

Abstract
Large sets of genotypes give rise to the same phenotype, because phenotypic expression is highly redundant. Accordingly, a population can accept mutations without altering its phenotype, as long as the genotype mutates into another one on the same set. By linking every pair of genotypes that are mutually accessible through mutation, genotypes organize themselves into neutral networks (NNs). These networks are known to be heterogeneous and assortative, and these properties affect the evolutionary dynamics of the population. By studying the dynamics of populations on NNs with arbitrary topology, we analyse the effect of assortativity, of NN (phenotype) fitness and of network size. We find that the probability that the population leaves the network is smaller the longer the time spent on it. This progressive ‘phenotypic entrapment’ entails a systematic increase in the overdispersion of the process with time and an acceleration in the fixation rate of neutral mutations. We also quantify the variation of these effects with the size of the phenotype and with its fitness relative to that of neighbouring alternatives.
Keywords: neutral evolution, homogeneous populations, assortative networks, mutation accumulation
1. Introduction
The relationship between genotype and phenotype, the ways in which this map conditions the adaptive dynamics of populations, or the imprints that life histories leave in the genomes of organisms are essential questions to be solved before a complete evolutionary theory can be achieved. Genotypes, which encode much of the information required to construct organisms, are occasionally affected by mutations that modify the phenotype, their visible expression and the target of natural selection. Many mutations are neutral instead [1], varying the regions of the space of genotypes that can be accessed by the population [2] and conditioning its evolvability [3] but leaving the phenotype unchanged. The relation between genotype and phenotype is not one-to-one, but many-to-many. In particular, genotypes encoding a specific phenotype may form vast, connected networks that often span the whole space of possible genotypes. The existence of these networks in the case of proteins was postulated by Maynard Smith [4] as a requirement for evolution by natural selection to occur. Subsequent research has shown that these networks do exist for functional proteins [5], for other macromolecules like RNA [6], and generically appear in simple models of the genotype–phenotype map-mimicking regulatory gene networks [7], metabolic reaction networks [8] or the self-assembly of protein quaternary structure [9].
Nevertheless, systematic explorations of the topological structure of neutral networks (NNs) have been undertaken only recently, despite the fact that some of the implications of NN structure on sequence evolution were identified long ago. For instance, Kimura's neutral theory [1] postulated that the number of neutral substitutions in a given time interval was Poisson distributed. That assumption had an underlying hypothesis that was not explicitly stated at the time, namely that the number of neutral mutations available to any genotype was constant, independent of the precise genotype, of time or of the expressed phenotype. In other words, NNs were assumed to be homogeneous in degree. A consequence was that the variance of the number of mutations accumulated should equal the mean, and the dispersion index (R, the ratio between the variance and the mean) must then be equal to 1. Very early, however, it was observed that R was significantly larger than 1 in almost all cases analysed [10–12]. The appearance of short bursts of rapid evolution was ascribed to episodes of positive Darwinian selection [12] that may reflect fluctuations in population size in quasi-neutral environments, where epistatic interactions would become relevant [13].
The fact is that NNs are highly heterogeneous. Some genotypes are brittle and easily yield a different phenotype under mutation. They have one or few neighbours within the NN. Other genotypes, instead, are robust and can stand a very large number of mutations while maintaining their biological or chemical function. The existence of variations in the degree of neutrality of genotypes (in their robustness) was soon put forth as a possible explanation for the overdispersion of the molecular clock [13]. Nowadays, the distributions of robustness of the genotypes in several different NNs have been measured, turning out to be remarkably broad [14–16]. The effects of fluctuating neutral spaces in the overdispersion of the molecular clock have been investigated in realistic models of evolution for proteins [17] and quasi-species [18], and also from a theoretical viewpoint [19].
In this contribution, we explore three features of NNs whose consequences in the rate of fixation of mutations have not been systematically investigated. They are (i) the correlations in neutrality between neighbouring genotypes, (ii) the degree of redundancy of phenotypes (the size of NN), and (iii) the fitness of the current phenotype in relation to accessible alternatives. First, the degree of neutrality is not randomly distributed in the NN. Thermodynamical arguments [20,21] and analyses of full NNs [22] indicate that genotypes tend to cluster as a function of their robustness, implying that NNs belong to the class of the so-called assortative networks. It is known that populations evolving on NNs tend to occupy maximally connected regions in order to minimize the number of mutations changing their phenotype [2]. In assortative networks, neutral drift entails a canalization towards mutation–selection equilibrium, progressively increasing the rate of fixation of neutral mutations through a dynamical process that we dub phenotypic entrapment. Second, there is abundant evidence coming from computational genotype–phenotype maps [9,23–24] and from empirical reconstruction of NNs [25] that the average robustness of a given phenotype grows with the size of its associated NN: here, we quantify the effect of a systematic difference in average degree on the probability of fixation of neutral mutations. Third, the difference in fitness between genotypes in the current NN and their mutational neighbours affects the probability that a mutation (be it neutral, beneficial or deleterious) gets fixed in the population, and with it, as we explicitly show, the rate of the molecular clock during intervals of strictly neutral drift.
With the former goals in mind, we develop an out-of-equilibrium formal framework to describe the dynamics of homogeneous, infinite populations on generic NNs. We demonstrate that the population keeps memory of its past history because, as time elapses, the likelihood that it visits genotypes of increasingly higher robustness augments in a precise way that we calculate. This is a consequence of assortativity and a dynamic manifestation of the ‘friendship paradox’ [26] described in social networks (your friends have more friends than you). As a result, the probability that the population leaves the network explicitly depends on the elapsed time. Further, the decline of this probability with time entails a systematic acceleration of the rate of accumulation of neutral mutations. The degree of entrapment is higher, the larger the NN and the broader the difference between the fitness of the current phenotype and that of accessible alternatives. These results are fairly general and have implications in the derivation of effective models of phenotypic change and in the calibration of molecular clocks.
2. Dynamical model
2.1. Description of neutral networks and populations
In the following sections, we employ terms related to dynamics on complex networks to describe the dynamical process undergone by a homogeneous population on an NN. Genotypes are the nodes of the network, and two nodes are linked if the corresponding genotypes can be mutually accessed through a single mutation. Links are all identical, so we assume that the mutational process affects all genotypes in the same way. The robustness of a node is a quantity proportional to its degree k, that is, to the number of links leading to other nodes within the NN (or the number of neutral mutations).
For the sake of analytical tractability, we use a mean-field-like description of the NN. This amounts to assuming that all nodes of the same degree (robustness) are equivalent with respect to the dynamics, and therefore the network can be characterized only through its degree distribution, p(k) (the probability that a node has degree k or the distribution of robustness values), and its nearest-neighbour degree distribution pnn(k|k′) (the probability that a degree k′ node's neighbour has degree k), where k, k′ = 1, … , z. We are assuming that there is a maximum degree z in the NN. This is always true if genotypes are sequences whose loci are taken from a finite alphabet (e.g. {0, 1}, nucleotides or amino acids). If there are A different types in the alphabet, sequences have length L, and mutations refer just to point mutations (the most widely studied mutational process), then z = (A − 1)L. Typically, thus, z ≫ 1.
The strength of correlation between the degree of neighbouring nodes is measured through the degree–degree correlation coefficient r. This is a quantity that characterizes the assortativity of a network and is defined as the Pearson correlation coefficient (cf. the covariance) between the degree of nodes and the degree of their neighbours. For a network with i = 1, … , M nodes, each with degree ki and whose neighbours have on average degree  , r is defined as
, r is defined as
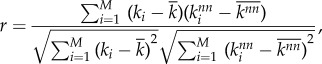 |
2.1 |
with 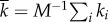 ,
, 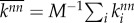 . The largest value of assortativity r = 1 is achieved when the average degree of neighbours of node i equals its degree,
. The largest value of assortativity r = 1 is achieved when the average degree of neighbours of node i equals its degree, 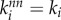 . Anticorrelations in degree yield negative values of r. By definition, −1 ≤ r ≤ 1.
. Anticorrelations in degree yield negative values of r. By definition, −1 ≤ r ≤ 1.
We assume that all genotypes in an NN have the same fitness, so evolution is strictly neutral within the network. We should only take into account non-neutral mutations that get fixed, that is, that move the population from a node in its current NN to a node of a different NN. As of neutral evolution, population dynamics distinguishes two basic regimes. If μ denotes the mutation rate and Ne the (effective [27]) population size, these correspond to low 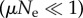 and high
and high 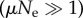 mutation rates. For high mutation rates, populations are heterogeneous, and we need an ensemble of individuals coupled through replication and mutation to account for their evolution. This is a complex situation where quasi-species dynamics comes into play, and one has to resort to computational simulations to describe its dynamics [18]. In this study, we work in the limit
mutation rates. For high mutation rates, populations are heterogeneous, and we need an ensemble of individuals coupled through replication and mutation to account for their evolution. This is a complex situation where quasi-species dynamics comes into play, and one has to resort to computational simulations to describe its dynamics [18]. In this study, we work in the limit 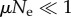 , where every mutation either gets fixed in or disappears from the population before a new mutation occurs, so the population is always homogeneous and its evolution is effectively described as a single random walk (RW) on the NN.
