

Abstract
Facioscapulohumeral muscular dystrophy (FSHD) is an incurable disease, characterized by skeletal muscle weakness and wasting. Genetically, FSHD is characterized by contraction or hypomethylation of repeat D4Z4 units on chromosome 4, which causes aberrant expression of the transcription factor DUX4 from the last repeat. Many genes have been implicated in FSHD pathophysiology, but an integrated molecular model is currently lacking. We developed a novel differential network methodology, Interactome Sparsification and Rewiring (InSpiRe), which detects network rewiring between phenotypes by integrating gene expression data with known protein interactions. Using InSpiRe, we performed a meta-analysis of multiple microarray datasets from FSHD muscle biopsies, then removed secondary rewiring using non-FSHD datasets, to construct a unified network of rewired interactions. Our analysis identified β-catenin as the main coordinator of FSHD-associated protein interaction signalling, with pathways including canonical Wnt, HIF1-α and TNF-α clearly perturbed. To detect transcriptional changes directly elicited by DUX4, gene expression profiling was performed using microarrays on murine myoblasts. This revealed that DUX4 significantly modified expression of the genes in our FSHD network. Furthermore, we experimentally confirmed that Wnt/β-catenin signalling is affected by DUX4 in murine myoblasts. Thus, we provide the first unified molecular map of FSHD signalling, capable of uncovering pathomechanisms and guiding therapeutic development.
Keywords: facioscapulohumeral muscular dystrophy, meta-analysis, β-catenin, differential networks, canonical Wnt signalling, DUX4
1. Introduction
Facioscapulohumeral muscular dystrophy (FSHD) is the third most common inheritable disease of skeletal muscle, yet, due to the relative longevity of patients, it is the most prevalent muscular dystrophy (approx. 12/100 000 [1,2]). Despite such prevalence, no curative therapy exists. Clinically, FSHD is characterized by asymmetric, skeletal muscle atrophy affecting specific muscle groups, often associated with features including retinal vasculature abnormalities and sensorineural hearing loss [1,3–5]. Approximately 95% of cases (FSHD1; OMIM158900) are associated with deletion of a number of 3.3 kb D4Z4 macrosatellite repeats on chromosome 4q35. Healthy individuals typically have between 11 and 100 such repeats, whereas FSHD1 patients have 1–10. Importantly, complete loss of D4Z4 units is not associated with FSHD. Each D4Z4 repeat contains an open reading frame for a transcription factor, double homeobox 4 (DUX4) [6,7]. Reduced D4Z4 repeat number leads to hypomethylation, which on a permissive genetic background supplying a polyadenylation signal, stabilizes DUX4 transcripts from the last D4Z4 repeat. In the remaining 5% of cases (FSHD2; OMIM158901), D4Z4 repeats are not contracted, but become hypomethylated, likely through mutations in chromatin-modifying genes such as SMCHD1 [8]. Again on the permissive genetic background with a polyadenylation signal, this leads to DUX4 transcription from the last D4Z4 repeat. Thus, aberrant DUX4 expression underlies FSHD pathogenesis in both FSHD1 and FSHD2 [9]. How DUX4 perturbs pathway interactions to produce the complex FSHD phenotype is unclear, with many genes and pathways implicated in FSHD. Indeed, the FSH Society FSHD International Research Consortium has annually set uncovering FSHD molecular networks as a top research priority.
Complex diseases are frequently examined by gene expression profiling using an arrayed platform of cDNA probes (microarray) and many datasets are published each year. These describe healthy and diseased biological processes, including FSHD and other muscular dystrophies and myopathies. However, their use is often limited to selecting genes for further examination based on differential expression, evidence from the literature or intuition. Systems biology and network concepts, particularly the conceptual framework of differential networks and network rewiring, allow higher order, unbiased analysis of such data [10,11]. These approaches involve integration of molecular profiles (especially gene or protein expression) with network models of protein interactions [12,13], to identify either hotspots of differential expression, or a change in local interaction patterns-network rewiring. Although many differential network algorithms exist [13,14], each has limitations.
Here, we developed a novel differential network algorithm based on information theoretic principles that we call Interactome Sparsification and Rewiring (InSpiRe). InSpiRe detects shifts in active pathway regimes, identifying proteins from the human interactome displaying a wide class of rewiring events between two phenotypes. This protein set is constructed into a sub-network, which is sparsified to describe pathways altered between phenotypes.
Here, we performed a meta-analysis with InSpiRe on multiple FSHD and healthy human control datasets to identify network rewiring. Rewiring associated with ageing, disuse atrophy, inflammation and muscle wasting was then subtracted to construct a unified FSHD-specific disease network. Our FSHD network is significantly more connected than expected by chance (99.5% of the network forms a single maximally connected component) demonstrating that the disease phenotype arises via a highly coordinated perturbation of signalling pathways.
Our analysis confirms previous findings on processes and signalling pathways perturbed in FSHD, such as myogenesis, oxidative stress sensitivity, actin cytoskeletal signalling, Wnt/β-catenin signalling and p53-mediated apoptosis [4,15–17]. Importantly, we also describe novel FSHD molecular mechanisms. Notably, local network measures revealed β-catenin at the centre of our network, as the main coordinator of FSHD-associated protein interaction signalling, and provides insight into the precise molecular determinants of oxidative stress sensitivity. To examine how much of our FSHD network can be explained by DUX4, we performed expression profiling using microarrays of primary satellite cell-derived murine myoblasts expressing DUX4. This revealed that expression of genes in our FSHD network is directly attributed to DUX4. A recent single dataset study demonstrated that DUX4 expression in human myoblasts perturbs a large proportion of genes which are differentially expressed in FSHD [18]. Our analysis further demonstrates in a meta-analysis setting that signalling network rewiring in FSHD, independent of atrophy, ageing and inflammation, can also be explained by DUX4, lending more support to the hypothesis that DUX4 expression drives pathology.
To determine whether DUX4 expression results in β-catenin signalling, we assayed readouts of the Wnt pathway and found significant DUX4-mediated activation of β-catenin signalling in mouse myoblasts. Thus, we provide the first integrated FSHD network, capable of explaining DUX4-driven pathomechanisms and informing development of therapeutics.
2. Results
2.1. Meta-analysis of facioscapulohumeral muscular dystrophy datasets using InSpiRe
InSpiRe is a differential network methodology we designed to extract a subset of the human protein interaction network containing proteins and interactions that are altered between two phenotypes described by expression data. The three steps of InSpiRe are summarized in figure 1, and explained in detail in Material and methods.
Figure 1.

An overview of the InSpiRe algorithm. Public databases are interrogated for expression data and protein–protein interaction data, as inputs to the algorithm. (a) Step 1 of InSpiRe: integration of expression data with the protein interaction network, via Pearson correlations, results in a weighted network for each phenotype. (b) Step 2 of InSpiRe: information theoretic measures detect rewiring hotspots between the two phenotypes. Differential (local) flux entropy detects shifts in active pathway regimes, Kullback–Leibler (KL) divergence detects rewiring events to which differential entropy is blind. (c) Step 3 of InSpiRe: sparsification of the neighbourhood of rewiring proteins, through consideration of differential correlations across phenotypes, results in a sparse network subset, containing proteins and interactions that are significantly rewiring between the two phenotypes.
Four human FSHD datasets were obtained from the GEO Database. GSE3307 [17,19] consisted of gene expression data from 14 skeletal muscle (nine biceps, five deltoid) biopsies from FSHD patients, and 14 skeletal muscle biopsies from healthy, matched control individuals. GSE10760 [5] contains gene expression data from vastus lateralis biopsies from 19 FSHD patients and 30 control individuals. Both GSE3307 and GSE10760 were profiled on the Affymetrix Human Genome U133A Array platform. GSE26145 [20] and GSE26061 [21] are exon array studies using the Affymetrix Human Exon 1.0 ST Array each profiling three myoblast and three myotube samples from either quadriceps, rhomboid or deltoid muscles of three FSHD patients and three control individuals. As only isolated cells were arrayed in GSE26145 [20] and GSE26061 [21], non-muscle gene expression was assumed to be low/negligible. These studies were employed in our meta-analysis to refine the larger datasets of primary muscle biopsies of GSE3307 [17,19] and GSE10760 [5]. All datasets were pre-processed and normalized as described in Material and methods.
