK activation; instead they induce delayed ERK activation that is dependent upon autocrine signaling by reactive oxygen species (ROS) generated in a Tpl2-dependent manner. These findings demonstrate a differential mechanism of ERK activation by diverse TLRs and also identify divergent signaling pathways emanating from the MyD88-dependent endosomal TLRs 7 and 9. Overall, our study provides a better understanding of signaling pathways utilized by major TLRs and also demonstrate a major role for Tpl2 in eliciting host protective immune responses, including the generation of antimicrobial reactive oxygen species.
EXPERIMENTAL PROCEDURES
Mice
Wild type (C57BL/6J), myd88−/−, ticamlps2/lps2, and ifnar1−/− mice were purchased from The Jackson Laboratory. Tpl2−/− mice backcrossed to C57B6/J were kindly provided by Dr. Philip Tsichlis (Tufts University) and Thomas Jefferson University. Femurs and tibiae from cd14−/− mice were generously provided by Dr. Donald Harn (University of Georgia). Femurs and tibiae from myd88/trif double-knock-out mice (21) were kindly provided by Dr. Alan Sher (NIAID, NIH). Animals were housed in sterile microisolator cages in the Central Animal Facility of the College of Veterinary Medicine. The Institutional Animal Care and Use Committee (IACUC) of the University of Georgia approved all animal experiments.
Generation of Bone Marrow-derived Cells
Bone marrow-derived macrophages (BMDMs) and dendritic cells (BMDCs) were generated from age- and sex-matched mice as described previously (16). The cells were cultured at a concentration of 2 × 106/ml in DMEM low glucose medium containing 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mm l-glutamine on sterile Petri dishes for 7 days at 37 °C supplemented with 10 ng/ml macrophage colony stimulating factor (M-CSF) (PeproTech). Fresh medium equal to half of the initial culture volume containing M-CSF was added on day 5 of the culture. On day 6, after removing the medium and washing the cells with PBS, the adherent cells were incubated with cell dissociation buffer (Invitrogen) for 10 min at 37 °C. The harvested cells were counted and replated in the same culture medium overnight before stimulation.
BMDCs and plasmacytoid DCs (pDCs) were generated by culture of bone marrow cells in complete RPMI (RPMI 1640 containing 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, 2 mm l-glutamine, and 50 μm 2-ME). Cells were cultured with 40 ng/ml GM-CSF (PeproTech) for 7 days or 100 ng/ml Flt3 ligand (PeproTech) for 10 days for BMDCs and pDCs, respectively. For BMDCs, nonadherent cells were harvested on day 7, and CD11c+ cells were isolated using CD11c microbeads and MACS columns (Miltenyi Biotec). The purity of the cell population was determined to be more than 95% by flow cytometry. CD11c+CD11b−B220+ pDCs were sorted using a Beckman Coulter MoFlo XDP cell sorter to >98% purity.
Peritoneal Exudate Cell Isolation
Mice were injected intraperitoneally with 1 ml of 3% Brewer thioglycollate medium to recruit macrophages. After 72 h, mice were sacrificed, and the peritoneal cavity was lavaged three times with 3 ml of sterile PBS to collect recruited cells. Cells were centrifuged at 1200 rpm for 10 min at room temperature and were resuspended in supplemented DMEM.
Antibodies and Other Reagents
The following antibodies were used for immunoblotting: Tpl2 (Cot M-20), ERK1 and ERK2, β-actin (Santa Cruz Biotechnology), phospho-ERK1/2 (Thr202/Tyr204), pIKKα/β (Ser176/180), IKKβ, pIκBα (Ser32), IκBα, pNFκBp105, NFκB-p65, and pSTAT1 (Tyr701) (Cell Signaling Technology). In some experiments cells were pretreated with 10 μg/ml cycloheximide (Sigma-Aldrich), 100 ng/ml pertussis toxin (Sigma-Aldrich), or 20 μm diphenyleneiodonium (DPI) (Sigma-Aldrich) for 30 min or 10 mm glutathione (GSH) (Sigma-Aldrich) for 10 min. To investigate ROS signaling, cells were treated with 1 or 10 mm H2O2 for 1 h. To block cytokine signaling 1 μg/ml of anti-TNFα (R&D Systems) or anti-IL-1β (BD Pharmingen) was used.
Cell Stimulation and Measurement of Cytokines
Bone marrow-derived cells or PECs at 1 × 106/ml concentration were stimulated with Pam3CSK4 (1 μg/ml), poly I:C (10 μg/ml), ultrapure LPS from Escherichia coli 0111:B4 (1 μg/ml), R848 (1 μg/ml), CpG ODN2395 (10–25 μg/ml, as noted), or CpG ODN1668 (0.5–1.5 μm). CpG ODN2395 was used in all experiments except Fig. 2C, in which CpG ODN1668 was used for comparison. All TLR ligands were purchased from Invivogen. Cell culture supernatants were collected at different time points after stimulation, and TNFα, IL-10, and IFNβ levels were measured by ELISA (eBioscience, PBL Interferon Source). Alternatively, BMDMs were directly stimulated with the following cytokines: rmTNFα (10 ng/ml; eBioscience), rmIL-1β (10 ng/ml; eBioscience), rmIFNα A (2000 IU/ml; R&D Systems), or rhIFNβ (10 ng/ml; Peprotech).
FIGURE 2.

Signaling by TLR3 and 9 fails to induce Tpl2-p58 or early ERK phosphorylation. A, BMDMs from WT mice were left untreated or stimulated with Pam3CSK4, poly I:C, LPS, R848, or CpG ODN for 15 min. B, BMDMs from WT and tpl2−/− mice were left untreated or stimulated with Pam3CSK4, poly I:C, LPS, R848, and CpG ODN for 1 h. C, BMDMs from WT mice were treated with 10 or 25 μg/ml poly I:C and CpG ODN or 1 μg/ml LPS for 15 min. (left panel) or stimulated with CpG-C ODNs (ODN2395), increasing doses of CpG-B ODNs (ODN1668) or LPS for 15 min (right panel). D, BMDMs from WT mice were left untreated or stimulated with poly I:C or LPS for 1 to 2 h. E, PMs, pDCs, or BMDCs from WT mice were left untreated or stimulated with poly I:C, LPS, or CpG ODN for 15 min. A–E, WCL were immunoblotted with antibodies recognizing Tpl2, pERK1/2, ERK1/2, and β-actin. Dashed line indicates that within a single exposure intervening lanes were removed. Data are representative of 2–4 independent experiments.
Analysis of mRNA Expression
BMDMs stimulated with various ligands were washed with PBS after collecting supernatants, and cell lysates were prepared using TRK lysis buffer (Omega Bio-Tek). RNA was extracted using a Total RNA Kit (Omega Bio-Tek). Real time PCR was performed after synthesizing cDNA using High capacity cDNA Reverse Transcription kit (Applied Biosystems). The expression of il12b (Mm00434174_m1), il6 (Mm00446190_m1), tnfα (Mm00443258_m1), il10 (Mm01288386_m1), ccl2 (Mm00441242_m1), ccl5 (Mm01302427_m1), nos2 (Mm00440502-m1), and actinb (4352341E-1112017) were determined by real-time PCR (Applied Biosystems). RT-PCR reactions were performed in microAmp Fast plates (Applied Biosystems) using SensiFAST Probe Hi-ROX kit (Bioline) and a StepOnePlus RT-PCR machine (Applied Biosystems). Relative gene expression levels were calculated by normalizing the Ct levels of target gene to both endogenous actin levels and an unstimulated WT control using the ΔΔCt method.