, where every mutation either gets fixed in or disappears from the population before a new mutation occurs, so the population is always homogeneous and its evolution is effectively described as a single random walk (RW) on the NN.
2.2. Dynamics
We describe the dynamical process that begins with the arrival of a random walker to the NN and ends when it leaves to a neighbouring NN. In particular, we aim at describing the entrapment effect that occurs while the walker remains within the NN. Let us mention, at this point, a fact that will become clear later on: because of frequent jumps in and out of NNs, populations will hardly spend a long enough time within a single NN to reach the equilibrium distribution. Therefore, the relevant process to describe is the transient dynamics that starts with arrival at the NN, instead of the asymptotic behaviour within the phenotype. For this reason, we require a good estimation of the initial probability distribution of arrival at any node of the NN.
The process starts as soon as the walker jumps to a node of the NN from outside. We assume that the initial jump is equally likely to occur through any link of the NN pointing outwards, so the probability that a node of degree k is chosen as the entry node should be proportional to the number of outbound links z − k and to the abundance of nodes of degree k. This yields the initial condition
| 2.2 |
The state i = 0 does not describe any node, but it proves convenient to introduce it to represent the exterior of the NN. Hence, P0(t) is the probability that the process ends at any time t > 0; the choice P0(0) = 0 means that the process begins as soon as it ‘leaves’ the exterior.
Because the mean-field description assumes that all nodes of the same degree are equivalent, the RW on the NN can be further described as a stochastic process X(t) taking values in the set {0, 1, … , z}. These values represent the degree of the node the walker is in at time t, with state 0 corresponding to the exterior of the NN, as just described. When the walker jumps to state 0 the phenotype changes, and other considerations are needed to follow up the evolution. For our purposes, the process ends at this point, so 0 is an absorbing state (the only one in this process).
The stochastic process just introduced is a continuous-time Markov chain with transition rate
| 2.3 |
Element Wkk′ of matrix W is the conditional probability that, given that a jump occurs when the process is at a node of degree k′, it ends up at a node of degree k. Within our mean-field description, this probability can be simply obtained as
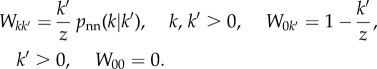 |
2.4 |
As mentioned above, the rationale for this choice is that z − k′ links of the node where the RW is located point outside the NN and k′ point inside, and if the process stays within the NN (probability k′/z), the probability that the node it jumps to has degree k is pnn(k|k′). The nearest-neighbour degree distribution pnn(k|k′) is zero if p(k′) = 0 and is normalized otherwise; therefore,
| 2.5 |
2.3. Leaving the neutral networks
Let us now discuss the meaning of the transition rates μk. If k > 0, the RW jumps to a node that belongs to the NN, and thus the mutation is neutral. Hence, μk = μ, because, in that case, the mutation rate is also the rate of fixation of neutral mutations in a population of arbitrary size [28]. On the other hand, μ0 implies a jump outside the NN, hence a phenotypic change. The new phenotype will have, in general, a different fitness, and the rate at which non-neutral mutations go to fixation in a population of effective size Ne is  , where f is the fitness of the current phenotype relative to that of the phenotype the process jumps to [27]. The ratio ϕ ≡ μ0/μ can take any value in the interval
, where f is the fitness of the current phenotype relative to that of the phenotype the process jumps to [27]. The ratio ϕ ≡ μ0/μ can take any value in the interval 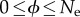 . The lower bound occurs, if purifying selection eliminates any phenotype other than the current one; the upper bound is the rate at which the population adopts a highly fitter new phenotype. Neutral changes of phenotype correspond to ϕ = 1.
. The lower bound occurs, if purifying selection eliminates any phenotype other than the current one; the upper bound is the rate at which the population adopts a highly fitter new phenotype. Neutral changes of phenotype correspond to ϕ = 1.
There is a simplification implicit in the expression of ϕ, though. If the neighbouring NNs are diverse in fitness values, ϕ should be replaced by an appropriate average over this diversity—weighted with the probabilities of jumping to these other NNs. In any case some factor, 0 < ϕ < Ne accounts for the differences in the fitness between the current NN and those of its neighbouring NNs. For the discussion to come, we do not need to be more specific about its precise form.
2.4. Master equation
The probability distribution of the process is Pk(t) ≡ Pr {X(t) = k}, which stands for the probability that at time t the process sits on a node of degree k = 1, … , z, or is outside the NN, if k = 0. This probability satisfies the master equation
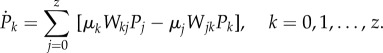 |
2.6 |
A few considerations transform (2.6) into a handier expression for further calculations. To begin with, we can set μ = 1, i.e. measure time in units of μ−1. Let us also introduce the support of p(k), i.e. the set 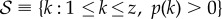 . Thus, any
. Thus, any 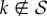 has p(k) = 0, pnn(j|k) = 0, and, given definition (2.2), Pk(0) = 0. Therefore, according to (2.4), for any
has p(k) = 0, pnn(j|k) = 0, and, given definition (2.2), Pk(0) = 0. Therefore, according to (2.4), for any 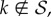 the master equation (2.6) becomes
the master equation (2.6) becomes 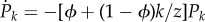 , whose solution is
, whose solution is  .
.
Because of this result, we can ignore in (2.6) any index 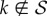 , that is, those degrees that are not represented by any node in the NN. Accordingly, if we introduce vector
, that is, those degrees that are not represented by any node in the NN. Accordingly, if we introduce vector 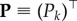 ,
,  and matrices
and matrices
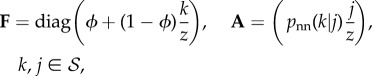 |
2.7 |
the master equation (2.6) simply becomes
| 2.8 |
3. Results
3.1. Time spent on the neutral networks
An important quantity characterizing the process is the probability that, after a time t, the population is still on the NN. This probability can be calculated as Q(t) ≡ 1 − P0(t) = 1 · P(t), where 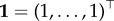 . For k = 0, equation (2.6) yields
. For k = 0, equation (2.6) yields 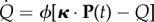 , where
, where 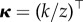 ,
, 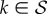 , a differential equation that can be rewritten as the integral equation
, a differential equation that can be rewritten as the integral equation
| 3.1 |
In appendix A, we prove that the eigenvalues αk of matrix F − A are all real and different. Therefore, denoting uk and vk the corresponding right and left eigenvectors, respectively, we can write
| 3.2 |
(Note that 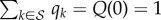 .)
.)
Equation (3.2) makes apparent that, in general, the lifetime distribution of the process before leaving the NN is not exponential, but displays many different time scales, as many as elements in the set {αk}. Asymptotically, 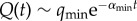 , with αmin the smallest eigenvalue. Thus, the rate at which the process leaves the NN tends to a minimum as time elapses—in other words, it gets more and more trapped within the NN. Eventually, mutation–selection equilibrium is reached, and the process can be described through a unique time scale, αmin.
, with αmin the smallest eigenvalue. Thus, the rate at which the process leaves the NN tends to a minimum as time elapses—in other words, it gets more and more trapped within the NN. Eventually, mutation–selection equilibrium is reached, and the process can be described through a unique time scale, αmin.
3.2. Mutation accumulation
Perhaps the most immediate consequence of this entrapment of the evolutionary process in an NN is that the observed rate at which neutral mutations accumulate increases with time. Together with the stochastic nature of the dynamics, the consequence is twofold: on the one hand, the molecular clock gets accelerated during those periods of strictly neutral evolution; on the other hand, overdispersion of the molecular clock varies non-monotonically with time, as we show in the following.
In order to analyse how mutations accumulate with time, we need to introduce Pk(m, t), the probability that the process is at time t at a node of degree k of the NN, having undergone exactly m mutations. Introducing 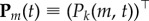 ,
, 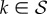 , the dynamic equation for this vector can be written as
, the dynamic equation for this vector can be written as
| 3.3 |
which is valid for all integers m if we assume Pm(t) = 0 for all m < 0. Obviously Pm(0) = P(0)δm,0, with P(0) the initial probability distribution (2.2).