Approximately 3500 genes were implicated per set by InSpiRe as rewiring between FSHD and control samples. Considering all FSHD data, the intersection of rewired sets consisted of a significantly large overlap of 829 genes (p < 10 × 10−5, based on randomly selecting genes from each dataset and assessing the size of overlap). Many genes in this intersection have been associated with FSHD, e.g. TP53 [16], JUNB [20,17], HIF1A [20], WNT3 [4], LMO3 [20], ANXA4 [5] and HSPB1 [22]. Gene Set Enrichment Analysis (GSEA) [23] on the intersection also implicated many FSHD associated processes, such as myogenesis [17] and regulation of the actin cytoskeleton [15], and pathways, including, p53 [16], Wnt [4], and VEGF [5] signalling.
2.2. Gene expression changes specific to facioscapulohumeral muscular dystrophy
Superimposed on network rewiring due to FSHD molecular mechanisms, are rewiring events due to non-specific changes. To identify genes implicated as rewiring specifically in FSHD, we also ran InSpiRe on two datasets each describing skeletal muscle gene expression during ageing (GSE5086 [24] and GSE9676 [25]), disuse atrophy (GSE5110 [26] and GSE8872 [27]) and other muscle diseases involving inflammation and wasting (GSE3307 [19], where juvenile dermatomyositis and limb-girdle muscular dystrophy type 2A datasets were independently analysed). Genes rewiring in these non-FSHD datasets were considered as secondary rewiring. Of the 829 InSpiRe identified genes, 273 were associated with ageing, 364 with disuse atrophy and 394 with other muscle diseases, identifying a core set of 164 genes specifically rewiring in FSHD (electronic supplementary material, table S1). For the final stage of InSpiRe, we considered the 164 high confidence genes and their direct neighbours in the protein interaction network (a complete network of 2866 genes). Statistical sparsification on the largest FSHD dataset (GSE10760) was performed to eliminate interactions that were not significantly altered between FSHD and control phenotypes, to generate the FSHD network.
2.3. The facioscapulohumeral muscular dystrophy network
Our FSHD network consists of 2616 proteins and 15 972 interactions, the majority of which form a single maximally connected component (2603/2616 genes). To evaluate the significance of certain properties of the FSHD network, we computed a distribution of random networks by performing 1000 random selections of 164 core genes and re-running the statistical sparsification on GSE10760 each time. This demonstrated that the FSHD network has significantly more interactions and genes than one would expect by chance (p = 0.04 and p = 0.034, respectively). Such network density implies that signalling dysregulation underlying FSHD is a coordinated perturbation of a large number of intersecting signalling pathways.
2.4. Dysregulation of β-catenin signalling is central to rewiring in facioscapulohumeral muscular dystrophy
To identify critical genes and pathways in our FSHD network, we employed local network measures. Betweenness centrality measures the number of shortest paths between any two genes passing through a given gene and can identify signalling bottlenecks. Genes in our network demonstrating high betweenness centrality are important for coordination of signal dysregulation in FSHD: the gene with the highest betweenness centrality is CTNNB1, encoding β-catenin. CTNNB1 is also highly connected in our network, with a degree of 73, supporting a role for this gene in numerous dysregulated interactions. To determine whether an increase in β-catenin activity is occurring in FSHD muscle, we considered the neighbourhood of CTNNB1 in the FSHD network (figure 2). β-catenin is highly correlated with its neighbours in the FSHD network across FSHD samples, but not across control samples, implying an increase in β-catenin activation in FSHD (figure 2).
Figure 2.

The neighbourhood of CTNNB1 in the FSHD network. Interactions are coloured proportional to the Pearson correlation in gene expression between connected genes across control samples (a) and FSHD samples (b). Red edges are negatively correlated, grey edges uncorrelated and green edges positively correlated. The thickness of edges is proportional to 1 − p, where 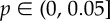 is the p-value of the statistical analysis performed to determine whether correlation in gene expression between connected edges is different between FSHD and control. Large nodes belong to the core set of 164 high confidence FSHD-specific rewiring genes. Large circles indicate proteins significantly rewiring between FSHD and control phenotypes, identified in the second stage of InSpiRe. There is a clear shift from predominantly uncorrelated to highly correlated between FSHD and controls, with an increased correlation between CTNNB1 and its interaction partners across FSHD samples as compared with controls. This is indicative of increased β-catenin activity. Note the increased positive correlation between CTNNB1 and HIF1A, CASP3 and CASP8.
is the p-value of the statistical analysis performed to determine whether correlation in gene expression between connected edges is different between FSHD and control. Large nodes belong to the core set of 164 high confidence FSHD-specific rewiring genes. Large circles indicate proteins significantly rewiring between FSHD and control phenotypes, identified in the second stage of InSpiRe. There is a clear shift from predominantly uncorrelated to highly correlated between FSHD and controls, with an increased correlation between CTNNB1 and its interaction partners across FSHD samples as compared with controls. This is indicative of increased β-catenin activity. Note the increased positive correlation between CTNNB1 and HIF1A, CASP3 and CASP8.
β-catenin is fundamental to canonical Wnt signalling, initiated when Wnt ligands interact with a heterodimeric Frizzled/LRP5/6 complex, which signals to DVL1/2, leading to stabilization of β-catenin and its nuclear translocation. In the nucleus, β-catenin interacts with TCF/LEF transcription factors that normally interact with Groucho, a transcriptional repressor. This interaction with β-catenin allows TCF/LEF to act as transcriptional activators to induce expression of target genes. To determine whether β-catenin is acting via its role in transcription, we queried our network for downstream targets of β-catenin signalling, i.e. TCF/LEF genes. Importantly, all members of the TCF/LEF family (TCF7, TCF7L1 (TCF-3), TCF7L2 (TCF-4) and LEF1) were involved in network rewiring in FSHD.
To analyse control of β-catenin activity via canonical WNT signalling, we queried our network for WNT, DVL and FZD family members [28]. This revealed WNT16, DVL1, DVL2 and FZD1 were rewired (figure 3).
Figure 3.

The Wnt pathway in the FSHD network. Interactions are coloured proportional to the Pearson correlation in gene expression between connected genes across control samples (a) and FSHD samples (b). Note the shift from predominantly uncorrelated links in controls to highly correlated links in FSHD, implying an activation of this Wnt pathway in FSHD.
These genes are connected in the FSHD network (except WNT16), indicating dysregulation of the β-catenin signalling pathway is contributing to FSHD pathogenesis. There is a significantly increased positive correlation in gene expression along the chain: FZD1 → DVL1 → CTNNB1 → TCF-3 (TCFL1) → c-Myc (MYC) in FSHD samples, implying an increased activation of this pathway (figure 3). Increased negative correlation between β-catenin expression and that of PITX2 and increased positive correlation between gene expression of PITX2 and LEF1 also occurred. β-catenin is also involved in numerous other processes including substantially altered correlations with CASP3 and CASP8 (interactions associated with apoptosis), and hypoxia inducible factor 1-α (HIF1-α) (figure 2).
2.5. Activation of HIF1-α signalling in facioscapulohumeral muscular dystrophy
HIF1-α is one of the most rewired of the 164 genes and increases in activity in FSHD (electronic supplementary material, figure S1), with many genes associated with HIF1-α signalling in the FSHD network, including, VHL, HSP90AA1, RBX1, RRAS, VEGFA, MAPK8, NCOA1, PIK3R3, SLC2A4, HIF1AN and TCEB2. HIF1-α signalling has recently been implicated in FSHD, due to identification of several downstream components of the pathway being differentially expressed [20].
2.6. TNF-α over-activation of reactive oxygen species induced JNK cell death pathways
Many genes involved in TNF-α over-activation of reactive oxygen species (ROS) induced JNK cell death pathways were in our FSHD network. These included MAP4K5, PARP2, JUNB (electronic supplementary material, figures S2–S4) TNFA, JUN, JUND, JNK1 and JNK3. MAP4K5 is a highly specific activator of JNK signalling [29] and displays a significantly increased (p = 0.0035) negative expression correlation with TNF-receptor-associated factor 2 (TRAF2) in FSHD samples. PARP2 is necessary for activating TNF-α-induced necrosis [30] and displays increased positive expression correlation with BRCA1 across FSHD samples, in an interaction associated with cell death [31]. JUNB displays significantly increased positive expression correlation with FOS (p = 1.5 × 10−8) and JUN (p = 0.044). JNK1 and JNK3 display clear shifts from predominantly uncorrelated in expression with neighbours in control samples, to highly correlated in FSHD samples, implying their increased activity in FSHD muscle.