Protein Analysis
Cell lysates were prepared in protein lysis buffer (0.5% Triton X-100, 50 mm Tris-HCl (pH 7.5), 300 mm NaCl, 2 mm EDTA, 0.4 mm Na3VO4, 2.5 mm aprotinin, 2.5 mm leupeptin, and 2.5 μm nitrophenyl p-guanidinobenzoate), and protein concentrations were measured using a BCA kit (Pierce). Approximately 20 μg of denatured proteins were separated on 4–12% gradient gels (Invitrogen) under reducing conditions and were transferred to PVDF membranes using the iBlot Gel Transfer system (Invitrogen). Membranes were probed with various antibobies followed by horseradish peroxidase-labeled secondary antibodies. Protein bands were visualized by enhanced chemiluminescent reagent (Lumigen) and Amersham Biosciences Hyperfilm ECL (GE Healthcare). In some experiments, images were acquired using a Fluorchem Hd2 imaging system (Alpha Innotech Corp.).
Confocal Microscopy
BMDMs plated on chamber slides (Lab-Tek) were treated with poly I:C (10 μg/ml), CpG ODN2395 (10 μg/ml) or LPS (1 μg/ml) for 1 h. Cells were washed and fixed in 3.7% paraformaldehyde solution at 37 °C for 30 min. Fixed cells were incubated in blocking buffer (1% BSA in PBS) for 30 min, followed by a 1 h incubation with primary antibody (NFκBp65, 1:100 dilution, Cell Signaling Technology) followed by a fluorochrome-conjugated secondary antibody and a DAPI nuclear counterstain. Images were acquired using a Nikon A1R confocal microscope.
Measurement of Intracellular ROS
1.5 × 105 BMDMs plated in clear bottom, white plates were washed, and 100 μl prewarmed (37 °C) assay medium containing HBSS, 100 μg/ml superoxide-specific lucigenin and stimuli were added. Cells were either left untreated or stimulated with poly I:C (25 μg/ml), CpG ODN2395 (25 μg/ml), LPS (1 μg/ml), or phorbol myristate acetate (PMA) (50 ng/ml). Chemiluminescence was followed for 60 min in a Varioskan Flash microplate luminometer. Integrated luminescence units indicative of superoxide production during the entire measurement were calculated and expressed as relative units (RU).
Statistical Analysis
Data are represented as the mean ± S.D. or S.E., as indicated. p values were determined by two-tailed Student's t test or paired Student's t test.
RESULTS
Early Induction of Proinflammatory Gene Expression and Secretion of TNFα and IL-10 Are Restricted to a Subset of TLRs
To better understand how Tpl2 regulates early innate responses induced by diverse TLR ligands, we measured the expression of various proinflammatory genes including il12b, il6, tnfα, ccl2, and ccl5 as well as anti-inflammatory il10 in WT and tpl2−/− BMDMs stimulated with Pam3CSK4 (TLR2), poly I:C (TLR3), LPS (TLR4), R848 (TLR7), and CpG (TLR9) for 1 h. Even at this early time point, induction of most genes was observed in response to TLR2, 4, and 7 ligands, but not in response to TLR3 and 9 ligands (Fig. 1A and data not shown). Of these cytokines, only TNFα and IL-10 were secreted by 1 h after stimulation (Fig. 1B and data not shown). TLR2, 4, and 7 stimulation induced high levels of TNFα and IL-10 production, and their secretion was significantly less or undetectable in Tpl2-deficient BMDMs (Fig. 1B). On the contrary, neither TLR3 nor 9 stimulation induced secretion of TNFα or IL-10, despite modest induction of both at the mRNA level. To determine whether cytokine secretion occurred with delayed kinetics in poly I:C- and CpG-treated cells, TNFα levels were measured at different time points. Significant induction of TNFα was observed in response to poly I:C and CpG by 2 h poststimulation, although the levels were less compared with LPS-treated cells (Fig. 1C). However, by 24 h, both CpG and poly I:C induced high levels of TNFα, comparable to that induced by LPS stimulation. TNFα secretion was significantly impaired in tpl2−/− cells at all time points confirming the critical role of Tpl2 in regulating TNFα production (Fig. 1C). This differential induction of early proinflammatory gene expression and secretion of TNFα and IL-10 suggested the possibility of immediate activation of an inflammatory signaling cascade downstream of a restricted set of TLRs.
FIGURE 1.

Expression of proinflammatory cytokines and early TNFα and IL-10 secretion are restricted to a subset of TLRs. A, BMDMs from WT and tpl2−/− mice were either left untreated or stimulated with Pam3CSK4, poly I:C, LPS, R848, or CpG ODN for 1 h. Cells were lysed after collecting supernatants, and il12b, tnfa, il10, and ccl2 mRNA expression were measured by real-time PCR relative to an actin control. B, TNFα and IL-10 secretion at 1 h were measured by ELISA. Values are relative to WT cells treated with LPS, which was considered 100% in individual experiments. C, BMDMs from WT and tpl2−/− mice were either left untreated or stimulated with poly I:C, LPS, or CpG ODN for 1, 2, 4, or 24 h. TNFα levels in cell culture supernatants were measured by ELISA. ** indicates p < 0.01, * indicates p < 0.05. For A-C, bars indicate mean ± S.E. from 3–4 independent experiments.
Signaling by TLR3 and 9 Fails to Induce Tpl2-p58 or Early ERK Phosphorylation
The MAP kinase ERK is activated in response to TLRs and mediates both transcriptional and post-transcriptional regulation of many immune mediators, including TNFα and IL-10 (14, 17). Tpl2 plays a critical role in TLR-mediated ERK activation and regulates TNFα production by promoting both nucleocytoplasmic transport of TNFα mRNA and processing of pre-TNFα by TNFα-converting enzyme (14, 22). Since early TNFα secretion was abolished in WT BMDMs in response to TLR3 and 9 ligands despite transcriptional induction, we investigated whether the Tpl2-ERK TNF-processing pathway was differentially activated by these TLRs. BMDMs from WT and tpl2−/− mice were stimulated with Pam3CSK4, poly I:C, LPS, R848 and CpG, and both Tpl2 activation and ERK phosphorylation were assessed by immunoblotting. We observed decreased mobility of Tpl2-p58, consistent with phosphorylation (10), at early time points following Pam3CSK4, LPS, and R848 stimulation (Fig. 2A). Tpl2-p58 was completely degraded by the proteosome within 1 h of stimulation with the same ligands that induced Tpl2 phosphorylation (Fig. 2B). Surprisingly, neither the TLR3 ligand poly I:C nor the TLR9 ligand CpG induced a mobility shift or Tpl2-p58 degradation (Fig. 2, A and B). Furthermore, the lack of Tpl2 degradation in response to TLR3 or TLR9 ligation was independent of the ligand dose or type of CpG ODN used, since we did not observe any Tpl2-p58 mobility shift in response to a high dose of poly I:C or CpG or in response to class B CpG ODN (Fig. 2C). Interestingly, no mobility shift or degradation of the Tpl2-p52 isoform occurred in response to any of these ligands. Consistent with previous studies (19), all TLR ligands, including poly I:C and CpG, induced strong ERK1/2 phosphorylation in WT BMDMs by 60 min (Fig. 2B), regardless of Tpl2 mobility shift or degradation. Notably, in WT BMDMs ERK phosphorylation by either CpG or poly I:C was delayed compared with that induced by Pam3CSK4, LPS, or R848 since it was evident at 60 min but not 15 min (Fig. 2, A and B). Stimulation of WT BMDMs with poly I:C over a longer time course up to 2 h also did not lead to Tpl2 degradation, despite maximal ERK activation by 1 h that was comparable to that induced by LPS, followed by a decline in ERK activation by 2 h (Fig. 2D). Consistent with an essential, non-redundant role for Tpl2 in activating ERK (19), none of these ligands induced ERK1/2 phosphorylation in Tpl2-deficient BMDMs (Fig. 2B). Thus, even though Tpl2-dependent ERK activation was observed following stimulation of all TLRs examined, only TLR2, 4, and 7 signaling induced Tpl2-p58 phosphorylation, degradation and early ERK1/2 activation. These findings demonstrate that Tpl2-p58 phosphorylation, degradation, and early ERK activation are restricted to a subset of TLRs.