In order to extract information on the mean and variance of the number of mutations accumulated at time t, it is useful to introduce the moment-generating function
| 3.4 |
so that
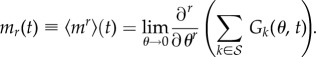 |
3.5 |
Multiplying equation (3.3) by emθ and summing on m, we obtain the dynamic equation for G(θ, t)
| 3.6 |
Setting θ = 0 shows that G(0, t) = P(t), the solution of (2.8), hence m0(t) = Q(t).
Differentiating (3.6) with respect to θ yields, for θ = 0,
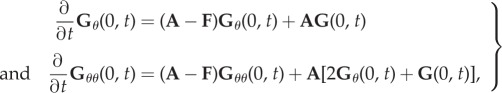 |
3.7 |
(subscripts θ denote partial derivatives).
Solving simultaneously equations (2.8), and (3.7), and using (3.5) yields m0(t) = Q(t), m1(t) and m2(t). We are not yet done, because these are not the quantities required to estimate magnitudes related to the molecular clock: actual measurements are always performed on extant populations, so we need the mean m(t) and variance ν(t) in the number of mutations conditioned on remaining within the NN at time t. These are obtained as
| 3.8 |
Correspondingly, the overdispersion of the molecular clock at time t should be computed as
| 3.9 |
4. Examples
In this section, we compare analytical and numerical results for the dynamics of a RW (representing a homogeneous population) on networks differing in size, fitness and topological features. Our aim is to better understand the quantitative effect of these variables on the dynamics of the population, paying particular attention to the time scales involved, to the change in overdispersion with time and to variations in the rate of fixation of mutations. We analyse a random network (with near zero assortativity and a well-defined average degree) as well as two highly assortative networks with either only two degrees or a constant degree distribution. Our final example comes from secondary structure RNA networks, which share several properties with other natural NN.
4.1. Random network
Despite its apparent simplicity, equation (2.8) cannot be analytically solved in general. An interesting exception is the case of a random network constructed by randomly drawing links between pairs of nodes. In this situation, the degree follows a Poisson distribution and, consequently, the average degree is well defined. Even this special case is exactly solvable only for ϕ = 1.
In a random network, there is no correlation between the degree of a given node and that of its nearest neighbours. If the network is infinitely large, its assortativity r = 0—though a residual assortativity will be typically obtained when the number of nodes is finite. This lack of correlation reflects in the probability that a node's neighbour has degree k only depends on its own degree. As a matter of fact, 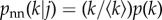 . (Here, and in what follows,
. (Here, and in what follows, 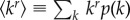 ). For this special case,
). For this special case,
| 4.1 |
and deriving an expression for Q(t) is straightforward (see appendix B). The resulting formula is
| 4.2 |
Despite the ease of this case, and the fact that the NN has no fitness advantage with respect to the average neighbouring NN (ϕ = 1), we can already observe what turns out to be a generic property of heterogeneous networks: the lifetime of the RW within the NN is not exponentially distributed. For random networks, there are only two time scales: the ‘standard’ one, μ−1, and a slower one, with characteristic time (1 − ρ)−1μ−1. The reason for this non-exponential behaviour—and for the equivalent effect that will arise in any heterogeneous network—is that the RW comes in the NN preferentially through nodes of low inner connectivity (high outer connectivity) and as time passes without leaving the NN it progressively moves towards the regions of higher connectivity (note that the mean degree of a node's neighbour is 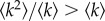 ), from which escaping is less probable. Thus, the first scale corresponds to walkers which leave the network in very few steps, whereas the second (slow) scale characterizes walkers that have spent some time in the NN.
), from which escaping is less probable. Thus, the first scale corresponds to walkers which leave the network in very few steps, whereas the second (slow) scale characterizes walkers that have spent some time in the NN.
Further insights can be gained by calculating the overdispersion (3.9) (see appendix B), whose expression for this case becomes
| 4.3 |
Figure 1 shows the probability Q(t) to remain on the network at time t, the overdispersion R(t) of the molecular clock with time and the mutation rate (measured as the time derivative of the mean number of mutations) for an Erdös–Renyi random network. In figure 1a, as well as in all other plots, Q(t) is represented as a function of the rescaled variable ϕt. The reason is that, as can be seen, there is a partial collapse of all Q(t) curves (but see the Discussion for forthcoming examples). In addition, the case ϕ = 0 is not included, because ϕ = 0 implies that the RW cannot leave the NN and, therefore, Q(t) = 1, for all t. For the Erdös–Renyi random networks, the fast time scale is barely visible, due to the fact that the RW becomes rapidly trapped. The slow time scale actually dominates the dynamics: for the values chosen, 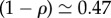 (case ϕ = 1) and the coefficient of the corresponding exponential in Q(t) is about 10 times larger than that of the fast time scale.
(case ϕ = 1) and the coefficient of the corresponding exponential in Q(t) is about 10 times larger than that of the fast time scale.
Figure 1.
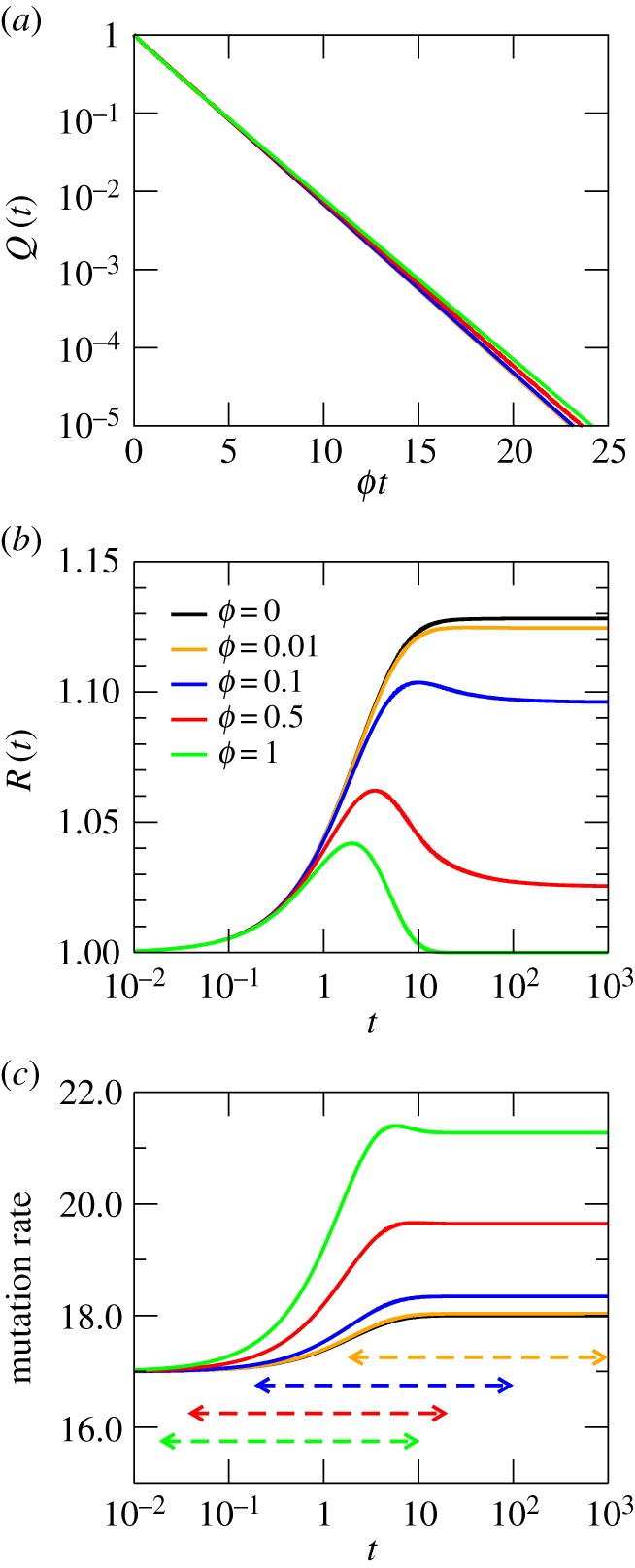
Dynamics of RWs on an Erdös–Renyi random network with M = 20 000 nodes and average degree 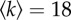 . (a) Probability Q(t) that the RW stays on the network after a time t. (b) Overdispersion of the network. (c) Mutation rate or acceleration of the molecular clock. The time interval through which the number of trajectories remaining on the network decreases from 99% to 1% is represented as a horizontal dashed line limited by two arrows, with the same colour as the ϕ curve they correspond to. The same code is used in subsequent corresponding panels for other networks. The measured assortativity for this finite network is r≈0.018.
. (a) Probability Q(t) that the RW stays on the network after a time t. (b) Overdispersion of the network. (c) Mutation rate or acceleration of the molecular clock. The time interval through which the number of trajectories remaining on the network decreases from 99% to 1% is represented as a horizontal dashed line limited by two arrows, with the same colour as the ϕ curve they correspond to. The same code is used in subsequent corresponding panels for other networks. The measured assortativity for this finite network is r≈0.018.