2.7. DUX4-driven gene expression mirrors facioscapulohumeral muscular dystrophy
We next determined how much network rewiring in FSHD is directly due to DUX4. We generated a panel of DUX4 constructs in the pMSCV-IRES-eGFP retroviral backbone [32]. In addition to full-length DUX4, we also analysed tMALDUX4—the putative splice variant initiating with the amino acids MAL and lacking the C-terminal domains [33]. We also fused tMALDUX4 to a VP16 transactivation domain from the human herpes simplex virus 1 VP16 protein, to generate tMALDUX4-VP16, a transcriptional activator of DUX4 targets. Conversely, to repress transcriptional targets of DUX4 via recruitment of a repressive complex, tMALDUX4 was also fused to the N-terminal of Drosophila melanogaster engrailed (residues 2–298) to create tMALDUX4-ERD. Also included was the single inverted truncated D4Z4 repeat centromeric to the D4Z4 arrays that encode DUX4c [34]. In all DUX4 constructs, DNA binding, and so target gene selection, is dictated by the homeodomains, which are common to all constructs. Altering the C-terminal region should only affect the degree of target gene activation, allowing both cross-validation and detection of target genes that are only weakly activated by DUX4. Thus, multiple DUX4 constructs containing the same DNA-binding domain robustly and independently confirm target gene selection.
To determine the confluence between our FSHD network and gene expression changes generated by DUX4, we performed a microarray to detect transcriptional changes induced by the DUX4 constructs compared to control retrovirus. Primary mouse satellite cells from three adult C57BL10 male mice were expanded, passaged and infected in parallel. RNA was prepared 20 h later to assay early gene expression changes caused by DUX4, before more non-specific changes associated with inhibition of myogenesis or cell death occur. Gene expression was analysed using Affymetrix GeneChip Mouse Gene 1.0 ST Arrays and was quality controlled and log-normalized as described in Material and methods.
Hierarchical clustering and principal component analysis (PCA) on the 1000 most variable probes demonstrated that the different constructs clustered as expected. Full-length DUX4 clustered with the strong transcriptional activator tMALDUX4-VP16, while DUX4c and tMALDUX4 displayed similar transcriptional profiles, distinct from those obtained for DUX4 and tMALDUX4-VP16 (figure 4). tMALDUX4-ERD, designed to negatively regulate DUX4 transcriptional targets, did not cluster with the other DUX4 constructs. All DUX4 constructs were distinct from control retrovirus. To determine whether DUX4 modifies pathways identified in our FSHD network, multivariate analysis was used to identify gene expression significantly perturbed by one or more DUX4 construct, using the limma package in R [35].
Figure 4.

Clustering of the DUX4 construct expressing mouse satellite cell-derived myoblast samples. PCA (a) and hierarchical clustering (b) on the 1000 most variable probes across the five DUX4 constructs and control RV show clustering of technical replicates, demonstrating reproducibility. Note also clustering of tMALDUX4-VP16 (black) with DUX4 (red); DUX4c (green) with tMALDUX4 (yellow) but separation of tMALDUX4-ERD (purple) and control (blue) from other DUX4 constructs.
Enrichment analysis was performed using Metacore (Genego), to identify biological processes and pathways (networks) modulated by genes perturbed by DUX4 constructs. This revealed considerable enrichment for Wnt signalling (p = 1 × 10−8, figure 5).
Figure 5.

Enrichment analysis of genes perturbed by DUX4 constructs in mouse satellite cell-derived myobalsts. (a) Biological processes enriched among genes perturbed by DUX4 constructs. (b) Biological pathways enriched among genes perturbed by DUX4 constructs. Note the significant enrichment for Wnt/β-catenin signalling among genes perturbed by DUX4 constructs.
Individual DUX4 constructs were also independently compared to control retrovirus and lists of significantly altered genes generated. As expected, Metacore (Genego) also identified significant enrichment of Wnt signalling in DUX4 perturbed genes (p = 7.99 × 10−9), which matches identification of β-catenin as critically rewiring in our FSHD network.
As several pathways identified as rewiring in our FSHD network involved transcription factors, we next determined which transcription factor targets were overrepresented among genes significantly perturbed by the DUX4 constructs. This analysis revealed several enriched transcriptional hubs perturbed by DUX4, of which nine of the top 11 matched to genes in our FSHD network. Consistent with our network identified pathways, among enriched hubs were HIF1-α, JUN and FOS.
To establish whether genes in the FSHD network are perturbed as a result of DUX4, we extracted all microarray probes mapping to genes with direct human orthologues in our FSHD network, creating a network probeset consisting of 1866 genes. We then performed a re-sampling procedure (see Material and methods) to determine whether expression of the network probeset was a significant biomarker of DUX4 expression. This analysis confirmed that mouse orthologues of genes in our human FSHD network are significantly modified by DUX4 (p = 1 × 10−5), and can be used as a biomarker of DUX4 expression.
2.8. Perturbed Wnt/β-catenin signalling in DUX4-expressing satellite cell-derived myoblasts
Several Wnt/β-catenin targets were identified as being altered in both the human FSHD network and the DUX4 microarray. These included Lgr5/6, Tcf3/4, Myf5 and Lef1. To validate this, we performed quantitative PCR (QPCR) on satellite cell-derived myoblasts from three C57Bl10 mice retrovirally infected with either DUX4 or control retroviral constructs and cultured for 24 or 48 h. At one or both time points, Lef1, Tcf4, Lgr5 and Myf5 were significantly increased in DUX4-infected samples, whereas Tcf3 and Lgr6 were significantly decreased by DUX4 (figure 6a), confirming that DUX4 alters this signalling cascade at a transcriptional level.
Figure 6.

DUX4 perturbs Wnt/β-catenin signalling. (a) QPCR of downstream targets of Wnt/β-catenin signalling Lef1, Tcf3, Tcf4, Lgr5, Lgr6 and Myf5 in mouse satellite cell-derived myoblasts, 24 or 48 h after infection with either DUX4 or control retroviral constructs. Data are mean ± s.e.m. from three mice, where an asterisk denotes a significant difference (p < 0.05) from infection with control retrovirus using a paired two-tailed Student's t-test. (b) TopFLASH/FopFLASH assay of immortalized C2C12 murine myoblasts transfected with either DUX4 or control plasmid, shows a significant increase in TopFLASH/FopFLASH luciferase activity with DUX4 expression (n = 4). Data are mean ± s.e.m. from four independent experiments where an asterisk denotes significance (p < 0.05) from control using a two-tailed Student's t-test.
2.9. DUX4 induces Wnt/β-catenin signalling in murine myoblasts
Wnt/β-catenin signalling involves the physical interaction of several proteins, and so transcriptional disruption of targets does not necessarily mean that Wnt/β-catenin signalling is altered. To confirm activation of Wnt/β-catenin signalling in DUX4-expressing myoblasts, we performed a TopFLASH/FopFLASH assay. The TopFLASH Tcf reporter plasmid contains six Tcf-binding sites, whereas in the FopFLASH reporter, these sites are mutated, upstream of a luciferase reporter. The relative ratio is a widely used assay for measuring canonical Wnt signalling [36]. Primary satellite cell-derived myoblasts can be difficult to transfect with high efficiency, so we switched to murine C2C12 myoblasts. These were co-transfected with either TopFLASH or FopFLASH plus Renilla, then passaged and transfected with DUX4 or control vector for 24 h. Relative luciferase activity measured from four independent experiments showed that DUX4 expression significantly increased the TopFLASH/FopFLASH ratio 2.36-fold (p = 0.04) and hence DUX4 expression activates Wnt/β-catenin signalling in myoblasts (figure 6b).