Although Tpl2 signaling is essential for TNFα production in macrophages, previous studies have reported distinct requirements for Tpl2 in response to different pattern recognition receptors in a cell-type specific manner (15, 16). To investigate whether differential Tpl2-ERK activation occurs in other cell types in a ligand-dependent manner, we assessed Tpl2-p58 shift and ERK phosphorylation in peritoneal macrophages (PMs), BMDCs, and pDCs. Similar to BMDMs, Tpl2-p58 mobility shift, degradation and ERK phosphorylation were observed in PMs in a stimulus-specific manner (Fig. 2E). Interestingly, complete degradation of both p52 and p58 isoforms of Tpl2 was observed within 15 min in pDCs treated with R848 (Fig. 2E). Similar to other cell types examined, CpG stimulation did not have any effect on Tpl2 in pDCs. In contrast, Tpl2-p58 mobility shift was not observed in BMDCs even upon LPS stimulation, despite early (15 min) ERK phosphorylation in response to both CpG and LPS stimulation (Fig. 2E). Collectively these studies demonstrate that the differential degradation of Tpl2 isoforms observed in response to LPS versus poly I:C or CpG in BMDMs is recapitulated in both PMs and pDCs. However, they also reveal cell type-specific differences in Tpl2 activation.
Either the MyD88 or TRIF Adaptor Protein Is Required for Activation of Tpl2-ERK Pathway during TLR4 Signaling
Ligand binding to TLRs initiates recruitment of specific adaptor molecules necessary for intracellular signal transduction. All TLRs except TLR3 transduce signals via the MyD88 adaptor, whereas TLR3 utilizes the TRIF adaptor (1). TLR4 is the only TLR that can transduce signals via both MyD88 (from the plasma membrane) and TRIF (from endosomes) (1). Lack of Tpl2 p58 phosphorylation by either TLR3 or 9 was surprising, especially because these two TLRs utilize different adaptor proteins for downstream signaling. To further investigate the role of TLR adaptors in Tpl2-ERK activation, we used cells deficient in MyD88 or TRIF adaptor proteins. WT, myd88−/−, and ticamLPS2/LPS2 (23) BMDMs were stimulated with different TLR ligands, and immunoblotting was performed to assess Tpl2 degradation and ERK activation. Tpl2 degradation was absent in myd88−/− macrophages in response to all TLR ligands tested except LPS, confirming the requirement for Myd88-dependent signaling in Tpl2-ERK activation during TLR2 and 7 signaling (Fig. 3A). Similar to myd88−/− cells, no impairment in Tpl2-p58 degradation was observed in ticamLPS2/LPS2 BMDMs in response to LPS (Fig. 3B). This pattern of Tpl2 degradation by ligands was also mirrored by the Tpl2-p58 mobility shift observed at 15 min. (data not shown). Consistent with the absolute requirement for either the MyD88 or TRIF adaptor protein for downstream signaling from TLR4 (23), Tpl2 mobility shift, degradation, and ERK activation were completely abrogated in the myd88−/−/trif−/− cells (Fig. 3C). Thus, signaling via either TLR adaptor protein is necessary for activation of the Tpl2-ERK pathway by TLR4.
FIGURE 3.

Activation of the Tpl2-ERK pathway by TLR4 requires either the MyD88 or TRIF adaptor protein and is regulated by the TLR4 co-receptor, CD14, and the tyrosine kinase Syk. A, BMDMs from WT and myd88−/− mice were stimulated with Pam3CSK4, poly I:C, LPS, R848, or CpG ODN for 1 h. B, BMDMs from WT and ticamlps2/lps2 mice were stimulated with poly I:C, LPS or R848 for 1 h. C, BMDMs from WT and myd88−/−/trif−/− mice were stimulated with LPS for 15, 30, and 60 min. D, WT and tpl2−/− BMDMs were treated with poly I:C or LPS for 24 h. IFNβ levels in cell culture supernatants were measured by ELISA. Bars indicate mean ± S.D. E, BMDMs from WT and cd14−/− mice were stimulated with poly I:C, LPS, or R848 for 1 h. F, BMDMs from WT mice were pretreated with piceatannol for 1 h before stimulating with LPS for 15 min. WCL were immunoblotted with antibodies recognizing Tpl2, pERK1/2, and ERK1/2. Data are representative of 2–4 independent experiments. ** indicates p < 0.01.
In myd88−/− BMDMs, LPS signals only via the TRIF adaptor, similar to TLR3 (23). Notably, we observed distinct capabilities of TLR3 and TLR4 to induce Tpl2 degradation in myd88−/− BMDMs, suggesting the possibility for different Tpl2-dependent consequences of TRIF signaling by these two ligands. To evaluate TRIF-dependent responses we measured the production of the TRIF-regulated cytokine, IFNβ, in response to poly I:C and LPS. While IFNβ levels were higher in Tpl2-deficient cells in response to LPS as previously reported (17), poly I:C-induced IFNβ was significantly reduced in tpl2−/− BMDMs compared with WT cells (Fig. 3D). Therefore, despite the lack of Tpl2-p58 mobility shift or degradation, there was still evidence of Tpl2-dependent regulation of poly I:C-mediated responses, suggesting a different mode of Tpl2 activation in response to this ligand.
Activation of the Tpl2-ERK Pathway by TLR4 Requires CD14 and the Tyrosine Kinase Syk
CD14 is a cell surface pattern recognition receptor that binds directly to LPS and serves as a co-receptor, delivering LPS to TLR4. Similar to tpl2−/− mice, cd14−/− mice, as well as Heedless mice harboring a mutation in the cd14 gene, are defective in TNFα production and consequently resistant to LPS-induced shock (24, 25). Importantly, CD14 co-receptor functions for TLR4 are dispensable at high LPS concentrations of greater than 100 ng/ml, and MyD88-dependent MAP kinase and NFκB activation occurs normally in cd14−/− cells (26). To investigate the contribution of CD14 to Tpl2-ERK signaling, WT and cd14−/− BMDMs were stimulated with TLR ligands, and Tpl2 activation was assessed. In contrast to WT cells treated with LPS, both phosphorylation-induced mobility shift and degradation of Tpl2-p58 were absent in cd14−/− cells (Fig. 3E and data not shown). These results demonstrate that, despite normal MyD88-dependent signaling, the TLR4 co-receptor CD14 was required for Tpl2 activation and degradation in response to LPS. Consistent with the lack of Tpl2 activation, ERK phosphorylation was abrogated in cd14−/− macrophages at earlier time points (data not shown), whereas similar levels of ERK phosphorylation were observed 1 h after stimulation (Fig. 3E). This delayed ERK activation in the absence of Tpl2 degradation was reminiscent of the phenotype observed in WT BMDMs treated with TLR3 and 9 ligands, suggesting an inability of cd14−/− BMDMs to induce early Tpl2-dependent ERK phosphorylation.