Figure 1b shows the typical behaviour of the overdispersion on heterogeneous NN: an initial increase up to a maximum followed by a decay to an asymptotic value. Interestingly, even for ϕ = 1 (corresponding to equation (4.3)), a 4% overdispersion is reached before it decays to its asymptotic value, a process that requires about 10 mutations (t ∼ 10 µ−1). As values of ϕ diminish, the asymptotic value of R(t) monotonically increases. Three features that will show up in the remaining examples as well should be highlighted: first, ϕ has a strong, non-trivial effect on overdispersion; second, it is in the transient where overdispersion reaches its largest value; third, the rate of accumulation of mutations increases with ϕ (it therefore diminishes the larger the difference between the fitness of the current NN and its neighbouring phenotypes).
In figure 1c, we can appreciate how the ticking rate of the molecular clock undergoes an acceleration from its initial value, eventually reaching a steady rate higher than the one it started off from. The change occurs at a time t ∼ 10 µ−1, which corresponds to the number of mutations accumulated. Note that, owing to the heterogeneity of the network, the initial mutation rate is already higher than μ. As we stated earlier, the stationary state is not attained in general. Now, we can quantify this statement by estimating the time t1%(ϕ) by which only 1% of the RWs remain on the network. This time can be obtained by numerically solving Q(t1%) = 0.01—it can be approximately read from figure 1a. In order to estimate the range of t values where typical trajectories on the NN should be found, we have calculated the time t99%(ϕ) as well, when only 1% of the trajectories have left the network. The interval (t99%(ϕ), t1%(ϕ),) is represented as a dashed line within two arrows in figure 1c (and all subsequent (c) panels). As can be seen, the maximum variation in R(t) and in the mutation rate is achieved within that time interval, whereas mutation–selection equilibrium is rarely attained. Note that t99%(ϕ) and t1%(ϕ) decrease with increasing ϕ. The acceleration of the molecular clock along neutral evolution, which is higher the smaller ϕ, is a feature that will reappear in all subsequent examples, and probably stands as the best illustration of the phenotypic entrapment produced by NN. Typically, most RWs will leave the network before reaching the asymptotic neutral ticking rate, and certainly before the RW has had time to explore a significant fraction of the NN [29].
4.2. Two-degree network
Consider now a network with M nodes such that p(k) = 1/2 for k = k1, k2 (k1 < k2), and 0 otherwise. The M/2 nodes of each degree are randomly connected between them, except for L links that connect (also randomly) nodes of the two different degrees. Accordingly, pnn(ki|ki) = 1 – 2L/(Mki) and pnn(kj|ki) = 1–2L/(Mki), for j≠i. The nearest-neighbour degree distribution clearly reflects, in this case, the trapping power of more connected regions: note that the probability of entering the group of high connectivity (k2) from that of low connectivity is higher than the probability of leaving it, pnn(k1|k2) < pnn(k2|k1). The asymmetry of pnn reflects the entrapment by progressively more connected regions of the NN.
In terms of the variables ℓ ≡ 2L/(Mz), κ = (k2 + k1)/2z and δ = (k2 − k1)/(2z), matrix A − F becomes
| 4.4 |
and the resulting probability (3.2) of remaining trapped within the NN at time t is
| 4.5 |
where
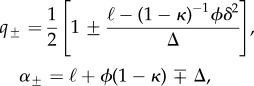 |
4.6 |
with 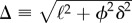 . Thus, the dynamics on a two-degree network is characterized by two time scales, with coefficients q± further determining the relative weight of each one in the process between arriving at the NN and achieving maximum entrapment.
. Thus, the dynamics on a two-degree network is characterized by two time scales, with coefficients q± further determining the relative weight of each one in the process between arriving at the NN and achieving maximum entrapment.
Despite the formal similarity of Q(t) with the solution for an Erdös–Renyi network, there are important quantitative differences between the two cases. Figure 2a illustrates the presence of the two time scales, here clearly visible (note the change in the horizontal scale with respect to figure 1a). The probability to stay on this two-degree network is also significantly higher after a fixed time, which speaks for longer trajectories in comparison with those on a random network. The average degree does not play an essential role in this behaviour, because it has value 16, slightly below the random network previously studied; this nonetheless, the two-degree network is more efficient at trapping the RW. Let us now explain why the curves for Q(t) collapse so nicely in this case. This is owing to the very low value of 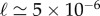 for the parameters chosen. As a result, the two time scales are well approximated as
for the parameters chosen. As a result, the two time scales are well approximated as 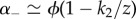 and
and 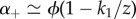 , thus nearly scaling with ϕ as long as
, thus nearly scaling with ϕ as long as 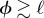 . Hence, the collapse of the Q(t) curves is a particular feature of this network and does not hold in general, as the next example will show.
. Hence, the collapse of the Q(t) curves is a particular feature of this network and does not hold in general, as the next example will show.
Figure 2.
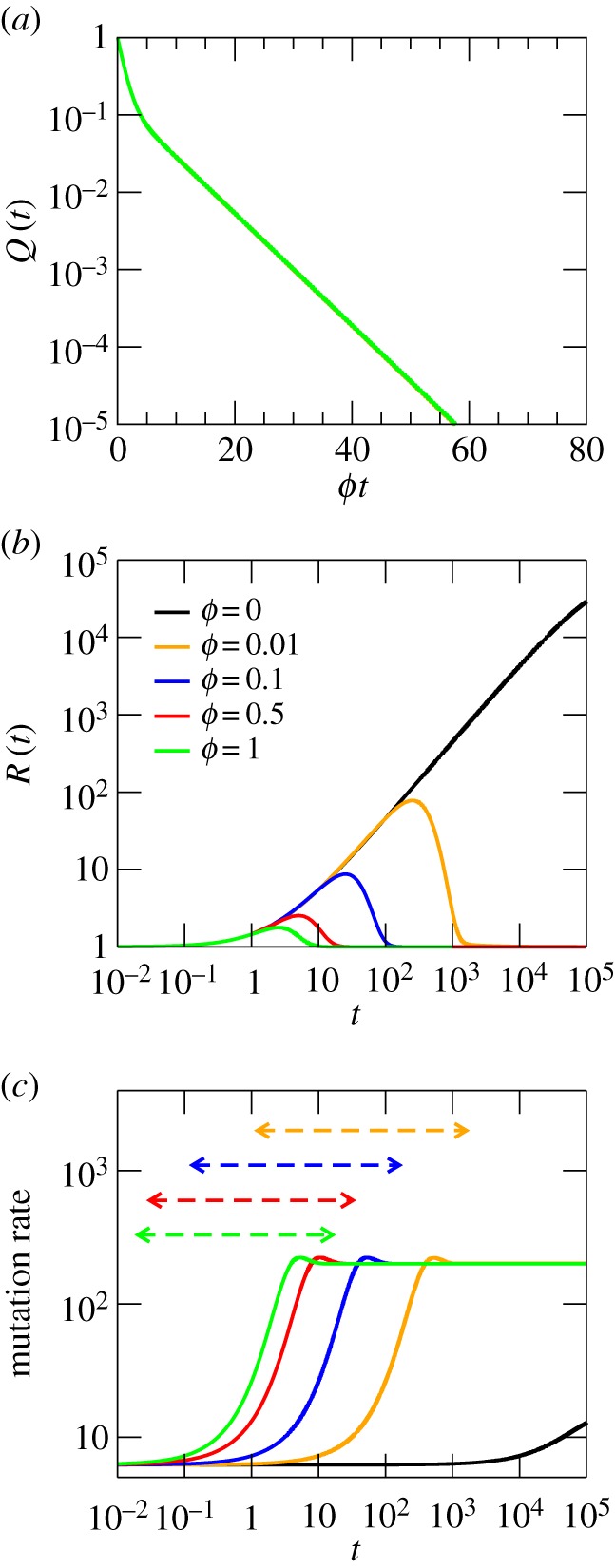
Dynamics of a RW on a two-degree network with k1 = 2, k2 = 30, L = 2 and M = 20 000 nodes. (a) Probability Q(t) that the RW stays on the network after a time t. (b) Overdispersion of the network. (c) Mutation rate. By construction, the assortativity is very high, r ≈ 0.99995.