3. Discussion
We have constructed the FSHD network: the first unified, unbiased map of network rewiring underlying FSHD. DUX4-driven transcriptional changes demonstrated that expression of genes in our FSHD network was significantly altered by DUX4. This is consistent with the growing consensus that FSHD1/2 is caused by aberrant DUX4 expression [9,18]. Importantly, our FSHD network is significantly more connected than a random network. Such connectivity allows analysis of crosstalk in FSHD pathological signalling, essential to guide development of therapeutics targeting the multiple symptoms of FSHD. Our network is derived from gene expression in muscle biopsies and myogenic cells, and thus provides clear insight into pathways perturbed in FSHD muscle. In addition however, we have also identified pathways which influence vasculogenesis and neurogenesis in general, suggesting that our results can also inform on extra-muscular symptoms of FSHD, including retinal telangiectasia and sensorineural hearing loss. Differential gene expression studies have generally implicated disjointed collections of genes associated with these symptoms (e.g. myogenesis [17] and vasculature growth factors [5]), and hypotheses have been proposed [4,37]. However, there has been no unbiased, data driven insight into how these pathways relate, let alone actually coordinate. Our results emphasize the importance of the development and application of network theoretic tools, such as InSpiRe, to identify pathomechanisms and therapeutic targets in complex diseases.
We compared InSpiRe to other commonly used network methodologies (electronic supplementary material): NetWalk [13] and GSEA on differentially expressed genes. NetWalk also uses a protein interaction network to identify candidate genes, but unlike InSpiRe, uses weighted random walks. Both NetWalk and InSpiRe displayed a significant consensus in the features identified across the four FSHD datasets (p = 1 × 10−5). This is in contrast to differential expression, which lacks consistency in the features identified across these datasets. Thus demonstrating the power of network-based methodologies over conventional differential expression. NetWalk performed similarly to InSpiRe in identifying genes which proved a significant classifier of the DUX4 construct microarray samples (p = 1 × 10−5) but generated a network with more genes and fewer interactions (3363 genes and 5651 interactions) than InSpiRe (2616 genes and 15 972 interactions). However, both NetWalk and GSEA on differentially expressed genes were inferior to InSpiRe in identifying functional annotations previously associated with FSHD (electronic supplementary material, figure S5).
There is increasing evidence that aberrant DUX4 expression due to changes in the number or methylation state of D4Z4 repeats on chromosome 4 underlies FSHD pathology [9]. Indeed, it was recently demonstrated that DUX4 drives over half the differentially expressed transcripts in FSHD [18]. However, the number of differentially expressed transcripts is very low in FSHD as compared with other pathologies [18,5,15] and it has been shown that a more subtle transcriptional dysregulation may better represent unifying features of FSHD [15]. Our FSHD network captures such subtle dysregulation and it was important to determine whether the network, derived from unbiased meta-analysis of multiple muscle biopsies from FSHD patients, could be controlled by DUX4. Performing microarray analysis to detect changes in gene expression after DUX4 constructs were expressed in muirne myoblasts demonstrated that DUX4 modifies expression of genes in our FSHD network. Moreover, we found significant enrichment of DUX4-mediated transcriptional changes for critical pathways identified in our network, including Wnt/β-catenin, HIF1-α and JNK signalling. This also indicates that many DUX4-mediated signalling changes are conserved between man and mouse, making mouse models useful for understanding FSHD pathology and as platforms for testing therapeutics [38].
CTNNB1 has the highest betweenness centrality in our FSHD network, identifying β-catenin as a bottleneck of FSHD-associated protein interaction signalling. Gene expression correlation between β-catenin and its interaction partners is also significantly increased in FSHD, implying increased activation. Moreover, the TCF/LEF transcription factors are present in our network, as well as several upstream components of the canonical Wnt pathway. Involvement of Wnt signalling in tissue-specific myogenesis and in retinal angiogenesis led to a recent hypothesis that Wnt/β-catenin signalling was important for FSHD pathomechanisms [4]. A role of Wnt/β-catenin signalling in controlling DUX4 expression and FSHD muscle cell apoptosis was also recently proposed [39]. Moreover, enhancing expression of β-catenin via administration of LiCl2 reduces apoptosis in a model of occulopharyngeal muscular dystrophy [40]. Our FSHD network, compiled algorithmically from multiple independent datasets, provides the detailed interactions of dysregulated β-catenin signalling. Furthermore, Wnt/β-catenin-related genes were enriched in our microarray of DUX4-driven transcriptional changes in mouse myoblasts and validated via QPCR. DUX4 also drives activation of the canonical Wnt pathway, as shown by the TopFLASH assay.
Interaction partners of β-catenin in our FSHD network reveal mechanisms by which Wnt/β-catenin signalling is involved in certain hallmark phenotypes of FSHD through pathway crosstalk. Among these is the characteristic oxidative stress sensitivity of FSHD muscle/myoblasts [41,42]. Our work implicates HIF1-α signalling as critically perturbed in FSHD and shows that HIF1A displays strong positive correlation with β-catenin in FSHD samples, providing mechanism for previous observations of involvement of downstream components of the HIF1-α pathway in FSHD [20]. HIF1-α binds β-catenin during hypoxia competitively with TCF-4 [43], which may both inhibit TCF4/β-catenin-driven cell proliferation and increase transcription of hypoxic response genes, including VEGF. We found that in FSHD, correlation in gene expression between β-catenin, and both HIF1A and TCF-4, significantly increases, resulting in elevated angiogenic genes such as VEGF, providing an explanation for the hallmark oxidative stress sensitivity in FSHD, as well as retinal vasculature abnormalities [4,5].
Actin cytoskeleletal signalling has also been implicated in FSHD pathology [15]. Our work shows perturbed crosstalk between such signalling and HIF1-α may contribute to FSHD. MAP2 is associated with microtubule stability and rewired in FSHD. In hypoxic cardiomyocytes, MAP2 is required for stabilization of the microtubule network, leading to suppression of pVHL and increased HIF1-α [44]. Interestingly via this mechanism, HIF1-α was upregulated at the protein level but not the mRNA level (in early stages of hypoxia) [44], emphasizing the power of our method for detecting events invisible to differential expression analysis of microarrays.
Another notable pathway perturbed in FSHD was ROS. The role of ROS in FSHD is well reported, and our FSHD network contains many genes in the ROS-mediated pathway, including TNF-α. DUX4 represses genes of the glutathione redox pathway, likely causing ROS accumulation in FSHD muscle [45], which may lead to increased TNF-α as part of a pro-inflammatory response [46]. Additionally, levels of TNF-α are negatively correlated with muscle endurance [42]. FSHD myoblasts undergo cell death in response to non-pathological levels of hydrogen peroxide [41] and other oxidative stress-inducing factors; while antioxidants inhibit DUX4-induced toxicity in FSHD myoblasts [45]. Thus, over-activation of ROS-mediated TNF-α-induced cell death pathways are potentially important pathomechanism in FSHD. TNF-α also stimulates ROS production via interaction with NADPH-oxidase, in a positive feedback loop [30]. Our results support this occurring in FSHD as RAC1 is a well-connected member of our FSHD network and a critical component of the NADPH complex, also capable of activating JNK cell death signalling. RAC1 is also regulated by non-canonical Wnt signalling in a manner dependent on MAP4K5 [47]. Our network unifies TNF-α, Wnt and JNK signalling in FSHD.
JNK signalling also plays an important role in oxidative stress-induced cell death, and we found that JNK signalling is more active in FSHD. JNK signalling can be controlled in many ways, including via TNF-α signalling. In this scenario, the AP-1 transcription factor target genes JUN, JUNB and FOS are specifically upregulated downstream of JNK. These genes are present in our FSHD network, moreover, all display a significant increase in positive gene expression correlation with one another in FSHD samples, indicating their co-regulation. This result is evidence of TNF-α-mediated JNK signalling in FSHD.
In the absence of NF-κB activity, prolonged JNK activation by TNF-α leads to apoptosis [48]. Owing to the increased apoptosis of FSHD muscle cells, we postulate that NF-κB may be less active. In addition to activating JNK cell death pathways in response to ROS, TNF-α also activates NF-κB survival signalling causing the production of the antioxidant MnSOD to suppress ROS and minimize JNK cell death signalling [30]. MnSOD is the only antioxidant downregulated in FSHD [42], suggesting that NF-κB may be less active in FSHD muscle. Our results provide evidence for this theory: NFKB1 encodes the DNA-binding subunit of the NF-κB transcription factor, which though present in our FSHD network, is relatively uncorrelated with its interaction partners in FSHD samples. This implies NFKB1 is inactive in FSHD muscle, and thus unable to repress the increased cell death via overactive JNK. Finally, we find crosstalk between JNK signalling and Wnt, in that all members of the PAR-1 gene family are in our FSHD network. This family was identified as dishevelled kinases, capable of simultaneously regulating Wnt activation of β-catenin signalling and JNK signalling [49]. Overactive JNK signalling has also been implicated in sensorineural hearing loss, an FSHD clinical phenotype [50]. Inhibitors of JNK signalling could partially mitigate the oxidative stress sensitivity of FSHD muscle cells, and D-JNKI-1 is currently in clinical trials for treatment of strokes [51].