CD14-mediated TLR4 endocytosis depends on the enzyme PLCγ2 and the tyrosine kinase Syk (26). Syk is known to regulate Tpl2 activation and release from NFκB1p105 in response to TNFα (27). Moreover, LPS-induced Syk phosphorylation is also CD14-dependent (26). This prompted us to investigate whether Syk is involved in Tpl2 activation by TLR4. The well-characterized Syk inhibitor piceatannol blocked LPS-induced mobility shift of Tpl2-p58 as well as ERK phosphorylation in a dose-dependent manner (Fig. 3). Notably, only early ERK phosphorylation by LPS was regulated by Syk, as piceatannol did not block LPS-induced ERK phosphorylation at 1 h (data not shown). These data confirmed the significance of CD14-dependent Syk activation, in addition to MyD88/TRIF signaling, in the regulation of Tpl2-ERK signaling by TLR4.
Early Induction of Tpl2-ERK Signaling Correlates with IKKβ Phosphorylation
One signaling event that regulates activation of Tpl2 and its release from inhibitory NFκB1p105 is IKKβ phosphorylation (5, 10). To investigate whether the observed differences in phosphorylation and degradation of Tpl2-p58 were due to differential activation of IKKβ, we examined phosphorylation of IKKβ and its downstream target IκBα in response to various TLR ligands. Consistent with Tpl2-p58 degradation, only TLR2, 4, and 7 stimulation induced phosphorylation of IKKβ early after stimulation (15 min) (Fig. 4A). A reduction in IKKβ mobility was also observed in response to the ligands that induced its phosphorylation. IKKβ immunoprecipitation followed by Western blotting confirmed that IKKβ was the IKK species being phosphorylated (data not shown). Since IKKβ is also known to regulate NFκB signaling, we examined whether NFκB activation is also differentially regulated by diverse TLR ligands. The phosphorylation of NFκB1-p105 as well as degradation of IκBα were observed in both poly I:C- and CpG-treated cells, although with different magnitude and kinetics compared with LPS (Fig. 4B). Moreover, NFκB-p65 was almost exclusively localized to the nuclei in cells treated with both poly I:C and CpG, confirming their activation of NFκB (Fig. 4C). Notably, although poly I:C and CpG induced phosphorylation of IKKβ by 30 - 60 min (Fig. 4B), we did not observe Tpl2-p58 mobility shift or degradation at later time points. Collectively, these findings support the conclusion that differential activation of IKKβ by diverse TLRs controls early induction of Tpl2-ERK signaling.
FIGURE 4.

Diverse TLRs differentially activate IKKβ. A, BMDMs from WT mice were left untreated or stimulated with Pam3CSK4, poly I:C, LPS, R848, and CpG ODN for 15 min. B, BMDMs from WT mice were left untreated or stimulated with poly I:C, CpG ODN, or LPS for 15 min, 30 min, or 1 h. A and B, WCL were immunoblotted with antibodies recognizing pIKKα/β, IKKβ, pIκBα, pNFκBp105, IκBα, and β-actin. Dashed line indicates that within a single exposure intervening lanes were removed. C, BMDMs from WT mice were left untreated or stimulated with poly I:C, CpG ODN, or LPS for 1 h. Nuclear translocation of NFκB-p65 was assessed by confocal microscopy. D, BMDMs from WT and cd14−/− mice were stimulated with poly I:C, LPS, or R848 for 30 min. E, BMDMs from WT mice were pretreated with piceatannol for 1 h before stimulating with LPS for 15 min. D–E, WCL were immunoblotted with antibodies recognizing pIKKα/β, IKKβ, pIκBα, IκBα, and ERK1/2. Data are representative of 2–4 independent experiments.
IKKβ phosphorylation in response to LPS was present in myd88−/− and ticamLPS2/LPS2 cells, but absent in myd88−/−/trif−/− and cd14−/− BMDMs, demonstrating that either one of the adaptors as well as CD14 is required for IKKβ activation (Fig. 4D and data not shown). In addition, IKKβ phosphorylation was significantly reduced in cells pretreated with the Syk inhibitor piceatannol (Fig. 4E). Under these conditions, IκBα degradation was not dramatically affected in piceatannol treated samples suggesting minimal off-target effects of the inhibitor (Fig. 4E). These results demonstrate that IKKβ is likely the convergence point through which TLR adaptor proteins, CD14 and Syk regulate the Tpl2-ERK pathway during TLR4 signaling.
TLR3 and 9 Activate ERK Indirectly via NADPH-oxidase-dependent Autocrine ROS Signaling
To explain TLR3- and 9-induced ERK activation in the absence of Tpl2-p58 phosphorylation, we considered the possibility of Tpl2-dependent ERK activation by autocrine signaling. Both TLR3- and 9-mediated ERK phosphorylation were abolished when BMDMs were treated with the translation inhibitor cycloheximide (CHX), suggesting that new protein synthesis was required for ERK activation by these TLRs (Fig. 5A). Since Tpl2 is also essential for TNFα- and IL-1β-mediated signal transduction leading to ERK phosphorylation (15), we investigated whether an indirect cytokine feedback loop via TNFα or IL-1 receptors was responsible for the delayed ERK activation observed in the absence of Tpl2 degradation. As expected, TNFα and IL-1β induced ERK phosphorylation in WT BMDMs (data not shown). However, unlike poly I:C or CpG, TNFα, but not IL-1β, also induced Tpl2-p58 degradation. Furthermore, neutralizing antibodies against TNFα and IL-1β did not inhibit delayed ERK phosphorylation in response to TLR3 and 9 ligands (data not shown). TLR3- and 9-induced ERK phosphorylation was also evident in ifnar1−/− BMDMs as well as in BMDMs pretreated with pertussis toxin, demonstrating that neither type 1 interferons nor chemokines are responsible for the indirect ERK phosphorylation in response to TLR3 and 9 ligands (data not shown).
FIGURE 5.

TLR3 and 9 activate ERK indirectly via NADPH-oxidase-dependent autocrine ROS signaling. WT BMDMs were pretreated (A) for 30 min with 10 μg/ml CHX, (B) for 30 min with 20 μm DPI, or (C) for 10 min with 10 mm GSH prior to 1 h stimulation with poly I:C, CpG ODN, or LPS. Dashed line indicates that within a single exposure intervening lanes were removed. WCL were immunoblotted with antibodies recognizing pERK1/2 and ERK1/2. Data are representative of three independent experiments. D, BMDMs from WT mice were either left untreated or stimulated with poly I:C, CpG ODN, or LPS for 1 h. Cells were lysed, and nos2 mRNA expression was measured by real-time PCR relative to an actin control. E, WT BMDMs were pretreated for 30 min with 20 μm DPI before stimulation with poly I:C, CpG ODN, or LPS for 2 h. TNFα levels in cell culture supernatants were measured by ELISA. Bars indicate mean ± S.E. from three independent experiments. * indicates p < 0.05.