Differences in overdispersion between the random and the two-degree network are still more remarkable. Figure 2b (note the logarithmic scale in the y-axis) represents R(t), showing an extraordinary increase in overdispersion with time, especially as ϕ decreases. Although not shown in the picture, R(t) eventually converges to 1, albeit the time required is huge. This is another important difference with the random network, where the overdispersion saturates at values R(t → ∞) > 1. This is due to the persistence of heterogeneity in the degree from the viewpoint of the RW. Whereas in the random network, the walker visits nodes of different degree once mutation–selection equilibrium has been reached, in the two-degree network the region of low connectivity is mostly invisible to the dynamics, trapped in the large cluster of high-connectivity nodes. Nonetheless, there is an enormous dispersion in the time required for a particular trajectory to become trapped; this fact explaining the growth of overdispersion, the delayed achievement of its maximum value and the slow convergence to equilibrium.
Similar differences can be observed in the mutation rate, depicted in figure 2c, compared with that for the Erdös–Renyi network. The acceleration of the molecular clock for this network is very large (over an order of magnitude with respect to the initial ticking rate). In addition, as ϕ decreases, the acceleration occurs at earlier times, an effect that was much milder in the Erdös–Renyi network. As a matter of fact, these curves collapse nicely when they are represented as a function of ϕt, as happened with Q(t). The asymptotic mutation rate is independent of ϕ, because, similar to what happens with overdispersion, the RW does not see any difference in the degrees visited at large times, even for different values of ϕ. The next example clarifies how ϕ affects the trapping strength of nodes with different degree, and the resulting variation in the asymptotic mutation rate.
4.3. Constant degree distribution network
This example shares some similarities with the previous one, but now nodes of all degrees between a minimum kmin = 2 and a maximum value appear in equal amounts. In addition, the fraction of links connecting nodes of different degrees is notably larger. Consider a network with a degree distribution p(k) = 1/50, 2 ≤ k ≤ 51, N = 20 000 nodes, and z = 52. In order to generate an assortative network, we proceed as follows. First, we assign an arbitrary index to the nodes (with no meaning whatsoever) and divide them up into blocks of 400(=20 000/50) consecutive nodes. We then assign degree 2 to the first block, degree 3 to the second block and so on until exhausting the nodes. Now, we randomly connect the nodes, in such a way that the probability to connect a pair of nodes at positions i and j is taken proportional to exp{−d/200}, where d = |i − j|. With this procedure, nodes with similar degree are preferentially connected, and high assortativity is ensured. It is not possible to derive any exact expression for Q(t) or R(t), though the results can be interpreted in the light of our previous examples.
Figure 3a shows the presence of many different time scales that become dominant at different times and cause a systematic bending of Q(t). The visibility of the different time scales is related to the presence of significant fractions of nodes of different degrees together with the canalization of the dynamics towards regions of increasingly higher connectivity—a consequence of assortativity. This behaviour differs from that of the two-degree network, where the curvature region was small and owing to the cross-over between the only two time scales involved. Overdispersion can reach very high values, even once mutation–selection equilibrium has been reached, and so does the mutation rate (figures 3b,c).
Figure 3.
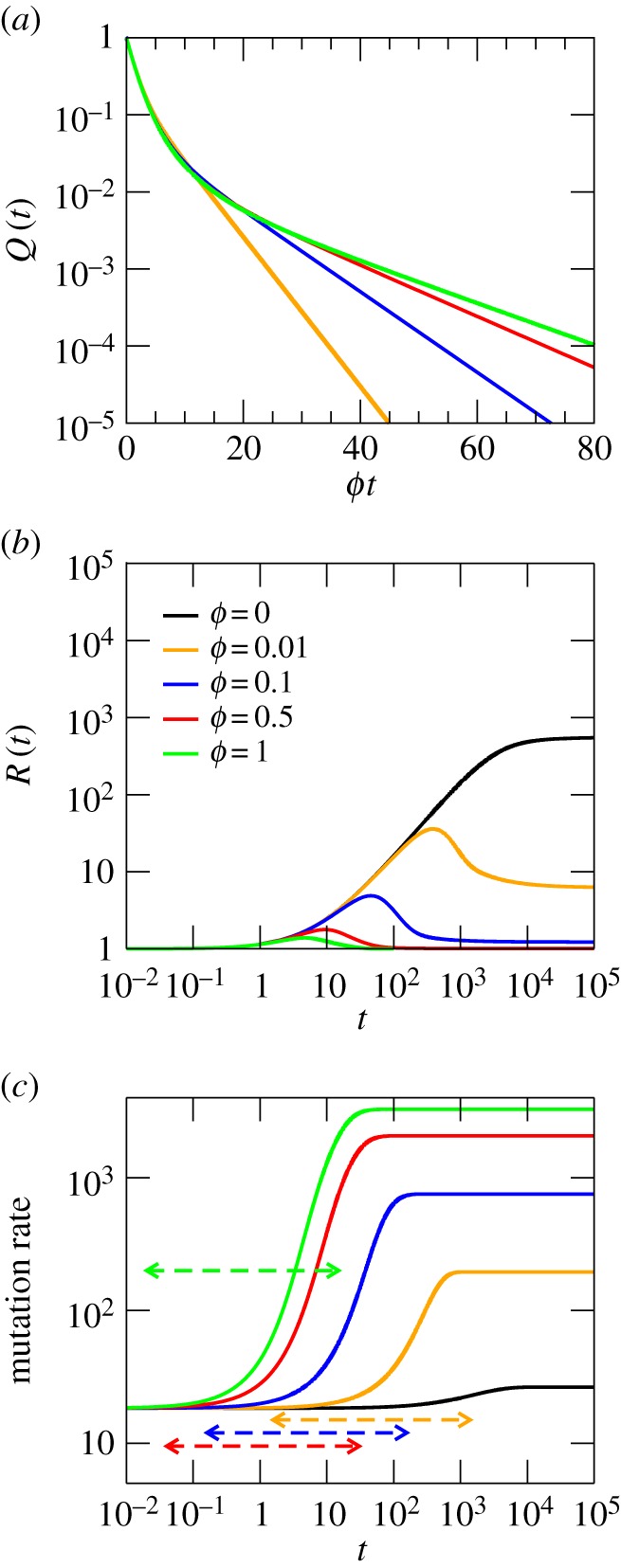
Dynamics of a RW on a network with a constant degree distribution, with parameters described in the main text. (a) Probability Q(t) that the RW stays on the network after a time t. (b) Overdispersion of the network. (c) Mutation rate. The assortativity is r ≈ 0.9997. For the sake of comparison, these plots share scale with figure 2.
In a network of this kind, the larger ϕ the less diverse is the set of different degrees visited by the RW in the asymptotic state. This responds to the fact that trajectories that survive for long times are preferentially concentrated in highly connected regions. As ϕ decreases, nodes of lower degree become more competent at trapping the dynamics, causing two effects. First, the asymptotic time scale αmin changes, decreasing in absolute value with ϕ. Second, differences between the trajectories are less pronounced the larger ϕ, such that the maximum in the overdispersion and the largest acceleration of the clock are reached at earlier times. Third, the asymptotic values of R(t) decrease with ϕ, and those of the mutation rate increase. At mutation–selection equilibrium, however, nodes with the highest connectivity cannot fully trap the RW, such that nodes of lower degree are visited frequently enough to yield R(t) > 1, attaining larger values the lower ϕ. In the asymptotic state, thus, the RW is more delocalized (in terms of the visited degree), the lower the value of ϕ. The fact that, at high ϕ, those RW remaining on the NN preferentially visit nodes of high degree is responsible for the larger ticking rates observed—reaching values orders of magnitude bigger than initially.
4.4. RNA networks
Secondary structure RNA NNs (here RNA networks for short) are a paradigm of the genotype–phenotype map, and many important properties on their topology and on genotype–phenotype mapping are known in detail. An RNA network is formed in principle by all the sequences that yield the same minimum free energy secondary structure. Usually, links are drawn between pairs of sequences in the set that differ in a single point mutation. In general, this leads to a number of disjoint connected components. Because dynamics are well defined on connected networks, we will assume that we work with subsets of the genotype–phenotype map that fulfil this condition. Among other properties known, there is a broad variability in the number of sequences that fold into a given phenotype for a fixed sequence length (several orders of magnitude) [6,30]; the average degree of an RNA network grows logarithmically with its size (number of genotypes leading to the same phenotype) [22], and RNA networks are highly assortative, with an index r that increases on average with size and approaches 1 [22]. In this section, we discuss the results obtained for RNA networks of sequences of length 12. The genotype–phenotype map for this case is fully known, with networks of sizes spanning four and a half orders of magnitude [22].