In conclusion, we have developed a novel, general algorithm, InSpiRe, to detect network rewiring from multiple gene expression datasets describing two phenotypes. Using InSpiRe, we performed the first meta-analysis of the FSHD transcriptome, creating an integrated FSHD network. We also assayed DUX4-driven transcriptional changes and demonstrated that expression of genes in our network is significantly altered by DUX4. Our FSHD network provides an unbiased, unifying molecular map of FSHD-associated protein interaction signalling, elucidating perturbed genes and pathways critical to pathomechanisms. Importantly, we identify and validate β-catenin as central to FSHD pathology. Our network provides insight into the crucial steps of dysregulated signalling in FSHD and so will inform design of well-targeted therapeutics: currently lacking for FSHD. We provide the FSHD network as a queryable interface (electronic supplementary material).
4. Material and methods
4.1. Construction of the protein interaction network
The protein interaction network was constructed as previously described [52] from integration of the Human Protein Interaction Network (www.pathwaycommons.org) [53], the Human Protein Reference Database [54], the National Cancer Institute Pathway Interaction Database, the Interactome (www.ebi.ac.uk/intact/) and the Molecular Interaction Database [55]. Redundant interactions were removed leaving only genes with a unique EntrezGene ID, within the maximally connected component. The protein interaction network contains 10 720 proteins and 152 889 documented interactions and post-translational modifications, including protein binding, complex formation, phosphorylation, ubiquitylation and sumoylation.
4.2. Public mRNA expression datasets
We ran InSpiRe on the following four datasets from the GEO Database [56]: GSE3307 [19] (14 FSHD samples, 14 controls), GSE10760 [5] (19 FSHD samples, 30 controls), both profiled on the Affymetrix Human Genome U133A Array platform. GSE26145 [20] and GSE26061 [21] expression studies of FSHD and control myoblasts and myotubes profiled on the Affymetrix Human Exon 1.0 ST Array, were also analysed.
A recent study GSE36398 [15] was excluded from our analysis as it reported substantially weaker gene expression changes between FSHD and control muscle biopsies compared with most previous studies [15]. It was considerably discordant with the other datasets analysed by InSpiRe in identification of rewired genes. Thus, given that the other studies displayed significant agreement, GSE36398 was omitted.
We also used human skeletal muscle studies into gene expression in muscle diseases other than FSHD (GSE3307 [19]), ageing (GSE5086 [24] and GSE9676 [25]) and atrophy (GSE5110 [26] and GSE8872 [27]).
4.3. Pre-processing and normalization of public datasets
Expression datasets underwent quality control using the Array QualityMetrics package in R [57]. This included analysis of RNA degradation and elimination of samples displaying clear signal saturation [58]. Datasets were log-normalized using robust multi-array average (RMA) and PCA performed to determine whether the dominant principal component correlated with condition being compared (e.g. disease status). PCA performed on the two larger FSHD studies (GSE3307 and GSE10760) revealed that the dominant principal component correlated with both disease status and age of patients sampled. For this reason, we additionally analysed datasets associated with age-dependent gene expression.
PCA on the age-dependent study, GSE5086 revealed that the dominant principal component correlated with age and separated into two groups corresponding to young and older samples. We used this clustering to classify samples as either younger or older patients. Non-FSHD muscle diseases analysed were selected from GSE3307 due to the large number of high-quality samples available.
GSE2614 and GSE26061 were suitable for integration by the ComBat function [59] in R which employs an empirical Bayes approach to eliminate batch effects and was recently demonstrated as superior to other microarray data integration methods [60]. The integrated dataset contained six FSHD myoblasts, six FSHD myotubes, six control myobalsts and six control myotubes. PCA was performed on the integrated dataset and the dominant principal component correlated with cell type and disease status but not with batch, implying a successful integration.
We extracted from datasets probes mapping to genes corresponding to proteins in our interaction network. For all microarray platforms considered, there were cases where multiple probes mapped to a single gene. As our protein interaction network does not take into account alternative splice variants of a given gene, we must assign a single value to each protein based on the expression data. There are multiple ways of achieving this, and for our methodology we computed the average expression across probes mapping to a single gene for each sample, assigning the resulting value to the gene. This approach has proved successful in our previous studies for identification of network rewiring in cancer [52] and cell differentiation [61]. For each dataset, proteins in the interaction network with no corresponding probe in the microarrays were deleted from the network, proteins with a degree of zero following this deletion were also removed. This resulted in a reduced protein interaction network for each dataset.
4.4. The interactome sparsification and rewiring algorithm
InSpiRe is a differential network methodology, consisting of three main steps (figure 1). An R-script for implementation of InSpiRe is provided in the electronic supplementary material.
4.5. Integration of mRNA expression data with the protein interaction network
The first step of InSpiRe is integration of expression data with interaction data (figure 1a). For each phenotype, we integrate expression data with the protein interaction network by assigning each interaction connecting two proteins, i and j, with a transformed Pearson correlation in gene expression profiles of proteins i and j across the samples corresponding to the given phenotype (denoted Cij). This results in a weighted network wij which is then transformed into a stochastic matrix Pij
| 4.1 |
where N(i) denotes the set of neighbours of protein i in the interactome.
We interpret row k in the matrix Pij as a probability distribution describing the interaction preferences of protein k. Note that 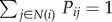 and Pij = 0 whenever i and j are not connected in the interaction network. For
and Pij = 0 whenever i and j are not connected in the interaction network. For 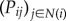 to be a probability distribution, we require that Pij ≥ 0, which is guaranteed if wij ≥ 0. The choice of the transformation of the Pearson correlation wij must be made carefully to ensure non-negativity and that interpretation of a high edge weight, indicative of an increased likelihood of interaction between connected proteins, is valid. We considered two possible transformations of which
to be a probability distribution, we require that Pij ≥ 0, which is guaranteed if wij ≥ 0. The choice of the transformation of the Pearson correlation wij must be made carefully to ensure non-negativity and that interpretation of a high edge weight, indicative of an increased likelihood of interaction between connected proteins, is valid. We considered two possible transformations of which 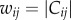 was superior (see the electronic supplementary material).
was superior (see the electronic supplementary material).
4.6. Detecting rewiring hotspots
The second stage of InSpiRe is utilization of information theoretic measures to detect rewiring between two phenotypes described by weighted networks constructed by stage 1 of InSpiRe (figure 1b). Two measures are employed to detect significant rewiring between two phenotypes: differential local flux entropy and Kullback–Leibler divergence.
Local flux entropy quantifies the disorder of a protein interaction distribution in a given phenotype and is based on Shannon entropy.
Given a stochastic matrix Pij describing a given phenotype, the local flux entropy of protein i is defined as
| 4.2 |
where ki is the degree of protein i in the interaction network.  is a measure of how close a protein's interaction distribution is to uniform. Values close to 0 imply protein i has a deterministic interaction distribution and values close to 1 suggest a uniform profile.
is a measure of how close a protein's interaction distribution is to uniform. Values close to 0 imply protein i has a deterministic interaction distribution and values close to 1 suggest a uniform profile.
We compute vectors describing local flux entropy of each protein in the interactome for two phenotypes. These are statistically analysed using the jack-knife technique (electronic supplementary material) to identify proteins with significantly different local flux entropies between phenotypes. Such proteins can be considered as altering heterogeneity in their interaction distributions across phenotypes.
Our previous studies on cancer and cell differentiation have revealed that proteins with lower entropy in a given phenotype can be interpreted as being more active in that phenotype [14,61,52]. Examples of interaction distribution changes which lead to either an increase or a decrease in local flux entropy are displayed in figure 1b.