Inhibition of protein synthesis is known to inhibit accumulation of ROS and oxidative stress by increasing cellular glutathione concentrations (28). In addition, several studies have demonstrated that ROS can induce activation of MAP kinases, including ERK (29, 30). Because both poly I:C and CpG are known inducers of ROS (31, 32), we tested the possibility that ERK was phosphorylated via a ROS-dependent mechanism in BMDMs stimulated with these ligands. Indeed, ERK phosphorylation in response to both poly I:C and CpG was completely abrogated in BMDMs pretreated with the NADPH oxidase (NOX) inhibitor diphenyleneiodonium (DPI), demonstrating that activation of ERK by TLR3 and 9 is dependent on NOX activity (Fig. 5B). Scavenging ROS with the antioxidant glutathione (GSH) also clearly reduced ERK phosphorylation in response to poly I:C and CpG, supporting the conclusion that autocrine ROS signaling is responsible for indirect, delayed ERK activation by TLR3 and 9 (Fig. 5C). Although DPI can also inhibit nitric-oxide synthase, the involvement of nitric oxide (NO) in the autocrine signaling loop was excluded because expression of nitric-oxide synthase 2 (NOS2), a secondary response gene that drives NO production (33), was not induced in poly I:C- and CpG-treated cells during the 1 h stimulation (Fig. 5D). If autocrine ROS signaling is responsible for ERK phosphorylation downstream of TLRs 3 and 9, then DPI treatment should also inhibit delayed ERK-dependent TNFα secretion observed in BMDMs treated with poly I:C and CpG (Fig. 1C). Indeed, DPI treatment significantly decreased TNFα secretion in response to poly I:C and CpG, but not LPS (Fig. 5E). Collectively, these data delineate a ROS-dependent autocrine loop that mediates ERK phosphorylation and TNFα secretion during TLR3 and 9 signaling.
Tpl2 Is Essential for ROS Production during TLR Signaling
The observation that inhibiting NADPH oxidase can abolish ERK phosphorylation in response to poly I:C and CpG was unexpected. Acidification of the endosomal compartment, a pre-requisite for both TLR3 and 9 signaling (34, 35), is coupled to rapid induction of ROS (32). Since ROS-mediated ERK phosphorylation is also Tpl2-dependent, it is necessary to determine whether Tpl2 regulates ROS production and/or signaling. The involvement of Tpl2 in ROS signaling was excluded since treatment with the oxidizing agent H2O2 induced ERK phosphorylation in both WT and tpl2−/− BMDMs (Fig. 6A). Despite inducing strong ERK phosphorylation, H2O2 did not induce Tpl2 degradation even at the non-physiological concentration of 10 mm (Fig. 6A). Having demonstrated that Tpl2 is dispensable for ROS-induced ERK phosphorylation, we next sought to determine whether Tpl2 regulates ROS production during TLR signaling. To determine whether Tpl2 regulates the generation of intracellular ROS, we measured superoxide production in WT and tpl2−/− BMDMs treated with various TLR ligands. As previously reported (31, 32, 36), poly I:C, CpG and LPS all induced ROS production (Fig. 6, B and C). As expected based on our previous results, superoxide generation in response to multiple TLR ligands was significantly less in Tpl2-deficient BMDMs compared with WT cells (Fig. 6, B and C). In contrast, there was no intrinsic defect in the capacity of tpl2−/− cells to generate ROS in response to phorbol myristate acetate (PMA), indicating a ligand-specific defect. This unanticipated role for Tpl2 in ROS production explains Tpl2- and ROS-dependent ERK phosphorylation observed during TLR3 and 9 signaling.
FIGURE 6.

Tpl2 is essential for ROS production during TLR signaling. A, BMDMs from WT mice were treated with H2O2 (1 or 10 mm) or LPS for 1 h. WCL were immunoblotted with antibodies recognizing Tpl2, pERK1/2, and ERK1/2. Data are representative of three independent experiments. B and C, BMDMs from WT and tpl2−/− mice were treated with poly I:C, LPS, CpG ODN, or PMA, and superoxide production was measured by a lucigenin-based chemiluminescence assay. Kinetics (B) and integrated luminescence data (C) from three independent experiments are represented. Bars indicate mean ± S.E. * indicates p < 0.05.
DISCUSSION
Activation of NFκB and MAP kinases are key features of all TLR signaling pathways initiating a proinflammatory response (3). In this study, we made several important discoveries regarding differential mechanisms activating a common inflammatory signaling cascade during TLR signaling as summarized in Fig. 7. First, we demonstrated an indirect, delayed mechanism of ERK activation by a subset of TLRs that limits early innate responses, including early TNFα and IL-10 secretion, to TLR3 and TLR9 ligands. This pathway is distinguished by the lack of Tpl2-p58 phosphorylation and degradation despite evidence of NFκB activation, including IκBα phosphorylation and degradation, and despite Tpl2-dependent biological responses to these ligands. Second, we delineated an inflammatory pathway controlled by CD14 and the tyrosine kinase Syk in the activation of the IKKβ-Tpl2-ERK axis during TLR4 signaling. Third, we identified a ROS-dependent autocrine loop responsible for the delayed, indirect ERK phosphorylation during TLR3 and 9 signaling. Finally, we demonstrated the critical role of Tpl2 in ROS generation during TLR signaling.
FIGURE 7.
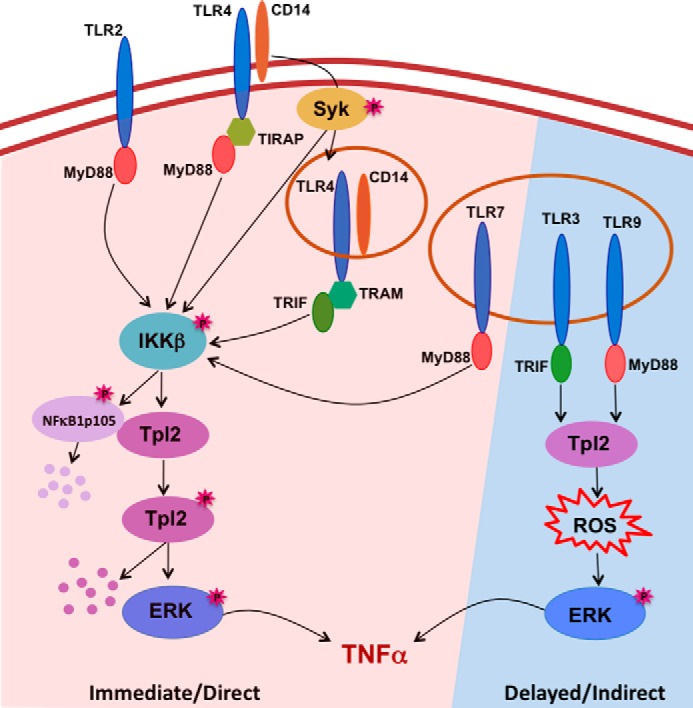
Model of Tpl2-ERK activation during TLR signaling. Stimulation of TLR2, 4 and 7 immediately activates the IKKβ-Tpl2-ERK inflammatory pathway. In addition to a TLR adaptor protein, activation of this pathway by TLR4 requires the TLR4 co-receptor CD14 and the tyrosine kinase Syk. Active IKKβ phosphorylates both NFκB1 and Tpl2-p58 and leads to release of active Tpl2, which in turn induces MEK-dependent ERK activation prior to Tpl2-p58 proteosomal degradation. ERK signaling facilitates processing and secretion of TNFα. TLR3 and 9 do not cause Tpl2-p58 phosphorylation-induced mobility shift, degradation, or early ERK activation. Instead, TLR3 and 9 induce delayed ERK phosphorylation via autocrine signaling by ROS, which is generated in a Tpl2-dependent manner. Consequently, TLR3 and 9 induce delayed secretion of innate TNFα compared with other TLRs.