We have chosen two connected NNs formed by genotypes that belong to different phenotypes. In dot–bracket notation, these are …((…..)) and (((…..))).; the first phenotype can be obtained from 14 675 different sequences which are grouped into four different connected subnetworks. As an example, we have chosen a connected component of size M = 1965. The second phenotype is more abundant, corresponding to the minimum free energy folded state of 142 302 sequences. As a second example, we have chosen one of its largest 17 connected subnetworks, of size M = 21 908. Curves for Q(t), R(t) and the mutation rate are plotted in figure 4. As can be seen, the behaviour of these networks is quantitatively comparable to the Erdös–Renyi random network. In RNA, there is a monotonic change in the slower time scale and in the equilibrium value of overdispersion with network size. This responds to the particular relationship between size and degree exhibited by secondary structure RNA networks, where 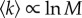 . The connection between the slower time scale and average degree can be made explicit in the calculations for random networks (equations (B 1) and (4.2)), and despite the high assortativity of RNA networks, it also shows up here. The degree distribution p(k) of RNA networks is very peaked for M small, though its variance increases with M faster than it does for a Poisson degree distribution [22]. Whether these differences lead, for realistic values of M, to significant departures from the behaviour of random networks remains to be seen.
. The connection between the slower time scale and average degree can be made explicit in the calculations for random networks (equations (B 1) and (4.2)), and despite the high assortativity of RNA networks, it also shows up here. The degree distribution p(k) of RNA networks is very peaked for M small, though its variance increases with M faster than it does for a Poisson degree distribution [22]. Whether these differences lead, for realistic values of M, to significant departures from the behaviour of random networks remains to be seen.
Figure 4.
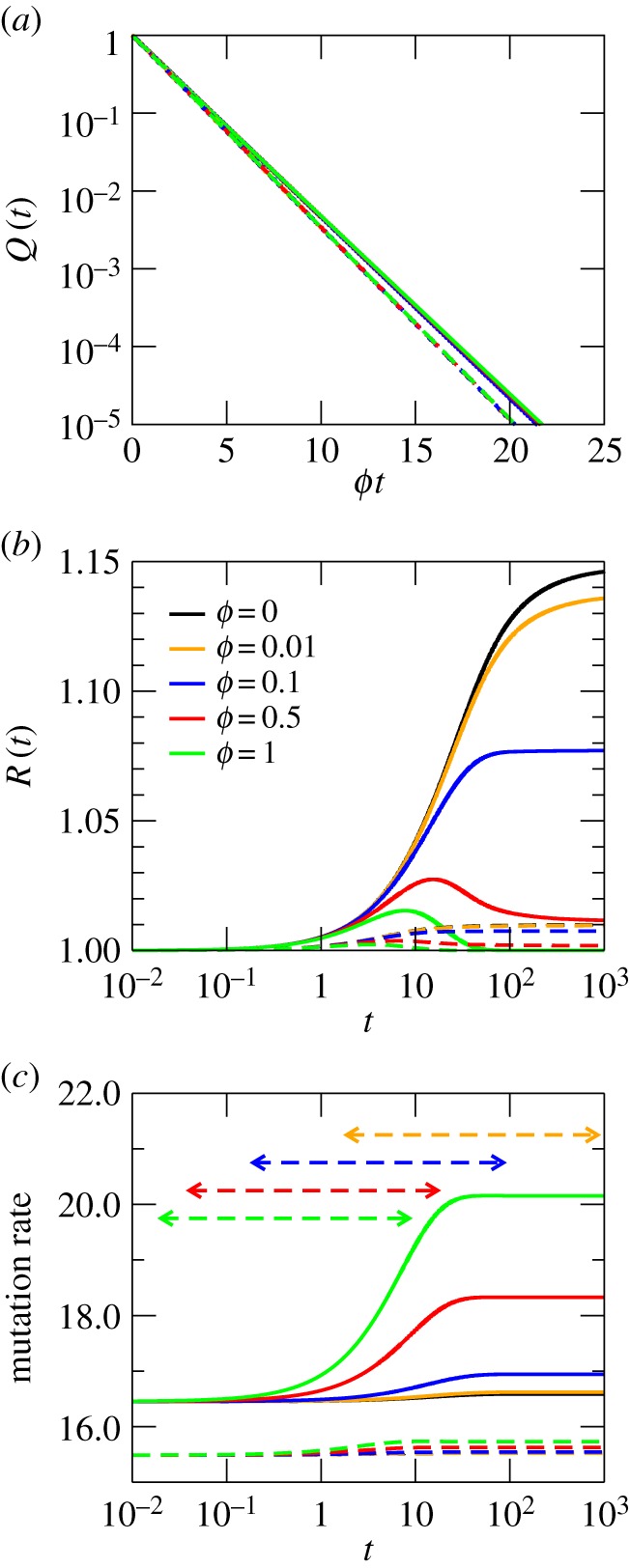
Dynamics of a RW on two different RNA secondary structure NNs with z = 36. Sizes are M = 1965 (dashed lines) and M = 21 908 (solid lines). (a) Probability Q(t) that the RW stays on the network after a time t. (b) Overdispersion of the network. (c) Mutation rate. Times t1%(ϕ) and t99%(ϕ) are only shown for the largest network. The assortativity is r > 0.90. For the sake of comparison, these plots share scale with figure 1.
5. Discussion
Certain features of the dynamics of homogeneous populations on NNs are generic, resulting from two ubiquitous topological properties: heterogeneity and assortativity. In general, the dynamical process that begins when the population enters the NN and finishes when mutation–selection equilibrium is achieved is characterized by as many different time scales as different degrees of robustness are found in the NN. These time scales successively show up along the transient and affect the process of mutation fixation. As time elapses, regions of increasingly higher degree are visited, causing entrapment of the population. The actual time spent on a NN should increase as the difference between the fitness of the current phenotype and that of neighbouring alternatives grows, because fitter phenotypes trap populations for longer times.
Some of the features here described directly affect evolutionary dynamics on realistic NNs and, therefore, quantitative properties of the actual mutation substitution process. We have used as an example RNA secondary structure NNs, because this is the only case we are aware of where exhaustive studies of the genotype–phenotype map encompassing the whole space of genotypes have been performed [22,31]. Still, the networks we may use to compute quantities such as overdispersion are limited by current computational power. The average size of an RNA network for sequences of length l grows as 0.673l3/22.1636l, which is an astronomically large number even for moderate values of l [6]. For instance, a natural tRNA 76 nt long should have a NN formed by over 1028 different sequences. There is partial evidence that, as the length of genotypes increases, the degree distribution of the corresponding NNs broadens, whereas assortativity grows with size [22]. If this is so, several different time scales should be visible and relevant in the dynamic process of mutation fixation in any realistic scenario. An additional example is provided by the NNs of natural proteins with known folds that have been computationally studied. Although the techniques used only permit a partial exploration of those immense NNs, the degree distributions obtained are remarkably broad [14]. There is sufficient evidence as well that protein NNs are highly assortative [20].
Network heterogeneity, together with stochasticity in the appearance and fixation of mutations, translates into potentially large values of R(t). In most cases, overdispersion changes non-monotonically, presenting a maximum value at intermediate times, before equilibrium is reached. The value of evolutionary time in our model yields an upper bound to the number of mutations accumulated up to time t, because we have defined time in units of μ−1, where μ represents the rate of attempted mutations. Therefore, typical trajectories accumulate between a few and a few tens of mutations before leaving the network, a range where the strongest variation in R(t) and mutation rates occurs. The non-monotonicity of R(t) has been observed in realistic models of protein evolution [14]. In an exhaustive analysis of thousands of protein sequences in several Drosophila species [32], it has been demonstrated that R(t) is time- and gene-dependent, and that it should be described as a function of a phylogeny's level of divergence. The former observations are in full qualitative agreement with our results.
For any time t, R(t) > 1 as long as genomes with different robustness (nodes with different degree) are visited with non-zero probability. There might be some extreme cases, such as the two-degree networks we have analysed, when only nodes with the same degree are visible to the population at mutation–selection equilibrium. In those special situations (and, trivially, if the network is homogeneous), R(t) = 1 is recovered, in agreement with Kimura's initial guess on the Poissonian nature of the substitution process. However, this is a very atypical situation that cannot be expected to occur in any realistic case. That network heterogeneity increases overdispersion had already been acknowledged before [19]. What had not been realized is the fact that this effect is stronger during transients; that transients are unavoidable, because evolution on NN begins preferentially in regions of low robustness; and that—depending on the value of ϕ—equilibrium may be virtually unreachable before a mutation hits a fitter phenotype, driving the process again far from equilibrium. Indeed, the number of mutations that have to be accumulated to attain equilibrium vary from slightly less than a hundred to a few thousands, a situation not reached by any typical trajectory unless ϕ is close to 1. It would be interesting to consider other mutational mechanisms, such as recombination, which might affect the nature of equilibrium and the length of the transient. For example, it has been shown that, under recombination, the mutation–selection equilibria of populations replicating on NNs display a higher degree of robustness (are thus more entrapped) than those corresponding to simple point mutations [29].