It is possible for a protein interaction distribution to be rewired without changing uniformity (figure 1b). Consequentially, we introduce a novel measure based upon Kullback–Leibler divergence, which though lacking the functional interpretation of local flux entropy, is sensitive to such rewiring.
The Kullback–Leibler divergence is defined as follows:
Given two probability distributions 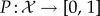 and
and 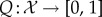 , provided
, provided 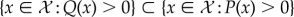 the Kullback–Leibler divergence between P and Q is given by
the Kullback–Leibler divergence between P and Q is given by
| 4.3 |
Note that 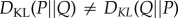 as
as 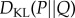 quantifies the expected number of bits required to describe a sample from the probability distribution P given one incorrectly assumes the sample follows the distribution Q. To compare distributions describing the interactions of proteins in control and pathological samples, choice of direction of Kullback–Leibler divergence is ambiguous, we will therefore use symmetrized Kullback–Leibler divergence defined by
quantifies the expected number of bits required to describe a sample from the probability distribution P given one incorrectly assumes the sample follows the distribution Q. To compare distributions describing the interactions of proteins in control and pathological samples, choice of direction of Kullback–Leibler divergence is ambiguous, we will therefore use symmetrized Kullback–Leibler divergence defined by
| 4.4 |
Given two phenotypes A and B described by stochastic matrices 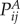 ,
, 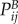 , the local symmetrized Kullback–Leibler divergence of protein i between phenotypes is
, the local symmetrized Kullback–Leibler divergence of protein i between phenotypes is
| 4.5 |
and
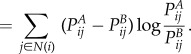 |
4.6 |
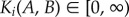 , values near 0 indicate that interaction distribution of protein i is similar across phenotypes, large values imply rewiring of protein i between phenotypes.
, values near 0 indicate that interaction distribution of protein i is similar across phenotypes, large values imply rewiring of protein i between phenotypes.
We employ the jack-knife technique (electronic supplementary material) to determine which proteins have a significantly non-zero Kullback–Leibler divergence (and therefore are rewiring) between two phenotypes.
4.7. Sparsification of relevant subset of protein interaction network
The final step of InSpiRe is sparsification of the rewiring subset of protein interactions (figure 1c). To create the relevant subset of the protein interaction network, proteins significantly rewiring between two phenotypes, as identified in step 2 of InSpiRe, are connected to their neighbours in the interactome. Interactions in this sub-network connecting proteins whose expression is not significantly differently correlated (assessed again via the jack-knife technique) between two phenotypes are deleted, sparsifying the network to leave only rewired interactions.
4.8. DUX4 constructs
Coding sequences for DUX4 and DUX4c were kindly received from Dr Stephen Tapscott and Dr Alexandra Belayew. tMALDUX4 was created by removing 75 amino acids from the C-terminus of DUX4 [33]. The stop codon of tMALDUX4 was removed and ligated to either a VP16 transactivation domain or engrailed repressor domain (ERD) to create tMALDUX4-VP16 and tMALDUX4-ERD, respectively. All inserts were ligated into the pMSCV-IRES-eGFP vector (Clonetech) [32].
Retroviruses-encoding DUX4 constructs and control pMSCV-IRES-eGFP were produced by transfecting HEK293T packaging cells with DUX4/pMSCV-IRES-eGFP constructs and a retroviral helper plasmid using Lipofectamine (Invitrogen).
4.9. Microarray
Satellite cells from three male eight-week-old C57BL/10 mice were isolated and cultured [62]. Satellite cell-derived myoblasts were expanded on Matrigel-coated plates for 6 days in DMEMGlutamax (Invitrogen) with 30% fetal bovine serum (PAA), 10% horse serum (Gibco), 1% chick embryo extract (ICN Flow), 10 ng ml−1 bFGF (Peprotech) and 1% penicillin/streptomycin (Sigma) at 37°C 5% CO2. Myoblasts were infected with DUX4, DUX4c, tMALDUX4, tMALDUX4-VP16, tMALDUX4-ERD or control pMSCV-IRES-eGFP retrovirus with 4 µg ml−1 Polybrene for 20 h, before RNA extraction using Qiagen RNeasy Kit and quantification on a Nanodrop ND-1000 spectrophotometer (Labtech). Gene expression analysis was performed using GeneChip Mouse Gene 1.0ST Array and GCS3000 microarray system (Affymetrix) by the King's Genomic Centre. Data were quality controlled and log-normalized using RMA, and processed in three independent batches, with batch effects compensated using the ComBat function in R citecombat. PCA on the 1000 most variable probes confirmed compensation, with the top principal components uncorrelated with batch (figure 4).
4.10. Re-sampling procedure to assess concordance between microarray and facioscapulohumeral muscular dystrophy network
To determine whether expression of genes in our FSHD network was significantly altered by DUX4, we first identified a probeset of mouse orthologues to 1866 genes in the FSHD network, to permit comparison with our murine satellite cell microarray of DUX4 construct expression. Our objective was to assess whether expression of genes in the network probeset was able to distinguish between the five DUX4 retroviral constructs better than would be expected by chance. If this is the case, then clustering of DUX4 constructs based on expression of the network probeset should be significantly better than that based on a random probeset of equivalent size.
We therefore performed a re-sampling procedure, evaluating 10 000 random probesets of 1866 genes from our microarray. For each random probeset, we performed a hierarchical clustering and enforced a six cluster solution, which we then compared to the optimal six cluster solution corresponding to the perfect separation of the six retroviral constructs. To compare cluster distributions, we used the Rand index, a function which assesses classification similarity on a unit scale. In this manner, we obtained a null distribution of Rand indices describing how well a random probeset may be expected to cluster the five DUX4 constructs. This null distribution then allowed us to calculate a p-value evaluating the hypothesis that expression of the network probeset clustered the DUX4 constructs better than could be expected by chance.
4.11. TopFLASH/FopFLASH
TopFLASH and FopFLASH Tcf reporter constructs (Millipore) contain either six complete (TopFLASH) or mutant (FopFLASH) Tcf-binding sites, upstream of a luciferase reporter gene [63]. C2C12 myoblasts cultured in DMEM + 10% FBS + 1% penicillin/streptomycin were transfected with either construct plus Renilla (transfection control) for 24 h before trypsinizing and transfected a second time in triplicates with either 100 ng DUX4 or control pMSCV-IRES-eGFP using Lipofectamine (Invitrogen). Myoblasts were then incubated in DMEM Glutamax (Invitrogen) + 2% horse serum (Gibco) + 1% penicillin/streptomycin (Sigma) for 24 h. Relative luciferase was measured using the Dual Luciferase Kit (Promega), β-catenin activity was calculated as luciferase/renilla, then TopFLASH/FopFLASH, and plotted as a fold change of the control. The experiment was performed on four independent occasions with statistics based on the combined average of each experiment.
4.12. Quantitative PCR
Satellite cell-derived myoblasts were infected with DUX4 and control retroviral constructs and RNA extracted as per the microarray samples at 24 and 48 h after infection. RNA was then reverse-transcribed using the Reverse Transcription Kit with genomic DNA wipe-out (Qiagen) and QPCR was performed on an Mx3005P QPCR system (Stratagene) with MESA Blue QPCR MasterMix Plus and ROX reference dye (Eurogentec). Expression was normalized relative to Tbp expression. Primers used were as follows: Lef1; F-TCATCACCTACAGCGACGAG, R-TGATGGGAAAACCTGGACAT. Lgr5; F-CCGCCAGTCTCCTACATCGCC, R-GCATTGTCATCTAGCCACAGGTGCC. Lgr6; F-CACACATCCCGGGACAGGCAT, R-GGGAGGAGAGCCCCTCAAGC. Tcf3; F-TCTCAAGCCGGTTCCCACAC, R-TTTCCGGGCAAGCTCATAGTATTT. Tcf4; F-TGCCGACTACAACAGGGACT, R-TGCTGGACTGTGGGATATGA. Myf5 F-TGAGGGAACAGGTGGAGAAC, R-AGCTGGACACGGAGCTTTTA. Tbp F-ATCCCAAGCGATTTGCTG, R-CCTGTGCACACCA.
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Acknowledgements
Statistical analysis was performed by C.R.S.B. with guidance from A.E.T. and S.S. The study was conceived by C.R.S.B., P.K. and P.S.Z. Experiments were performed by P.K., L.M. and C.R.S.B. C.R.S.B. and P.S.Z wrote the manuscript with contributions from L.M., A.E.T., S.S., R.O., and P.K.