Activation of ERK in response to diverse TLR ligands and the critical role of Tpl2 in transducing ERK activation signals are well documented (19). Our results are in agreement with previous studies demonstrating that stimulation of all major TLRs induce Tpl2-dependent ERK activation in BMDMs. Consistent with the data reported by Kaiser et al., we observed delayed ERK phosphorylation in both CpG- and poly I:C-treated cells (17). We further linked this reduced ERK activation to lack of Tpl2 phosphorylation and degradation.
Conflicting reports regarding CpG-induced ERK phosphorylation in different cell types exist (17, 37, 38). Our findings clarify a controversy and demonstrate that TLR3 and 9 signaling do not directly couple to ERK activation. Instead, the observed ERK phosphorylation by TLR3 and 9 is due to NADPH oxidase-dependent autocrine ROS signaling. The significance of ROS as second messengers during innate immune responses and in regulating the production of various inflammatory mediators is well appreciated (39). For example, ROS-dependent activation of MAP3K5/ASK-1 and MAP kinase p38 was shown to be necessary for TLR4 mediated innate responses (36). In addition to ROS-mediated ERK phosphorylation during TLR3 and 9 signaling, our study identified Tpl2 as a critical regulator of ROS production during TLR signaling. The requirement of Tpl2 in ROS production may contribute in part to the defective induction of IL-1β in tpl2−/− macrophages, since ROS is important for IL-1β expression in response to LPS (29). The signaling events linking Tpl2 to NOX enzymes are currently unknown. Therefore, further studies are needed to determine the precise mechanisms by which Tpl2 regulates ROS production.
Tpl2-p58 mobility shift and degradation, while excellent predictors of Tpl2-dependent MEK/ERK activation, are poor indicators of overall Tpl2 biological activity. For example, Tpl2 is required for TNF processing and secretion in response to both poly I:C and CpG (Fig. 1C), both of which fail to induce Tpl2-p58 phosphorylation-induced mobility shift, degradation or early ERK activation. Tpl2 is also required for normal IFNβ production in response to poly I:C (Fig. 3D). Similarly, IL-1β also utilizes Tpl2 to transduce signals, but fails to induce Tpl2-p58 degradation (data not shown). These findings raise the possibility of phosphorylation-independent functions for Tpl2-p58 or Tpl2-p52 isoforms in cell signaling. Thr290 phosphorylation occurs only on the Tpl2 p58 isoform, whereas both p52 and p58 isoforms undergo phosphorylation on Ser400 in LPS-treated macrophages (12). Despite the fact that no functional differences between Tpl2-p58 and p52 have been reported so far, it is tempting to speculate that Tpl2-p52 transduces signals from receptors that do not induce IKKβ-mediated Thr290 phosphorylation and p58 degradation, such as poly I:C, CpG, and IL-1β.
Cell type-specific requirements for Tpl2 in transducing TLR signals have been demonstrated previously (15, 16). However cell type-specific differences in Tpl2 phosphorylation is a novel finding. While LPS induced Tpl2-p58 Thr290 phosphorylation and mobility shift in macrophages, a decrease in Tpl2-p58 mobility was not observed in BMDCs. This difference in Tpl2 activation could account for the partial requirement of Tpl2 for TNFα secretion in BMDCs compared with BMDMs (16). Notably, cell type-specific differences between BMDMs and BMDCs in the requirement for CD14 during TLR4 signaling have been reported (26). Unlike macrophages and BMDCs, both isoforms of Tpl2 were completely degraded in pDCs early after stimulation, further supporting the uniqueness of signaling pathways in pDCs (17).
CD14 is a GPI-anchored protein without intrinsic signaling potential (40), however CD14 functions are necessary for Myd88-independent signaling by TLR4 (25). In their elegant study demonstrating the role of CD14 in TLR4 endocytosis, Zanoni et al. commented that all LPS responses actually initiate with CD14 (26). Our data confirming the necessity of CD14 in IKKβ-Tpl2-ERK signaling support their placement of CD14 as the “king of all LPS responses” although either one of the TLR adaptor proteins is also necessary for this response. An inflammatory endocytosis pathway regulated by Syk was proposed for endocytosed receptors like TLR4, Dectin-1 and FcγRI (26). Interestingly, a recent study reported Tpl2-mediated ERK activation during FcγR signaling (41). Thus, regulation of IKKβ-Tpl2-ERK signaling by CD14 and Syk supports the existence of this proposed inflammatory pathway.
Although differences in biological responses upon TLR7 and 9 stimulation have been reported (42, 43), the molecular basis for these differences has remained enigmatic. Herein, we demonstrate the direct coupling of TLR7, but not TLR9, to the IKKβ-Tpl2-ERK signaling pathway. To our knowledge, differences between TLR7 and 9 signaling per se have not been demonstrated. This finding was surprising, as both of these endosomal TLRs transduce signals via the same MyD88 adaptor (44). However, a recent study did report differences in UNC93B1-mediated trafficking of TLR7 and 9 (45). Identification of discrete trafficking pathways suggests the possibility of distinct signaling compartments for TLR7 and 9 that may correlate with their activation of distinct signaling cascades and cellular responses. Since cell surface expression of TLR3 has been reported (46, 47), a trafficking route similar to that of TLR9 was proposed for this receptor. Hence, the differential activation of the IKKβ-Tpl2-ERK pathway could correlate with the involvement of distinct signaling compartments for these endosomal TLRs.
In addition to the new insights into TLR signaling pathways, our findings have many implications regarding the role of Tpl2 in innate immune responses during infections. We and others have previously demonstrated the critical role of Tpl2 in host defense against intracellular bacteria like Listeria monocytogenes and Mycobacterium tuberculosis (16, 48). Defective ROS production in tpl2−/− mice may contribute to decreased bacterial clearance, increased susceptibility to infection or altered redox-sensitive signaling (39, 49). In addition to its bactericidal functions, ROS is also required for RIG-I-mediated antiviral responses (50). Moreover, direct and immediate activation of Tpl2 and ERK during TLR7 signaling suggests that Tpl2 is likely to play a preferential role in host defense against RNA viruses that trigger TLR7. In this regard, a recent study reported increased replication of vesicular stomatitis virus (VSV) in Tpl2-deficient mouse embryonic fibroblasts (51). This is especially interesting because, in addition to TLR7, VSV is known to signal via the CD14-TLR4 axis, and increased replication of VSV was also reported in macrophages from CD14 mutant mice (25). These findings suggest a role for Tpl2 in controlling virus replication and warrant further studies to assess the contribution of Tpl2 in antiviral host responses. Overall, our study provides a better understanding about key events that distinguish signal transduction by diverse TLRs and further underscores the significance of Tpl2 in eliciting host protective immune responses against diverse pathogens.
Acknowledgments
We thank Rebecca Kirkland for excellent technical assistance. We also thank Julie Nelson and the Center for Tropical and Emerging Global Diseases Flow Cytometry Core Facility for cell sorting and Dr. Barbara Reaves and the CVM Cytometry Core Facility for confocal microscopy. We would also like to acknowledge UGA's Veterinary Medicine Central Animal Facility for animal care.