There might be some natural systems where the accumulation of mutations, and therefore the transient time before equilibrium is achieved, proceeds at a pace significantly higher than that in most genes or proteins, namely RNA viruses, viroids and miRNAs. These three systems are characterized by high mutation rates and, in RNA viruses and miRNAs, a correlation between selection for thermostability and increased robustness to mutations has been detected (a phenomenon called plastogenetic congruence) [33,34]. Viroids, which share several structural properties with miRNAs, also increase robustness along evolution [35] and present a limited effect of mutations on their structure [36]. The higher robustness of these systems with respect to randomized counterparts is an indirect evidence of phenotypic entrapment and, therefore, of populational states closer to mutation–selection equilibrium. Their corresponding phylogenies might be excellent testing beds for the theoretical results here derived.
Evolutionary dynamics on heterogeneous and assortative networks with phenotype-dependent size has important consequences regarding the molecular clock [37]. First, there is an increase in the rate at which mutations are fixed as time elapses, with an occasional maximum in its value during the transient. When the population first enters the network, the probability that it stays is relatively low. However, if it remains, the neutral mutation that is consequently fixed does it in a typical time that depends on the robustness of the current genotype. Because robustness systematically increases along the transient in assortative networks, the age of a population which has accumulated few neutral mutations will be systematically underestimated if a regularly ticking molecular clock is assumed. This prediction is in agreement with observations of a steady increase in R(t) along phylogenies [32] where, moreover, a gene-dependent R(t) was identified. Second, the ticking rate of the clock depends on ϕ, that is, on the fitness value of the current phenotype (or NN) relative to neighbouring phenotypes: the smaller ϕ, the higher the trapping power of the network, a situation that causes larger overdispersion and lower asymptotic mutational rates. Finally, even sequences of the same length may belong to NNs that differ in size in several orders of magnitude. The ticking rate of the clock is unavoidably dependent on robustness, which in its turn is a function of phenotype size in all genotype–phenotype maps studied [9,22–25]. Reliable estimations of NN sizes, at hand with appropriate computational algorithms [38], should aid in the calibration of neutral evolutionary rates. This information could be combined with knowledge of R(t) in homologous genes with the same function (thus presumably characterized by the same NN). Finally, the R(t) and the NN sizes of different genes in diverging lineages could be compared in order to disentangle time-dependent changes in R(t) from variation owing to phenotype sizes, extending studies such as those in reference [32].
The results here obtained do not contradict observations revealing a decrease in the ticking rate of the molecular clock along phylogenetic branches [39,40], because most fixed mutations are in those cases subjected to selection. In this sense, our model addresses a partial aspect of phylogenies, namely strictly neutral evolution. A tractable model of how NNs of different fitness intermingle in the space of genomes may bring about the formulation of mean-field dynamical models, such as the one here presented, that simultaneously take into account neutral drift and selection without discarding the complex architecture of the space of genotypes. Actually, it is plausible that the integration of several different structural features of phenotypes may clarify why the molecular clock depends so strongly on specific cases analysed [41]. Indeed, there are several mechanisms known to affect the uniformity of rates of molecular evolution [42]. In the light of the results here presented, also the particular topology of NNs, the degree of adaptation of populations and the abundance of the current phenotype should be taken into account when calibrating molecular clocks.
Acknowledgements
The authors are indebted to Ricardo Azevedo and Ugo Bastolla for constructive criticism on the manuscript.
Appendix A. Solving for Q(t) in general
According to equation (3.1), to obtain Q(t), we first need to compute P. Except for very specific choices of pnn(k|j) and ϕ, the master equation (2.8) needs to be solved numerically. We prove here that matrix F − A has s (the cardinal of set 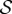 ) different real eigenvalues—which justifies formula (3.1).
) different real eigenvalues—which justifies formula (3.1).
Let us define M(x) ≡ F − xA, where x is a complex variable. Standard perturbation theory [43] tells us that the eigenvalues of M(x) form a set of analytic functions of x with constant cardinality, except for a finite set of singular points in the complex plane—where at least two of the eigenvalues coalesce. But M(0) = diag(ϕ + (1 − ϕ)k/z), 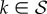 , so it has s different real eigenvalues (s being the cardinal of
, so it has s different real eigenvalues (s being the cardinal of 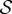 ), provided ϕ ≠ 1. Therefore, M(x) will also have s different eigenvalues—hence will be diagonalizable—except when x is one of those singular points. Now, x = 1 will not be a singular point for almost any probability distributions p(k|j). But if p(k|j) happens to be one of those singular distributions, then the analysis to come is still valid for
), provided ϕ ≠ 1. Therefore, M(x) will also have s different eigenvalues—hence will be diagonalizable—except when x is one of those singular points. Now, x = 1 will not be a singular point for almost any probability distributions p(k|j). But if p(k|j) happens to be one of those singular distributions, then the analysis to come is still valid for 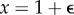 , for arbitrarily small
, for arbitrarily small  (because the singular points are isolated in the complex plane). Thus, we can eventually take the limit
(because the singular points are isolated in the complex plane). Thus, we can eventually take the limit 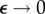 without changing the conclusions.
without changing the conclusions.
We now show that all eigenvalues of M(x) are real. To that aim let us introduce the (non-singular) matrix D = diag((z/k)p(k)),  . Then, the elements of T(x) ≡ M(x)D are [(1 − ϕ) + ϕz/k]p(k)δkj − xpnn(k, j), where pnn(k, j) is the fractions of links of the NN joining two nodes of degrees k and j. Hence, T(x) is a symmetric matrix and therefore all its eigenvalues are real. But then so are the eigenvalues of M(x), because M(x) = T(x)D−1, which has exactly the same eigenvalues as the symmetric matrix D
−
1/2T(x)D−1/2.
. Then, the elements of T(x) ≡ M(x)D are [(1 − ϕ) + ϕz/k]p(k)δkj − xpnn(k, j), where pnn(k, j) is the fractions of links of the NN joining two nodes of degrees k and j. Hence, T(x) is a symmetric matrix and therefore all its eigenvalues are real. But then so are the eigenvalues of M(x), because M(x) = T(x)D−1, which has exactly the same eigenvalues as the symmetric matrix D
−
1/2T(x)D−1/2.
Let uj and vj, 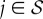 , be the right and left eigenvectors of M(1) corresponding to the eigenvalue αj. They verify the orthogonality condition vk · uj ∝ δkj. Then, the solution of
, be the right and left eigenvectors of M(1) corresponding to the eigenvalue αj. They verify the orthogonality condition vk · uj ∝ δkj. Then, the solution of  can be obtained as
can be obtained as
| A1 |
Substituting this result into (3.1) yields (3.2).
Appendix B. Q(t) and R(t) for random networks
To obtain Q(t), we left-multiply (2.8) by 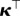 . This leads to
. This leads to
| B1 |
whose solution is simply
| B2 |
Substituting into (3.1) finally yields (4.2).
As for overdispersion, from equations (3.7), we can derive
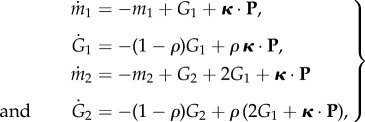 |
B3 |
with the initial condition m1(0) = m2(0) = G1(0) = G2(0) = 0. Combining the former two equations and the latter two, we obtain
| B4 |
which, given the null initial conditions, yield Gi = ρmi. Therefore, the four equations (B 3) reduce to
 |
B5 |
Using (B 2), this linear system can be readily solved as
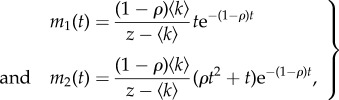 |
B6 |
from which overdispersion can be obtained as (4.3).
Funding statement
The authors acknowledge the financial support of the Spanish Ministerio de Economía y Competitividad under projects FIS2011-27569 and PRODIEVO (FIS2011-22449), and of Comunidad de Madrid under project MODELICO-CM (S2009/ESP-1691).