Funding statement
C.R.S.B. is supported by a CoMPLEX PhD studentship, the British Heart Foundation (SP/08/004) and the FSH Society Shack Family and Friends research grant (FSHS-82013–06). L.M. is funded by a Muscular Dystrophy Campaign studentship (RA4/817). S.S. is supported by the Royal Society. A.E.T. is supported by a Heller Research Fellowship. The Zammit laboratory is additionally supported by the Medical Research Council and Association Française contre les Myopathies, together with BIODESIGN (262948–2) from EU FP7.
Conflict of interests
The authors declare that they have no conflict of interest.
References
- 1.Orrell RW. 2011. Facioscapulohumeral dystrophy and scapuloperoneal syndromes. Handb. Clin. Neurol. 101, 167–180. ( 10.1016/B978-0-08-045031-5.00013-X) [DOI] [PubMed] [Google Scholar]
- 2.Deenen JC, Arnts H, van der Maarel SM, Padberg GW, Verschuuren JJ, Bakker E, Weinreich SS, Verbeek AL, van Engelen BG. 2014. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology 83, 1056–1059. ( 10.1212/WNL.0000000000000797) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brouwer OF, Padberg GW, Ruys CJ, Brand R, de Laat JA, Grote JJ. 1991. Hearing loss in facioscapulohumeral muscular dystrophy. Neurology 41, 1878–1881. ( 10.1212/WNL.41.12.1878) [DOI] [PubMed] [Google Scholar]
- 4.Fitzsimons RB. 2011. Retinal vascular disease and the pathogenesis of facioscapulohumeral muscular dystrophy. A signalling message from Wnt? Neuromuscul. Disord. 21, 263–271. ( 10.1016/j.nmd.2011.02.002) [DOI] [PubMed] [Google Scholar]
- 5.Osborne RJ, Welle S, Venance SL, Thornton CA, Tawil R. 2007. Expression profile of FSHD supports a link between retinal vasculopathy and muscular dystrophy. Neurology 68, 569–577. ( 10.1212/01.wnl.0000251269.31442.d9) [DOI] [PubMed] [Google Scholar]
- 6.Dixit M, et al. 2007. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc. Natl Acad. Sci. USA 104, 18 157–18 162. ( 10.1073/pnas.0708659104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gabriels J, et al. 1999. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3kb element. Gene 236, 25–32. ( 10.1016/S0378-1119(99)00267-X) [DOI] [PubMed] [Google Scholar]
- 8.Lemmers RJ, et al. 2012. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 44, 1370–1374. ( 10.1038/ng.2454) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tawil R, van der Maarel SM, Tapscott SJ. 2014. Facioscapulohumeral dystrophy: the path to consensus on pathophysiology. Skeletal Muscle 4, 12 ( 10.1186/2044-5040-4-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Califano A. 2011. Rewiring makes the difference. Mol. Syst. Biol. 7, 463 ( 10.1038/msb.2010.117) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ideker T, Krogan NJ. 2012. Differential network biology. Mol. Syst. Biol. 8, 565 ( 10.1038/msb.2011.99) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim YA, Wuchty S, Przytycka TM. 2011. Identifying causal genes and dysregulated pathways in complex diseases. PLoS Comput. Biol. 7, e1001095 ( 10.1371/journal.pcbi.1001095) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Komurov K, White MA, Ram PT. 2010. Use of data-biased random walks on graphs for the retrieval of context-specific networks from genomic data. PLoS Comput. Biol. 6, e1000889 ( 10.1371/journal.pcbi.1000889) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teschendorff AE, Severini S. 2010. Increased entropy of signal transduction in the cancer metastasis phenotype. BMC Syst. Biol. 4, 104 ( 10.1186/1752-0509-4-104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rahimov F, King OD, Leung DG, Bibat GM, Emerson JCP, Kunkel LM, Wagner KR. 2012. Transcriptional profiling in facioscapulohumeral muscular dystrophy to identify candidate biomarkers. Proc. Natl Acad. Sci. USA 109, 16 234–16 239. ( 10.1073/pnas.1209508109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wallace LM, Garwick SE, Mei W, Belayew A, Coppee F, Ladner KJ, Guttridge D, Yang J, Harper SQ. 2011. DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Ann. Neurol. 69, 540–552. ( 10.1002/ana.22275) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Winokur ST, Chen YW, Masny PS, Martin JH, Ehmsen JT, Tapscott SJ, van der Maarel SM, Hayashi Y, Flanigan KM. 2003. Expression profiling of FSHD muscle supports a defect in specific stages of myogenic differentiation. Hum. Mol. Genet. 12, 2895–2907. ( 10.1093/hmg/ddg327) [DOI] [PubMed] [Google Scholar]
- 18.Yao Z, Snider L, Balog J, Lemmers RJ, Van Der Maarel SM, Tawil R, Tapscott SJ. 2014. DUX4-induced gene expression is the major molecular signature in FSHD skeletal muscle. Hum. Mol. Genet. 23, 5342–5352. ( 10.1093/hmg/ddu251) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bakay M, et al. 2006. Nuclear envelope dystrophies show a transcriptional fingerprint suggesting disruption of Rb-MyoD pathways in muscle regeneration. Brain 129, 996–1013. ( 10.1093/brain/awl023) [DOI] [PubMed] [Google Scholar]
- 20.Tsumagari K, Chang SC, Lacey M, Baribault C, Chittur SV, Sowden J, Tawil R, Crawford GE, Ehrlich M. 2011. Gene expression during normal and FSHD myogenesis. BMC Med. Genomics 4, 67 ( 10.1186/1755-8794-4-67) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheli S, Francois S, Bodega B, Ferrari F, Tenedini E, Roncaglia E, Ferrari S, Ginelli E, Meneveri R. 2011. Expression profiling of FSHD-1 and FSHD-2 cells during myogenic differentiation evidences common and distinctive gene dysregulation patterns. PLoS ONE 6, e20966 ( 10.1371/journal.pone.0020966) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Celegato B, et al. 2006. Parallel protein and transcript profiles of FSHD patient muscles correlate to the D4Z4 arrangement and reveal a common impairment of slow to fast fibre differentiation and a general deregulation of MyoD-dependent genes. Proteomics 6, 5303–5321. ( 10.1002/pmic.200600056) [DOI] [PubMed] [Google Scholar]
- 23.Subramanian A, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15 545–15 550. ( 10.1073/pnas.0506580102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zahn JM, et al. 2006. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet. 2, e115 ( 10.1371/journal.pgen.0020115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Welle S, Tawil R, Thornton CA. 2008. Sex-related differences in gene expression in human skeletal muscle. PLoS ONE 3, e1385 ( 10.1371/journal.pone.0001385) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Urso ML, Scrimgeour AG, Chen YW, Thompson PD, Clarkson PM. 2006. Analysis of human skeletal muscle after 48 h immobilization reveals alterations in mRNA and protein for extracellular matrix components. J. Appl. Physiol. 101, 1136–1148. ( 10.1152/japplphysiol.00180.2006) [DOI] [PubMed] [Google Scholar]
- 27.Chen YW, Gregory CM, Scarborough MT, Shi R, Walter GA, Vandenborne K. 2007. Transcriptional pathways associated with skeletal muscle disuse atrophy in humans. Physiol. Genomics 31, 510–520. ( 10.1152/physiolgenomics.00115.2006) [DOI] [PubMed] [Google Scholar]
- 28.Gordon MD, Nusse R. 2006. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 281, 22 429–22 433. ( 10.1074/jbc.R600015200) [DOI] [PubMed] [Google Scholar]
- 29.Kyriakis JM, Avruch J. 2012. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol. Rev. 92, 689–737. ( 10.1152/physrev.00028.2011) [DOI] [PubMed] [Google Scholar]
- 30.Morgan MJ, Liu ZG. 2010. Reactive oxygen species in TNFalpha-induced signaling and cell death. Mol. Cells 30, 1–12. ( 10.1007/s10059-010-0105-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Helleday T. 2011. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol. Oncol. 5, 387–393. ( 10.1016/j.molonc.2011.07.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zammit PS, Relaix F, Nagata Y, Ruiz AP, Collins CA, Partridge TA, Beauchamp JR. 2006. Pax7 and myogenic progression in skeletal muscle satellite cells. J. Cell Sci. 119, 1824–1832. ( 10.1242/jcs.02908) [DOI] [PubMed] [Google Scholar]