This work was supported by startup funds (to W. T. W.) by the Office of the Vice President for Research at the University of Georgia.
- TLR
- Toll-like receptor
- BMDMs
- bone marrow-derived macrophages
- BMDCs
- bone marrow-derived dendritic cells
- CHX
- cycloheximide
- DPI
- diphenyleneiodonium
- GSH
- glutathione
- IKKβ
- inhibitor of κB kinase-β
- MCSF
- macrophage colony stimulating factor
- MyD88
- myeloid differentiation primary response gene 88
- NOX
- NADPH oxidase
- PAMPs
- pathogen-associated molecular patterns
- pDCs
- plasmacytoid dendritic cells
- PMs
- peritoneal macrophages
- ROS
- reactive oxygen species
- TACE
- TNFα-converting enzyme
- Tpl2
- tumor progression locus 2
- TRIF
- TIR-domain-containing adapter-inducing interferon-β
- VSV
- vesicular stomatitis virus
- WCL
- whole cell lysates.
REFERENCES
- 1. Kawai T., Akira S. (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384 [DOI] [PubMed] [Google Scholar]
- 2. Iwasaki A., Medzhitov R. (2004) Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5, 987–995 [DOI] [PubMed] [Google Scholar]
- 3. Kawai T., Akira S. (2007) Signaling to NF-κB by Toll-like receptors. Trends Mol. Med. 13, 460–469 [DOI] [PubMed] [Google Scholar]
- 4. Banerjee A., Gerondakis S. (2007) Coordinating TLR-activated signaling pathways in cells of the immune system. Immunol. Cell Biol. 85, 420–424 [DOI] [PubMed] [Google Scholar]
- 5. Cho J., Melnick M., Solidakis G. P., Tsichlis P. N. (2005) Tpl2 (tumor progression locus 2) phosphorylation at Thr290 is induced by lipopolysaccharide via an Iκ-B Kinase-β-dependent pathway and is required for Tpl2 activation by external signals. J. Biol. Chem. 280, 20442–20448 [DOI] [PubMed] [Google Scholar]
- 6. Perkins N. D. (2007) Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Molecular cell biology 8, 49–62 [DOI] [PubMed] [Google Scholar]
- 7. Makris A., Patriotis C., Bear S. E., Tsichlis P. N. (1993) Genomic organization and expression of Tpl-2 in normal cells and Moloney murine leukemia virus-induced rat T-cell lymphomas: activation by provirus insertion. J. Virol. 67, 4283–4289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aoki M., Hamada F., Sugimoto T., Sumida S., Akiyama T., Toyoshima K. (1993) The human cot proto-oncogene encodes two protein serine/threonine kinases with different transforming activities by alternative initiation of translation. J. Biol. Chem. 268, 22723–22732 [PubMed] [Google Scholar]
- 9. Waterfield M. R., Zhang M., Norman L. P., Sun S. C. (2003) NF-κB1/p105 regulates lipopolysaccharide-stimulated MAP kinase signaling by governing the stability and function of the Tpl2 kinase. Mol. Cell 11, 685–694 [DOI] [PubMed] [Google Scholar]
- 10. Beinke S., Robinson M. J., Hugunin M., Ley S. C. (2004) Lipopolysaccharide activation of the TPL-2/MEK/extracellular signal-regulated kinase mitogen-activated protein kinase cascade is regulated by IκB kinase-induced proteolysis of NF-κB1 p105. Mol. Cell. Biol. 24, 9658–9667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roget K., Ben-Addi A., Mambole-Dema A., Gantke T., Yang H. T., Janzen J., Morrice N., Abbott D., Ley S. C. (2012) IκB kinase 2 regulates TPL-2 activation of extracellular signal-regulated kinases 1 and 2 by direct phosphorylation of TPL-2 serine 400. Mol. Cell. Biol. 32, 4684–4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Robinson M. J., Beinke S., Kouroumalis A., Tsichlis P. N., Ley S. C. (2007) Phosphorylation of TPL-2 on serine 400 is essential for lipopolysaccharide activation of extracellular signal-regulated kinase in macrophages. Mol. Cell. Biol. 27, 7355–7364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cho J., Tsichlis P. N. (2005) Phosphorylation at Thr-290 regulates Tpl2 binding to NF-κB1/p105 and Tpl2 activation and degradation by lipopolysaccharide. Proc. Natl. Acad. Sci. U.S.A. 102, 2350–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dumitru C. D., Ceci J. D., Tsatsanis C., Kontoyiannis D., Stamatakis K., Lin J. H., Patriotis C., Jenkins N. A., Copeland N. G., Kollias G., Tsichlis P. N. (2000) TNF-α induction by LPS is regulated post-transcriptionally via a Tpl2/ERK-dependent pathway. Cell 103, 1071–1083 [DOI] [PubMed] [Google Scholar]
- 15. Das S., Cho J., Lambertz I., Kelliher M. A., Eliopoulos A. G., Du K. Y., Tsichlis P. N. (2005) Tpl2/Cot signals activate ERK, JNK, and NF-κB in a cell-type and stimulus-specific manner. J. Biol. Chem. 280, 23748–23757 [DOI] [PubMed] [Google Scholar]
- 16. Mielke L. A., Elkins K. L., Wei L., Starr R., Tsichlis P. N., O'Shea J. J., Watford W. T. (2009) Tumor Progression Locus 2 (Map3k8) Is Critical for Host Defense against Listeria monocytogenes and IL-1β Production. J. Immunol. 183, 7984–7993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kaiser F., Cook D., Papoutsopoulou S., Rajsbaum R., Wu X., Yang H. T., Grant S., Ricciardi-Castagnoli P., Tsichlis P. N., Ley S. C., O'Garra A. (2009) TPL-2 negatively regulates interferon-beta production in macrophages and myeloid dendritic cells. J. Exp. Med. 206, 1863–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eliopoulos A. G., Dumitru C. D., Wang C. C., Cho J., Tsichlis P. N. (2002) Induction of COX-2 by LPS in macrophages is regulated by Tpl2-dependent CREB activation signals. EMBO J. 21, 4831–4840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Banerjee A., Gugasyan R., McMahon M., Gerondakis S. (2006) Diverse Toll-like receptors utilize Tpl2 to activate extracellular signal-regulated kinase (ERK) in hemopoietic cells. Proc. Natl. Acad. Sci. U.S.A. 103, 3274–3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang H. T., Papoutsopoulou S., Belich M., Brender C., Janzen J., Gantke T., Handley M., Ley S. C. (2012) Coordinate regulation of TPL-2 and NF-κB signaling in macrophages by NF-κB1 p105. Mol. Cell. Biol. 32, 3438–3451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Melo M. B., Kasperkovitz P., Cerny A., Könen-Waisman S., Kurt-Jones E. A., Lien E., Beutler B., Howard J. C., Golenbock D. T., Gazzinelli R. T. (2010) UNC93B1 mediates host resistance to infection with Toxoplasma gondii. PLoS pathogens 6, e1001071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rousseau S., Papoutsopoulou M., Symons A., Cook D., Lucocq J. M., Prescott A. R., O'Garra A., Ley S. C., Cohen P. (2008) TPL2-mediated activation of ERK1 and ERK2 regulates the processing of pre-TNFα in LPS-stimulated macrophages. J. Cell Sci. 121, 149–154 [DOI] [PubMed] [Google Scholar]
- 23. Hoebe K., Du X., Georgel P., Janssen E., Tabeta K., Kim S. O., Goode J., Lin P., Mann N., Mudd S., Crozat K., Sovath S., Han J., Beutler B. (2003) Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 424, 743–748 [DOI] [PubMed] [Google Scholar]