References
- 1.Kimura M. 1968. Evolutionary rate at the molecular level. Nature 217, 624–626. ( 10.1038/217624a0) [DOI] [PubMed] [Google Scholar]
- 2.van Nimwegen E, Crutchfield JP, Huynen M. 1999. Neutral evolution of mutational robustness. Proc. Natl Acad. Sci. USA 96, 9716–9720. ( 10.1073/pnas.96.17.9716) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Draghi JA, Parsons TL, Plotkin JB. 2011. Epistasis increases the rate of conditionally neutral substitution in an adapting population. Genetics 187, 1139–1152. ( 10.1534/genetics.110.125997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith JM. 1970. Natural selection and the concept of a protein space. Nature 225, 563–564. ( 10.1038/225563a0) [DOI] [PubMed] [Google Scholar]
- 5.Lipman DJ, Wilbur WJ. 1991. Modelling neutral and selective evolution of protein folding. Proc. R. Soc. Lond. B 245, 7–11. ( 10.1098/rspb.1991.0081) [DOI] [PubMed] [Google Scholar]
- 6.Schuster P, Fontana W, Stadler PF, Hofacker IL. 1994. From sequences to shapes and back: a case study in RNA secondary structures. Proc. R. Soc. Lond. B 255, 279–284. ( 10.1098/rspb.1994.0040) [DOI] [PubMed] [Google Scholar]
- 7.Ciliberti S, Martin OC, Wagner A. 2007. Innovation and robustness in complex regulatory gene networks. Proc. Natl Acad. Sci. USA 104, 13 595–13 596. ( 10.1073/pnas.0705396104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodrigues JFM, Wagner A. 2009. Evolutionary plasticity and innovations in complex metabolic reaction networks. PLoS Comput. Biol. 5, e1000613 ( 10.1371/journal.pcbi.1000613) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greenbury SF, Johnston IG, Louis AA, Ahnert SE. 2014. A tractable genotype–phenotype map for the self-assembly of protein quaternary structure. JRSI 11, 20140249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kimura M, Ohta T. 1971. On the rate of molecular evolution. J. Mol. Evol. 1, 1–17. ( 10.1007/BF01659390) [DOI] [PubMed] [Google Scholar]
- 11.Langley CH, Fitch WM. 1974. An examination of the constancy of the rate of molecular evolution. J. Mol. Evol. 3, 161–177. ( 10.1007/BF01797451) [DOI] [PubMed] [Google Scholar]
- 12.Gillespie JH. 1984. The molecular clock may be an episodic clock. Proc. Natl Acad. Sci. USA 81, 8009–8013. ( 10.1073/pnas.81.24.8009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takahata N. 1987. On the overdispersed molecular clock. Genetics 116, 169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bastolla U, Porto M, Roman HE, Vendruscolo M. 2003. Connectivity of neutral networks, overdispersion, and structural conservation in protein evolution. J. Mol. Biol. 56, 243–254. [DOI] [PubMed] [Google Scholar]
- 15.Aguirre J, Buldú JM, Manrubia SC. 2009. Evolutionary dynamics on networks of selectively neutral genotypes: effects of topology and sequence stability. Phys. Rev. E 80, 066112 ( 10.1103/PhysRevE.80.066112) [DOI] [PubMed] [Google Scholar]
- 16.Wagner A. 2011. The origins of evolutionary innovations. Oxford, UK: Oxford University Press. [Google Scholar]
- 17.Bastolla U, Roman HE, Vendruscolo M. 1999. Neutral evolution of model proteins: diffusion in sequence space and overdispersion. JTB 200, 49–64. ( 10.1006/jtbi.1999.0975) [DOI] [PubMed] [Google Scholar]
- 18.Wilke CO. 2004. Molecular clock in neutral protein evolution. BMC Genet. 5, 25 ( 10.1186/1471-2156-5-25) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raval A. 2007. Molecular clock on a neutral network. Phys. Rev. Lett. 99, 138104 ( 10.1103/PhysRevLett.99.138104) [DOI] [PubMed] [Google Scholar]
- 20.Bornberg-Bauer E, Chan HS. 1999. Modeling evolutionary landscapes: Mutational stability, topology, and superfunnels in sequence space. Proc. Natl Acad. Sci. USA 96, 10 689–10 694. ( 10.1073/pnas.96.19.10689) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wuchty S, Fontana W, Hofacker IL, Schuster P. 1999. Complete suboptimal folding of RNA and the stability of secondary structures. Biopolymers 49, 145–165. () [DOI] [PubMed] [Google Scholar]
- 22.Aguirre J, Buldú JM, Stich M, Manrubia SC. 2011. Topological structure of the space of phenotypes: the case of RNA neutral networks. PLoS ONE 6, e26324 ( 10.1371/journal.pone.0026324) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Helling R, Tang C, Wingreen N. 1996. Emergence of preferred structures in a simple model of protein folding. Science 273, 666–669. ( 10.1126/science.273.5275.666) [DOI] [PubMed] [Google Scholar]
- 24.Bloom JD, Raval A, Wilke CO. 2007. Thermodynamics of neutral protein evolution. Genetics 175, 255–266. ( 10.1534/genetics.106.061754) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dall'Olio GM, Bertranpetit J, Wagner A, Laayouni H. 2014. Human genome variation and the concept of genotype networks. PLoS ONE 9, e107347 ( 10.1371/journal.pone.0107347) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feld SL. 1991. Why your friends have more friends than you do. Am. J. Sociol. 96, 1464–1477. ( 10.1086/229693) [DOI] [Google Scholar]
- 27.Ewens WJ. 2004. Mathematical population genetics, 2nd edn New York, NY: Springer. [Google Scholar]
- 28.Gillespie JH. 1994. The causes of molecular evolution. New York, NY: Oxford University Press. [Google Scholar]
- 29.Szöllősi GJ, Derényi I. 2008. The effect of recombination on the neutral evolution of genetic robustness. Math. Biosci. 214, 58–62. ( 10.1016/j.mbs.2008.03.010) [DOI] [PubMed] [Google Scholar]
- 30.Stich M, Briones C, Manrubia SC. 2008. On the structural repertoire of pools of short, random RNA sequences. J. Theor. Biol. 252, 750–763. ( 10.1016/j.jtbi.2008.02.018) [DOI] [PubMed] [Google Scholar]
- 31.Grüner W, Giegerich R, Strothmann D, Reidys C, Weber J, Hofacker IL, Stadler PF, Schuster P. 1996. Analysis of RNA sequence structure maps by exhaustive enumeration. I. Neutral networks. Monatshefte F. Chem. 127, 355–374. ( 10.1007/BF00810881) [DOI] [Google Scholar]
- 32.Bedford T, Hartl DL. 2008. Overdispersion of the molecular clock: temporal variation of gene-specific substitution rates in drosophila. Mol. Biol. Evol. 25, 1631–1638. ( 10.1093/molbev/msn112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Domingo-Calap P, Pereira-Gómez M, Sanjuán R. 2010. Selection for thermostability can lead to the emergence of mutational robustness in an RNA virus. J. Evol. Biol. 23, 2453–2460. ( 10.1111/j.1420-9101.2010.02107.x) [DOI] [PubMed] [Google Scholar]
- 34.Szöllősi GJ, Derényi I. 2009. Congruent evolution of genetic and environmental robustness in micro-RNA. Mol. Biol. Evol. 26, 867–874. ( 10.1093/molbev/msp008) [DOI] [PubMed] [Google Scholar]
- 35.Sanjuán R, Forment J, Elena SF. 2006. In silico predicted robustness of viroids RNA secondary structures. I. The effect of single mutations. Mol. Biol. Evol. 23, 1427–1436. ( 10.1093/molbev/msl005) [DOI] [PubMed] [Google Scholar]
- 36.Manrubia SC, Sanjuán R. 2013. Shape matters: effect of point mutations on RNA secondary structure. Adv. Comp. Syst. 16, 1250052 ( 10.1142/S021952591250052X) [DOI] [Google Scholar]
- 37.Zuckerkandl E, Pauling L. 1965. Evolutionary divergence and convergence in proteins. In Evolving genes and proteins (eds Bryson V, Vogel H.), pp. 97–166. New York, NY: Academic Press. [Google Scholar]
- 38.Jörg T, Martin OC, Wagner A. 2008. Neutral network sizes of biological RNA molecules can be computed and are atypically large. BMC Bioinform. 9, 464 ( 10.1186/1471-2105-9-464) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pereira SL, Baker AJ. 2006. A mitogenomic timescale for birds detects variable phylogenetic rates of molecular evolution and refutes the standard molecular clock. Mol. Biol. Evol. 23, 1731–1740. ( 10.1093/molbev/msl038) [DOI] [PubMed] [Google Scholar]
- 40.Ho SYW, Lanfear R, Bromham L, Phillips MJ, Soubrier J, Rodrigos AG, Cooper A. 2011. Time-dependent rates of molecular evolution. Mol. Ecol. 20, 3087–3101. ( 10.1111/j.1365-294X.2011.05178.x) [DOI] [PubMed] [Google Scholar]
- 41.Kumar S. 2005. Molecular clocks: four decades of evolution. Nat. Rev. Genet. 6, 654–662. ( 10.1038/nrg1659) [DOI] [PubMed] [Google Scholar]
- 42.Liò P, Goldman N. 1998. Models of molecular evolution and phylogeny. Genome Res. 8, 1233–1244. [DOI] [PubMed] [Google Scholar]
- 43.Kato T. 1980. Perturbation theory for linear operators. Heidelberg, Germany: Springer. [Google Scholar]