- 33.Snider L, et al. 2010. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 6, e1001181 ( 10.1371/journal.pgen.1001181) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bosnakovski D, Lamb S, Simsek T, Xu Z, Belayew A, Perlingeiro R, Kyba M. 2008. DUX4c, an FSHD candidate gene, interferes with myogenic regulators and abolishes myoblast differentiation. Exp. Neurol. 214, 87–96. ( 10.1016/j.expneurol.2008.07.022) [DOI] [PubMed] [Google Scholar]
- 35.Smyth GK. 2004. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, 1544–6115. ( 10.2202/1544-6115.1027) [DOI] [PubMed] [Google Scholar]
- 36.Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destree O, Clevers H. 1996. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell 86, 391–399. ( 10.1016/S0092-8674(00)80112-9) [DOI] [PubMed] [Google Scholar]
- 37.Lefkowitz DL, Lefkowitz SS. 2005. Fascioscapulohumeral muscular dystrophy: a progressive degenerative disease that responds to diltiazem. Med. Hypotheses 65, 716–721. ( 10.1016/j.mehy.2005.04.035) [DOI] [PubMed] [Google Scholar]
- 38.Krom YD, et al. 2013. Intrinsic epigenetic regulation of the D4Z4 macrosatellite repeat in a transgenic mouse model for FSHD. PLoS Genet. 9, e1003415 ( 10.1371/journal.pgen.1003415) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Block GJ, Narayanan D, Amell AM, Petek LM, Davidson KC, Bird TD, Tawil R, Moon RT, Miller DG. 2013. Wnt/beta-catenin signaling suppresses DUX4 expression and prevents apoptosis of FSHD muscle cells. Hum. Mol. Genet. 22, 4661–4672. ( 10.1093/hmg/ddt314) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abu-Baker A, Laganiere J, Gaudet R, Rochefort D, Brais B, Neri C, Dion PA, Rouleau GA. 2013. Lithium chloride attenuates cell death in oculopharyngeal muscular dystrophy by perturbing Wnt/beta-catenin pathway. Cell Death Dis. 4, e821 ( 10.1038/cddis.2013.342) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barro M, Carnac G, Flavier S, Mercier J, Vassetzky Y, Laoudj-Chenivesse D. 2010. Myoblasts from affected and non-affected FSHD muscles exhibit morphological differentiation defects. J. Cell Mol. Med. 14, 275–289. ( 10.1111/j.1582-4934.2008.00368.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turki A, et al. 2012. Functional muscle impairment in facioscapulohumeral muscular dystrophy is correlated with oxidative stress and mitochondrial dysfunction. Free Radic. Biol. Med. 53, 1068–1079. ( 10.1016/j.freeradbiomed.2012.06.041) [DOI] [PubMed] [Google Scholar]
- 43.Kaidi A, Williams AC, Paraskeva C. 2007. Interaction between β-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat. Cell Biol. 9, 210–217. ( 10.1038/ncb1534) [DOI] [PubMed] [Google Scholar]
- 44.Teng M, Jiang XP, Zhang Q, Zhang JP, Zhang DX, Liang GP, Huang YS. 2012. Microtubular stability affects pVHL-mediated regulation of HIF-1alpha via the p38/MAPK pathway in hypoxic cardiomyocytes. PLoS ONE 7, e35017 ( 10.1371/journal.pone.0035017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bosnakovski D, et al. 2008. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J. 27, 2766–2779. ( 10.1038/emboj.2008.201) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakao N, Kurokawa T, Nonami T, Tumurkhuu G, Koide N, Yokochi T. 2008. Hydrogen peroxide induces the production of tumor necrosis factor-alpha in RAW 264.7 macrophage cells via activation of p38 and stress-activated protein kinase. Innate Immun. 14, 190–196. ( 10.1177/1753425908093932) [DOI] [PubMed] [Google Scholar]
- 47.Shi CS, Huang NN, Harrison K, Han SB, Kehrl JH. 2006. The mitogen-activated protein kinase kinase kinase kinase GCKR positively regulates canonical and noncanonical Wnt signaling in B lymphocytes. Mol. Cell Biol. 26, 6511–6521. ( 10.1128/MCB.00209-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu J, Lin A. 2005. Role of JNK activation in apoptosis: a double-edged sword. Cell Res. 15, 36–42. ( 10.1038/sj.cr.7290262) [DOI] [PubMed] [Google Scholar]
- 49.Sun TQ, Lu B, Feng JJ, Reinhard C, Jan YN, Fantl WJ, Williams LT. 2001. PAR-1 is a dishevelled-associated kinase and a positive regulator of Wnt signalling. Nat. Cell Biol. 3, 628–636. ( 10.1038/35083016) [DOI] [PubMed] [Google Scholar]
- 50.Wang J, Van De Water TR, Bonny C, de Ribaupierre F, Puel JL, Zine A. 2003. A peptide inhibitor of c-Jun N-terminal kinase protects against both aminoglycoside and acoustic trauma-induced auditory hair cell death and hearing loss. J. Neurosci. 23, 8596–8607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao Y, Spigolon G, Bonny C, Culman J, Vercelli A, Herdegen T. 2012. The JNK inhibitor D-JNKI-1 blocks apoptotic JNK signaling in brain mitochondria. Mol. Cell Neurosci. 49, 300–310. ( 10.1016/j.mcn.2011.12.005) [DOI] [PubMed] [Google Scholar]
- 52.West J, Bianconi G, Severini S, Teschendorff AE. 2012. Differential network entropy reveals cancer system hallmarks. Sci. Rep. 2, 802 ( 10.1038/srep00802) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cerami EG, Gross BE, Demir E, Rodchenkov I, Babur O, Anwar N, Schultz N, Bader GD, Sander C. 2011. Pathway commons, a web resource for biological pathway data. Nucleic Acids Res. 39, D685–D690. ( 10.1093/nar/gkq1039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prasad TS, Kandasamy K, Pandey A. 2009. Human protein reference database and human proteinpedia as discovery tools for systems biology. Methods Mol. Biol. 577, 67–79. ( 10.1007/978-1-60761-232-2_6) [DOI] [PubMed] [Google Scholar]
- 55.Licata L, et al. 2012. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 40, D857–D861. ( 10.1093/nar/gkr930) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barrett T, et al. 2013. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 41, D991–D995. ( 10.1093/nar/gks1193) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kauffmann A, Gentleman R, Huber W. 2009. arrayQualityMetrics—a bioconductor package for quality assessment of microarray data. Bioinformatics 25, 415–416. ( 10.1093/bioinformatics/btn647) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hsiao LL, Jensen RV, Yoshida T, Clark KE, Blumenstock JE, Gullans SR. 2002. Correcting for signal saturation errors in the analysis of microarray data. Biotechniques 32, 330–336. [DOI] [PubMed] [Google Scholar]
- 59.Johnson WE, Li C, Rabinovic A. 2007. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. ( 10.1093/biostatistics/kxj037) [DOI] [PubMed] [Google Scholar]
- 60.Chen C, Grennan K, Badner J, Zhang D, Gershon E, Jin L, Liu C. 2011. Removing batch effects in analysis of expression microarray data: an evaluation of six batch adjustment methods. PLoS ONE 6, e17238 ( 10.1371/journal.pone.0017238) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Banerji CRS, Miranda-Saavedra D, Severini S, Widschwendter M, Enver T, Zhou JX, Teschendorff AE. 2013. Cellular network entropy as the energy potential in Waddington's differentiation landscape. Sci. Rep. 3, 3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moyle LA, Zammit PS. 2014. Isolation, culture and immunostaining of skeletal muscle fibres to study myogenic progression in satellite cells. Methods Mol. Biol. 1210, 63–78. ( 10.1007/978-1-4939-1435-7_6) [DOI] [PubMed] [Google Scholar]
- 63.Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. 1997. Constitutive transcriptional activation by a β-catenin-Tcf complex in APC-/- colon carcinoma. Science 275, 1784–1787. ( 10.1126/science.275.5307.1784) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.