- 24. Moore K. J., Andersson L. P., Ingalls R. R., Monks B. G., Li R., Arnaout M. A., Golenbock D. T., Freeman M. W. (2000) Divergent response to LPS and bacteria in CD14-deficient murine macrophages. J. Immunol. 165, 4272–4280 [DOI] [PubMed] [Google Scholar]
- 25. Jiang Z., Georgel P., Du X., Shamel L., Sovath S., Mudd S., Huber M., Kalis C., Keck S., Galanos C., Freudenberg M., Beutler B. (2005) CD14 is required for MyD88-independent LPS signaling. Nat. Immunol. 6, 565–570 [DOI] [PubMed] [Google Scholar]
- 26. Zanoni I., Ostuni R., Marek L. R., Barresi S., Barbalat R., Barton G. M., Granucci F., Kagan J. C. (2011) CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell 147, 868–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eliopoulos A. G., Das S., Tsichlis P. N. (2006) The tyrosine kinase Syk regulates TPL2 activation signals. J. Biol. Chem. 281, 1371–1380 [DOI] [PubMed] [Google Scholar]
- 28. Ratan R. R., Murphy T. H., Baraban J. M. (1994) Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J. Neurosci. 14, 4385–4392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hsu H. Y., Wen M. H. (2002) Lipopolysaccharide-mediated reactive oxygen species and signal transduction in the regulation of interleukin-1 gene expression. J. Biol. Chem. 277, 22131–22139 [DOI] [PubMed] [Google Scholar]
- 30. Son Y., Cheong Y. K., Kim N. H., Chung H. T., Kang D. G., Pae H. O. (2011) Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal Transduction 792639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang C. S., Kim J. J., Lee S. J., Hwang J. H., Lee C. H., Lee M. S., Jo E. K. (2013) TLR3-triggered reactive oxygen species contribute to inflammatory responses by activating signal transducer and activator of transcription-1. J. Immunol. 190, 6368–6377 [DOI] [PubMed] [Google Scholar]
- 32. Yi A. K., Tuetken R., Redford T., Waldschmidt M., Kirsch J., Krieg A. M. (1998) CpG motifs in bacterial DNA activate leukocytes through the pH-dependent generation of reactive oxygen species. J. Immunol. 160, 4755–4761 [PubMed] [Google Scholar]
- 33. Buxadé M., Lunazzi G., Minguillón J., Iborra S., Berga-Bolaños R., Del Val M., Aramburu J., López-Rodriguez C. (2012) Gene expression induced by Toll-like receptors in macrophages requires the transcription factor NFAT5. J. Exp. Med. 209, 379–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Park B., Brinkmann M. M., Spooner E., Lee C. C., Kim Y. M., Ploegh H. L. (2008) Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nat. Immunol. 9, 1407–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leonard J. N., Ghirlando R., Askins J., Bell J. K., Margulies D. H., Davies D. R., Segal D. M. (2008) The TLR3 signaling complex forms by cooperative receptor dimerization. Proc. Natl. Acad. Sci. U.S.A. 105, 258–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matsuzawa A., Saegusa K., Noguchi T., Sadamitsu C., Nishitoh H., Nagai S., Koyasu S., Matsumoto K., Takeda K., Ichijo H. (2005) ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat. Immunol. 6, 587–592 [DOI] [PubMed] [Google Scholar]
- 37. Yi A. K., Krieg A. M. (1998) Rapid induction of mitogen-activated protein kinases by immune stimulatory CpG DNA. J. Immunol. 161, 4493–4497 [PubMed] [Google Scholar]
- 38. Hacker H., Mischak H., Hacker G., Eser S., Prenzel N., Ullrich A., Wagner H. (1999) Cell type-specific activation of mitogen-activated protein kinases by CpG-DNA controls interleukin-12 release from antigen-presenting cells. EMBO J. 18, 6973–6982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ray P. D., Huang B. W., Tsuji Y. (2012) Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 24, 981–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wright S. D., Ramos R. A., Tobias P. S., Ulevitch R. J., Mathison J. C. (1990) CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 249, 1431–1433 [DOI] [PubMed] [Google Scholar]
- 41. Kyrmizi I., Ioannou M., Hatziapostolou M., Tsichlis P. N., Boumpas D. T., Tassiulas I. (2013) Tpl2 kinase regulates FcγR signaling and immune thrombocytopenia in mice. J. Leukocyte Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Christensen S. R., Shupe J., Nickerson K., Kashgarian M., Flavell R. A., Shlomchik M. J. (2006) Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 25, 417–428 [DOI] [PubMed] [Google Scholar]
- 43. Fukui R., Saitoh S., Kanno A., Onji M., Shibata T., Ito A., Onji M., Matsumoto M., Akira S., Yoshida N., Miyake K. (2011) Unc93B1 restricts systemic lethal inflammation by orchestrating Toll-like receptor 7 and 9 trafficking. Immunity 35, 69–81 [DOI] [PubMed] [Google Scholar]
- 44. Kawai T., Akira S. (2007) TLR signaling. Sem. Immunol. 19, 24–32 [DOI] [PubMed] [Google Scholar]
- 45. Lee B. L., Moon J. E., Shu J. H., Yuan L., Newman Z. R., Schekman R., Barton G. M. (2013) UNC93B1 mediates differential trafficking of endosomal TLRs. eLife 2, e00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Qi R., Hoose S., Schreiter J., Sawant K. V., Lamb R., Ranjith-Kumar C. T., Mills J., San Mateo L., Jordan J. L., Kao C. C. (2010) Secretion of the human Toll-like receptor 3 ectodomain is affected by single nucleotide polymorphisms and regulated by Unc93b1. J. Biol. Chem. 285, 36635–36644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pohar J., Pirher N., Benčina M., Manček-Keber M., Jerala R. (2013) The role of UNC93B1 protein in surface localization of TLR3 receptor and in cell priming to nucleic acid agonists. J. Biol. Chem. 288, 442–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McNab F. W., Ewbank J., Rajsbaum R., Stavropoulos E., Martirosyan A., Redford P. S., Wu X., Graham C. M., Saraiva M., Tsichlis P., Chaussabel D., Ley S. C., O'Garra A. (2013) TPL-2-ERK1/2 signaling promotes host resistance against intracellular bacterial infection by negative regulation of type I IFN production. J. Immunol. 191, 1732–1743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Corcoran A., Cotter T. G. (2013) Redox regulation of protein kinases. FEBS J. 280, 1944–1965 [DOI] [PubMed] [Google Scholar]
- 50. Soucy-Faulkner A., Mukawera E., Fink K., Martel A., Jouan L., Nzengue Y., Lamarre D., Vande Velde C., Grandvaux N. (2010) Requirement of NOX2 and reactive oxygen species for efficient RIG-I-mediated antiviral response through regulation of MAVS expression. PLoS pathogens 6, e1000930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schmid S., Sachs D., Tenoever B. R. (2014) Mitogen-activated Protein Kinase-mediated Licensing of Interferon Regulatory Factor 3/7 Reinforces the Cell Response to Virus. J. Biol. Chem. 289, 299–311 [DOI] [PMC free article] [PubMed] [Google Scholar]